REVIEW: Proteasome System of Protein Degradation and Processing
A. V. Sorokin*, E. R. Kim, and L. P. Ovchinnikov
Institute of Protein Research, Russian Academy of Sciences, 142290 Pushchino, Moscow Region, Russia; E-mail: sorokin@vega.protres.ru; ovchinn@vega.protres.ru* To whom correspondence should be addressed.
Received February 5, 2009
In eukaryotic cells, degradation of most intracellular proteins is realized by proteasomes. The substrates for proteolysis are selected by the fact that the gate to the proteolytic chamber of the proteasome is usually closed, and only proteins carrying a special “label” can get into it. A polyubiquitin chain plays the role of the “label”: degradation affects proteins conjugated with a ubiquitin (Ub) chain that consists at minimum of four molecules. Upon entering the proteasome channel, the polypeptide chain of the protein unfolds and stretches along it, being hydrolyzed to short peptides. Ubiquitin per se does not get into the proteasome, but, after destruction of the “labeled” molecule, it is released and labels another molecule. This process has been named “Ub-dependent protein degradation”. In this review we systematize current data on the Ub–proteasome system, describe in detail proteasome structure, the ubiquitination system, and the classical ATP/Ub-dependent mechanism of protein degradation, as well as try to focus readers’ attention on the existence of alternative mechanisms of proteasomal degradation and processing of proteins. Data on damages of the proteasome system that lead to the development of different diseases are given separately.
KEY WORDS: proteasome, Ub, ubiquitin, degradation, processingDOI: 10.1134/S000629790913001X
Abbreviations: Ub, ubiquitin; 19S RP, 19S regulatory particle; 20S CP, 20S core particle, core proteasome; 26S PR, 26S proteasome.
Living organisms carry from thousands through scores of thousands of
protein-coding genes and a far greater number of proteins encoded by
them. Diverse studies have been devoted to analyzing protein synthesis,
but the reverse process of protein degradation has long remained
outside the scope of proper attention. R. Schoenheimer was a pioneer in
studying protein degradation. In 1942 he published the results of his
studies using the “labeling” of molecules with radioactive
isotopes, according to which proteins in animals are constantly
synthesized and degraded [1]. As known, proteins
differ greatly from each other in lifetime, and the lifetime of protein
molecules in an organism depend on their role. So, some structural
proteins can remain unchanged for many years, whereas regulatory
proteins are frequently required only for a few minutes to trigger a
certain process and after completing their function they should be
destroyed. In the course of time, cells accumulate a large amount of
aberrantly folded and oxidized protein that should be also eliminated
somehow. Degradation of faulty proteins and the proteins that
“have done their part” should be selective and accomplished
in isolated compartments, so that structural components of the cell and
proteins required for it will remain undamaged.
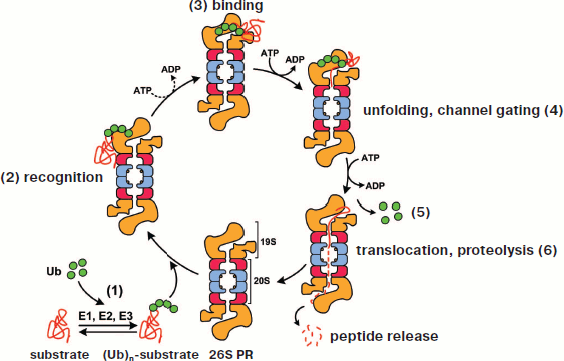
In a eukaryotic cell, one of the compartments for protein processing is the lysosome. However, proteolysis in lysosomes is a nonspecific process. In higher eukaryotes, only membrane-associated proteins and alien proteins captured during endocytosis (viral, bacterial, etc.) are destroyed in lysosomes. Degradation of the vast majority (80-90%) of intracellular proteins is realized by the 26S proteasome (26S PR) [2, 3]. In this case, the isolated compartment is the internal proteolytic cavity of its core portion (20S CP) (the 20S proteasome), which has several peptidase centers. The selection of substrates for proteolysis is assured by the fact that the gate to the 20S proteasome is usually closed and only proteins having a special “label” can get in. The polyubiquitin (polyUb) chain plays the role of “label”: degraded are proteins conjugated with polyUb consisting of at least four ubiquitin (Ub) monomers. Upon entering the proteasome channel, the polypeptide chain of the protein unfolds and stretches along the channel, being hydrolyzed to short peptides (3-25 amino acid residues), which are released from the opposite ending of the channel [4-6]. Ubiquitin per se does not get into the proteasome, and after destruction of the “labeled” molecule it is released and labels another molecule. This process has been named the “ubiquitin-dependent degradation of protein” (Fig. 1; see color insert). The discoverers of this phenomenon — A. Ciechanover, A. Hershko, and I. Rose — were awarded the Nobel Prize in 2004.
This scheme of Ub-dependent protein degradation by proteasomes was corroborated by various researchers. At the same time, by the end of the 1990s there accumulated sufficiently numerous data evidencing that proteasomes can destroy proteins in another Ub-independent way. More than that, it became evident that the proteasome can regulate not only the amount of proteins but also their functions: in some cases proteins are not hydrolyzed to short peptides but undergo limited proteolysis (processing), as a result of which the protein functions can change significantly. In this review, we will focus special attention on the two latter “non-canonical” functions of the proteasome.Fig. 1. Simplified model of ubiquitin-dependent degradation of proteins by proteasomes. A conventional model of ubiquitin-dependent degradation of proteins by a proteasome includes the following stages. 1) Formation of the protein–substrate conjugate with ubiquitin proceeds in several steps and is mediated by enzymes E1, E2, and E3. 2) Subunit Rpn10 of the proteasome regulatory particle recognizes the quaternary structure of the polyubiquitin chain. 3) Subunits Rpn1 and Rpn2 of the proteasome regulatory particle bind the substrate. 4) Subunit ATPases of the regulatory particle unfold the substrate. The interaction of ATPases Rpt2 and Rpt5 with α-subunits of the core proteasome causes the gating of the channel. 5) Subunit Rpn11 of the regulatory particle and deubiquitinating enzymes Uch37 and Usp14/Ubp6 detach ubiquitin from the substrate. 6) Translocation of the substrate polypeptide chain into the proteolytic chamber of the proteasome is facilitated by subunit ATPases of the regulatory particle, and hydrolysis of the peptide bonds is accomplished by protease subunits (β1, β2, and β5; catalytic centers are designated by circles). The size of released peptides varies from three to 25 amino acid residues. Designations: Ub, ubiquitin; (Ub)n-substrate, polyubiquitinated protein substrate; 26S PR, 26S isoform of proteasome; 20S, 20S core proteasome (CP); 19S, 19S regulatory particle (RP).
PROTEASOME STRUCTURE
26S Proteasome
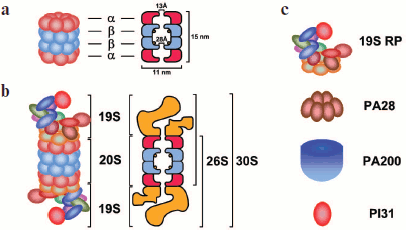
The proteasome that accomplishes Ub-dependent degradation of proteins consists of two basic subcomplexes: the core 20S proteasome (20S CP, about 700 kDa) and the PA700 activator or the 19S regulatory particle (19S RP, about 900 kDa). The 20S CP contains protease subunits, while the 19S RP includes subunits capable of binding the polyUb chains and the substrate, as well as isopeptidases cleaving Ub and ATPases that unfold the substrate and deliver it to the core proteasome channel [7]. The 19S RP can dock at the 20S CP either from one or both ends, as a consequence of which the 26S and 30S proteasomes are formed, respectively. However, the term “30S proteasome” is practically not used, and the name “26S proteasome” has been accepted to designate both isoforms. In addition to the 19S RP, the structure of the 26S proteasome can include alternative regulatory particles: PA28α/β (or 11S REG), PA28γ (or REGγ), PA200, PI31, etc. (Fig. 2; see color insert). There also occur asymmetric isoforms of the 26S proteasome containing different regulatory particles at the ends of the 20S CP. Moreover, proteasome isoforms were revealed in which regulatory particles are substituted by a multisubunit protein complex PC530 or signalosome COP9 [8]. The structure and functions of proteasome subcomplexes are analyzed in detail below.
Core 20S ProteasomeFig. 2. Schematic representation of the structure of 20S and 26S proteasomes and regulatory particles. a) The 20S proteasome (20S CP) is a 700-kDa barrel consisting of four rings arranged in a stack. It includes 28 subunits, which are products of two groups of homologous genes (α and β). The two external rings consist of only α-subunits, whereas the two internal rings consist of β-subunits. The functions of α- and β-subunits are different: α-subunits form a physical barrier restricting the access of proteins to the internal proteolytic chamber, and β-subunits are responsible for proteolytic activity of the proteasome (catalytic centers are designated by circles). The α-subunits also interact with regulatory complexes, which affect the functions of the 26S proteasome. The gate formed by α-subunits opens only on activation of the proteasome, and its diameter is 13 Å. (The 20S proteasome from Thermoplasma acidophilum has dimensions of 148 × 113 Å, and the dimensions of the bovine 20S proteasome are 150 × 115 Å.) b) The 19S regulatory particle (19S RP) associates itself with one or both ends of the 20S CP, as a result of which the 26S and 30S proteasomes are respectively formed. The 19S RP can be substituted by alternative regulatory particles or other multisubunit protein complexes. c) regulatory complexes. 19S RP is PA700 or the 19S regulatory particle. In mammals it consists of 18 core subunits. PA28 is PA28 regulatory particle. Two isoforms exist: PA28α/β and PA28γ. PA28α/β is regulator 11S REG which is a heterohexamer consisting of α- and β-subunits. PA28γ is regulator REGγ which is a hexamer consisting of γ-subunits. PA200 is regulator PA200 consisting of one 200-kDa protein having an asymmetric dome-like shape. PI31 is regulator PI31 consisting of one proline-rich 30-kDa protein.
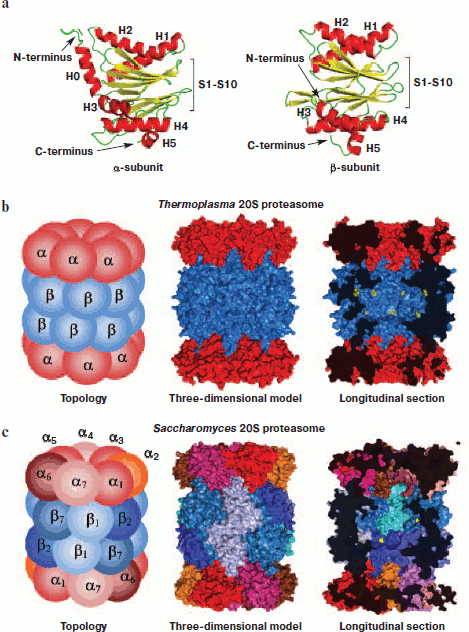
Molecular structure. Prokaryotic and eukaryotic 20S proteasomes consist of 28 subunits (Table 1). A prokaryotic proteasome contains 14 copies of identical α-subunits and 14 copies of identical β-subunits (Fig. 3; see color insert). A eukaryotic proteasome carries two copies of seven different α-subunits and two copies of seven different β-subunits. In addition to the constitutive 20S proteasome, in mammals there is also an immunoproteasome, the assembly of which within the cell begins after its stimulation by γ-interferon. This cytokine triggers the synthesis of three additional proteasomal subunits – β1i, β2i, and β5i – which in the course of assembling are incorporated instead of constitutively synthesized subunits β1, β2, and β5 [9-11]. It is accepted that in contrast to the constitutive proteasome, the immunoproteasome generates peptides that are then used during antigen presentation [12-14].
Table 1. Nomenclature of 20S proteasome
subunits

Note: γ-Interferon-induced subunits of immunoproteasomes are given
in bold type. Different names of the same subunit are separated by a
slash.
The quaternary structure of 20S CP is the same in bacteria, archaea, and eukaryotes including mammals: α- and β-subunits each form two heptamer rings arranged in a stack. The external rings contain only α-subunits, and the internal two rings have only β-subunits [15]. The spatial structure of all proteasomal subunits is the same, which follows from high homology of the amino acid sequence of α- and β-subunits. The three-dimensional packing of subunits represents two antiparallel five-stranded β-sheets (S1-S10) that are located between two α-helices from one side (H1 and H2) and three α-helices from the other side (H3, H4, and H5). The main difference between α- and β-subunits is an additional N-terminal α-helix (H0, 35 amino acid residues) in α-subunits (Fig. 3a).Fig. 3. Molecular arrangement of the 20S proteasome. a) Ribbon drawing of α- and β-subunits of the Thermoplasma acidophilum proteasome. Subunits are represented in the same orientation demonstrating high spatial homology. The main distinction is an additional N-terminal α-helix (H0) in α-subunit. b) Topology and three-dimensional model of the Thermoplasma acidophilum proteasome. The proteasome consists of 14 copies of identical α-subunits and 14 copies of identical β-subunits. in longitudinal section, threonine residues in the catalytic centers are shown by yellow color. In the given projection, seven of the 14 are seen. c) Topology and three-dimensional model of the Saccharomyces cerevisiae proteasome. The proteasome consists of two copies of seven different α-subunits and two copies of seven different β-subunits. in longitudinal section, threonine residues in catalytic centers are shown by yellow color. In the given projection, three of the six are seen. The figures were drawn with the PyMOL program using PDB 1PMA, 1JD2, and 1G0U datasets.
There are three compartments within the proteasome: two external cavities (“antechambers”) and one internal proteolytic chamber. The volume of an antechamber of the Thermoplasma acidophilum 20S proteasome is about 59 nm3, and that of the proteolytic chamber is about 84 nm3, which can readily accommodate a globular protein of ~70 kDa [16]. The distance from the outside of the 20S proteasome to the catalytic centers is ~70 Å, which correlates with the length of an unfolded peptide of about 20 amino acid residues.
The proteasome is ascribed to the class of N-terminal nucleophilic hydrolases (NTN hydrolases). The N-terminal threonine of β-subunits is vital for catalysis, and its substitution by serine leads to lower hydrolysis efficiency [17]. In prokaryotes, all of the 14 β-subunits are identical and consequently the proteasome contains 14 protease centers (Fig. 3b). In eukaryotes, three of the seven β-subunits have threonine-protease catalytic centers of diverse substrate specificity, i.e. every proteasome has six protease centers (Fig. 3c). It is known that subunit β1 has caspase-like activity (hydrolyzes the peptide bond after negatively charged amino acid residues), and subunit β2 has trypsin-like activity (hydrolyzes the peptide bond mostly after positively charged amino acid residues), whereas subunit β5 has chymotrypsin-like activity (hydrolyzes the peptide bond after large hydrophobic amino acid residues) [18, 19].
All the proteinase centers are facing the internal proteolytic chamber formed by β-subunits, and the substrate can access to them via the gate formed by α-subunits (Fig. 3) [20, 21]. Besides the three main types of proteinase centers, the existence of two additional centers have been reported: (i) one hydrolyzing the peptide bond following branched chain amino acid residues (BrAAP, branched chain amino acid peptidase) and (ii) the other hydrolyzing the peptide bond after small neutral amino acid residues (SNAAP, small neutral amino acid peptidase) [22]. However, the results of subsequent studies did not support this conclusion [23]. Based on structural data, it has been proposed recently that there exists a SNAAP catalytic center in subunit β7, but no experimental corroboration or rejection has followed yet [24].
The substrate is translocated into the proteolytic chamber via the gate formed by α-subunits. In the T. acidophilum proteasome, the gate diameter is ~13 Å. Unfortunately, in the available structure it is impossible to see the 12 N-terminal residues of α-subunits facing the channel (Fig. 4; see color insert) [25]. This region has no fixed structure and does not close the channel to the proteolytic chamber. The T. acidophilum 20S proteasome is active in cleaving peptides, but in this case the ATP-dependent activator PAN is required for cleaving proteins [26, 27]. In eukaryotes substrate access into the proteolytic chamber is limited: the gate opens only upon activation of the proteasome (Fig. 4). The gate size shows that it can transmit only α-helices with small side chains or β-hairpins, i.e. peptides or unfolded proteins [20, 28]. But the experimental data suggest that the gate can widen up to 20 Å, which allows concurrent passage of three unfolded polypeptide chains [29].
Gating of the 20S CP channel. Activation of ATP-dependent proteases is frequently connected with allosteric regulation of their proteolytic centers. In the case of the eukaryotic 20S proteasome, there is no structural evidence for the existence of such regulation. Therefore, activation of the proteasome is accepted to be connected with the opening of the gate in 20S CP, which provides for substrate access to the catalytic centers. The available model of the mechanism of gate opening is based on data on conformational rearrangements in the N-terminal regions of the α-subunits forming the gate [30-33]. Such changes in the conformation can occur (i) upon interaction with regulatory particles [34-36], (ii) upon interaction with substrates [37, 38], and (iii) upon treatment with low concentrations of SDS, polylysine, etc. [15, 39].Fig. 4. The 20S proteasome entrance. a) Three-dimensional model of a ring of α-subunits of the 20S proteasome (top view). In the Thermoplasma acidophilum proteasome, 12 N-terminal residues of α-subunits are not ordered and are not given in the presented structure (they should be facing the channel). The three-dimensional structure of the “closed”, “open”, and “theoretically open” conformations of the Saccharomyces cerevisiae proteasome. The “closed” conformation corresponds to the conformation of the wild-type latent proteasome. The “open” conformation corresponds to the conformation of the α3ΔN proteasome (the proteasome without nine N-terminal residues in subunit α3). The “theoretically open” conformation was obtained from the three-dimensional structure of the “closed” conformation by removing nine N-terminal residues in each α-subunit. b) Ribbon drawing of α-subunits of the 20S proteasome (top view). The topology of α-subunits of the Saccharomyces cerevisiae 20S proteasome is shown in the “closed” conformation. The figures were drawn with the PyMOL program using PDB 1PMA, 1JD2, and 1G0U datasets.
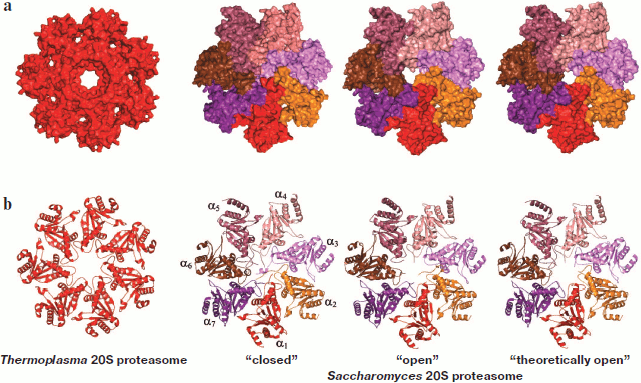
The key role in the gate opening belongs to subunit α3, more exactly to its N-terminal residues. The N-terminus of subunit α3 is unique in its packing: when compared to N-termini of other α-subunits it protrudes most of all into the channel crossing the axis of the pseudoseven-beam symmetry and forms contacts with every α-subunit [30] (Fig. 4b). Deletion of nine N-terminal residues of subunit α3 of the Saccharomyces cerevisiae proteasome (below this mutant proteasome will be called α3ΔN) results in destabilization of the packing of N-termini of the other α-subunits and opening of the gate to a diameter comparable with that of the gate in the T. acidophilum proteasome [20] or the “theoretically open” proteasome of S. cerevisiae (in the structural model, nine N-terminal residues in all α-subunits are deleted) (Fig. 4b). This is sufficient for the proteasome to become active in cleaving short peptides rather than proteins, unfolding of the latter requiring a regulatory particle. The same N-terminal deletion in the subunit α7 (α7ΔN) does not cause remarkable enhancement of the peptidase activity of the proteasome, i.e. does not cause large rearrangements in the packing of residues forming the gate and does not lead to its opening [40]. It was demonstrated that the α3ΔN mutation does not affect the stability of the 26S proteasome. Moreover, the peptidase activity of the α3ΔN 26S proteasome does not differ greatly from that of the wild-type 26S proteasome [7]. These facts suggest that the docking of 19S RP to 20S CP results in changes in the gate structure that are similar to those for the α3ΔN mutation. This means that besides involvement in recognition, unfolding, and translocation of the substrate, 19S RP operates as an “opener” of the gate. The role of regulatory particles in the functioning of the proteasome will be described in detail below.
Assembling of the 20S proteasome. Escherichia coli is devoid of the proteasome and therefore it is an ideal system for expression of proteasomal subunits and studying proteasome assembly. Coexpression of the T. acidophilum proteasome α- and β-subunits in E. coli results in the assembly of an active proteasome in it. If α-subunits are expressed without β-subunits, they can form single or coupled heptamer rings. The N-terminal α-helix (H0, 35 amino acid residues) is responsible for assembly of α-subunits into rings. It was shown that α-subunits lacking this helix cannot assemble into rings [41]. In contrast to α-subunits, β-subunits cannot form rings by themselves, rather remaining in a monomer state that is proteolytically inactive. Most likely, β-subunits assemble into rings on the α-subunit rings, thus forming a “half-proteasome”. Two such “half-proteasomes” assemble into a pre-20S proteasome. A mature 20S proteasome is formed upon cleavage of the N-terminal propeptide (eight residues) from every β-subunit. However, a propeptide is not necessarily required for assembling of the 20S proteasome: it can be formed from a mixture of α-subunits and processed β-subunits [42, 43] (Fig. 5; see color insert).
The process of assembly of a eukaryotic proteasome is more intricate and less studied. As in the case with the T. acidophilum proteasome, H0 α-helices of α-subunits are required for assembly of a eukaryotic proteasome [44]. It is known that like the α-subunit of the T. acidophilum proteasome, the subunit α7 of the human proteasome can form double ring-like structures upon expression in E. coli [45]. The neighboring subunits (α1 and α6) do not form rings upon their individual expression in E. coli, but are involved in ring-like structures when expressed together with subunit α7. In this case, the mutual arrangement of subunits in such rings is quite diverse. This shows that not each α-subunit “possesses” information about its position in the ring [46].Fig. 5. Model of 20S proteasome assembly. a) Scheme of assembly of the proteasome from the archaeon Thermoplasma acidophilum. b) Scheme of assembly of the eukaryotic proteasome. Intermediates that are not stable and cannot be found in vivo are given in parentheses. See details in the text.
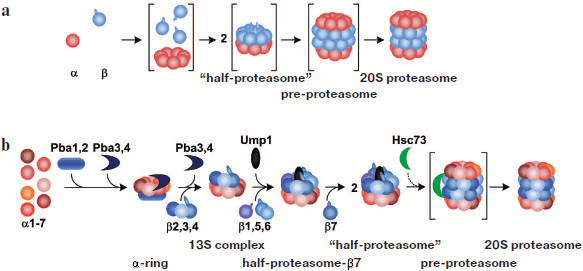
Analysis of assembly intermediates of a eukaryotic proteasome is complicated because they are very unstable and heterogeneous. The involvement in proteasome maturation of several specific chaperones also adds complexity [47]. The assembly of the S. cerevisiae proteasome begins with the formation of a ring of α-subunits (an α-ring) with involvement of heterodimer Pba1–Pba2 (in S. cerevisiae) or PAC1–PAC2 (in mammals). These chaperones interact with an intermediate that contains all α-subunits except α3 and α4 [48-50]. Pba1–Pba2 or PAC1–PAC2 dimers attach to the external side of the α-ring (facing the regulatory particles) and prevent the following assembly of the proteasome from immature intermediates. Chaperones remain attached to α-rings until complete assembly of 20S CP, after which they very likely degrade under the action of the proteasome or are removed by 19S RP or another regulatory particles [48]. Correct assembly of an α-ring accomplished with the participation of another pair of chaperones, Pba3–Pba4/PAC3–PAC3 [51-53]. In the absence of these chaperones the subunit α4 is embedded into the α-ring in the place of the subunit α3, which significantly reduces the efficiency of assembly of the entire proteasome [51]. Dimer Pba3–Pba4/PAC3–PAC3 remains bound to the inner side of the α-ring (facing the β-ring) [53] prior to attaching subunits β2, β3, and β4. This intermediate including the α-ring, subunits β2, β3, and β4 and Pba1–Pba2/PAC1–PAC2 is called the “13S complex” [10, 54]. It cannot form dimers, which may be connected with the fact that in this case only subunit β4 of the subunits β2, β3, and β4 can be involved in the formation of contacts. A special feature of the assembly of the immunoproteasome is that at the stage of the “13S complex”, in addition to β2i, β3, and β4, subunit βli is also attached [10].
With the assistance of the proteasome maturation factor Ump1 (underpinning maturation of proteasome), subunits β1, β5, and β6 are also attached to this complex. The result is a “pre-half-proteasome” (or “half-proteasome minus β7”) [50, 55, 56]. After attaching subunit β7, a “15S half-proteasome” or a “half-proteasome” is formed. Only after this dimerization of half-proteasomes, i.e. assembly of a “preproteasome” (or immature 20S proteasome), can take place [56]. It is just the subunit β7 that plays a key role in dimerization: its extended C-terminus is embedded in the channel between subunits β1 and β2 of the other half-proteasome, which results in a strong coupling of the two half-proteasomes. Removal of the C-terminus of subunit β7 greatly reduces the efficiency of preproteasome assembly. Besides, the C-terminus of subunit β7 stabilizes the conformation of subunit β1, which is required for processing this catalytic subunit [56, 57].
A mammalian preproteasome assembled in vitro is described as a 650-kDa complex with sedimentation coefficient of 16S. Chaperone Hsc73 was found within it. It was demonstrated that on incubation of the preproteasome with ATP, Hsc73 dissociates and a higher-molecular-weight complex is formed. However, even so the processing of propeptides of β-subunits is not observed. It is probable that additional factors are necessary for final maturation [54].
Both the assembly of the preproteasome and its maturation (cleavage of β-subunit propeptides) require the Ump1 factor. Five of the seven β-subunits (β1, β2, β5, β6, and β7) have N-terminal propeptides. They are not conservative in sequences and differ greatly in length. Their deletion does not affect the correct positioning of β-subunits in the proteasome [58, 59]. However, they are required for the folding of β-subunits and their more efficient embedding into the proteasome, i.e. they perform a chaperone-like function. Only deletion of the propeptide of subunit β5 is lethal [60]. This propeptide is not vital for embedding of subunit β5 per se into the proteasome: the proteasome is assembled even with the processed subunit β5 [55]. However, only in the presence of non-processed subunit β5 the Ump1 factor occupies the right position in the half-proteasome, which is necessary for processing of all β-subunits in the preproteasome [61]. The Ump1 factor itself degrades upon cleavage of the propeptides of β-subunits: it is not found either in the mature 20S proteasome or in the 26S proteasome. In the end, the final maturation of 20S CP involves the regulatory particle PA200/Blm10 [56, 62].
19S Regulatory Particle
The 19S regulatory particle (19S RP) is the key regulatory component of the 26S proteasome. It is responsible for the recognition of polyubiquitinated proteins and hence provides for selectivity of the substrate degradation. The 19S RP is involved in opening the gate of the 20S CP, the substrate unfolding, and its advancing into the proteolytic chamber. The 19S RP can attach to the 20S CP from one or both ends forming RP1CP- and RP2CP-isoforms of the 26S proteasome, respectively. In S. cerevisiae, the proteasome is mainly in the complex with two 19S RP (the RP2CP-isoform) [63]; in mammals the ratio of 19S RP and 20S CP is lower and therefore the cells contain a significant amount of free 20S CP as well as RP1CP-isoforms of the 26S proteasome [64].
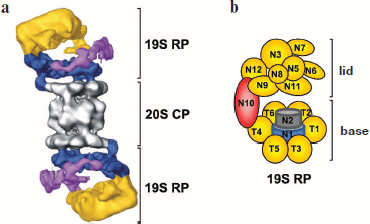
Molecular structure of 19S RP. The 19S RP isolated from various organisms have a similar subunit composition and consist at least of 17 core subunits. Six subunits are ATPases of the AAA-superfamily, members of which are found within many multisubunit complexes such as translocators, transporters, and proteases [65, 66]. These regulatory particle subunits are homologous and are designated as Rpt (Regulatory particle tripleA-ATPase). The other subunits are designated as Rpn (Regulatory particle non-ATPase) (Table 2). Six Rpt subunits and three Rpn subunits (Rpn1, Rpn2, and Rpn10) form the base of the 19S RP; subunits Rpn3, Rpn5, Rpn6, Rpn7, Rpn8, Rpn9, Rpn11, and Rpn12 form the lid of the 19S RP (Fig. 6; see color insert). In S. cerevisiae the base contains an additional Rpn13 subunit [67] and in mammals the S5b subunit [68]. In some isoforms of mammalian 26S PR the hRpn13 subunit is found within the 19S RP [69]. Besides eight basic subunits of the lid (Rpn3, 5, 6, 7, 8, 9, 11, and 12), sometimes loosely or temporarily associated subunits p28/gankyrin, p27, and Sem1/Rpn15 are observed [70-72], and in S. cerevisiae also an additional Son1/Rpn4 subunit [73].
Table 2. Nomenclature of proteasome 19S
regulatory particle subunits
Note: Different names of subunit are separated by slash.
X-Ray studies of the 19S RP are complicated due to a very high motility of the complex and a heterogeneous set of subunits, some of which are associated rather weakly [74]. A low-resolution three-dimensional model of the 19S RP was obtained by electron microscopy (Fig. 6a) [75, 76]. Biochemical methods established 20 intersubunit contacts in the Caenorhabditis elegans 26S RP [77], 40 such contacts in the S. cerevisiae 26S PR [30, 74, 78-83], and 114 contacts in the human 26S PR [84]. The spatial structure is known only for subunits Rpn13 and S5a. According to computer modeling of the structure, subunits Rpn1 and Rpn2 have the packing of an α-helical toroid [85, 86]. Rpn1 and Rpn2 are stacked and enclosed in a ring of six Rpt-subunits [87]. It is likely that accessory Ub-binding proteins are attached to the base. Simplified topology of core subunits of the 19S RP is shown in Fig. 6b.Fig. 6. Molecular arrangement of the 19S regulatory particle. a) A low-resolution three-dimensional model of the 19S RP was obtained by electron microscopy. In 19S RP, subunits of the base, base arm, and lid are given in different colors. The figure is a modified version from [76]. b) Topology of subunits of 19S RP. See details in text.
Functions of the 19S RP. Binding with 20S CP and gate opening. The main role in the interaction of the 19S RP with the 20S CP and the gate opening is given to C-terminal peptides of subunits Rpt2 and Rpt5 [20, 88-90]. It is believed that the attachment of the 19S RP to the 20S CP is accompanied by conformational rearrangements in N-terminal regions of α-subunits leading to changes in the gate structure, i.e. switching to an open conformation. To bind the 19S RP to the 20S CP and open the gate, ATP hydrolysis should be used [91]. Recent data show that in addition to subunits Rpt2 and Rpt5, subunits Rpn1 and Rpn2 are involved in the gate opening [92].
Recognition of ubiquitin. The principal subunit of the 19S RP that ensures recognition of the polyubiquitinated substrate is Rpn10 [93]. Rpn10 binds to Ub because of the presence of a Ub-binding site in the C-terminal hydrophobic cluster containing motif LALAL [94]. The isolated Rpn10 subunit binds with the same efficiency various forms of polyUb in which individual Ub molecules are linked via Lys6, Lys11, or Lys48, whereas the entire 26S PR binds only the “correct” polymeric Ub formed via Lys48 [95]. This indicates the existence of mechanisms providing not only efficient but correct binding of the substrate. Probably the correct recognition of the quaternary structure of the polyUb chain and the specificity of the substrate binding is due to loosely associated accessory proteins such as Rad23 (hHR23a, hHR23b) and Dsk2 (hPLIC) [96-98]. Experiments with S. cerevisiae on deleting the Ub-binding site in Rpn10 demonstrated that phenotypically such mutants do not differ from the wild type [96]. This shows that Rpn10 is not the only subunit in the proteasome that can bind the ubiquitin chain. The binding of the polyubiquitinated substrate apparently involves also subunits Rpn1, Rpn2, Rpn13, and Rpt5 [77, 79, 99, 100].
Binding of substrate. Subunits of the 19S RP base are believed to play the critical role in substrate binding. The existence of leucine-rich repeats (LRR) allows subunits Rpn1 and Rpn2 to be efficiently involved in non-specific protein–protein interactions [101]. By analogy with regulatory domains of simple ATP-dependent proteases, Rpt-subunits can directly bind to substrates and so retain the substrate on the proteasome [102, 103]. apparently, such retention of the substrate by the 19S RP base is required for its unfolding and correct orientation for translocation into the 20S PR channel. With aberrant orientation, Ub conjugated with the substrate would not permit it to enter the channel.
Unfolding and translocation. ATP is required not only for the assembly of the 26S proteasome but also for substrate degradation [104]. Hydrolysis of ATP is necessary for conformational rearrangements of the proteasomal subunits occurring upon substrate unfolding and probably for its translocation into the 20S CP proteolytic chamber. The unfolding of the substrate is especially necessary since the dimension of the gate of the 20S CP is too small to allow a protein with a developed tertiary structure to pass through it.
It is proposed that the binding and hydrolysis of ATP on Rpt-subunits triggers the cycle of high- and low-affinity states of the proteasome relative to the substrate, which is required for retaining the substrate in an unfolded state [88]. The mechanism of translocation remains unclear, but it is accepted that from the mechanical point of view the substrate unfolding is closely connected with the translocation of the unfolded part of the substrate via the gate into the 20S CP proteolytic chamber. Experiments with the archaeal proteasome and PAN (proteasome-activating nucleotidase, which is an analog of eukaryotic 19S RP) demonstrated that the translocation of substrates is ATP-dependent [31].
Deubiquitination. Upon degradation of the substrate or just after it, the Ub attached to the substrate is released. The hydrolysis of the isopeptide link between the substrate and the Ub molecule is catalyzed by the Zn2+-dependent metallopeptidase activity of subunit Rpn11 of the 19S RP lid. This reaction is ATP-dependent [105]. Some enzymes associated with the proteasome—Ubp6 (ubiquitin-binding protein 6) and UCH37 (ubiquitin C-terminal hydrolase 37)—can function in a similar or additional way, for example, deubiquitinate conjugates of certain proteins to prevent their degradation or depolymerize the Ub cleaved from the substrate to replenish the pool of free Ub [106-108]. Doa4 is another deubiquitinating enzyme (in contrast to Ubp6 and UCH37, it is more loosely associated with the 26S PR) that also plays an important role in reutilization of Ub [109].
Assembly of 19S RP and 26S PR. In in vitro experiments, the 26S proteasome can dissociate into the 19S RP and 20S CP and reassociate in an ATP-dependent manner [110, 111]. However, it is still unclear just how the 19S regulatory particle is assembled. One of the reasons for this is incomplete knowledge of the structure of 19S RP per se and its heterogeneity in the set of subunits. There are two possible pathways of assembly: (i) the 19S RP is assembled from individual subunits separate of the 20S CP and only then it is attached to the 20S CP, or (ii) the 19S RP is assembled on the 20S CP. The assembly of the 19S RP on the 20S CP is supported by the fact that the central part of the 19S RP base consisting of Rpn1 and Rpn2 can be attached to the surface of the α-ring of the 20S CP and thus plays the role of a “germ” of the 19S RP assembly [92]. This “germ” serves as a site for gathering of other base subunits, but such a complex (20S CP/19S RP base) is very unstable [112]. At the next stage, the lid of the 19S RP is attached and a mature 26S PR is generated with a completely formed 19S RP. It is thought that the 19S RP lid is assembled independently of the base. This is corroborated by the fact that within the cells two types of stable intermediates of the lid assembly are found: (i) the germ from Rpn5, Rpn8, Rpn9, and Rpn11 (lidrpn6-1), or (ii) that from Rpn5, Rpn6, Rpn8, Rpn9, and Rpn11 (lidrpn7-3) [82, 113]. All experiments studying the 19S RP assembly were performed on mutant lines of S. cerevisiae. All attempts to find assembly intermediates in wild-type cells have failed, perhaps because the 19S RP assembly in the cell proceeds extremely rapidly [112].
As mentioned above, in the in vitro experiments the 26S proteasome can dissociate into 19S RP and 20S CP and reassemble again. It appeared that in vivo the 26S proteasome can be assembled both de novo and, by analogy with the in vitro experiments, from the 20S CP and the already assembled 19S RP [114-116]. In accord with the experimental data, in the cell there is dynamic equilibrium between the 20S CP and the 26S PR that can be shifted depending on conditions in the cell [40].
A great variety of additional proteins, among which there are many chaperones, are found in the complex with the 26S PR [117-119]. Evidently, as in the case with the 20S CP, they can be involved in the assembly of the 26S PR. Such a variety of proteins reflect, first of all, the dynamic character of the proteasome complex, peculiarities of intracellular distribution, or substrate specificity. The involvement of only some of these proteins in the proteasome assembly was corroborated. chaperone Hsp90, which is one of such proteins, is required not only for assembly of the 26S PR, but also for maintaining the proteasome in a stable state. Thus, upon inactivation of this protein in the cell, the 26S PR dissociates into the 19S RP and the 20S CP [120]. The nuclear protein Nod1 is involved in the assembly of the 26S PR in growing cells and in the transportation of the 20S CP to the nucleus [121].
Alternative ATP-Independent Regulatory Particles
in addition to the 19S RP (PA700), eukaryotic cells contain a great number of proteins that can interact with α-rings of the 20S CP forming alternative isoforms of the proteasome (Fig. 2). Many of these proteins affect the proteolytic activity of the proteasome, but the physiological functions of such alternative complexes are not understood yet [122]. In contrast to the 19S RP, these alternative regulators are not ATPases and do not bind polyUb chains, i.e. they regulate Ub-independent substrate degradation by the proteasome.
PA28. The family of PA28 regulators discovered in mammals and in some other eukaryotes, excluding yeasts, consists of two cognate complexes: PA28α/β (or 11S REG) and PA28γ (or REGγ). The first complex activates the peptidase activity of the 20S CP increasing the efficiency of hydrolysis of some peptides a hundred times, but it does not affect the degradation of folded proteins [91, 123, 124].
PA28α/β is a heteroheptamer consisting of α- and β-subunits that form a ring attached to one or both ends of the 20S CP. The content of α- and β-subunits and consequently PA28α/β increases in cells as a result of stimulation by γ-interferon [36, 125]. It was also found that eukaryotes have a cognate complex PA28γ (or Ki antigen, or REGγ), which is a homoheptamer. Molecular peculiarities of the interaction of regulator PA28 with 20S CP and the mechanism of activation of the proteolytic activity of the proteasome were studied on PA26 from Trypanosoma brucei, which is a homolog of PA28. Upon binding of homoheptamer PA26 with helices H0 and H1 of α-subunits from the 20S CP, N-terminal regions of α-subunits closing the channel are rearranged: they are drawn into the central chamber of PA26 and open the gate of the 20S CP [126]. Hence, these structural rearrangements in the α-ring gate increase the peptidase activity of the proteasome relative to peptides due to facilitation of their penetration into the proteolytic chamber of the 20S CP.
PA200 (or Blm10, or Blm3). Regulator PA200 consists of one 200-kDa protein of an asymmetric bell-like shape attached to one or both ends of the 20S CP [127]. It was shown in in vitro experiments that PA200 activates hydrolysis of short peptides but not folded proteins [128, 129]. As mentioned above, PA200 is implicated in maturation of the 20S CP [56, 62]. One of its possible functions is the involvement of the proteasome in enzymatic complexes participating in DNA reparation: PA200 is a peculiar linker between the 26S proteasome and proteins recognizing damaged DNA regions. Due to this, the proteasome is attracted to the damaged DNA regions and destroys chromatin proteins there, thus exposing the damaged DNA for reparation enzymes [127].
Ecm29. Ecm29 is a 200-kDa protein which is the prominent among proteins found in the complex with the S. cerevisiae 26S PR. It was demonstrated that in vitro it interacts both with 19S RP and 20S CP, acting as a stabilizer of 26S PR. When this protein is eliminated, the S. cerevisiae 26S PR can disintegrate into the 19S RP and 20S CP in an ATP-independent manner [117]. It is impossible to find mammalian Ecm29 in complex with 20S CP. It is of interest that its content in organs varies greatly: a high content in brain, testicles, and lungs; it is practically absent in the liver, kidneys, heart, and pancreas [130]. It is believed that Ecm29 serves as an adapter via which the 26S proteasome is localized to endosomes, centrosomes, and endoplasmic reticulum (EPR), i.e. intracellular compartments with a high level of Ub-independent degradation of proteins [130].
PI31. PI31 is a proline-rich 30-kDa protein. It competes with other regulatory proteins (PA28 and PA200) for binding with 20S CP, i.e. conjugates with α-rings of 20S CP. In contrast to these proteins, it is unable to form a stable complex with the proteasome and probably modulates proteasome functions only at definite moments [131]. It was shown that in vitro PI31 inhibits chymotrypsin- and caspase-like activity and stimulates trypsin-like activity of the proteasome. This protein localizes mainly to the nuclear membrane and the EPR. It is believed that PI31 is not involved in usual ATP/Ub-dependent degradation of proteins, but is involved in biogenesis and functioning of the immunoproteasome [122, 132].
PC530. It is a 530-kDa multisubunit regulatory complex discovered in starfish [133]. PC530 is involved in gametogenesis and fertilization. The most important physiological functions of the proteasome with the PC530 regulator are the removal of defective sperm in the epididymis and the elimination of paternal mitochondria in fertilized eggs [134].
Signalosome COP9 (or CSN). The regulatory complex COP9 (430-kDa) consists of eight subunits (CSN1-8) that are highly homologous to core subunits of the 19S RP lid. Signalosome COP9 was initially identified as a repressor of photomorphogenesis in Arabidopsis thaliana [135]. Later it was discovered in numerous eukaryotes, where it participates in many cell processes including those at the stage of embryonic development [136]. It is generally accepted that COP9 interacts with cullin-containing E3 ubiquitin ligases and is required for their accurate functioning. Such E3 ligases are activated by a Ub-like NEDD8 modification: after conjugating to NEDD8, the E3 ligase becomes more active in attracting E2 enzymes (ubiquitination of proteins and functions of enzymes E1, E2, and E3 in this process will be described in detail below) [137]. The reaction of conjugation to NEDD8 is called “neddylation”. The function of COP9 is antagonistic to NEDDylation: the CSN5 subunit of signalosome COP9 possesses an isopeptidase activity that removes NEDD8 from cullin-containing E3 ligases, thus regulating the assembly and activity of ligases of this class [138]. In addition, COP9 can bind to protein kinases and deubiquitinating enzymes and regulate their degradation [136]. Signalosome COP9 also has deubiquitinating activity [139-141].
Other regulators. The human immunodeficiency virus protein Tat and hepatitis B virus protein X interact with the proteasome and suppress its peptidase activity [142, 143]. As reckoned, these proteins not only compete with other regulatory particles for the binding with the 20S CP, but being bound to it they keep the proteasome gate closed. Thus, the proteins serve as immunosuppressors preventing antigen presentation upon viral infection.
The multisubunit proteasome inhibitor CF-2 is found in human blood. It is a 240-kDa homooctamer of the 35-kDa protein ALAD (6-aminolevulinic acid dehydratase) involved in heme biosynthesis [144]. Physiological functions of this protein are not sufficiently studied. Most likely distortions in its functions are one of the reasons of acute intermittent porphyria [145].
By interacting with the 26S PR, protein E7 of the human papilloma virus (HPV) 16 enhances the efficiency of degradation of intracellular protein of retinoblastoma (Rb protein). A reduction of the Rb protein level causes transformation to cancer cells. Apparently, just this circumstance explains the oncogenicity of the high-risk HPV protein E7 [146].
Alanine/proline-rich peptide Pr39 is an uncompetitive inhibitor of the 26S PR that does not influence the binding of other regulators with the 20S CP. It interacts with subunit α7 of the 20S CP and causes structural changes that lead to substrate-specific inhibition of the proteasome relative to proteins IκBα and HIF-1α [147].
UBIQUITIN-DEPENDENT PROTEOLYSIS
Ubiquitin-dependent protein degradation has two main stages: (i) attaching the “label” (a polyubiquitin chain) to the substrate protein, and (ii) cleaving the polyubiquitinated substrate protein by the 26S proteasome with release of free Ub. The latter is mediated by deubiquitinating enzymes (DUBs). The conjugation of Ub, an evolutionarily highly conserved 76-amino acid protein, to the substrate protein occurs by means of a three-stage cascade reaction.
Ubiquitination/Deubiquitination System
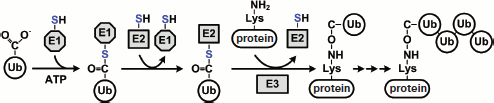
In eucaryotes, most proteasome substrates are polyubiquitinated. Polyubiquitination is performed by cascade reactions catalyzed by three enzymes: E1, E2, and E3. Figure 7 (see color insert) shows the ubiquitination process in a simplified way. In the first stage, Ub-activating enzyme E1 with the use of ATP activates Ub, during which a high-energy thiol ester intermediate (E1-S~Ub) is formed. Then one of the Ub-carrier enzymes E2 (UBC) via the formation of one more intermediate (E2-S~Ub) carries the activated Ub to ligase E3, which is bound specifically to the substrate. In the case of RING-domain-containing E3 ligases, Ub is delivered directly by the ligase to the substrate. In the case of HECT-domain-containing E3 ligases, Ub is delivered to the substrate via the formation of an additional intermediate (E3-S~Ub). After connecting the first Ub to the substrate, E3 ligase attaches sequentially another score of Ub molecules to the first Ub molecule on the Lys residue. In some cases, polyubiquitination involves additional U-box-domain-containing E3 ligases (also called E4 ligases) [148]. As a rule, the C-terminus of Ub forms an isopeptide bond with the ε-amino group of Lys in the substrate molecule, but in some cases Ub can conjugate via the N-terminus of the substrate or via a cysteine side chain [149-151]. The minimal signal of degradation for the proteasome is a chain of four Ub molecules connected in series by an isopeptide link between the C-terminus of one molecule and Lys48 of another molecule [95]. The system of ubiquitination in mammals contains several hundreds of various enzymes including one E1 enzyme, about 50 E2 enzymes, and about 500 E3 ligases. E3 ligases are vital for Ub-dependent proteasomal degradation of proteins since they provide for specificity of polyubiquitination of the substrate.
Before the substrate gets into the proteolytic chamber of the proteasome, Ub should be eliminated from it. The reaction of deubiquitination is performed by deubiquitinating enzymes (DUBs). All known DUBs are cysteine proteases that specifically hydrolyze the isopeptide chain just after the C-terminal residue of Ub (Gly76). In mammals there are nearly 100 deubiquitinating enzymes and, as mentioned above (section “Functions of the 19S RP”), at least four of them (Rpn11, Ubp6, UCH37, Doa4) are components of the 26S PR or are frequently found in a complex with it. Based on the data of their molecular mass, homology of amino acid sequence, and catalytically significant residues in the peptidase center, they are separated into two large subfamilies: UCHs (ubiquitin COOH-terminal hydrolases) and USPs (or UBPs, ubiquitin-specific proteases). UCH enzymes are in general small proteins (20-30 kDa) cleaving Ub from short and non-structured polypeptide chains. Like all cysteine proteases, in the peptidase center they contain a catalytic triad of amino acid residues (Cys, His, and Asp) and an additional conservative Glu residue. USP enzymes are a more heterogeneous group of proteins (30-100 kDa) that cleave the isopeptide link both between Ub and the substrate and between adjacent Ub molecules. Proteins of this family also have a catalytic triad of amino acid residues (Cys, His, and Asp) in the peptidase center [152, 153]. Besides these two DUBs families, some minor ones can also be separated: OUTs (otubain proteases), MJDs (Machado–Joseph disease proteases), and JAMM/MPN [154]. The recently discovered OUT family is not yet numerous. It includes otubain 1 and 2, cezanne, and A20. All of these contain a catalytic OUT-domain with three residues (Cys, His, and Asp). Protein ataxin-3 causing the Machado–Joseph neurodegenerative disease belongs to the MJD family. This protease also has the Cys, His, and Asp catalytic triad. Subunit Rpn11 of 19S RP belongs to the family of JAMM/MPN-proteases, which are Zn2+-dependent metallopeptidases.Fig. 7. Stages of polyubiquitination of substrate protein. Ubiquitin (Ub) is activated by enzyme E1 and translocated to enzyme E2. In the last stage, E3 ligase conjugates Ub to the substrate protein. The protein must be polyubiquitinated for Ub-dependent protein degradation by the proteasome. See details in text.
Ubiquitin
Ubiquitin is a 76-amino-acid-residue protein with a well-studied α/β packing (Fig. 8; see color insert). It is highly conservative in eukaryotes, but is absent in bacteria and archaea. In eucaryotes, several genes encode Ub. It is frequent that Ub is synthesized as an inactive polyubiquitin precursor in which the number of monoubiquitin repeats can differ in different organisms. Some genes encode one copy of Ub linked to ribosomal proteins L40 and S27a [155]. Processing by deubiquitinating enzymes is required to activate Ub (expose the C-terminal Gly residue).
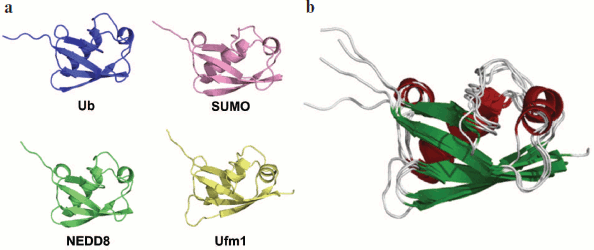
Most often Ub is attached to substrates by forming an isopeptide link between the C-terminal Gly of Ub and the ε-amino group of Lys in the substrate molecule. Ubiquitin forms various types of modifications. The most simple is monoubiquitination, i.e. attachment of one Ub molecule to the protein [156]. Monoubiquitination is a predominant regulatory modification. Attachment of Ub to histones and transcription factors can regulate transcription [157]. Monoubiquitination of proteins PCNA and FANCD2 plays an important role in DNA reparation [158-160]. Attachment of monoubiquitin to different surface cell receptors is a signal for their endocytosis and subsequent degradation in lysosomes [156].Fig. 8. Structure of Ub and Ubl proteins. a) Ribbon drawing of Ub, NEDD8, SUMO, and Ufm1. All of them have a common secondary structure ββααββαβ and β-grasp (ubiquitin-like) three-dimensional packing. b) Superposing ribbon drawings of Ub and Ubl proteins. The figure demonstrates high spatial homology. The figure is modified from [154].
Another modification—multiubiquitination or multiple monoubiquitination—is characterized by the ability of several Lys residues on the substrate to form conjugates with single Ub molecules. Such a modification causes endocytosis of the substrate and its subsequent degradation in lysosomes [161].
Ubiquitin molecules can conjugate between each other forming different variants of chains. The attachment of such ubiquitin chains to the substrate is polyubiquitination [162]. In the Ub molecule per se there are seven Lys residues (Lys6, Lys11, Lys27, Lys29, Lys33, Lys48, and Lys63). It is assumed that all of them can be involved in chain formation. polyubiquitin chains formed via Lys48 and Lys63 occur more frequently and are well studied [163, 164]. Lys48-polyubiquitin chains are most often, though not always, a signal for proteasomal degradation of the substrate [165]. Lys63-polyubiquitin chains participate in regulation of endocytosis, DNA reparation, and protein kinase activation [166]; in in vitro experiments they can cause substrate degradation [167]. Polyubiquitin chains formed via Lys6, Lys11, Lys27, Lys29, and Lys33 are quite rare and their functions are not explicit [168, 169].
Ubiquitin-like proteins. Since the discovery of Ub in the middle of the 1970s, several related proteins have been found. They are separated into two classes: proteins with a Ub-like domain (UDP, ubiquitin-domain proteins) and Ub-like modifiers (Ubl, ubiquitin-like modifiers). The UDP proteins are highly homologous to Ub in their amino acid sequence and similar in three-dimensional structure, but they do not form conjugates with proteins. They serve as adapters attaching to Ub or Ubl proteins [170]. The Ubl proteins are not only homologous to Ub in the amino acid sequence and three-dimensional structure (Fig. 8), but they also have residue Gly at their C-terminus, which allows them to form conjugates with proteins. Analogously to Ub, they (i) attach to the substrate via cascade reactions and (ii) are synthesized as inactive precursors that are processed by specific proteases [154]. Below we describe some of the Ubl proteins.
SUMO (Small ubiquitin-like modifier). This modifier is involved in the regulation of various cell processes such as nuclear transport, transcription, DNA reparation and replication, apoptosis, and protein stabilization [171-173]. In S. cerevisiae, SUMO is encoded by one gene—Smt3 [174]; in vertebrates, four homologous genes have been found—SUMO-1, SUMO-2 (sentrin-3, SMT3A), SUMO-3 (Sentrin-2, SMT3B), and SUMO-4 [175]. Like ubiquitination, the attachment of SUMO to the substrate (sumoylation) proceeds via the formation of an isopeptide link between the C-terminal residue Gly in the SUMO molecule and the ε-amino acid group of a Lys residue in the substrate molecule. However, the Lys residue by which sumoylation occurs usually lies in the ψΚxE consensus (where ψ is a large hydrophobic residue and x is a random residue). The sumoylation proceeds as cascade reactions with the help of Uba2-Aos1 enzymes in S. cerevisiae or SAE1-SAE2 enzymes in vertebrates (enzyme E1), Ubc9 (enzyme E2), and Siz1, Siz2, and Mtm21 (enzyme E3).
NEDD8 (Neuronal-precursor cell-expressed developmentally down-regulated protein 8). This modifier suppresses expression of a set of genes in neuronal precursors during brain development. NEDD8 is a 9-kDa protein the amino acid sequence of which is 60% identical to that of Ub. It has its own enzymes E1 (APPBP1-Uba), E2 (Ubc12), and E3 (Dcn1) for attaching to substrates [176]. Known substrates include cullins, p53, and Mdm2 [177].
ISG15 (IFN-stimulated gene 15). The content of this modifier in the cell increases greatly when induced by interferon [178]. In contrast to Ub and other Ub-like modifiers, it consists of two domains, each of which is 30% identical to Ub and has a similar three-dimensional structure. This modifier is involved in immune response regulation, cell growth, and differentiation [179, 180]. The attachment of ISG15 is also mediated by cascade reactions involving protein UbeL1 (enzyme E1). Most probably the subsequent stages of the reaction are performed by enzymes E2 and E3 of the ubiquitination system. Substrates of this modifier are proteins STATq1, Serpina3G/Sp12A, JAK1, MAPK3/ERK1, PLCγ1, EIF2AK2/PKR, MX1/MXA, and RIG-1.
Atg (autophagy). When studying mutant strains of S. cerevisiae with distortions in autophagy, two Ub-like modifiers—Atg8 and Atg12—were found. In mammals homologous modifiers were discovered: Atg12 and orthologs of Atg8—proteins LC3, GABARAP, and GATE-16 [181]. In mammals such modifiers are involved in regulation of autophagy in neurodegenerative, neuromuscular, and oncological diseases, as well as in bacterial and viral infections. In the case of Atg8, the substrate of such modification is the phospholipid PE (phosphatidylethanolamine), and the reaction of conjugation is mediated by enzymes Atg7 (E1) and Atg3 (E2). Conjugation of Atg8 with PE is absolutely necessary for normal autophagy [182].
FAT10 (F-adjacent transcript-10). This 18-kDa protein is encoded by the gene of the major histocompatibility complex and is induced by TNFα and γ-interferon. It consists of two Ub-like domains, one of which can directly bind to the 26S PR and thus mediate Ub-independent degradation of proteins [183].
Ufm1 (Ubiquitin-fold modifier 1). This modifier is only 16% identical to Ub, but it has a similar spatial structure [184, 185]. Biological functions of this modification are not yet established. Conjugation of Ufm1 with the substrate is mediated by enzymes Uba5 (E1) and Ufc1 (E2).
NON-CLASSICAL PROTEASOME PROTEOLYSIS
The well-studied classical Ub-dependent proteasomal degradation of proteins requires polyubiquitination of the substrate. It proceeds with expenditure of the energy of ATP, and its products are short peptides. During recent years much experimental data have accumulated on deviations from this classical degradation pathway. Thus, proteins can be subjected to (i) Ub-independent degradation [186], (ii) ATP-independent degradation [187], (iii) degradation by the core latent 20S PR rather than the 26S PR [188], and (iv) processing rather than complete degradation to small peptides [189]. upon degradation of a particular substrate, most frequently only one of the mentioned deviations is observed (usually it is independence from Ub), occasionally two (for example, independence from Ub and ATP), and quite rarely when all the deviations can be observed during proteolysis of one protein [37]. Let us analyze in detail the Ub-independent degradation and processing.
Ubiquitin-Independent Degradation
The vast majority of examples of non-classical proteasomal degradation are associated with the Ub-independent degradation of proteasome substrates. Upon degradation by the 26S proteasome, the only distinction from the classical mechanism is that another protein plays the role of Ub, or the signal for degradation is contained in the sequence of the protein substrate per se. In the case of the 20S proteasome, in addition to the proteasome recognition of the substrate, the problems of substrate unfolding and opening of the gate into the proteasome cavity should be solved somehow. Ornithine decarboxylase (ODC) [190], α-synuclein [188], p21Cip1 [191], tau [192], DHFR [193], RPN4 [194], p53 and p73 [195], HIF-1α 1, Rb [197], p105 subunit of NF-κB [198], pertussis toxin [199], NFAT5 [200], kinase Aurora-A [201], pp89 [202], KLF5 [203], hepatitis C virus (HCV) F protein [204], c-Jun [205], calmodulin (CaM) [206], troponin C [207], and oxidized proteins [208] are proteins that can undergo Ub-independent degradation. Below we consider some examples.
Non-structured/damaged proteins. Most proteins degraded by the proteasome in a Ub-independent manner are non-structured due to some reason. (1) In accord with the genome analysis, about one third of all eukaryotic proteins have long regions with disordered secondary structure [209]. Due to their activity, regulatory proteins are involved in interactions with different ligands (proteins, DNAs, RNAs, membranes, etc.). It is frequent that one regulator interacts with several specific ligands. Such a variety of its intermolecular links is possible only with high plasticity of the three-dimensional structure. Therefore, it is not surprising that very many regulatory proteins are natively disordered and short living. Probably, their disorder that in the long run leads to rapid Ub-independent degradation is an additional level of regulation of the content of such proteins within the cell depending on environmental conditions [210]. (2) Another reason for protein disorder may be its damage. Proteins in the cell are constantly subjected to spontaneous nonenzymatic modifications (deamination of Asn residues, isomerization of Asp residues, oxidation), which not only inactivate them but also decrease their lifetime. Such modifications often result in protein unfolding and, as a consequence, in enlarging of hydrophobic surface, which in turn leads to Ub-independent recognition and degradation by the 20S proteasome [211]. It was shown in experiments in vitro that the 26S proteasome is inactive in degradation of oxidized and unfolded proteins even in the presence of Ub and ATP [208]. Unfolded proteins do not require Ub for binding with the proteasome. In this case, the function of Ub is fulfilled by hydrophobic regions of the unfolded chain. For their degradation, no ATP hydrolysis is required for the task of unfolding. In many cases they can be processed without the 19S RP because of their ability to open the gate into the proteasome cavity.
ODC. Ornithine decarboxylase (ODC) was the first protein for which Ub-independent degradation was demonstrated. Degradation of ODC is ATP-dependent and regulated by protein AZ1 (antizyme 1) [190, 212]. ODC is a short living protein, but upon interaction with AZ1 the rate of its degradation increases greatly. The C-terminus of ODC is an instability element: it is responsible for the interaction of ODC with the proteasome (probably with the Rpt5 subunit). It is assumed that in the absence of AZ1 ornithine decarboxylase forms dimers in which these C-termini are masked [213]. It was shown that the formation of the ODC/AZ1 complex enhances the exposure of the C-terminus and, as a result, increases the efficiency of proteasomal recognition [214].
p21Cip1. The p21Cip1 inhibitor of cyclin-dependent kinase is a crucial regulatory protein, and its content in the cell is controlled by a complicated multilevel regulation. It is an unstable protein that is processed by the proteasome. Its stability is regulated both by phosphorylation and by interaction with other proteins [215-217]. In the cell, an insignificant part of p21Cip1 is ubiquitinated, and the portion of the ubiquitinated protein grows in the presence of proteasome inhibitors. This corroborates the Ub-dependent degradation of p21Cip1. Nonetheless, in a series of studies devoted to the stability of non-ubiquitinatable p21Cip1 analog (in which all Lys residues are substituted for Arg and the N-terminus is blocked with a tag preventing N-terminal ubiquitination) it has been reported that such a modified protein remains unstable and undergoes regulated degradation similar to the wild-type protein [213]. These data demonstrate that ubiquitination of p21Cip1 is not a necessary condition for its degradation, i.e. its proteolysis can be also Ub-independent. PA28γ-containing proteasomes contribute significantly to the degradation of p21Cip1. So p21Cip1 is greatly stabilized in cells depleted of PA28γ by RNA interference or in PA28γ-knockout embryonic rat fibroblast cells [187, 218]. However, in some cell lines PA28γ is not required at all for degradation of p21Cip1, and upon elimination of PA28γ by RNA interference it is impossible to achieve complete stabilization of p21Cip1 [187, 218]. This shows that other proteasome isoforms participate in p21Cip1 degradation. In particular, data of in vivo and in vitro experiments demonstrate that the C-terminal part of p21Cip1 can interact directly with subunit α7 of the 20S CP [219]. It is also shown in the cited paper that wild-type p21Cip1, but not a mutant isoform depleted of the C-terminal part, can be degraded under the action of the 20S CP in vitro. A critical role in recognition and degradation of p21Cip1 caused by PA28γ-containing proteasomes is also given to its C-terminal part [187, 218]. As concerns the problem of penetration into the proteolytic chamber, as in the case with damaged proteins p21Cip1, being a non-structured protein [220], can itself open the gate of the 20S proteasome [38]. It was shown that the degradation of p21Cip1 caused by the 26S proteasome in vitro is ATP-independent [38, 111]. The sole explanation of the ATP-independent degradation can be the ability of unfolded substrates to penetrate into the proteolytic chamber of the 20S CP via an open gate due to passive diffusion [111].
p53. Transcription factor p53 is an important coordinator of the cell response to the DNA-damaging stress in most if not in all cells. Depending on conditions and cell type, the response to stress can be interruption of the cell cycle, apoptosis, or aging [221]. Distortions in the functions of p53 are found in more than 50% of cancer cases. It is known that in non-transformed cells under normal conditions p53 is labile and degrades quite rapidly. As a result of stress, enhanced synthesis and stabilization of p53 is observed in the cells. For p53, both Ub-independent and Ub-dependent degradation pathways have been shown. The Ub-dependent mechanism will be briefly described in section “Proteasomes and medicine”. Herein we will analyze the Ub-independent mechanisms. At present two mechanisms of Ub-independent degradation of p53 are well studied.
E6-dependent Ub-independent degradation of p53. High-risk human papilloma virus (types 16 and 18) express protein E6 that binds to p53 and causes its degradation [222]. Two binding sites of protein E6 and the p53 molecule are localized: one is in the DNA-binding domain of p53 (amino acid residues 66-326) and the other in the C-terminal part (amino acid residues 376-384). The binding of E6 to the C-terminal region of p53 occurs without mediators, while the binding of E6 to the DNA-binding domain of p53 is mediated by protein E6-AP (E6-associated protein). After the formation of the E6/p53 or E6/E6-AP/p53 complex, protein p53 undergoes Ub-independent degradation by the proteasome [223]. However, it is still unclear what proteasome complex (20S or 26S) is involved in the degradation of p53 and in what way E6 presents p53 to the proteasome. It is known that E6 can cause Ub-dependent degradation of p53 as well. This mechanism also involves protein E6-AP. This protein is a cell ligase E3 specific for p53, but under normal conditions it does not ubiquitinate p53. The participation of E6 and E6-AP in the Ub-dependent degradation of p53 is described in more detail in the section “Proteasomes and Medicine”.
NQO1-inhibited Ub-independent degradation of p53. Protein NQO1 (NAD(P)H quinone oxidoreductase 1) is an enzyme regulating the quinone content in the cell. A considerable part of this protein is complexed with the 20S but not the 26S proteasome. NQO1 is able to interact with p53, and in addition, NQO1, p53, and 20S are found in the triple complex [195]. It was demonstrated that the interaction of NQO1 with the 20S proteasome is independent of NADH and is not destroyed by dicumarol (a competitive inhibitor of NQO1). In contrast, the binding of NQO1 to p53 is intensified in the presence of NADH and destroyed by an addition of dicumarol or other inhibitors of NQO1. It was hypothesized that the free protein NQO1 or that located on the 20S proteasome binds to p53 in the presence of NADH, stabilizes it, and locally raises its concentration near the 20S proteasome. It is assumed that the catalytic activity of NQO1 is not vital for stabilization of p53 [224]. In the absence of NADH, p53 dissociates from NQO1 and is destroyed by the 20S proteasome. However, the mechanism of recognition and processing of p53 by the proteasome remains unclear. It has been proposed that NQO1 plays the role of a peculiar gate-keeper that is able to open the gate into the proteolytic chamber of the proteasome for p53, p73α, ODC, and other nonstructural proteins [225].
Rb. Like p107 and p130, oncosuppressive Rb proteins are members of a multigene family. These proteins play a crucial role in such cell processes as cell cycle control, response to DNA-damaging stress, DNA replication and reparation, and cell differentiation and aging [226]. The content of these proteins in the cell is controlled due to their proteasomal degradation. Both Ub-dependent and Ub-independent degradation mechanisms have been described.
pp71-induced Ub-independent degradation of Rb. Several proteins encoded by DNA-containing viruses that induce proteasomal degradation of Rb are known. They are human papilloma virus protein E7 [227], Epstein–Barr virus protein EBNA3C [228], hepatitis C virus protein NS5B [229], and human cytomegalovirus protein pp71 [230]. It was shown that proteins E7, EBNA3C, and NS5B induce enhanced ubiquitination of Rb and as a result its Ub-dependent degradation. Protein pp71 causes Ub-independent degradation of Rb, but the molecular mechanism of this is not sufficiently studied.
Mdm2-induced Ub-independent degradation of Rb. Oncoprotein Mdm2 is overexpressed in many types of human cancer, and its C-terminal region is a RING-domain-containing ligase E3. It was demonstrated that Mdm2 binds specifically to the hypophosphorylated protein Rb and induces its proteasomal degradation [231, 232]. There are two contradicting points of view on the mechanism of the Mdm2-induced degradation of Rb. (1) Mdm2 enhances ubiquitination of Rb, which suggests the Ub-dependent mechanism of Rb degradation in the cell [232]. (2) Mdm2 does not induce ubiquitination of Rb in the cell, and in such case the proteasomal degradation of Rb proceeds by the Ub-independent pathway [231]. The second mechanism is supported by the following data: Rb is more sensitive to the 20S than to the 26S proteasome; in experiments on gel filtration of cell extracts, Rb is coeluted with the 20S and not the 26S proteasome. Mdm2 and Rb bind to subunit α7 of the proteasome. The RING-domain of Mdm2 and the C-pocket of Rb are responsible for the binding to the subunit α7; both are necessary for the Mdm2-induced degradation of Rb in vivo. In the absence of the RING-domain, Mdm2 is unable to promote/stabilize the interaction of Rb with the proteasome subunit α7. All these data indicate a possibility of Ub-independent degradation of Rb in the cell. However, it remains unclear why one group of researchers observes ubiquitination of Rb while the other does not.
Processing
As mentioned above, the classical mechanism of protein degradation by the 26S proteasome includes stages of polypeptide chain unfolding and translocation into the proteolytic chamber. It is believed that the substrate penetrates with one of its termini into the proteolytic chamber of the 20S CP via the gate, and the degradation occurs processively from the terminus (exoproteolytically). However, it was found that the p50 subunit of transcription factor NF-κB is formed from the p105 precursor cotranslationally by a proteasome-dependent pathway. Based on the fact that the processed p50 subunit contained the N-terminal region of the p105 precursor and the C-terminal region was linked to the ribosome, a hypothesis was offered on the ability of the proteasome to perform endoproteolysis, i.e. to disrupt the polypeptide chain of the substrate significantly far away from its termini [189]. During this, it was proposed that the growing polypeptide chain folds like a hairpin, which slides into the proteolytic chamber of the proteasome. Such an opportunity was demonstrated in experiments in vitro on degradation of barnase whose polypeptide chain was crosslinked by disulfide links. It was shown in the cited paper that the gate of the 20S CP can widen to 20 Å, which allows three polypeptide chains to concurrently pass through it [29]. Later the possibility of endoproteolysis was demonstrated directly in in vitro experiments on degradation of model substrates of the proteasome. The substrates were proteins α-synuclein and p21Cip1, to the C- and/or N-termini of which GFP protein had been attached using a gene-engineering method. α-synuclein and p21Cip1 are unstructured proteins able to be cleaved by the 20S and 26S PR in an Ub-independent pathway. In contrast, GFP is a structured molecule resistant to proteasomal degradation. It was shown in the above experiments that both in GFP-α-synuclein and GFP-p21Cip1 (in which the N-terminus of unstructured proteins is protected from initiation of degradation) and in α-synuclein-GFP and p21Cip1-GFP (in which the C-terminus of unstructured proteins is protected from initiation of degradation), the 20S and 26S proteasomes can degrade the unstructured part (α-synuclein or p21Cip1) leaving GFP intact. This demonstrated that the substrate can be degraded by the proteasome from either end of the polypeptide chain. In spite of the protection of both their termini from initiation of degradation, α-synuclein and p21Cip1 in the substrates (GFP-α-synuclein-GFP and GFP-p21Cip1-GFP) had also undergone proteasomal degradation, whereas GFP remained intact. This points to the endoproteolytic character of the degradation of these model substrates and suggests limited degradation or processing [38]. The publication of that experimental paper let to the belief that the alternative Ub-independent proteasomal degradation is not an artifact and initiated consideration of the 20S proteasome as a valid enzyme able to regulate the functions of proteins in the cell via their degradation or processing.
NF-κB p105. NF-κB is a family of dimeric transcription factors. By controlling the expression of a wide range of genes, they are implicated in regulation of the immune response, reparation reactions, and apoptosis. The NF-κB family consists of five members: p50, p52, p65/RelA, c-rel, and RelB [233]. p50 and p52 are formed as a result of processing from precursors p105 and p100, respectively. As mentioned above, the p50 subunit is the N-terminal part of the p105-precursor. In accord with experimental data, processing of p105 can be performed both by the Ub-dependent pathway by the 26S proteasome [234] and by the ATP-/Ub-independent pathway by the 20S proteasome [198].
It was first assumed that the formation of p50 from the p105-precursor occurs cotranslationally [189]. According to this hypothesis, p50 is formed not from a completely synthesized p105-precursor but cotranslationally. Upon translation of the p105 mRNA, the ribosome makes a pause in the region of the triplet corresponding to amino acid residue 530. At this moment, the proteasome docks at the synthesized polypeptide and performs limited endoproteolysis. As a result, the C-terminal region is completely degraded and the N-terminal (amino acid residues 1-433) remains intact. If the ribosome does not make a pause, a full-size polypeptide p105 is synthesized, which is no longer able to be processed into p50. This protein functions as an inhibitor of the activity of a variety of other proteins: it binds to proteins Tpl2, FLIP, LYL1, and ZUD5 and subunits NF-κB [235-238]. The action of definite stimuli activates IκB kinase β, which phosphorylates p105 at residues Ser927 and Ser932, thus triggering its proteasomal degradation [239].
Later there appeared data showing that in vitro p50 can be formed as a result of processing caused by the action of the 20S proteasome from a completely synthesized p105 precursor. It was demonstrated in experiments in vivo that processing occurs independently of translation and does not require ubiquitination of p105 [198]. Experiments with a number of deletion mutations of p105 demonstrated that (i) the processing begins from endoproteolysis in the unstructured region of p105 corresponding to amino acid residues 430-530, and (ii) the element that prevents complete degradation of the protein is region GRR (glycine-rich region, amino acid residues 365-430); elimination of this region leads to complete degradation of p105. It remains unclear why GRR prevents complete degradation.
YB-1. YB-1 is a DNA/RNA-binding nuclear-cytoplasmic protein of animal cells that is involved in practically all DNA- and mRNA-dependent processes. It enhances the resistance of cells to ionizing radiation and genotoxic xenobiotics, serves as a marker of both multiple drug resistance and cancer, and can cause or suppress oncogenic transformation of cells. Multiple functions of YB-1 in the cell are dependent on its amount, activity, and intracellular distribution [240].
In in vitro experiments, it was shown that YB-1 is cleaved into two fragments by the 20S proteasome in the ATP-/Ub-independent manner. The cleavage is performed by the caspase-like activity of the 20S CP in the peptide link after residue Glu219. The same cleavage of YB-1 by the proteasome is observed in cancer cells after their treatment by some therapeutic DNA-damaging agents. However, in cells it is possible to find only a large N-terminal fragment of YB-1 (most likely the C-terminal region undergoes further degradation) [37]. A similar cleavage was also revealed in endothelial cells treated with thrombin, but the protease responsible for the processing was not identified [241]. After the proteasome processing, the N-terminal fragment of YB-1 (amino acid residues 1-219) is translocated into the cell nucleus [37]. The translocation into the nucleus of the truncated YB-1 can be explained by the presence of a nuclear localization signal (amino acid residues 183-205) in it. The intact protein contains also another signal, i.e. the cytoplasmic retention signal (amino acid residues 247-290) that is localized in the cleaved C-terminal part of the molecule. This signal predominates over the signal of nuclear localization and masks it, which defines the cytoplasmic localization of the intact protein [242]. It was elucidated that accumulation of the processed YB-1 in nuclei correlates with the appearance in the cells of resistance to DNA-damaging reagents. At the present time, we do not know whether processing of the YB-1 protein is required only for regulating its nuclear–cytoplasmic distribution or for modulating its functional activity as well. Molecular mechanisms triggering the Ub-independent processing of YB-1 have not yet been clarified.
Besides the ATP-/Ub-independent processing, YB-1 can undergo a complete Ub-dependent degradation. Initially it was found that cells treated with the proteasome inhibitor accumulate ubiquitinated YB-1 [37], and later E3 ligases involved in the process were also revealed. One of them was the SCFFBX33 E3 ligase consisting of proteins Skp1, Cul1, and the F-box of protein FBX33, which is a substrate-recognizing component of this multisubunit enzyme [243]. Another E3 ligase specific to YB-1 is the RING-domain-containing protein RBBP6 (retinoblastoma binding protein 6) [244]. In all probability, the amount of YB-1 in the cell is strictly controlled by the Ub-dependent degradation, whereas the Ub-independent processing of YB-1 is triggered in particular conditions and directed to regulate intracellular distribution and/or modulation of YB-1 activities.
eIF4G and eIF3a. Translation initiation of cell and viral mRNAs in eukaryotic cells occurs with the help of special proteins—translation initiation factors. It has been noted repeatedly that upon isolation of these factors from rabbit reticulocyte lysates some of them are subjected to proteolytic fragmentation, which remarkably affects their activity. Most affected by such fragmentation are subunits eIF4G of factor eIF4F and eIF3a of factor eIF3. An addition of proteasome inhibitors to the cell lysate upon fractionation of proteins prevented their fragmentation, based on which it was concluded that the proteasome participates in their processing. This conclusion was confirmed in experiments in vitro. It was shown that the 20S proteasome is able to cleave eIF4G and eIF3a in the Ub-independent manner [245]. The cleavage of factors eIF4F and eIF3 by the 20S proteasome has an inhibiting effect on the translation of mRNAs that are dependent on these factors. It is known that some cell and viral mRNAs are “less strict” and can be effectively involved in translation even in the absence of some initiation factors [246, 247]. Based on these data, the authors of the cited paper proposed that the proteasomal processing of proteins eIF4G and eIF3a is one of the mechanisms of translation regulation in the cell [245].
PROTEASOME REGULATION
The 26S proteasome is responsible for the regulated proteolysis of many intracellular proteins, it per se always being controlled. The relative content of the proteasome and its localization in the cell change dynamically and adjust in accord with the cell requirements and particular stress conditions. The proteasome is constantly assembled and disassembled, and its subunits are targets for a great number of posttranslational modifications [248]. As follows from this, the proteasome functioning is continuously interacting with a variety of proteins, which stabilize it, regulate its activity, assist in recognizing substrates and in utilizing Ub, etc. Let us briefly characterize the basic mechanisms of regulation of the Ub-dependent degradation.
Heterogeneity of Proteasomes in the Cell
As known, the 26S PR consists of the 20S CP and regulatory particles which can dock at one or both termini of the 20S CP. Heterogeneity of regulatory particles and the number of combinations in which they can attach to the 20S CP provide a whole set of proteasomes with different functions. The content of regulatory particles in the cell and correspondingly the functions of proteasomes can change greatly (see section “Proteasome Structure”). The immunoproteasome is another striking example of regulation of proteasome functions by changing its subunit composition. As shown above, the stimulation of cells by γ-interferon activates the synthesis of three proteasomal subunits (β1i, β2i, and β5i), which during proteasome assembly are inserted instead of constitutively synthesized subunits β1, β2, and β5. The peptides generated by the immunoproteasome are not subjected to further degradation by cell peptidases and are used for antigen presentation. Moreover, the content of individual proteasomal subunits can depend on the gender of the organism [249] or, as in the case of Rpn10, proteasomal subunits may be synthesized as tissue-specific isoforms appearing as a result of alternative splicing of their mRNA precursors [250].
Synthesis of Proteasomal Subunits
Mechanisms of synthesis regulation of proteasomal subunits are studied insufficiently, and at present only several examples of such regulation are known. Proteasomal subunits are synthesized in stoichiometric amounts, which confirms the coordinated control of the expression of proteasomal genes.
In S. cerevisiae the system of regulation at the transcription level has been discovered. It was found that promoters of genes of proteasomal subunits and a number of other genes (their number exceeding 700) have regulatory element PACE (proteasome activation control element) that is recognized by the transcription factor Son1. In earlier studies, this protein was found in complex with proteasomal subunits; therefore it was named Rpn4. Rpn4/Son1 binds to the PACE element in promoters of proteasomal genes and activates their transcription. This increases the proteasome content in the cell. Under normal conditions, Rpn4 is short living. Its half-life is about 2 min, and the degradation proceeds under the action of newly synthesized proteasomes by the Ub-independent mechanism [251]. Under stress, when a great number of proteasomes in the cell is required or when their activity is suppressed by mutations in subunits or by inhibitors, the degradation of Rpn4 slows down and the half-life increases several-fold [194]. The mechanism of regulation of the synthesis of proteasomal subunits by Rpn4 is corroborated by an elegant experiment in which the synthesis of Rpn4 resistant to proteasomal degradation resulted in a significant increase in the proteasome content [252].
It is known that the overall level of expression of proteasomal subunits in higher eukaryotes depends on the cell proliferation stage. So the synthesis of proteasomes in proliferating leucosis cells is more active compared to non-dividing cells at the final stage of differentiation [253]. Enhanced synthesis of proteasomal subunits was found in cells treated with proteasomal inhibitors [254]. The mechanisms of proteasomal synthesis regulation in higher eukaryotes are not studied yet, but it is evident that they differ from the above-described mechanism in yeasts, since neither any ortholog of Rpn4 nor PACE elements in promoters of proteasomal genes have been found in higher eukaryotic cells.
Localization of Proteasomes
Proteasomes in eukaryotic cells are localized both in the cytoplasm and in the nucleus. For example, in hepatocytes 17% of the overall number of proteasomes is in the nucleus, 14% is linked to endoplasmic reticulum, and the remaining portion is found in the cytoplasmic matrix; the proteasomal content in the nuclei of lung epithelium cells is 51% [255]. This indicates that the nuclear–cytoplasmic distribution of proteasomes can be tissue-specific. The distribution of proteasomes between the cytoplasm and the nucleus changes remarkably during embryogenesis. In spermatozoids and ovules, proteasomes are concentrated in the cytoplasm, at early stages of subdivision they translocate to the nucleus, and by the blastocyst stage the intracellular localization of proteasomes is close to their distribution in somatic cells [256, 257]. Besides, intracellular distribution of proteasomes changes dynamically in accord with the cell cycle phase [258]. The cell cycle is regulated by two basic biochemical mechanisms. This is protein phosphorylation–dephosphorylation and proteolysis of proteins implicated in the regulation of the cell cycle (for example, cyclins and cyclin-dependent kinases) [259]. Proteasomal degradation of cyclins in the nucleus is a necessary condition for the normal course of the cell cycle [260].
The mechanisms of regulation of intracellular distribution of proteasomes are not sufficiently studied. Some proteasomal subunits contain nuclear localization signals (NLS) [261-263]. Many subunits are phosphorylated [248]. At least six proteasomal subunits are phosphorylated on Tyr residues, and this phosphorylation might be involved in regulation of proteasome redistribution between the nucleus and the cytoplasm [262, 264, 265]. In some recent papers it is reported that proteasomes can be localized not only in the cell, but also outside it. Proteasomes have been found in blood serum of both normal subjects and patients with different malignant diseases (leukemia, myeloma, carcinoma, etc.) [266-268]. Based on the fact that the proteasome level in the blood serum of oncology patients was much higher, the authors concluded that this might originate from their increased secretion by tumor cells [266]. It was also demonstrated that the level of proteasome synthesis in leucosis cells is far greater than in normal blood cells [253]. An increased proteasome level in the blood serum was also found in patients with different autoimmune diseases, and at present this is used as one of the clinical markers of disease development (see section “Proteasomes and Medicine”). The molecular mechanisms of proteasome secretion are still little studied.
Modification of Proteasomal Subunits
In the cell, many proteins experience various structural changes as a result of cotranslational and posttranslational modifications. More than 100 different modifications have been described; however, the role of most of them is not clear. Some modifications are random and in all probability have no functional significance, but there are those that are vital for the cell because they are strictly controlled by specific enzymes. It has been demonstrated for subunits of 26S PR that they can be subjected to phosphorylation, N-acetylation, N-terminal processing, cleavage by caspases, N-myristoylation, O-glycosylation, S-glutathionylation, alkylation, and oxidation of sulfur-containing amino acid residues (see review [248]). Some of these modifications can modulate proteasome functions. Let us consider examples of such regulation.
Phosphorylation. Phosphorylation of the 20S PR by protein kinase A in vitro (in subunits α1, α2, α3, β2, β3, and β7) enhances the chymotrypsin- and caspase-like activities [269]. Data are available according to which phosphorylation of subunit α2 on Tyr120 is required for nuclear localization of the proteasome [265]. Phosphorylation of subunits α3 and α7 of the 20S CP can affect the attachment of regulatory particles to the ends of the 20S CP [270, 271]. In particular, phosphorylation by casein kinase II (CKII) in subunit α7 stabilizes the complex of 20S CP with 19S RP, whereas dephosphorylation enhances the binding of 20S CP to PA28α/β [272]. It has been found recently that about half of all phosphorylated residues of the proteasome are in consensuses for mitogen-activated and cyclin-dependent kinases, i.e. phosphorylation of the proteasome and consequently its functions can change greatly depending on the cell cycle phase [119].
N-Terminal processing. Five of the seven β-subunits (β1, β2, β5, β6, and β7) of the 20S proteasome have N-terminal propeptide sequences. They are required for both the proteasome assembly and the protection of Thr1 residues in potential catalytic centers from acetylation and inactivation [273]. Premature cleavage of propeptide sequences from subunits β1, β2, and β5 leads to inactivation of catalytic centers and decreases the efficiency of 20S CP assembly.
N-Myristoylation. Using mass-spectral analysis, it was discovered that subunit Rpt2 of the 19S RP undergoes myristoylation in the N-terminal residue Gly. It is known that such a modification influences protein–protein interactions and the interaction of proteins with membranes. It was assumed that myristoylation of subunit Rpt2 also controls the proteasome interaction with proteins and membranes [119]. But this has not been supported yet by experimental results.
S-Glutathionylation. It was shown that glutathionylation of subunits of the 20S proteasome from S. cerevisiae on Cys residues inactivates the chymotrypsin- and trypsin-like activities rather than the caspase-like activity of the proteasome [274].
O-Glycosylation. Many Rpn, Rpt, and α- and β-subunits of the proteasome are glycosylated. Since glycosylation and phosphorylation is experienced by the same residues of the polypeptide chain, ser and thr residues, their influence on the functions being frequently antagonistic, the switching from one type of modification to another type can rapidly modulate the proteasome activity. As known, the ATPase subunit Rpt2 of the 19S RP is glycosylated in vivo and in vitro. Such a modification results in inhibiting the ATPase activity and, as a consequence, decreasing the proteolysis efficiency [275].
Cleavage by caspases. upon apoptosis, some subunits of the 20S CP as well as subunits Rpn2, Rpn10, and Rpt5 in human cells undergo cleavage by caspase 3. Subunits Rpn2 and Rpn10 play an important role in attaching the 19S RP base to the lid. Moreover, Rpn10 is the main subunit implicated in the recognition of polyubiquitinated substrates of the 26S PR. It is logical that their cleavage leads to suppression of the protein degradation and accumulation of ubiquitinated proteins, including proapoptotic ones [276].
Adaptor Proteins
The polyUb chain can be directly bound to proteasomal subunits (described in detail in “19S Regulatory Particle”). Nevertheless, the family of adaptor polyUb-binding proteins UbL-UbA plays a great role in regulation of the interaction of the polyubiquitinated substrate with the proteasome. These proteins interact with the proteasome by their N-terminal domain UbL (Ub-like domain) and with the ubiquitinated substrate by the C-terminal domain UbA (Ub-association domain). It was shown that proteins UbL-UbA can have both diverse and overlapping substrate specificity [97, 99, 277]. UbL-UbA proteins include, for example, proteins Rad23, Dsk2, and Ddi1 [67]. The specificity of recognition of substrates by these adaptor proteins can provide for a local increase in the proteasome concentration in regions where it is necessary to process proteins promptly. Thus, e.g. under DNA-damaging stress, protein Rad23 binds to protein Rad4 implicated in DNA reparation, and thus attracts the proteasome to damaged DNA sites involving it in protein degradation in the chromatin sites required for effective DNA reparation [278].
Some E3 ligases can play the role of an adaptor between the proteasome and the substrate. They can interact with the proteasome directly or via accessory proteins. In this case, E3 ligase can provide for an efficient substrate-specific degradation in two ways: directly, by increasing locally the concentration of the specific substrate, or mediated, by lengthening the polyUb chain of the specific substrate and thus enhancing its affinity to the proteasome [98].
For some substrates, the binding with the proteasome is possible only in the presence of the complex Cdc48/p97 attached to the proteasome [279]. Cdc48 is an ATP-dependent chaperone consisting of six identical subunits. It can interact with E3 ligases and deubiquitinating enzymes and can bind substrates directly or via a great family of adaptor proteins UbX-UbA. Besides recognizing specific substrates, the Cdc48 complex participates in their unfolding [98].
PROTEASOMES AND MEDICINE
Practically all intracellular processes including cell cycle control, transcription, translation regulation, cell response to stress, etc. are controlled by the Ub–proteasome system. An impressive number of key regulatory proteins in the cell are eliminated or processed by the proteasome. They include cyclins, inhibitors of cyclin-dependent kinases, phosphatases, kinases, and transcription and translation factors. Numerous examples of the involvement of proteasomes in the regulation of cell processes are described in detail in the review of Konstantinova et al. [248]. The important biological role of the proteasome system implies that it should be unavoidably involved in pathophysiological processes causing the development of malignant, autoimmune, and neurodegenerative diseases. Let us briefly analyze the changes in the proteasome system that lead to the development of various diseases.
Cancer
At present, several ways are known how the proteasome system can be involved in the development of malignant tumors. In general, the development of cancer can be caused by stabilization of an oncoprotein or destabilization of a tumor suppressor.
p53. The multifunctional protein p53 is one of the most thoroughly studied tumor suppressors. It is implicated in a wide range of cell processes including apoptosis induction, stimulation of DNA reparation, cell cycle arrest, and regulation of basic metabolism [280]. Today several mechanisms are known to regulate the activity of p53 in the cell. Many are somehow related to the degradation of this protein by the 26S or 20S proteasomes. Several E3 ligases are known for p53: Mdm2, E6-AP/E6, COP1, Pirh2, ARF-BP1, Topors, and CHIP [281-288]. Mutations in these enzymes, disturbance of their activity, as well as duplication of their genes or disturbance of regulation of their expression can lead to malignant degeneration of cells as a result of inadequate activity of p53. This can be readily demonstrated on Mdm2. In some types of tumors, such disturbances as gene amplification [289, 290], enhanced protein synthesis [291-293], and incorrect splicing of pre-mRNA of Mdm2 [294, 295] were found. Besides, in the promoter of the human Mdm2 gene (HDM2), single nucleotide polymorphism was discovered: either thymine or guanine (SNP309T or SNP309G) can be located in position 309 of the first intron. The substitution of T by G leads to a tighter binding of transcription factor Sp1 to the intron DNA, as a result of which the level of transcription from the Mdm2 gene increases [296]. According to the data of some researchers, this polymorphism does not at all affect the probability of cancer development [297-299]. But the results obtained by other researchers show that the homo- or heterozygote state of SNP309G correlates with higher risk of malignant tumor development [300, 301], poor prognosis for survival outcome [302], and earlier beginning of the disease [296, 303, 304]. It should be noted that Mdm2 can also promote the formation of tumors by a mechanism independent of p53 [305].
When analyzing genes the expression of which is enhanced in hepatocellular carcinoma, the protein gankyrin was found, its content being higher in all tumor samples analyzed. In an independent study, this protein was identified as component p28/PSMD10 of the 19S proteasomal regulatory particle and as a protein specifically interacting with ATPase S6b/Tbp7/PSMC4 [306]. It was demonstrated that gankyrin can associate with protein Mdm2 and promote its interaction with p53. Gankyrin enhances the ability of Mdm2 to mono- and polyubiquitinate p53; as a consequence, the increase in the content of this protein is accompanied by enhanced Mdm2-dependent degradation of p53 [307]. Furthermore, it was proposed that gankyrin can deliver polyubiquitinated p53 in the complex with Mdm2 directly to the proteasome due to the binding with S6b [308].
In addition to Mdm2, some tumors are characterized also by a higher content of other E3 ligases specific to p53. So, a higher amount of ARF-BP1 E3 ligase is connected with the development of colorectal cancer [309], the expression of COP1 E3 ligase is enhanced in many ovary and breast carcinomas [284], and Pirh2 is enhanced in lung tumors [310].
Enhanced degradation of p53 plays a vital role in the development of cervix cancer caused by human high-oncogenic-risk papilloma viruses (types 16 and 18). It was found that the content of p53 in such tumors is significantly lower, but at the same time in contrast to many nonviral cervix carcinomas, they contain for the most part a non-mutation gene of p53 [311]. Detailed investigations demonstrated that protein E6 of the papilloma virus can form a complex with cell proteins p53 and E6-AP (E6-associated protein) [312-314]. E6-AP is an E3 ligase that as a rule does not interact with p53 [315]. E6 acts as a mediator that combines E6-AP and p53 into a single complex; within this complex E6-AP can ubiquitinate p53 [316]. One can say that in this case the E6-AP/E6 complex plays the role of the E3 ligase.
p27Kip1. Protein p27Kip1 is an inhibitor of the activity of Cdk2/CyclinA and Cdk2/CyclinE complexes and thus prevents the passage of cells to S phase of the cell cycle [317]. In response to the mitogenic stimuli, p27 is ubiquitinated and subjected to proteasomal degradation. E3 ligase specific to p27 is an SCFSkp2 complex consisting of proteins Skp1, Cul1, and Skp2, the latter being a substrate-recognizing component of the complex [318, 319]. The recognition and effective ubiquitination of p27 with the SCFSkp2 complex also require the small accessory protein Cks1 [320, 321].
Being a negative regulator of cell division, p27 has properties of a tumor suppressor. A decreased content of p27 is a specific feature of cancer tumors of different origin [322-324]. As antagonists of p27, Skp2, and Cks1 are oncoproteins, their enhanced synthesis correlates with a low level of p27 and poor prognosis for the patient [322, 324-329]. Skp2 recognizes a number of tumor suppressors in addition to p27, namely inhibitors of cyclin-dependent kinases p21Cip1, p57, and p130 [330-332] and transcription factor FOXO1 [333]. Therefore, an oncogenic function of SKP2 independent of p27 can be postulated.
pVHL. Another example of how a mutation of one component of the ubiquitinating complex can be connected with the development of malignant tumors is von Hippel–Lindau disease. This is a hereditary disease caused by autosomal dominant mutation in one of the alleles of gene VHL that encodes a subunit of the multimeric E3 ligase. The disease occurs as frequently as one case per 35,000 subjects [334]. Such patients have a higher predisposition to the development of some types of tumors such as pheochromocytomas, hemangioblastomas of the central nervous system, clear-cell renal carcinomas, and retinal capillary angiomas. The development of these tumors per se begins as a somatic inactivation of the wild-type allele or after its mutation. Characteristic features of tumors developing in von Hippel–Lindau disease are a high degree of their vascularization and hyperexpression of proteins that are usually synthesized only at hypoxia, such as VEGF (vascular endothelial growth factor) [335, 336].
Protein pVHL, a product of gene VHL, is a substrate-recognizing component of a multimeric E3 ligase [337-341]. Besides pVHL, this multisubunit complex contains also proteins cullin 2A, Rbx/Hrt1, elongin B, and elongin C [342-344]. The most studied and best-known substrate of this enzyme is HIF1α that is a subunit of heterodimeric transcription factor HIF-1.
The heterodimeric transcription factor HIF-1 plays a key role in the cell response to hypoxia. It consists of two subunits: one of the three versions of HIFα and HIFβ1 [345]. A sufficient amount of HIFβ1 is constantly present in the cells, whereas HIFα is rapidly degraded in the presence of oxygen. Under hypoxia, HIFα is stabilized and, together with HIFβ1, forms a functional transcription factor HIF-1, which by binding with the consensus sequence HRE (hypoxia-responsive element) activates transcription of some genes involved in angiogenesis, cell cycle control, glucose metabolism, and apoptosis [345]. the mechanism of such oxygen-dependent regulation of the HIF-1 level in the cell has been disclosed lately. It was shown that in the presence of oxygen, HIF1α is rapidly ubiquitinated and cleaved by the proteasome [346]. This degradation was dependent on the ODD (oxygen-dependent degradation) domain that is recognized by pVHL. To facilitate the recognition of HIF1α by protein pVHL, it is important that residues Pro402 and especially Pro564 in HIF1α were hydroxylated. Such hydroxylation is performed by special prolylhydroxylases in the presence of oxygen [347-351]. Under conditions of hypoxia this reaction is virtually absent; therefore, the level of HIF1α rises. The same occurs on mutations of the VHL gene. The high extent of tumor vascularization in von Hippel–Lindau disease may be explained by active HIF-1-dependent transcription. However, it is known that far from all cells with increased level of HIF-1 undergo cancer transformation. This gives grounds to believe that protein pVHL has additional functions not related to HIF-1 [334].
BRCA1/BARD1. Protein BRCA1 (breast cancer type 1 susceptibility protein) is a recognized suppressor of breast and ovary tumors. Gene BRCA1 is frequently inactive as a result of mutations in the case of both inheritable and sporadic cancers [352, 353]. In addition, the BRCA1 dysfunction correlates with the basal phenotype of tumor cells and poor prognosis for survival [354, 355].
BRCA1 contains a RING domain in the N-terminal part of the molecule and forms a complex with the structurally related protein BARD1 (BRCA1-associated RING-domain protein 1), which also has a RING-domain [356]. BARD1 mutations occur also, though less frequently than BRCA1 mutations, in breast and ovary cancers [357]. The BRCA1/BARD1 complex has E3 ligase activity and is capable of autoubiquitination that enhances its stability and activity [358-360]. There are some indirect data showing that the functions of BRCA1 and BARD1 as tumor suppressors is realized via the activity of their heterodimeric complex and that during this of importance is the integrity of domains involved in ubiquitination [358, 361, 362]. recently this has been directly demonstrated on murine models, but it has remained unclear whether the E3 ligase activity of the BRCA1/BARD1 complex is essential for suppression [363]. At present, several BRCA1/BARD1 substrates are known including histones, γ-tubulin, phosphorylated RNA-polymerase II, estrogen receptor, and others [360, 364-368]. Nonetheless, it is not clear yet the interaction with what substrates mediate the antitumor effect of BRCA1/BARD1.
Liddle Syndrome
This inheritable autosomal dominant disorder was first described by Liddle in the 1960s. Its symptoms are severe hypertension at an early age, hypokalemia, and metabolic alkalosis caused by excess reabsorption of sodium ions in kidneys with low secretion of aldosterone and rennin [369, 370]. It was found that this disease is caused by dysregulation of the epithelial Na+-channel (ENaC), which consists of three homologous subunits α-, β-, and γ-ENaC [371]. The activity of ENaC is mostly regulated upon its inclusion into the cytoplasmic membrane. It was demonstrated that nonsense and missense mutations in C-terminal parts of β- and γ-ENaC involving the conservative proline-rich motif cause an increase of the ENaC amount in the membrane and development of Liddle syndrome [372-378]. It became clear that PY-motifs in the C-terminal parts of ENaC subunits are binding sites of E3 ligases from the NEDD4 family that regulate negatively the activity of ENaC [379-383]. Though ENaC can be inhibited by several members of this family, the most significant regulator in vivo is NEDD4-2 [384]. It is assumed that ligase NEDD4-2 binds to ENaC on the cytoplasmic membrane and ubiquitinates it. This results in the release of ENaC subunits from the membrane and their subsequent degradation [385, 386]. The membrane always has two pools of ENaC: intact channels and channels α- and γ-subunits of which have undergone limited proteolysis in the region of the extracellular domain. Cleaved channels are more active, and the open-state probability in them is higher. It is noteworthy that NEDD4-2 acts in such a way that first the more active ENaC are removed from the membrane [387].
Neurodegenerative Disorders
Neurodegenerative disorders include diseases the main feature of which is the loss of functions and extinction of brain and spinal cord cells. A specific feature of these diseases is the formation of large intracellular aggregates consisting of irregularly folded protein molecules. In many cases, aggregates are found to contain Ub and proteasomes [388-391]. The role of the Ub–proteasome system in pathogenesis of neurodegenerative diseases has not been clarified yet. It is not clear whether inclusion bodies are toxic for the cell. Initially it was proposed that the reason for the formation of inclusion bodies in neurodegenerative disorders is impairment of the Ub–proteasome system functioning. Indeed, several teams of researchers demonstrated that proteasomal inhibitors cause the formation of inclusion bodies and apoptotic death of neurons in model systems [392-394]. Moreover, it appeared that the enzymatic activity of proteasomes is reduced in neurons afflicted with Parkinson’s disease [395]. On the other hand, it is known that aggregated proteins per se can impair proteasome functioning [396-398]. It was established just to a certain extent of accuracy that there is a correlation between the disease development and dysfunction of the Ub–proteasome system for only some types of inheritable Parkinson’s and Alzheimer’s diseases. Let us consider one such case.
At the present time we know six mutations of genes that can cause inheritable Parkinson’s disease. They are α-synuclein, parkin, UCH-L1, DJ-1, PINK 1, and LRRK2 [399-403]. The protein parkin is an E3 ligase containing two RING-domains and an Ubl domain, due to which it can bind to the Rpn10 subunit of the proteasome [404-407]. Mutations reducing the ligase activity of parkin lead to the disease development. Currently several parkin substrates are known, including O-glycosylated α-synuclein, CDCrel-1, and Pael receptor [404, 406, 408]. Though the available data suggest that parkin plays a quite important role in the pathogenesis of inheritable Parkinson’s disease, it is not clear whether the accumulation of any of its substrates is toxic for neurons.
Viral Diseases
Viruses have elaborated various strategic pathways to avoid recognition by the host immune system and multiply effectively. Some of these pathways are somehow connected with the Ub–proteasome system.
HCMV (human cytomegalovirus). HCMV and its related viruses prevent recognition of infected cells by cytotoxic lymphocytes. These viruses induce reverse transport of glycosylated heavy chains of MHC-I proteins (major histocompatibility complex class I) from EPR back to the cytosol, where they are deglycosylated, polyubiquitinated, and undergo proteasomal degradation. Responsible for these processes are transmembrane viral glycoproteins US-2 and US-11 differing from each other by the mechanism of transportation of heavy chains of MHC-I [409-413]. To provide efficient US-11- and US-2-dependent transportation of heavy chains of MHC-I, the presence of the functional system of ubiquitination is required; however, it is known that in the case of US-11-dependent transportation heavy chains are not acceptors of ubiquitin. The US-11-dependent degradation of heavy chains of MHC-I requires proteins which when normal are activated if EPR is overloaded with irregularly folded proteins. US-11 triggers a signal pathway that leads to the synthesis of these proteins [414, 415].
Furthermore, HCMV inhibits the process of antigen presentation in one more way. It is known that peptide ligands for MHC-I molecules are produced by immunoproteasomes. Specific subunits of immunoproteasomes are synthesized in response to cell stimulation by γ-interferon. Protein M27 of cytomegalovirus inhibits STAT2, which is a component of the signal pathway beginning from the interferon receptor and thus prevents the formation of immunoproteasomes [416].
Herpes viruses. Murine γ-herpes virus 68 and Kaposi’s sarcoma-associated with herpes virus (herpes virus 8) also impair the antigen presentation in infected cells, but at a later stage than cytomegalovirus. Protein MK3 of γ-herpes virus 68 and proteins K3 and K5 of herpes virus 8 are E3 ligases that ubiquitinate cytoplasmic regions of MHC-I molecules and other molecules vital for immune recognition, such as ICAM-1 and B7-2 [417-420]. Ubiquitination of these molecules results in their rapid release from the membrane by means of clathrin-dependent endocytosis and cleavage in lysosomes.
EBV (Epstein–Barr virus). The Epstein–Barr virus protein EBNA1 (Epstein–Barr virus (EBV)-encoded nuclear antigen 1) is the only one of its nine proteins that is always found in all EBV-associated tumors. Cytotoxic lymphocytes do not recognize cells in which EBNA1 is synthesized. This is connected with the presence of elongated Gly-Ala-repeats in the N-terminal part of EBNA1. These prevent the cleavage of the protein by the immunoproteasome [421, 422]. As a result, EBNA1 fragments are not present on the membrane of the infected cell in complex with MHC-I. It is assumed that Gly-Ala-repeats impede the polypeptide chain translocation into the catalytic cavity of the proteasome, therefore the proteolysis of proteins with such repeats is incomplete [423, 424].
Paramyxoviruses. The JAK-STAT-signaling pathway (Janus kinase–signal transducers and activators of transcription) is the central one in protecting the host organism from viral infections; that is why the action of some viruses is aimed at suppression of its activity. So, the mumps virus protein V (as well as proteins V of its related human parainfluenza virus type 2 and virus SV5) initiates the assembly of a complex E3 ligase consisting of protein V and a number of cell proteins. Such a complex enzyme ubiquitinates STAT1 and STAT3 in the case of the mumps virus, STAT2 in the case of the parainfluenza virus, and STAT1 in the case of SV5 [425-427]. After ubiquitination, these proteins are cleaved by the proteasome.
Hepatitis C virus. There are examples of how viral proteins can directly associate with the components of the proteasome and change its proteolytic activity. Thus, protein NS3 of hepatitis C virus interacts with the LMT7 subunit of the immunoproteasome and reduces its trypsin-like and caspase-like activities [428].
Inflammatory and Autoimmune Diseases
The Ub–proteasome system plays an essential role in the mechanisms of immune protection of the organism. First, it takes part in the antigen processing in antigen-presenting cells. Second, it regulates the transmission of signals from T-cell antigen receptors and the costimulatory CD28 molecule. And third, it is involved in activation of NF-κB, which is the key regulator of the activity of genes of many inflammatory cytokines, adhesion molecules, and immune system receptors. Let us consider the latter function of the Ub–proteasome system in more detail.
The Ub–proteasome system activates hetero- and homodimeric transcription factors of the NF-κB family in two stages. At first, the proteasome performs Ub-dependent processing of phosphorylated precursors p105 and p100 with the formation of active subunits of transcription factors p50 (NF-κB1) and p52 (NF-κB2). Active factors are retained by inhibitors of NF-κB (IκBs) in the cytoplasm. After the signal-induced phosphorylation and ubiquitination, IκBs are cleaved by the proteasome and the released factors are translocated to the nucleus, where they activate the transcription of corresponding genes [429]. It is believed that the pathologic activation of NF-κB is a cause of many inflammatory diseases.
The presence in blood of increased numbers of proteasomes accompanies some autoimmune and oncological diseases [268, 430, 431]. Furthermore, the presence of antibodies to proteasome components and regulatory particle PA28 is detected in blood of patients with systemic lupus erythematosus, myositis, disseminated sclerosis, and Sjogren syndrome [432-435]. The functional significance of these autoantibodies has not been established yet, though it was demonstrated that they can block activation of the proteasome by regulatory particle PA28. This gives grounds to suggest that antibodies against the proteasome are able to somehow regulate its activity [436].
As known, pathological activation of T helpers 2 (Th2) leads to the development of asthma and allergy symptoms. Activation of Th2 causes the JunB-dependent transcription of genes of some interleukins (IL-4, IL-5, IL-9, IL-10, IL-13) [437, 438]. The E3 ligase Itch plays a vital role in the development of immunologic tolerance of T cells. After phosphorylation by kinase JNK1, Itch is activated and ubiquitinates JunB, which is then cleaved by the proteasome [439]. It is known that mice lacking gene Itch suffer various types of allergy [440].
Rheumatoid arthritis is an autoimmune disease characterized by chronic inflammation of the synovial tissue and bone and cartilage damage. It was demonstrated that activation of NF-κB is implicated in pathogenesis of rheumatoid arthritis [441, 442].
Proteasomal inhibitors are considered as potential remedies for autoimmune and inflammatory diseases, acting chiefly by inhibiting NF-κB. The problem of using inhibitors of the Ub–proteasome system in medicine is discussed in more detail below.
Inhibitors of the Ubiquitin–Proteasome System as Potential Remedies
As mentioned above, the disturbance of functioning of any components of the Ub–proteasome system can cause different disorders. Therefore, the search for specific inhibitors of this system seems to be an attractive direction. However, it is necessary to take into account that the Ub–proteasome system is constantly involved in events vital for the cell and that, as a result of its inhibition, the normal sequence of a vast number of processes would be violated.
Less specific inhibitors of the Ub–proteasome system are inhibitors of the enzymatic activity of the 20S proteasome and E1. The target of most of the used inhibitors of the 20S proteasome is suppression of its chymotrypsin-like activity. This is connected, first, with the fact that the blocking of just this activity leads to the greatest reduction in the level of protein cleavage. And second, inhibitors of such activity are usually quite hydrophobic molecules and can therefore easily get into the cell, in contrast to inhibitors of other catalytic centers of the proteasome that contain charged regions [443]. In accord with the chemical structure, the major part of inhibitors is short peptides carrying chromophores that interact with catalytic residues in the active center. Peptide boronates are highly specific inhibitors of the 20S proteasome [443].
At a rather prolonged action, inhibitors of the 20S proteasome are toxic for cells and cause their death as a result of apoptosis, the proliferating cells being typically more sensitive to these substances [444-447]. Taking into consideration this circumstance and the antiangiogenic effect of inhibitors, it can be assumed that these substances should be effective in coping with oncologic diseases [445, 448]. Bortezomib, which is one of the proteasomal inhibitors (PS-341, pyrazinylcarbonyl-Phe-Leu-boronate, Velcade), is now efficiently used to this end.
Bortezomib is a strong inhibitor of chymotrypsin-like and partly of trypsin-like activity of the proteasome. When the antitumor activity of bortezomib in vitro and in vivo had been described, its clinical trials began [449, 450]. It was clarified that bortezomib is largely ineffective as a monotherapy in treating solid tumors, but it gives incredibly good results in multiple myeloma and other hematologic disorders [451-456].
At present, a search is in progress for novel proteasomal inhibitors that would have higher efficiency than bortezomib and its derivatives. For example, the highly specific inhibitor salinosporamide A (NPI-0052) was recently isolated from marine bacteria. In contrast to bortezomib, it can irreversibly bind to all catalytic centers of the proteasome [457]. It was demonstrated in in vitro studies that NPI-0052 is a better inhibitor of the proteasome and NF-κB than bortezomib when used at equal concentrations, and also a stronger stimulator of apoptosis of isolated lymphocytes from patients with chronic lymphocytic leukemia [458, 459]. Moreover, NPI-0052 induces apoptosis of multiple myeloma cells resistant to bortezomib and other drugs [460]. Clinical trials of this substance have begun.
A more intricate approach than the use of inhibitors of the 20S proteasome is the search and application of inhibitors of E3 ligases and specific interactions of the substrate and E3. At this point, several approaches can be separated. First, peptides corresponding to the substrate region to which E3 ligase is bound can be used as inhibitors of E3. So, it was shown that the phosphopeptide appropriate to the N-terminal region of IκB protects the entire protein from proteasomal degradation, and a microinjection of this phosphopeptide into cells prevents activation of NF-κB in them [461].
Second, it is possible to search for small molecules that will specifically inhibit active centers of E3 or sites of their binding to the substrate. The choice and efficient modification of such molecules are much simpler if the structures of substrate–E3 complexes are known. Let us analyze the results of using such an approach on the example of searching for inhibitors of the p53–Mdm2 interaction. As a consequence of screening of a large library of small molecules, it became possible to identify family HLI-98 whose members inhibited autoubiquitination of Mdm2 in vitro. Although these molecules inhibited other E3 ligases as well, and at high concentrations even E2, they were able to cause apoptosis of transformed cells almost without any toxic effect on normal cells [462]. This study demonstrated clearly that inhibition of E3, in particular Mdm2, is a quite promising advance in cancer treatment.
At the moment, several more specific low molecular weight inhibitors of the p53–Mdm2 interaction are known. First nutlins were found. They are derivatives of cis-imidazoline, which are able to displace p53 from the complex with Mdm2 [463]. Nutlins occupy the hydrophobic pocket of Mdm2, where side chains of three amino acids of p53 are usually located, and prevent the latter from binding to Mdm2. Treatment with nutlins leads to the accumulation of p53 and products of genes activated by it (e.g. p21 and p27) in cells. Nutlin-3 causes apoptosis of cancer cells from the wild-type p53, and in normal cells it induces growth deceleration maintaining their viability [463]. However, later it was found that in addition to p53–Mdm2, nutlins inhibit other protein–protein interactions as well. So, protein HIF1α binds to the same region of Mdm2 as p53, and thus nutlin-3 impairs this interaction too [464]. Nutlins can also have a therapeutic effect on cancer cells with mutant p53: in such cells they cause enhanced sensitivity to many remedies [465]. At present properties of some other inhibitors of the p53–Mdm2 interaction (RITA, MI-63) are investigated, and their effects prove to be similar to that of nutlin [466-468].
Let us give some more examples of action of low molecular weight inhibitors of E3-ligases. Peptide aptamers binding human papilloma virus protein E6 cause apoptotic elimination of HPV16-positive cancer cells without any effect on normal cells [469]. CpdA (Compound A), a quite recently identified inhibitor of complex ligase SCFSkp2, prevents incorporation of Skp2 into the composition of the enzyme. CpdA causes cell cycle arrest and SCFSkp2- and p27-dependent cell death affecting chiefly cancer cells [470].
On the whole, the therapeutic use of reagents modulating the activity of the Ub–proteasome system seems to be rather promising. Currently they are used first of all for cancer treatment, but with account of the fact that distortions in the well coordinated functioning of the Ub–proteasome system occur in other diseases as well, it can be suggested that in the near future the range of use of inhibitors of this system in medicine will be widened.
CONCLUSION
The 20S proteasome was initially detected in 1968 on electron micrographs of human erythrocyte lysate. Because of the cylindrical shape, this structure was named “cylindrin”, but at that time its functions were unclear [471]. In the end of the 1970s and the beginning of the 1980s, there appeared reports describing high molecular weight proteases of a cylindrical shape with molecular mass of 600-700 kDa. These proteases consisted of several subunits with molecular mass from 24 to 28 kDa and hydrolyzed substrates following hydrophobic and positively and negatively charged amino acids, i.e. were multispecific [472-475]. These two properties served to name the discovered enzyme “multicatalytic proteinase”. At approximately the same time, but in another pathway of investigations, subunits were found that were called “prosomes” or the 19S RNP (19S ribonucleoprotein) [476]. In their size and subunit composition they greatly resembled the multicatalytic proteinase, but it was assumed that they were associated with mRNA and were involved in the regulation of translation [477]. However, later everything fell into place; it was shown that the “prosome” and “multicatalytic proteinase” are identical subunits [478]. At the same time, a new name for these particles appeared (it reflected their proteolytic essence and compact structure)—the 20S proteasome. A quarter of a century after the discovery of the proteasome it became clear that the proteasome is the basis of a complex multicomponent cellular machine for utilizing exhausted proteins in the cell, which was called the “ubiquitin–proteasome system”. During that period remarkable progress in studying the structure and functioning of the proteasome, in particular of the Ub–proteasome system as a whole, was achieved. The importance of the disclosure of the Ub–proteasome system was corroborated by the fact that in 2004 its authors were awarded the Nobel Prize in Chemistry. However, even now there are many questions on the functioning of the system.
Herein we have systemized the currently available data on the Ub–proteasome system. We have described in detail the proteasome structure, ubiquitination system, classic ATP/Ub-dependent mechanism of protein degradation, and have focused attention on the existence of alternative mechanisms of proteasome-mediated degradation and protein processing. Separately are given data on disturbances of the proteasome system that cause the development of various disorders. We hope that this review will be of interest for a wide range of researchers.
This work was supported by the Russian Foundation for Basic Research (No. 07-04-00403) and by the Presidium of the Russian Academy of Sciences (Programs on Molecular and Cell Biology and basic Science to Medicine).
REFERENCES
1.Schoenheimer, R. (1942) The Dynamic State of
Body Constituents, Harvard University Press, Cambridge, MA.
2.Rock, K. L., Gramm, C., Rothstein, L., Clark, K.,
Stein, R., Dick, L., Hwang, D., and Goldberg, A. L. (1994) Cell,
78, 761-771.
3.Craiu, A., Gaczynska, M., Akopian, T., Gramm, C.
F., Fenteany, G., Goldberg, A. L., and Rock, K. L. (1997) J. Biol.
Chem., 272, 13437-13445.
4.Kisselev, A. F., Akopian, T. N., and Goldberg, A.
L. (1998) J. Biol. Chem., 273, 1982-1989.
5.Kisselev, A. F., Akopian, T. N., Woo, K. M., and
Goldberg, A. L. (1999) J. Biol. Chem., 274,
3363-3371.
6.Glickman, M. H., and Ciechanover, A. (2002)
Physiol. Rev., 82, 373-428.
7.Groll, M., and Huber, R. (2003) Int. J. Biochem.
Cell Biol., 35, 606-616.
8.Dahlmann, B. (2005) Essays Biochem.,
41, 31-48.
9.Frentzel, S., Pesold-Hurt, B., Seelig, A., and
Kloetzel, P. M. (1994) J. Mol. Biol., 236, 975-981.
10.Nandi, D., Woodward, E., Ginsburg, D. B., and
Monaco, J. J. (1997) EMBO J., 16, 5363-5375.
11.Griffin, T. A., Nandi, D., Cruz, M., Fehling, H.
J., Kaer, L. V., Monaco, J. J., and Colbert, R. A. (1998) J. Exp.
Med., 187, 97-104.
12.Kloetzel, P. M., Soza, A., and Stohwasser, R.
(1999) Biol. Chem., 380, 293-297.
13.Kloetzel, P. M. (2001) Nat. Rev. Mol. Cell.
Biol., 2, 179-187.
14.Yewdell, J. W., Schubert, U., and Bennink, J. R.
(2001) J. Cell Sci., 114, 845-851.
15.Coux, O., Tanaka, K., and Goldberg, A. L. (1996)
Annu. Rev. Biochem., 65, 801-847.
16.Baumeister, W., Walz, J., Zuhl, F., and
Seemuller, E. (1998) Cell, 92, 367-380.
17.Kisselev, A. F., Songyang, Z., and Goldberg, A.
L. (2000) J. Biol. Chem., 275, 14831-14837.
18.Heinemeyer, W., Fischer, M., Krimmer, T.,
Stachon, U., and Wolf, D. H. (1997) J. Biol. Chem., 272,
25200-25209.
19.Arendt, C. S., and Hochstrasser, M. (1997)
Proc. Natl. Acad. Sci. USA, 94, 7156-7161.
20.Groll, M., Bajorek, M., Kohler, A., Moroder, L.,
Rubin, D. M., Huber, R., Glickman, M. H., and Finley, D. (2000) Nat.
Struct. Biol., 7, 1062-1067.
21.Kohler, A., Bajorek, M., Groll, M., Moroder, L.,
Rubin, D. M., Huber, R., Glickman, M. H., and Finley, D. (2001)
Biochimie, 83, 325-332.
22.Orlowski, M., Cardozo, C., and Michaud, C. (1993)
Biochemistry, 32, 1563-1572.
23.Dick, T. P., Nussbaum, A. K., Deeg, M.,
Heinemeyer, W., Groll, M., Schirle, M., Keilholz, W., Stevanovic, S.,
Wolf, D. H., Huber, R., Rammensee, H. G., and Schild, H. (1998) J.
Biol. Chem., 273, 25637-25646.
24.Unno, M., Mizushima, T., Morimoto, Y., Tomisugi,
Y., Tanaka, K., Yasuoka, N., and Tsukihara, T. (2002) Structure,
10, 609-618.
25.Lowe, J., Stock, D., Jap, B., Zwickl, P.,
Baumeister, W., and Huber, R. (1995) Science, 268,
533-539.
26.Zwickl, P., Ng, D., Woo, K. M., Klenk, H. P., and
Goldberg, A. L. (1999) J. Biol. Chem., 274,
26008-26014.
27.Navon, A., and Goldberg, A. L. (2001) Mol.
Cell, 8, 1339-1349.
28.Forster, A., and Hill, C. P. (2003) Trends
Cell Biol., 13, 550-553.
29.Lee, C., Prakash, S., and Matouschek, A. (2002)
J. Biol. Chem., 277, 34760-34765.
30.Groll, M., Ditzel, L., Lowe, J., Stock, D.,
Bochtler, M., Bartunik, H. D., and Huber, R. (1997) Nature,
386, 463-471.
31.Benaroudj, N., Zwickl, P., Seemuller, E.,
Baumeister, W., and Goldberg, A. L. (2003) Mol. Cell, 11,
69-78.
32.Smith, D. M., Kafri, G., Cheng, Y., Ng, D., Walz,
T., and Goldberg, A. L. (2005) Mol. Cell, 20,
687-698.
33.Smith, D. M., Chang, S. C., Park, S., Finley, D.,
Cheng, Y., and Goldberg, A. L. (2007) Mol. Cell, 27,
731-744.
34.Glickman, M. H., Rubin, D. M., Coux, O., Wefes,
I., Pfeifer, G., Cjeka, Z., Baumeister, W., Fried, V. A., and Finley,
D. (1998) Cell, 94, 615-623.
35.DeMartino, G. N., and Slaughter, C. A. (1999)
J. Biol. Chem., 274, 22123-22126.
36.Rechsteiner, M., Realini, C., and Ustrell, V.
(2000) Biochem. J., 345, Pt. 1, 1-15.
37.Sorokin, A. V., Selyutina, A. A., Skabkin, M. A.,
Guryanov, S. G., Nazimov, I. V., Richard, C., Th’ng, J., Yau, J.,
Sorensen, P. H., Ovchinnikov, L. P., and Evdokimova, V. (2005) EMBO
J., 24, 3602-3612.
38.Liu, C. W., Corboy, M. J., DeMartino, G. N., and
Thomas, P. J. (2003) Science, 299, 408-411.
39.Tanaka, K., Yoshimura, T., and Ichihara, A.
(1989) J. Biochem., 106, 495-500.
40.Bajorek, M., Finley, D., and Glickman, M. H.
(2003) Curr. Biol., 13, 1140-1144.
41.Zwickl, P., Kleinz, J., and Baumeister, W. (1994)
Nat. Struct. Biol., 1, 765-770.
42.Seemuller, E., Lupas, A., and Baumeister, W.
(1996) Nature, 382, 468-471.
43.Grziwa, A., Maack, S., Puhler, G., Wiegand, G.,
Baumeister, W., and Jaenicke, R. (1994) Eur. J. Biochem.,
223, 1061-1067.
44.Seelig, A., Multhaup, G., Pesold-Hurt, B.,
Beyreuther, K., and Kloetzel, P. M. (1993) J. Biol. Chem.,
268, 25561-25567.
45.Gerards, W. L., Enzlin, J., Haner, M., Hendriks,
I. L., Aebi, U., Bloemendal, H., and Boelens, W. (1997) J. Biol.
Chem., 272, 10080-10086.
46.Gerards, W. L., de Jong, W. W., Bloemendal, H.,
and Boelens, W. (1998) J. Mol. Biol., 275, 113-121.
47.Rosenzweig, R., and Glickman, M. H. (2008)
Biochem. Soc. Trans., 36, 807-812.
48.Hirano, Y., Hendil, K. B., Yashiroda, H., Iemura,
S., Nagane, R., Hioki, Y., Natsume, T., Tanaka, K., and Murata, S.
(2005) Nature, 437, 1381-1385.
49.Le Tallec, B., Barrault, M. B., Courbeyrette, R.,
Guerois, R., Marsolier-Kergoat, M. C., and Peyroche, A. (2007) Mol.
Cell, 27, 660-674.
50.Li, X., Kusmierczyk, A. R., Wong, P., Emili, A.,
and Hochstrasser, M. (2007) EMBO J., 26, 2339-2349.
51.Yashiroda, H., Mizushima, T., Okamoto, K.,
Kameyama, T., Hayashi, H., Kishimoto, T., Niwa, S., Kasahara, M.,
Kurimoto, E., Sakata, E., Takagi, K., Suzuki, A., Hirano, Y., Murata,
S., Kato, K., Yamane, T., and Tanaka, K. (2008) Nat. Struct. Mol.
Biol., 15, 228-236.
52.Hirano, Y., Hayashi, H., Iemura, S., Hendil, K.
B., Niwa, S., Kishimoto, T., Kasahara, M., Natsume, T., Tanaka, K., and
Murata, S. (2006) Mol. Cell, 24, 977-984.
53.Kusmierczyk, A. R., Kunjappu, M. J., Funakoshi,
M., and Hochstrasser, M. (2008) Nat. Struct. Mol. Biol.,
15, 237-244.
54.Schmidtke, G., Schmidt, M., and Kloetzel, P. M.
(1997) J. Mol. Biol., 268, 95-106.
55.Ramos, P. C., Hockendorff, J., Johnson, E. S.,
Varshavsky, A., and Dohmen, R. J. (1998) Cell, 92,
489-499.
56.Marques, A. J., Glanemann, C., Ramos, P. C., and
Dohmen, R. J. (2007) J. Biol. Chem., 282,
34869-34876.
57.Ramos, P. C., Marques, A. J., London, M. K., and
Dohmen, R. J. (2004) J. Biol. Chem., 279,
14323-14330.
58.Schmidtke, G., Kraft, R., Kostka, S., Henklein,
P., Frommel, C., Lowe, J., Huber, R., Kloetzel, P. M., and Schmidt, M.
(1996) EMBO J., 15, 6887-6898.
59.Schmidt, M., Zantopf, D., Kraft, R., Kostka, S.,
Preissner, R., and Kloetzel, P. M. (1999) J. Mol. Biol.,
288, 117-128.
60.Chen, P., and Hochstrasser, M. (1996)
Cell, 86, 961-972.
61.Heinemeyer, W., Ramos, P. C., and Dohmen, R. J.
(2004) Cell. Mol. Life Sci., 61, 1562-1578.
62.Fehlker, M., Wendler, P., Lehmann, A., and
Enenkel, C. (2003) EMBO Rep., 4, 959-963.
63.Russell, S. J., Steger, K. A., and Johnston, S.
A. (1999) J. Biol. Chem., 274, 21943-21952.
64.Brooks, P., Fuertes, G., Murray, R. Z., Bose, S.,
Knecht, E., Rechsteiner, M. C., Hendil, K. B., Tanaka, K., Dyson, J.,
and Rivett, J. (2000) Biochem. J., 346, Pt. 1,
155-161.
65.Beyer, A. (1997) Protein Sci., 6,
2043-2058.
66.Ogura, T., and Wilkinson, A. J. (2001) Genes
Cells, 6, 575-597.
67.Verma, R., Chen, S., Feldman, R., Schieltz, D.,
Yates, J., Dohmen, J., and Deshaies, R. J. (2000) Mol. Biol.
Cell, 11, 3425-3439.
68.Deveraux, Q., Jensen, C., and Rechsteiner, M.
(1995) J. Biol. Chem., 270, 23726-23729.
69.Qiu, X. B., Ouyang, S. Y., Li, C. J., Miao, S.,
Wang, L., and Goldberg, A. L. (2006) EMBO J., 25,
5742-5753.
70.Hori, T., Kato, S., Saeki, M., DeMartino, G. N.,
Slaughter, C. A., Takeuchi, J., Toh-e, A., and Tanaka, K. (1998)
Gene, 216, 113-122.
71.Watanabe, T. K., Saito, A., Suzuki, M., Fujiwara,
T., Takahashi, E., Slaughter, C. A., DeMartino, G. N., Hendil, K. B.,
Chung, C. H., Tanahashi, N., and Tanaka, K. (1998) Genomics,
50, 241-250.
72.Sone, T., Saeki, Y., Toh-e, A., and Yokosawa, H.
(2004) J. Biol. Chem., 279, 28807-28816.
73.Fujimuro, M., Tanaka, K., Yokosawa, H., and
Toh-e, A. (1998) FEBS Lett., 423, 149-154.
74.Fu, H., Reis, N., Lee, Y., Glickman, M. H., and
Vierstra, R. D. (2001) EMBO J., 20, 7096-7107.
75.Walz, J., Erdmann, A., Kania, M., Typke, D.,
Koster, A. J., and Baumeister, W. (1998) J. Struct. Biol.,
121, 19-29.
76.Da Fonseca, P. C., and Morris, E. P. (2008) J.
Biol. Chem., 283, 23305-23314.
77.Davy, A., Bello, P., Thierry-Mieg, N., Vaglio,
P., Hitti, J., Doucette-Stamm, L., Thierry-Mieg, D., Reboul, J.,
Boulton, S., Walhout, A. J., Coux, O., and Vidal, M. (2001) EMBO
Rep., 2, 821-828.
78.Uetz, P., Giot, L., Cagney, G., Mansfield, T. A.,
Judson, R. S., Knight, J. R., Lockshon, D., Narayan, V., Srinivasan,
M., Pochart, P., Qureshi-Emili, A., Li, Y., Godwin, B., Conover, D.,
Kalbfleisch, T., Vijayadamodar, G., Yang, M., Johnston, M., Fields, S.,
and Rothberg, J. M. (2000) Nature, 403, 623-627.
79.Cagney, G., Uetz, P., and Fields, S. (2001)
Physiol. Genom., 7, 27-34.
80.Ho, Y., Gruhler, A., Heilbut, A., Bader, G. D.,
Moore, L., Adams, S. L., Millar, A., Taylor, P., Bennett, K.,
Boutilier, K., Yang, L., Wolting, C., Donaldson, I., Schandorff, S.,
Shewnarane, J., Vo, M., Taggart, J., Goudreault, M., Muskat, B.,
Alfarano, C., Dewar, D., Lin, Z., Michalickova, K., Willems, A. R.,
Sassi, H., Nielsen, P. A., Rasmussen, K. J., Andersen, J. R., Johansen,
L. E., Hansen, L. H., Jespersen, H., Podtelejnikov, A., Nielsen, E.,
Crawford, J., Poulsen, V., Sorensen, B. D., Matthiesen, J.,
Hendrickson, R. C., Gleeson, F., Pawson, T., Moran, M. F., Durocher,
D., Mann, M., Hogue, C. W., Figeys, D., and Tyers, M. (2002)
Nature, 415, 180-183.
81.Santamaria, P. G., Finley, D., Ballesta, J. P.,
and Remacha, M. (2003) J. Biol. Chem., 278,
6687-6695.
82.Isono, E., Saito, N., Kamata, N., Saeki, Y., and
Toh-e, A. (2005) J. Biol. Chem., 280, 6537-6547.
83.Sharon, M., Taverner, T., Ambroggio, X. I.,
Deshaies, R. J., and Robinson, C. V. (2006) PLoS Biol.,
4, e267.
84.Chen, C., Huang, C., Chen, S., Liang, J., Lin,
W., Ke, G., Zhang, H., Wang, B., Huang, J., Han, Z., Ma, L., Huo, K.,
Yang, X., Yang, P., He, F., and Tao, T. (2008) Proteomics,
8, 508-520.
85.Kajava, A. V. (2002) J. Biol. Chem.,
277, 49791-49798.
86.Kajava, A. V., Gorbea, C., Ortega, J.,
Rechsteiner, M., and Steven, A. C. (2004) J. Struct. Biol.,
146, 425-430.
87.Rosenzweig, R., and Glickman, M. H. (2008)
Nat. Struct. Mol. Biol., 15, 218-220.
88.Rubin, D. M., Glickman, M. H., Larsen, C. N.,
Dhruvakumar, S., and Finley, D. (1998) EMBO J., 17,
4909-4919.
89.Kohler, A., Cascio, P., Leggett, D. S., Woo, K.
M., Goldberg, A. L., and Finley, D. (2001) Mol. Cell, 7,
1143-1152.
90.Gillette, T. G., Kumar, B., Thompson, D.,
Slaughter, C. A., and DeMartino, G. N. (2008) J. Biol. Chem.,
283, 31813-31822.
91.Ma, C. P., Slaughter, C. A., and DeMartino, G. N.
(1992) J. Biol. Chem., 267, 10515-10523.
92.Rosenzweig, R., Osmulski, P. A., Gaczynska, M.,
and Glickman, M. H. (2008) Nat. Struct. Mol. Biol., 15,
573-580.
93.Baboshina, O. V., and Haas, A. L. (1996) J.
Biol. Chem., 271, 2823-2831.
94.Fu, H., Sadis, S., Rubin, D. M., Glickman, M.,
van Nocker, S., Finley, D., and Vierstra, R. D. (1998) J. Biol.
Chem., 273, 1970-1981.
95.Thrower, J. S., Hoffman, L., Rechsteiner, M., and
Pickart, C. M. (2000) EMBO J., 19, 94-102.
96.Van Nocker, S., Sadis, S., Rubin, D. M.,
Glickman, M., Fu, H., Coux, O., Wefes, I., Finley, D., and Vierstra, R.
D. (1996) Mol. Cell. Biol., 16, 6020-6028.
97.Elsasser, S., Chandler-Militello, D., Muller, B.,
Hanna, J., and Finley, D. (2004) J. Biol. Chem., 279,
26817-26822.
98.Elsasser, S., and Finley, D. (2005) Nat. Cell
Biol., 7, 742-749.
99.Verma, R., Oania, R., Graumann, J., and Deshaies,
R. J. (2004) Cell, 118, 99-110.
100.Husnjak, K., Elsasser, S., Zhang, N., Chen, X.,
Randles, L., Shi, Y., Hofmann, K., Walters, K. J., Finley, D., and
Dikic, I. (2008) Nature, 453, 481-488.
101.Lupas, A., Baumeister, W., and Hofmann, K.
(1997) Trends Biochem. Sci., 22, 195-196.
102.Braun, B. C., Glickman, M., Kraft, R.,
Dahlmann, B., Kloetzel, P. M., Finley, D., and Schmidt, M. (1999)
Nat. Cell Biol., 1, 221-226.
103.Strickland, E., Hakala, K., Thomas, P. J., and
DeMartino, G. N. (2000) J. Biol. Chem., 275,
5565-5572.
104.Hershko, A., Leshinsky, E., Ganoth, D., and
Heller, H. (1984) Proc. Natl. Acad. Sci. USA, 81,
1619-1623.
105.Yao, T., and Cohen, R. E. (2002) Nature,
419, 403-407.
106.Verma, R., Aravind, L., Oania, R., McDonald, W.
H., Yates, J. R., 3rd, Koonin, E. V., and Deshaies, R. J. (2002)
Science, 298, 611-615.
107.Amerik, A. Y., and Hochstrasser, M. (2004)
Biochim. Biophys. Acta, 1695, 189-207.
108.Guterman, A., and Glickman, M. H. (2004) J.
Biol. Chem., 279, 1729-1738.
109.Papa, F. R., Amerik, A. Y., and Hochstrasser,
M. (1999) Mol. Biol. Cell, 10, 741-756.
110.Eytan, E., Ganoth, D., Armon, T., and Hershko,
A. (1989) Proc. Natl. Acad. Sci. USA, 86, 7751-7755.
111.Liu, C. W., Li, X., Thompson, D., Wooding, K.,
Chang, T. L., Tang, Z., Yu, H., Thomas, P. J., and DeMartino, G. N.
(2006) Mol. Cell, 24, 39-50.
112.Isono, E., Nishihara, K., Saeki, Y., Yashiroda,
H., Kamata, N., Ge, L., Ueda, T., Kikuchi, Y., Tanaka, K., Nakano, A.,
and Toh-e, A. (2007) Mol. Biol. Cell, 18, 569-580.
113.Isono, E., Saeki, Y., Yokosawa, H., and Toh-e,
A. (2004) J. Biol. Chem., 279, 27168-27176.
114.Kurucz, E., Ando, I., Sumegi, M., Holzl, H.,
Kapelari, B., Baumeister, W., and Udvardy, A. (2002) Biochem.
J., 365, 527-536.
115.Babbitt, S. E., Kiss, A., Deffenbaugh, A. E.,
Chang, Y. H., Bailly, E., Erdjument-Bromage, H., Tempst, P., Buranda,
T., Sklar, L. A., Baumler, J., Gogol, E., and Skowyra, D. (2005)
Cell, 121, 553-565.
116.Kiss, P., Szabo, A., Hunyadi-Gulyas, E.,
Medzihradszky, K. F., Lipinszki, Z., Pal, M., and Udvardy, A. (2005)
Biochem. J., 391, 301-310.
117.Leggett, D. S., Hanna, J., Borodovsky, A.,
Crosas, B., Schmidt, M., Baker, R. T., Walz, T., Ploegh, H., and
Finley, D. (2002) Mol. Cell, 10, 495-507.
118.Schmidt, M., Hanna, J., Elsasser, S., and
Finley, D. (2005) Biol. Chem., 386, 725-737.
119.Wang, X., Chen, C. F., Baker, P. R., Chen, P.
L., Kaiser, P., and Huang, L. (2007) Biochemistry, 46,
3553-3565.
120.Imai, J., Maruya, M., Yashiroda, H., Yahara,
I., and Tanaka, K. (2003) EMBO J., 22, 3557-3567.
121.Tone, Y., Tanahashi, N., Tanaka, K., Fujimuro,
M., Yokosawa, H., and Toh-e, A. (2000) Gene, 243,
37-45.
122.Rechsteiner, M., and Hill, C. P. (2005)
Trends Cell Biol., 15, 27-33.
123.Dubiel, W., Pratt, G., Ferrell, K., and
Rechsteiner, M. (1992) J. Biol. Chem., 267,
22369-22377.
124.Stohwasser, R., Salzmann, U., Giesebrecht, J.,
Kloetzel, P. M., and Holzhutter, H. G. (2000) Eur. J. Biochem.,
267, 6221-6230.
125.Groettrup, M., Soza, A., Eggers, M., Kuehn, L.,
Dick, T. P., Schild, H., Rammensee, H. G., Koszinowski, U. H., and
Kloetzel, P. M. (1996) Nature, 381, 166-168.
126.Whitby, F. G., Masters, E. I., Kramer, L.,
Knowlton, J. R., Yao, Y., Wang, C. C., and Hill, C. P. (2000)
Nature, 408, 115-120.
127.Ustrell, V., Hoffman, L., Pratt, G., and
Rechsteiner, M. (2002) EMBO J., 21, 3516-3525.
128.Schmidt, M., Haas, W., Crosas, B., Santamaria,
P. G., Gygi, S. P., Walz, T., and Finley, D. (2005) Nat. Struct.
Mol. Biol., 12, 294-303.
129.Iwanczyk, J., Sadre-Bazzaz, K., Ferrell, K.,
Kondrashkina, E., Formosa, T., Hill, C. P., and Ortega, J. (2006) J.
Mol. Biol., 363, 648-659.
130.Gorbea, C., Goellner, G. M., Teter, K., Holmes,
R. K., and Rechsteiner, M. (2004) J. Biol. Chem., 279,
54849-54861.
131.Tanahashi, N., Kawahara, H., Murakami, Y., and
Tanaka, K. (1999) Mol. Biol. Rep., 26, 3-9.
132.Zaiss, D. M., Standera, S., Kloetzel, P. M.,
and Sijts, A. J. (2002) Proc. Natl. Acad. Sci. USA, 99,
14344-14349.
133.Tanaka, E., Takagi Sawada, M., Morinaga, C.,
Yokosawa, H., and Sawada, H. (2000) Arch. Biochem. Biophys.,
374, 181-188.
134.Sakai, N., Sawada, M. T., and Sawada, H. (2004)
Int. J. Biochem. Cell Biol., 36, 776-784.
135.Wei, N., and Deng, X. W. (1992) Plant
Cell, 4, 1507-1518.
136.Schwechheimer, C. (2004) Biochim. Biophys.
Acta, 1695, 45-54.
137.Deshaies, R. J. (1999) Annu. Rev. Cell Dev.
Biol., 15, 435-467.
138.Lyapina, S., Cope, G., Shevchenko, A., Serino,
G., Tsuge, T., Zhou, C., Wolf, D. A., Wei, N., and Deshaies, R. J.
(2001) Science, 292, 1382-1385.
139.Zhou, C., Wee, S., Rhee, E., Naumann, M.,
Dubiel, W., and Wolf, D. A. (2003) Mol. Cell, 11,
927-938.
140.Groisman, R., Polanowska, J., Kuraoka, I.,
Sawada, J., Saijo, M., Drapkin, R., Kisselev, A. F., Tanaka, K., and
Nakatani, Y. (2003) Cell, 113, 357-367.
141.Liu, C., Powell, K. A., Mundt, K., Wu, L.,
Carr, A. M., and Caspari, T. (2003) Genes Dev.,
17, 1130-1140.
142.Seeger, M., Ferrell, K., Frank, R., and Dubiel,
W. (1997) J. Biol. Chem., 272, 8145-8148.
143.Hu, Z., Zhang, Z., Doo, E., Coux, O., Goldberg,
A. L., and Liang, T. J. (1999) J. Virol., 73,
7231-7240.
144.Guo, G. G., Gu, M., and Etlinger, J. D. (1994)
J. Biol. Chem., 269, 12399-12402.
145.Grünberg-Etkovitz, N., Greenbaum, L.,
Grinblat, B., and Malik, Z. (2006) Biochim. Biophys. Acta,
1762, 819-827.
146.Boyer, S. N., Wazer, D. E., and Band, V. (1996)
Cancer Res., 56, 4620-4624.
147.Gaczynska, M., Osmulski, P. A., Gao, Y., Post,
M. J., and Simons, M. (2003) Biochemistry, 42,
8663-8670.
148.Koegl, M., Hoppe, T., Schlenker, S., Ulrich, H.
D., Mayer, T. U., and Jentsch, S. (1999) Cell, 96,
635-644.
149.Breitschopf, K., Bengal, E., Ziv, T., Admon,
A., and Ciechanover, A. (1998) EMBO J., 17,
5964-5973.
150.Ben-Saadon, R., Fajerman, I., Ziv, T., Hellman,
U., Schwartz, A. L., and Ciechanover, A. (2004) J. Biol. Chem.,
279, 41414-41421.
151.Cadwell, K., and Coscoy, L. (2005)
Science, 309, 127-130.
152.D’Andrea, A., and Pellman, D. (1998)
Crit. Rev. Biochem. Mol. Biol., 33, 337-352.
153.Chung, C. H., and Baek, S. H. (1999)
Biochem. Biophys. Res. Commun., 266, 633-640.
154.Ha, B. H., and Kim, E. E. (2008) BMB
Rep., 41, 435-443.
155.Catic, A., Sun, Z. Y., Ratner, D. M., Misaghi,
S., Spooner, E., Samuelson, J., Wagner, G., and Ploegh, H. L. (2007)
EMBO J., 26, 3474-3483.
156.Hicke, L., and Dunn, R. (2003) Annu. Rev.
Cell Dev. Biol., 19, 141-172.
157.Muratani, M., and Tansey, W. P. (2003) Nat.
Rev. Mol. Cell. Biol., 4, 192-201.
158.Hoege, C., Pfander, B., Moldovan, G. L.,
Pyrowolakis, G., and Jentsch, S. (2002) Nature, 419,
135-141.
159.Stelter, P., and Ulrich, H. D. (2003)
Nature, 425, 188-191.
160.Gregory, R. C., Taniguchi, T., and
D’Andrea, A. D. (2003) Semin. Cancer Biol., 13,
77-82.
161.Haglund, K., Di Fiore, P. P., and Dikic, I.
(2003) Trends Biochem. Sci., 28, 598-603.
162.Pickart, C. M., and Fushman, D. (2004) Curr.
Opin. Chem. Biol., 8, 610-616.
163.Varadan, R., Walker, O., Pickart, C., and
Fushman, D. (2002) J. Mol. Biol., 324, 637-647.
164.Varadan, R., Assfalg, M., Haririnia, A., Raasi,
S., Pickart, C., and Fushman, D. (2004) J. Biol. Chem.,
279, 7055-7063.
165.Hershko, A., and Ciechanover, A. (1998)
Annu. Rev. Biochem., 67, 425-479.
166.Wu, C. J., Conze, D. B., Li, T., Srinivasula,
S. M., and Ashwell, J. D. (2006) Nat. Cell Biol., 8,
398-406.
167.Hofmann, R. M., and Pickart, C. M. (2001) J.
Biol. Chem., 276, 27936-27943.
168.Peng, J., Schwartz, D., Elias, J. E., Thoreen,
C. C., Cheng, D., Marsischky, G., Roelofs, J., Finley, D., and Gygi, S.
P. (2003) Nat. Biotechnol., 21, 921-926.
169.Kim, I., and Rao, H. (2006) Sci. STKE,
2006, pe18.
170.Jentsch, S., and Pyrowolakis, G. (2000)
Trends Cell Biol., 10, 335-342.
171.Girdwood, D., Bumpass, D., Vaughan, O. A.,
Thain, A., Anderson, L. A., Snowden, A. W., Garcia-Wilson, E., Perkins,
N. D., and Hay, R. T. (2003) Mol. Cell, 11,
1043-1054.
172.Seeler, J. S., and Dejean, A. (2003) Nat.
Rev. Mol. Cell. Biol., 4, 690-699.
173.Muller, S., Ledl, A., and Schmidt, D. (2004)
Oncogene, 23, 1998-2008.
174.Meluh, P. B., and Koshland, D. (1995) Mol.
Biol. Cell, 6, 793-807.
175.Bohren, K. M., Nadkarni, V., Song, J. H.,
Gabbay, K. H., and Owerbach, D. (2004) J. Biol. Chem.,
279, 27233-27238.
176.Hochstrasser, M. (1998) Genes Dev.,
12, 901-907.
177.Xirodimas, D. P., Saville, M. K., Bourdon, J.
C., Hay, R. T., and Lane, D. P. (2004) Cell, 118,
83-97.
178.Korant, B. D., Blomstrom, D. C., Jonak, G. J.,
and Knight, E., Jr. (1984) J. Biol. Chem., 259,
14835-14839.
179.Malakhova, O. A., Yan, M., Malakhov, M. P.,
Yuan, Y., Ritchie, K. J., Kim, K. I., Peterson, L. F., Shuai, K., and
Zhang, D. E. (2003) Genes Dev., 17, 455-460.
180.Zhao, C., Denison, C., Huibregtse, J. M., Gygi,
S., and Krug, R. M. (2005) Proc. Natl. Acad. Sci. USA,
102, 10200-10205.
181.Tanida, I., Ueno, T., and Kominami, E. (2004)
Int. J. Biochem. Cell Biol., 36, 2503-2518.
182.Ohsumi, Y. (2001) Nat. Rev. Mol. Cell.
Biol., 2, 211-216.
183.Raasi, S., Schmidtke, G., and Groettrup, M.
(2001) J. Biol. Chem., 276, 35334-35343.
184.Komatsu, M., Chiba, T., Tatsumi, K., Iemura,
S., Tanida, I., Okazaki, N., Ueno, T., Kominami, E., Natsume, T., and
Tanaka, K. (2004) EMBO J., 23, 1977-1986.
185.Sasakawa, H., Sakata, E., Yamaguchi, Y.,
Komatsu, M., Tatsumi, K., Kominami, E., Tanaka, K., and Kato, K. (2006)
Biochem. Biophys. Res. Commun., 343, 21-26.
186.Orlowski, M., and Wilk, S. (2003) Arch.
Biochem. Biophys., 415, 1-5.
187.Li, X., Amazit, L., Long, W., Lonard, D. M.,
Monaco, J. J., and O’Malley, B. W. (2007) Mol. Cell,
26, 831-842.
188.Tofaris, G. K., Layfield, R., and Spillantini,
M. G. (2001) FEBS Lett., 509, 22-26.
189.Lin, L., DeMartino, G. N., and Greene, W. C.
(1998) Cell, 92, 819-828.
190.Bercovich, Z., Rosenberg-Hasson, Y.,
Ciechanover, A., and Kahana, C. (1989) J. Biol. Chem.,
264, 15949-15952.
191.Jin, Y., Lee, H., Zeng, S. X., Dai, M. S., and
Lu, H. (2003) EMBO J., 22, 6365-6377.
192.David, D. C., Layfield, R., Serpell, L.,
Narain, Y., Goedert, M., and Spillantini, M. G. (2002) J.
Neurochem., 83, 176-185.
193.Amici, M., Sagratini, D., Pettinari, A.,
Pucciarelli, S., Angeletti, M., and Eleuteri, A. M. (2004) Arch.
Biochem. Biophys., 422, 168-174.
194.Ju, D., and Xie, Y. (2004) J. Biol.
Chem., 279, 23851-23854.
195.Asher, G., Tsvetkov, P., Kahana, C., and Shaul,
Y. (2005) Genes Dev., 19, 316-321.
196.Kong, X., Lin, Z., Liang, D., Fath, D., Sang,
N., and Caro, J. (2006) Mol. Cell. Biol., 26,
2019-2028.
197.Ying, H., and Xiao, Z. X. (2006) Cell
Cycle, 5, 506-508.
198.Moorthy, A. K., Savinova, O. V., Ho, J. Q.,
Wang, V. Y., Vu, D., and Ghosh, G. (2006) EMBO J., 25,
1945-1956.
199.Pande, A. H., Moe, D., Jamnadas, M., Tatulian,
S. A., and Teter, K. (2006) Biochemistry, 45,
13734-13740.
200.Ito, T., Fujio, Y., Takahashi, K., and Azuma,
J. (2007) J. Biol. Chem., 282, 1152-1160.
201.Lim, S. K., and Gopalan, G. (2007) Biochem.
J., 403, 119-127.
202.Voigt, A., Salzmann, U., Seifert, U., Dathe,
M., Soza, A., Kloetzel, P. M., and Kuckelkorn, U. (2007) Biochem.
Biophys. Res. Commun., 355, 549-554.
203.Chen, C., Zhou, Z., Guo, P., and Dong, J. T.
(2007) FEBS Lett., 581, 1124-1130.
204.Yuksek, K., Chen, W. L., Chien, D., and Ou, J.
H. (2009) J. Virol., 83, 612-621.
205.Jariel-Encontre, I., Pariat, M., Martin, F.,
Carillo, S., Salvat, C., and Piechaczyk, M. (1995) J. Biol.
Chem., 270, 11623-11627.
206.Tarcsa, E., Szymanska, G., Lecker, S.,
O’Connor, C. M., and Goldberg, A. L. (2000) J. Biol.
Chem., 275, 20295-20301.
207.Benaroudj, N., Tarcsa, E., Cascio, P., and
Goldberg, A. L. (2001) Biochimie, 83, 311-318.
208.Grune, T., Reinheckel, T., and Davies, K. J.
(1997) FASEB J., 11, 526-534.
209.Fink, A. L. (2005) Curr. Opin. Struct.
Biol., 15, 35-41.
210.Wright, P. E., and Dyson, H. J. (1999) J.
Mol. Biol., 293, 321-331.
211.Davies, K. J. (2001) Biochimie,
83, 301-310.
212.Murakami, Y., Matsufuji, S., Kameji, T.,
Hayashi, S., Igarashi, K., Tamura, T., Tanaka, K., and Ichihara, A.
(1992) Nature, 360, 597-599.
213.Jariel-Encontre, I., Bossis, G., and
Piechaczyk, M. (2008) Biochim. Biophys. Acta, 1786,
153-177.
214.Zhang, M., Pickart, C. M., and Coffino, P.
(2003) EMBO J., 22, 1488-1496.
215.Blagosklonny, M. V., Wu, G. S., Omura, S., and
el-Deiry, W. S. (1996) Biochem. Biophys. Res. Commun.,
227, 564-569.
216.Kim, G. Y., Mercer, S. E., Ewton, D. Z., Yan,
Z., Jin, K., and Friedman, E. (2002) J. Biol. Chem., 277,
29792-29802.
217.Jascur, T., Brickner, H., Salles-Passador, I.,
Barbier, V., El Khissiin, A., Smith, B., Fotedar, R., and Fotedar, A.
(2005) Mol. Cell, 17, 237-249.
218.Chen, X., Barton, L. F., Chi, Y., Clurman, B.
E., and Roberts, J. M. (2007) Mol. Cell, 26, 843-852.
219.Touitou, R., Richardson, J., Bose, S.,
Nakanishi, M., Rivett, J., and Allday, M. J. (2001) EMBO J.,
20, 2367-2375.
220.Kriwacki, R. W., Hengst, L., Tennant, L., Reed,
S. I., and Wright, P. E. (1996) Proc. Natl. Acad. Sci. USA,
93, 11504-11509.
221.Vogelstein, B., Lane, D., and Levine, A. J.
(2000) Nature, 408, 307-310.
222.Zur Hausen, H. (2000) J. Natl. Cancer
Inst., 92, 690-698.
223.Camus, S., Menendez, S., Cheok, C. F.,
Stevenson, L. F., Lain, S., and Lane, D. P. (2007) Oncogene,
26, 4059-4070.
224.Anwar, A., Dehn, D., Siegel, D., Kepa, J. K.,
Tang, L. J., Pietenpol, J. A., and Ross, D. (2003) J. Biol.
Chem., 278, 10368-10373.
225.Asher, G., Reuven, N., and Shaul, Y. (2006)
Bioessays, 28, 844-849.
226.Classon, M., and Harlow, E. (2002) Nat. Rev.
Cancer, 2, 910-917.
227.Scheffner, M., and Whitaker, N. J. (2003)
Semin. Cancer Biol., 13, 59-67.
228.Knight, J. S., Sharma, N., and Robertson, E. S.
(2005) Proc. Natl. Acad. Sci. USA, 102, 18562-18566.
229.Munakata, T., Nakamura, M., Liang, Y., Li, K.,
and Lemon, S. M. (2005) Proc. Natl. Acad. Sci. USA, 102,
18159-18164.
230.Kalejta, R. F., Bechtel, J. T., and Shenk, T.
(2003) Mol. Cell. Biol., 23, 1885-1895.
231.Sdek, P., Ying, H., Chang, D. L., Qiu, W.,
Zheng, H., Touitou, R., Allday, M. J., and Xiao, Z. X. (2005) Mol.
Cell, 20, 699-708.
232.Uchida, C., Miwa, S., Kitagawa, K., Hattori,
T., Isobe, T., Otani, S., Oda, T., Sugimura, H., Kamijo, T., Ookawa,
K., Yasuda, H., and Kitagawa, M. (2005) EMBO J., 24,
160-169.
233.Neumann, M., and Naumann, M. (2007) FASEB
J., 21, 2642-2654.
234.Coux, O., and Goldberg, A. L. (1998) J.
Biol. Chem., 273, 8820-8828.
235.Beinke, S., Deka, J., Lang, V., Belich, M. P.,
Walker, P. A., Howell, S., Smerdon, S. J., Gamblin, S. J., and Ley, S.
C. (2003) Mol. Cell. Biol., 23, 4739-4752.
236.Li, Z., Zhang, J., Chen, D., and Shu, H. B.
(2003) Biochem. Biophys. Res. Commun., 309, 980-985.
237.Zhang, J., Xu, L. G., Han, K. J., and Shu, H.
B. (2004) J. Biol. Chem., 279, 17819-17825.
238.Ferrier, R., Nougarede, R., Doucet, S.,
Kahn-Perles, B., Imbert, J., and Mathieu-Mahul, D. (1999)
Oncogene, 18, 995-1005.
239.Lang, V., Janzen, J., Fischer, G. Z., Soneji,
Y., Beinke, S., Salmeron, A., Allen, H., Hay, R. T., Ben-Neriah, Y.,
and Ley, S. C. (2003) Mol. Cell. Biol., 23, 402-413.
240.Skabkin, M. A., Liabin, D. N., and Ovchinnikov,
L. P. (2006) Mol. Biol. (Moscow), 40, 620-633.
241.Stenina, O. I., Poptic, E. J., and DiCorleto,
P. E. (2000) J. Clin. Invest., 106, 579-587.
242.Jurchott, K., Bergmann, S., Stein, U., Walther,
W., Janz, M., Manni, I., Piaggio, G., Fietze, E., Dietel, M., and
Royer, H. D. (2003) J. Biol. Chem., 278, 27988-27996.
243.Lutz, M., Wempe, F., Bahr, I., Zopf, D., and
von Melchner, H. (2006) FEBS Lett., 580, 3921-3930.
244.Chibi, M., Meyer, M., Skepu, A. G., Rees, D.
J., Moolman-Smook, J. C., and Pugh, D. J. (2008) J. Mol. Biol.,
384, 908-916.
245.Baugh, J. M., and Pilipenko, E. V. (2004)
Mol. Cell, 16, 575-586.
246.Jackson, R. J. (2005) Biochem. Soc.
Trans., 33, 1231-1241.
247.Qin, X., and Sarnow, P. (2004) J. Biol.
Chem., 279, 13721-13728.
248.Konstantinova, I. M., Tsimokha, A. S., and
Mittenberg, A. G. (2008) Int. Rev. Cell Mol. Biol., 267,
59-124.
249.Ma, J., Katz, E., and Belote, J. M. (2002)
Insect Mol. Biol., 11, 627-639.
250.Kawahara, H., Kasahara, M., Nishiyama, A.,
Ohsumi, K., Goto, T., Kishimoto, T., Saeki, Y., Yokosawa, H., Shimbara,
N., Murata, S., Chiba, T., Suzuki, K., and Tanaka, K. (2000) EMBO
J., 19, 4144-4153.
251.Xie, Y., and Varshavsky, A. (2001) Proc.
Natl. Acad. Sci. USA, 98, 3056-3061.
252.Ju, D., Wang, L., Mao, X., and Xie, Y. (2004)
Biochem. Biophys. Res. Commun., 321, 51-57.
253.Kumatori, A., Tanaka, K., Inamura, N., Sone,
S., Ogura, T., Matsumoto, T., Tachikawa, T., Shin, S., and Ichihara, A.
(1990) Proc. Natl. Acad. Sci. USA, 87, 7071-7075.
254.Meiners, S., Heyken, D., Weller, A., Ludwig,
A., Stangl, K., Kloetzel, P. M., and Kruger, E. (2003) J. Biol.
Chem., 278, 21517-21525.
255.Rivett, A. J., Palmer, A., and Knecht, E.
(1992) J. Histochem. Cytochem., 40, 1165-1172.
256.Wojcik, C., Benchaib, M., Lornage, J., Czyba,
J. C., and Guerin, J. F. (2000) Mol. Hum. Reprod., 6,
331-336.
257.Wojcik, C., Benchaib, M., Lornage, J., Czyba,
J. C., and Guerin, J. F. (2000) Int. J. Androl., 23,
169-177.
258.Lafarga, M., Fernandez, R., Mayo, I., Berciano,
M. T., and Castano, J. G. (2002) Glia, 38, 313-328.
259.Nurse, P. (2000) Cell, 100,
71-78.
260.Johnson, D. G., and Walker, C. L. (1999)
Annu. Rev. Pharmacol. Toxicol., 39, 295-312.
261.Nederlof, P. M., Wang, H. R., and Baumeister,
W. (1995) Proc. Natl. Acad. Sci. USA, 92,
12060-12064.
262.Wang, H. R., Kania, M., Baumeister, W., and
Nederlof, P. M. (1997) Eur. J. Cell Biol., 73,
105-113.
263.Sorokin, A. V., Kim, E. R., and Ovchinnikov, L.
P. (2007) Biochemistry (Moscow), 72,
1439-1457.
264.Tanaka, K., Yoshimura, T., Tamura, T.,
Fujiwara, T., Kumatori, A., and Ichihara, A. (1990) FEBS Lett.,
271, 41-46.
265.Benedict, C. M., Ren, L., and Clawson, G. A.
(1995) Biochemistry, 34, 9587-9598.
266.Wada, M., Kosaka, M., Saito, S., Sano, T.,
Tanaka, K., and Ichihara, A. (1993) J. Lab. Clin. Med.,
121, 215-223.
267.Feist, E., Brychcy, M., Hausdorf, G., Hoyer,
B., Egerer, K., Dorner, T., Kuckelkorn, U., and Burmester, G. R. (2007)
Ann. Rheum. Dis., 66, 5-11.
268.Jakob, C., Egerer, K., Liebisch, P., Turkmen,
S., Zavrski, I., Kuckelkorn, U., Heider, U., Kaiser, M., Fleissner, C.,
Sterz, J., Kleeberg, L., Feist, E., Burmester, G. R., Kloetzel, P. M.,
and Sezer, O. (2007) Blood, 109, 2100-2105.
269.Zong, C., Gomes, A. V., Drews, O., Li, X.,
Young, G. W., Berhane, B., Qiao, X., French, S. W., Bardag-Gorce, F.,
and Ping, P. (2006) Circ. Res., 99, 372-380.
270.Bose, S., Mason, G. G., and Rivett, A. J.
(1999) Mol. Biol. Rep., 26, 11-14.
271.Bose, S., Brooks, P., Mason, G. G., and Rivett,
A. J. (2001) Biochem. J., 353, 291-297.
272.Bose, S., Stratford, F. L., Broadfoot, K. I.,
Mason, G. G., and Rivett, A. J. (2004) Biochem. J., 378,
177-184.
273.Arendt, C. S., and Hochstrasser, M. (1999)
EMBO J., 18, 3575-3585.
274.Demasi, M., Silva, G. M., and Netto, L. E.
(2003) J. Biol. Chem., 278, 679-685.
275.Zhang, F., Su, K., Yang, X., Bowe, D. B.,
Paterson, A. J., and Kudlow, J. E. (2003) Cell, 115,
715-725.
276.Sun, X. M., Butterworth, M., MacFarlane, M.,
Dubiel, W., Ciechanover, A., and Cohen, G. M. (2004) Mol. Cell,
14, 81-93.
277.Medicherla, B., Kostova, Z., Schaefer, A., and
Wolf, D. H. (2004) EMBO Rep., 5, 692-697.
278.Schauber, C., Chen, L., Tongaonkar, P., Vega,
I., Lambertson, D., Potts, W., and Madura, K. (1998) Nature,
391, 715-718.
279.Meyer, H. H., Wang, Y., and Warren, G. (2002)
EMBO J., 21, 5645-5652.
280.Chumakov, P. M. (2007) Biochemistry
(Moscow), 72, 1399-1421.
281.Haupt, Y., Maya, R., Kazaz, A., and Oren, M.
(1997) Nature, 387, 296-299.
282.Huibregtse, J. M., Scheffner, M., and Howley,
P. M. (1993) Mol. Cell. Biol., 13, 775-784.
283.Scheffner, M., Huibregtse, J. M., Vierstra, R.
D., and Howley, P. M. (1993) Cell, 75, 495-505.
284.Dornan, D., Bheddah, S., Newton, K., Ince, W.,
Frantz, G. D., Dowd, P., Koeppen, H., Dixit, V. M., and French, D. M.
(2004) Cancer Res., 64, 7226-7230.
285.Leng, R. P., Lin, Y., Ma, W., Wu, H., Lemmers,
B., Chung, S., Parant, J. M., Lozano, G., Hakem, R., and Benchimol, S.
(2003) Cell, 112, 779-791.
286.Chen, D., Kon, N., Li, M., Zhang, W., Qin, J.,
and Gu, W. (2005) Cell, 121, 1071-1083.
287.Rajendra, R., Malegaonkar, D., Pungaliya, P.,
Marshall, H., Rasheed, Z., Brownell, J., Liu, L. F., Lutzker, S.,
Saleem, A., and Rubin, E. H. (2004) J. Biol. Chem., 279,
36440-36444.
288.Esser, C., Scheffner, M., and Hohfeld, J.
(2005) J. Biol. Chem., 280, 27443-27448.
289.Oliner, J. D., Kinzler, K. W., Meltzer, P. S.,
George, D. L., and Vogelstein, B. (1992) Nature, 358,
80-83.
290.Forslund, A., Zeng, Z., Qin, L. X., Rosenberg,
S., Ndubuisi, M., Pincas, H., Gerald, W., Notterman, D. A., Barany, F.,
and Paty, P. B. (2008) Mol. Cancer Res., 6, 205-211.
291.Bueso-Ramos, C. E., Manshouri, T., Haidar, M.
A., Yang, Y., McCown, P., Ordonez, N., Glassman, A., Sneige, N., and
Albitar, M. (1996) Breast Cancer Res. Treat., 37,
179-188.
292.Lianes, P., Orlow, I., Zhang, Z. F., Oliva, M.
R., Sarkis, A. S., Reuter, V. E., and Cordon-Cardo, C. (1994) J.
Natl. Cancer Inst., 86, 1325-1330.
293.Lim, K. P., Sharifah, H., Lau, S. H., Teo, S.
H., and Cheong, S. C. (2005) Oncol. Rep., 14,
963-968.
294.Lukas, J., Gao, D. Q., Keshmeshian, M., Wen, W.
H., Tsao-Wei, D., Rosenberg, S., and Press, M. F. (2001) Cancer
Res., 61, 3212-3219.
295.Bartel, F., Taubert, H., and Harris, L. C.
(2002) Cancer Cell, 2, 9-15.
296.Bond, G. L., Hu, W., Bond, E. E., Robins, H.,
Lutzker, S. G., Arva, N. C., Bargonetti, J., Bartel, F., Taubert, H.,
Wuerl, P., Onel, K., Yip, L., Hwang, S. J., Strong, L. C., Lozano, G.,
and Levine, A. J. (2004) Cell, 119, 591-602.
297.Alhopuro, P., Ylisaukko-Oja, S. K., Koskinen,
W. J., Bono, P., Arola, J., Jarvinen, H. J., Mecklin, J. P., Atula, T.,
Kontio, R., Makitie, A. A., Suominen, S., Leivo, I., Vahteristo, P.,
Aaltonen, L. M., and Aaltonen, L. A. (2005) J. Med. Genet.,
42, 694-698.
298.Sotamaa, K., Liyanarachchi, S., Mecklin, J. P.,
Jarvinen, H., Aaltonen, L. A., Peltomaki, P., and de la Chapelle, A.
(2005) Clin. Cancer Res., 11, 6840-6844.
299.Zainuddin, N., Berglund, M., Wanders, A., Ren,
Z. P., Amini, R. M., Lindell, M., Kanduri, M., Roos, G., Rosenquist,
R., and Enblad, G. (2009) Leuk. Res., 33, 60-66.
300.Lind, H., Zienolddiny, S., Ekstrom, P. O.,
Skaug, V., and Haugen, A. (2006) Int. J. Cancer, 119,
718-721.
301.Bond, G. L., Menin, C., Bertorelle, R.,
Alhopuro, P., Aaltonen, L. A., and Levine, A. J. (2006) J. Med.
Genet., 43, 950-952.
302.Han, J. Y., Lim, H. S., Yoo, Y. K., Shin, E.
S., Park, Y. H., Lee, S. Y., Lee, J. E., Lee, D. H., Kim, H. T., and
Lee, J. S. (2007) Cancer, 110, 138-147.
303.Bougeard, G., Baert-Desurmont, S., Tournier,
I., Vasseur, S., Martin, C., Brugieres, L., Chompret, A., Bressac-de
Paillerets, B., Stoppa-Lyonnet, D., Bonaiti-Pellie, C., and Frebourg,
T. (2006) J. Med. Genet., 43, 531-533.
304.Menin, C., Scaini, M. C., de Salvo, G. L.,
Biscuola, M., Quaggio, M., Esposito, G., Belluco, C., Montagna, M.,
Agata, S., D’Andrea, E., Nitti, D., Amadori, A., and Bertorelle,
R. (2006) J. Natl. Cancer Inst., 98, 285-288.
305.Bouska, A., Lushnikova, T., Plaza, S., and
Eischen, C. M. (2008) Mol. Cell. Biol., 28,
4862-4874.
306.Dawson, S., Apcher, S., Mee, M., Higashitsuji,
H., Baker, R., Uhle, S., Dubiel, W., Fujita, J., and Mayer, R. J.
(2002) J. Biol. Chem., 277, 10893-10902.
307.Higashitsuji, H., Itoh, K., Sakurai, T., Nagao,
T., Sumitomo, Y., Masuda, T., Dawson, S., Shimada, Y., Mayer, R. J.,
and Fujita, J. (2005) Cancer Cell, 8, 75-87.
308.Higashitsuji, H., Liu, Y., Mayer, R. J., and
Fujita, J. (2005) Cell Cycle, 4, 1335-1337.
309.Yoon, S. Y., Lee, Y., Kim, J. H., Chung, A. S.,
Joo, J. H., Kim, C. N., Kim, N. S., Choe, I. S., and Kim, J. W. (2005)
Biochem. Biophys. Res. Commun., 326, 7-17.
310.Duan, W., Gao, L., Druhan, L. J., Zhu, W. G.,
Morrison, C., Otterson, G. A., and Villalona-Calero, M. A. (2004) J.
Natl. Cancer Inst., 96, 1718-1721.
311.Scheffner, M., Munger, K., Byrne, J. C., and
Howley, P. M. (1991) Proc. Natl. Acad. Sci. USA, 88,
5523-5527.
312.Scheffner, M., Werness, B. A., Huibregtse, J.
M., Levine, A. J., and Howley, P. M. (1990) Cell, 63,
1129-1136.
313.Huibregtse, J. M., Scheffner, M., and Howley,
P. M. (1991) EMBO J., 10, 4129-4135.
314.Thomas, M., Pim, D., and Banks, L. (1999)
Oncogene, 18, 7690-7700.
315.Beer-Romero, P., Glass, S., and Rolfe, M.
(1997) Oncogene, 14, 595-602.
316.Huibregtse, J. M., Scheffner, M., and Howley,
P. M. (1993) Mol. Cell. Biol., 13, 4918-4927.
317.Sherr, C. J., and Roberts, J. M. (1999)
Genes Dev., 13, 1501-1512.
318.Carrano, A. C., Eytan, E., Hershko, A., and
Pagano, M. (1999) Nat. Cell Biol., 1, 193-199.
319.Sutterluty, H., Chatelain, E., Marti, A.,
Wirbelauer, C., Senften, M., Muller, U., and Krek, W. (1999) Nat.
Cell Biol., 1, 207-214.
320.Ganoth, D., Bornstein, G., Ko, T. K., Larsen,
B., Tyers, M., Pagano, M., and Hershko, A. (2001) Nat. Cell
Biol., 3, 321-324.
321.Spruck, C., Strohmaier, H., Watson, M., Smith,
A. P., Ryan, A., Krek, T. W., and Reed, S. I. (2001) Mol. Cell,
7, 639-650.
322.Lahav-Baratz, S., Ben-Izhak, O., Sabo, E.,
Ben-Eliezer, S., Lavie, O., Ishai, D., Ciechanover, A., and Dirnfeld,
M. (2004) Mol. Hum. Reprod., 10, 567-572.
323.Slotky, M., Shapira, M., Ben-Izhak, O., Linn,
S., Futerman, B., Tsalic, M., and Hershko, D. D. (2005) Breast
Cancer Res., 7, R737-744.
324.Hershko, D. D., and Shapira, M. (2006)
Cancer, 107, 668-675.
325.Kudo, Y., Kitajima, S., Sato, S., Miyauchi, M.,
Ogawa, I., and Takata, T. (2001) Cancer Res., 61,
7044-7047.
326.Huang, H. Y., Kang, H. Y., Li, C. F., Eng, H.
L., Chou, S. C., Lin, C. N., and Hsiung, C. Y. (2006) Clin. Cancer
Res., 12, 487-498.
327.Inui, N., Kitagawa, K., Miwa, S., Hattori, T.,
Chida, K., Nakamura, H., and Kitagawa, M. (2003) Biochem. Biophys.
Res. Commun., 303, 978-984.
328.Kitajima, S., Kudo, Y., Ogawa, I., Bashir, T.,
Kitagawa, M., Miyauchi, M., Pagano, M., and Takata, T. (2004) Am. J.
Pathol., 165, 2147-2155.
329.Yokoi, S., Yasui, K., Mori, M., Iizasa, T.,
Fujisawa, T., and Inazawa, J. (2004) Am. J. Pathol., 165,
175-180.
330.Yu, Z. K., Gervais, J. L., and Zhang, H. (1998)
Proc. Natl. Acad. Sci. USA, 95, 11324-11329.
331.Kamura, T., Hara, T., Kotoshiba, S., Yada, M.,
Ishida, N., Imaki, H., Hatakeyama, S., Nakayama, K., and Nakayama, K.
I. (2003) Proc. Natl. Acad. Sci. USA, 100,
10231-10236.
332.Tedesco, D., Lukas, J., and Reed, S. I. (2002)
Genes Dev., 16, 2946-2957.
333.Huang, H., Regan, K. M., Wang, F., Wang, D.,
Smith, D. I., van Deursen, J. M., and Tindall, D. J. (2005) Proc.
Natl. Acad. Sci. USA, 102, 1649-1654.
334.Kim, W. Y., and Kaelin, W. G. (2004) J.
Clin. Oncol., 22, 4991-5004.
335.Igarashi, H., Esumi, M., Ishida, H., and Okada,
K. (2002) Cancer, 95, 47-53.
336.Stratmann, R., Krieg, M., Haas, R., and Plate,
K. H. (1997) J. Neuropathol. Exp. Neurol., 56,
1242-1252.
337.Lisztwan, J., Imbert, G., Wirbelauer, C.,
Gstaiger, M., and Krek, W. (1999) Genes Dev., 13,
1822-1833.
338.Iwai, K., Yamanaka, K., Kamura, T., Minato, N.,
Conaway, R. C., Conaway, J. W., Klausner, R. D., and Pause, A. (1999)
Proc. Natl. Acad. Sci. USA, 96, 12436-12441.
339.Maxwell, P. H., Wiesener, M. S., Chang, G. W.,
Clifford, S. C., Vaux, E. C., Cockman, M. E., Wykoff, C. C., Pugh, C.
W., Maher, E. R., and Ratcliffe, P. J. (1999) Nature,
399, 271-275.
340.Tanimoto, K., Makino, Y., Pereira, T., and
Poellinger, L. (2000) EMBO J., 19, 4298-4309.
341.Cockman, M. E., Masson, N., Mole, D. R.,
Jaakkola, P., Chang, G. W., Clifford, S. C., Maher, E. R., Pugh, C. W.,
Ratcliffe, P. J., and Maxwell, P. H. (2000) J. Biol. Chem.,
275, 25733-25741.
342.Duan, D. R., Pause, A., Burgess, W. H., Aso,
T., Chen, D. Y., Garrett, K. P., Conaway, R. C., Conaway, J. W.,
Linehan, W. M., and Klausner, R. D. (1995) Science, 269,
1402-1406.
343.Pause, A., Lee, S., Worrell, R. A., Chen, D.
Y., Burgess, W. H., Linehan, W. M., and Klausner, R. D. (1997) Proc.
Natl. Acad. Sci. USA, 94, 2156-2161.
344.Kamura, T., Koepp, D. M., Conrad, M. N.,
Skowyra, D., Moreland, R. J., Iliopoulos, O., Lane, W. S., Kaelin, W.
G., Jr., Elledge, S. J., Conaway, R. C., Harper, J. W., and Conaway, J.
W. (1999) Science, 284, 657-661.
345.Patiar, S., and Harris, A. L. (2006) Endocr.
Relat. Cancer, 13, Suppl. 1, S61-75.
346.Huang, L. E., Gu, J., Schau, M., and Bunn, H.
F. (1998) Proc. Natl. Acad. Sci. USA, 95, 7987-7992.
347.Bruick, R. K., and McKnight, S. L. (2001)
Science, 294, 1337-1340.
348.Ivan, M., Kondo, K., Yang, H., Kim, W.,
Valiando, J., Ohh, M., Salic, A., Asara, J. M., Lane, W. S., and
Kaelin, W. G., Jr. (2001) Science, 292, 464-468.
349.Jaakkola, P., Mole, D. R., Tian, Y. M., Wilson,
M. I., Gielbert, J., Gaskell, S. J., Kriegsheim, A., Hebestreit, H. F.,
Mukherji, M., Schofield, C. J., Maxwell, P. H., Pugh, C. W., and
Ratcliffe, P. J. (2001) Science, 292, 468-472.
350.Masson, N., Willam, C., Maxwell, P. H., Pugh,
C. W., and Ratcliffe, P. J. (2001) EMBO J., 20,
5197-5206.
351.Yu, F., White, S. B., Zhao, Q., and Lee, F. S.
(2001) Proc. Natl. Acad. Sci. USA, 98, 9630-9635.
352.Gayther, S. A., Russell, P., Harrington, P.,
Antoniou, A. C., Easton, D. F., and Ponder, B. A. (1999) Am. J. Hum.
Genet., 65, 1021-1029.
353.Staff, S., Isola, J., and Tanner, M. (2003)
Cancer Res., 63, 4978-4983.
354.Sorlie, T., Tibshirani, R., Parker, J., Hastie,
T., Marron, J. S., Nobel, A., Deng, S., Johnsen, H., Pesich, R.,
Geisler, S., Demeter, J., Perou, C. M., Lonning, P. E., Brown, P. O.,
Borresen-Dale, A. L., and Botstein, D. (2003) Proc. Natl. Acad. Sci.
USA, 100, 8418-8423.
355.Turner, N. C., Reis-Filho, J. S., Russell, A.
M., Springall, R. J., Ryder, K., Steele, D., Savage, K., Gillett, C.
E., Schmitt, F. C., Ashworth, A., and Tutt, A. N. (2007)
Oncogene, 26, 2126-2132.
356.Wu, L. C., Wang, Z. W., Tsan, J. T., Spillman,
M. A., Phung, A., Xu, X. L., Yang, M. C., Hwang, L. Y., Bowcock, A. M.,
and Baer, R. (1996) Nat. Genet., 14, 430-440.
357.Thai, T. H., Du, F., Tsan, J. T., Jin, Y.,
Phung, A., Spillman, M. A., Massa, H. F., Muller, C. Y., Ashfaq, R.,
Mathis, J. M., Miller, D. S., Trask, B. J., Baer, R., and Bowcock, A.
M. (1998) Hum. Mol. Genet., 7, 195-202.
358.Hashizume, R., Fukuda, M., Maeda, I.,
Nishikawa, H., Oyake, D., Yabuki, Y., Ogata, H., and Ohta, T. (2001)
J. Biol. Chem., 276, 14537-14540.
359.Chen, A., Kleiman, F. E., Manley, J. L., Ouchi,
T., and Pan, Z. Q. (2002) J. Biol. Chem., 277,
22085-22092.
360.Mallery, D. L., Vandenberg, C. J., and Hiom, K.
(2002) EMBO J., 21, 6755-6762.
361.Joukov, V., Chen, J., Fox, E. A., Green, J. B.,
and Livingston, D. M. (2001) Proc. Natl. Acad. Sci. USA,
98, 12078-12083.
362.Baer, R., and Ludwig, T. (2002) Curr. Opin.
Genet. Dev., 12, 86-91.
363.Shakya, R., Szabolcs, M., McCarthy, E., Ospina,
E., Basso, K., Nandula, S., Murty, V., Baer, R., and Ludwig, T. (2008)
Proc. Natl. Acad. Sci. USA, 105, 7040-7045.
364.Xia, Y., Pao, G. M., Chen, H. W., Verma, I. M.,
and Hunter, T. (2003) J. Biol. Chem., 278, 5255-5263.
365.Starita, L. M., Machida, Y., Sankaran, S.,
Elias, J. E., Griffin, K., Schlegel, B. P., Gygi, S. P., and Parvin, J.
D. (2004) Mol. Cell. Biol., 24, 8457-8466.
366.Starita, L. M., Horwitz, A. A., Keogh, M. C.,
Ishioka, C., Parvin, J. D., and Chiba, N. (2005) J. Biol. Chem.,
280, 24498-24505.
367.Eakin, C. M., Maccoss, M. J., Finney, G. L.,
and Klevit, R. E. (2007) Proc. Natl. Acad. Sci. USA, 104,
5794-5799.
368.Starita, L. M., and Parvin, J. D. (2006)
Cancer Biol. Ther., 5, 137-141.
369.Reinstein, E., and Ciechanover, A. (2006)
Ann. Intern. Med., 145, 676-684.
370.Debigare, R., and Price, S. R. (2003) Am. J.
Physiol. Renal Physiol., 285, F1-8.
371.Snyder, P. M. (2005) Endocrinology,
146, 5079-5085.
372.Shimkets, R. A., Warnock, D. G., Bositis, C.
M., Nelson-Williams, C., Hansson, J. H., Schambelan, M., Gill, J. R.,
Jr., Ulick, S., Milora, R. V., Findling, J. W., et al. (1994)
Cell, 79, 407-414.
373.Firsov, D., Schild, L., Gautschi, I., Merillat,
A. M., Schneeberger, E., and Rossier, B. C. (1996) Proc. Natl. Acad.
Sci. USA, 93, 15370-15375.
374.Schild, L., Lu, Y., Gautschi, I., Schneeberger,
E., Lifton, R. P., and Rossier, B. C. (1996) EMBO J., 15,
2381-2387.
375.Hansson, J. H., Schild, L., Lu, Y., Wilson, T.
A., Gautschi, I., Shimkets, R., Nelson-Williams, C., Rossier, B. C.,
and Lifton, R. P. (1995) Proc. Natl. Acad. Sci. USA, 92,
11495-11499.
376.Tamura, H., Schild, L., Enomoto, N., Matsui,
N., Marumo, F., and Rossier, B. C. (1996) J. Clin. Invest.,
97, 1780-1784.
377.Inoue, J., Iwaoka, T., Tokunaga, H., Takamune,
K., Naomi, S., Araki, M., Takahama, K., Yamaguchi, K., and Tomita, K.
(1998) J. Clin. Endocrinol. Metab., 83, 2210-2213.
378.Furuhashi, M., Kitamura, K., Adachi, M.,
Miyoshi, T., Wakida, N., Ura, N., Shikano, Y., Shinshi, Y., Sakamoto,
K., Hayashi, M., Satoh, N., Nishitani, T., Tomita, K., and Shimamoto,
K. (2005) J. Clin. Endocrinol. Metab., 90, 340-344.
379.Staub, O., Dho, S., Henry, P., Correa, J.,
Ishikawa, T., McGlade, J., and Rotin, D. (1996) EMBO J.,
15, 2371-2380.
380.Staub, O., Gautschi, I., Ishikawa, T.,
Breitschopf, K., Ciechanover, A., Schild, L., and Rotin, D. (1997)
EMBO J., 16, 6325-6336.
381.Goulet, C. C., Volk, K. A., Adams, C. M.,
Prince, L. S., Stokes, J. B., and Snyder, P. M. (1998) J. Biol.
Chem., 273, 30012-30017.
382.Abriel, H., Loffing, J., Rebhun, J. F., Pratt,
J. H., Schild, L., Horisberger, J. D., Rotin, D., and Staub, O. (1999)
J. Clin. Invest., 103, 667-673.
383.Harvey, K. F., Dinudom, A., Cook, D. I., and
Kumar, S. (2001) J. Biol. Chem., 276, 8597-8601.
384.Snyder, P. M., Steines, J. C., and Olson, D. R.
(2004) J. Biol. Chem., 279, 5042-5046.
385.Zhou, R., Patel, S. V., and Snyder, P. M.
(2007) J. Biol. Chem., 282, 20207-20212.
386.Wiemuth, D., Ke, Y., Rohlfs, M., and McDonald,
F. J. (2007) Biochem. J., 405, 147-155.
387.Knight, K. K., Olson, D. R., Zhou, R., and
Snyder, P. M. (2006) Proc. Natl. Acad. Sci. USA, 103,
2805-2808.
388.Sieradzan, K. A., Mechan, A. O., Jones, L.,
Wanker, E. E., Nukina, N., and Mann, D. M. (1999) Exp. Neurol.,
156, 92-99.
389.Ii, K., Ito, H., Tanaka, K., and Hirano, A.
(1997) J. Neuropathol. Exp. Neurol., 56, 125-131.
390.Kuzuhara, S., Mori, H., Izumiyama, N.,
Yoshimura, M., and Ihara, Y. (1988) Acta Neuropathol.,
75, 345-353.
391.Ciechanover, A., and Brundin, P. (2003)
Neuron, 40, 427-446.
392.Sawada, H., Kohno, R., Kihara, T., Izumi, Y.,
Sakka, N., Ibi, M., Nakanishi, M., Nakamizo, T., Yamakawa, K.,
Shibasaki, H., Yamamoto, N., Akaike, A., Inden, M., Kitamura, Y.,
Taniguchi, T., and Shimohama, S. (2004) J. Biol. Chem.,
279, 10710-10719.
393.Rideout, H. J., Larsen, K. E., Sulzer, D., and
Stefanis, L. (2001) J. Neurochem., 78, 899-908.
394.Sun, F., Anantharam, V., Zhang, D.,
Latchoumycandane, C., Kanthasamy, A., and Kanthasamy, A. G. (2006)
Neurotoxicology, 27, 807-815.
395.McNaught, K. S., and Jenner, P. (2001)
Neurosci. Lett., 297, 191-194.
396.Bence, N. F., Sampat, R. M., and Kopito, R. R.
(2001) Science, 292, 1552-1555.
397.Zhang, N. Y., Tang, Z., and Liu, C. W. (2008)
J. Biol. Chem., 283, 20288-20298.
398.Emmanouilidou, E., Stefanis, L., and Vekrellis,
K. (2008) Neurobiol. Aging, Epub ahead of print, DOI:
10.1016/j.neurobiolaging.2008.07.008.
399.Polymeropoulos, M. H., Lavedan, C., Leroy, E.,
Ide, S. E., Dehejia, A., Dutra, A., Pike, B., Root, H., Rubenstein, J.,
Boyer, R., Stenroos, E. S., Chandrasekharappa, S., Athanassiadou, A.,
Papapetropoulos, T., Johnson, W. G., Lazzarini, A. M., Duvoisin, R. C.,
Di Iorio, G., Golbe, L. I., and Nussbaum, R. L. (1997) Science,
276, 2045-2047.
400.Kitada, T., Asakawa, S., Hattori, N.,
Matsumine, H., Yamamura, Y., Minoshima, S., Yokochi, M., Mizuno, Y.,
and Shimizu, N. (1998) Nature, 392, 605-608.
401.Liu, Y., Fallon, L., Lashuel, H. A., Liu, Z.,
and Lansbury, P. T., Jr. (2002) Cell, 111, 209-218.
402.Valente, E. M., Abou-Sleiman, P. M., Caputo,
V., Muqit, M. M., Harvey, K., Gispert, S., Ali, Z., Del Turco, D.,
Bentivoglio, A. R., Healy, D. G., Albanese, A., Nussbaum, R.,
Gonzalez-Maldonado, R., Deller, T., Salvi, S., Cortelli, P., Gilks, W.
P., Latchman, D. S., Harvey, R. J., Dallapiccola, B., Auburger, G., and
Wood, N. W. (2004) Science, 304, 1158-1160.
403.Bonifati, V., Rizzu, P., van Baren, M. J.,
Schaap, O., Breedveld, G. J., Krieger, E., Dekker, M. C., Squitieri,
F., Ibanez, P., Joosse, M., van Dongen, J. W., Vanacore, N., van
Swieten, J. C., Brice, A., Meco, G., van Duijn, C. M., Oostra, B. A.,
and Heutink, P. (2003) Science, 299, 256-259.
404.Zhang, Y., Gao, J., Chung, K. K., Huang, H.,
Dawson, V. L., and Dawson, T. M. (2000) Proc. Natl. Acad. Sci.
USA, 97, 13354-13359.
405.Chung, K. K., Zhang, Y., Lim, K. L., Tanaka,
Y., Huang, H., Gao, J., Ross, C. A., Dawson, V. L., and Dawson, T. M.
(2001) Nat. Med., 7, 1144-1150.
406.Shimura, H., Schlossmacher, M. G., Hattori, N.,
Frosch, M. P., Trockenbacher, A., Schneider, R., Mizuno, Y., Kosik, K.
S., and Selkoe, D. J. (2001) Science, 293, 263-269.
407.Sakata, E., Yamaguchi, Y., Kurimoto, E.,
Kikuchi, J., Yokoyama, S., Yamada, S., Kawahara, H., Yokosawa, H.,
Hattori, N., Mizuno, Y., Tanaka, K., and Kato, K. (2003) EMBO
Rep., 4, 301-306.
408.Imai, Y., Soda, M., Inoue, H., Hattori, N.,
Mizuno, Y., and Takahashi, R. (2001) Cell, 105,
891-902.
409.Wiertz, E. J., Jones, T. R., Sun, L., Bogyo,
M., Geuze, H. J., and Ploegh, H. L. (1996) Cell, 84,
769-779.
410.Kikkert, M., Hassink, G., Barel, M., Hirsch,
C., van der Wal, F. J., and Wiertz, E. (2001) Biochem. J.,
358, 369-377.
411.Shamu, C. E., Flierman, D., Ploegh, H. L.,
Rapoport, T. A., and Chau, V. (2001) Mol. Biol. Cell, 12,
2546-2555.
412.Furman, M. H., Loureiro, J., Ploegh, H. L., and
Tortorella, D. (2003) J. Biol. Chem., 278,
34804-34811.
413.Hassink, G. C., Barel, M. T., van Voorden, S.
B., Kikkert, M., and Wiertz, E. J. (2006) J. Biol. Chem.,
281, 30063-30071.
414.Tirosh, B., Iwakoshi, N. N., Lilley, B. N.,
Lee, A. H., Glimcher, L. H., and Ploegh, H. L. (2005) J. Virol.,
79, 2768-2779.
415.Barel, M. T., Hassink, G. C., van Voorden, S.,
and Wiertz, E. J. (2006) Mol. Immunol., 43,
1258-1266.
416.Khan, S., Zimmermann, A., Basler, M.,
Groettrup, M., and Hengel, H. (2004) J. Virol., 78,
1831-1842.
417.Coscoy, L., and Ganem, D. (2000) Proc. Natl.
Acad. Sci. USA, 97, 8051-8056.
418.Coscoy, L., Sanchez, D. J., and Ganem, D.
(2001) J. Cell Biol., 155, 1265-1273.
419.Boname, J. M., and Stevenson, P. G. (2001)
Immunity, 15, 627-636.
420.Coscoy, L., and Ganem, D. (2001) J. Clin.
Invest., 107, 1599-1606.
421.Levitskaya, J., Coram, M., Levitsky, V., Imreh,
S., Steigerwald-Mullen, P. M., Klein, G., Kurilla, M. G., and Masucci,
M. G. (1995) Nature, 375, 685-688.
422.Levitskaya, J., Sharipo, A., Leonchiks, A.,
Ciechanover, A., and Masucci, M. G. (1997) Proc. Natl. Acad. Sci.
USA, 94, 12616-12621.
423.Zhang, M., and Coffino, P. (2004) J. Biol.
Chem., 279, 8635-8641.
424.Heessen, S., Leonchiks, A., Issaeva, N.,
Sharipo, A., Selivanova, G., Masucci, M. G., and Dantuma, N. P. (2002)
Proc. Natl. Acad. Sci. USA, 99, 1532-1537.
425.Andrejeva, J., Young, D. F., Goodbourn, S., and
Randall, R. E. (2002) J. Virol., 76, 2159-2167.
426.Ulane, C. M., Rodriguez, J. J., Parisien, J.
P., and Horvath, C. M. (2003) J. Virol., 77,
6385-6393.
427.Ulane, C. M., Kentsis, A., Cruz, C. D.,
Parisien, J. P., Schneider, K. L., and Horvath, C. M. (2005) J.
Virol., 79, 10180-10189.
428.Khu, Y. L., Tan, Y. J., Lim, S. G., Hong, W.,
and Goh, P. Y. (2004) Biochem. J., 384, 401-409.
429.Van Waes, C. (2007) Clin. Cancer Res.,
13, 1076-1082.
430.Egerer, K., Kuckelkorn, U., Rudolph, P. E.,
Ruckert, J. C., Dorner, T., Burmester, G. R., Kloetzel, P. M., and
Feist, E. (2002) J. Rheumatol., 29, 2045-2052.
431.Lavabre-Bertrand, T., Henry, L., Carillo, S.,
Guiraud, I., Ouali, A., Dutaud, D., Aubry, L., Rossi, J. F., and
Bureau, J. P. (2001) Cancer, 92, 2493-2500.
432.Feist, E., Dorner, T., Kuckelkorn, U.,
Schmidtke, G., Micheel, B., Hiepe, F., Burmester, G. R., and Kloetzel,
P. M. (1996) J. Exp. Med., 184, 1313-1318.
433.Feist, E., Kuckelkorn, U., Dorner, T., Donitz,
H., Scheffler, S., Hiepe, F., Kloetzel, P. M., and Burmester, G. R.
(1999) Arthritis Rheum., 42, 697-702.
434.Mayo, I., Arribas, J., Villoslada, P., Alvarez
DoForno, R., Rodriguez-Vilarino, S., Montalban, X., De Sagarra, M. R.,
and Castano, J. G. (2002) Brain, 125, 2658-2667.
435.Matsushita, M., Takasaki, Y., Takeuchi, K.,
Yamada, H., Matsudaira, R., and Hashimoto, H. (2004) J.
Rheumatol., 31, 252-259.
436.Brychcy, M., Kuckelkorn, U., Hausdorf, G.,
Egerer, K., Kloetzel, P. M., Burmester, G. R., and Feist, E. (2006)
Arthritis Rheum., 54, 2175-2183.
437.Li, B., Tournier, C., Davis, R. J., and
Flavell, R. A. (1999) EMBO J., 18, 420-432.
438.Hartenstein, B., Teurich, S., Hess, J.,
Schenkel, J., Schorpp-Kistner, M., and Angel, P. (2002) EMBO J.,
21, 6321-6329.
439.Gao, M., Labuda, T., Xia, Y., Gallagher, E.,
Fang, D., Liu, Y. C., and Karin, M. (2004) Science, 306,
271-275.
440.Venuprasad, K., Elly, C., Gao, M.,
Salek-Ardakani, S., Harada, Y., Luo, J. L., Yang, C., Croft, M., Inoue,
K., Karin, M., and Liu, Y. C. (2006) J. Clin. Invest.,
116, 1117-1126.
441.Tsao, P. W., Suzuki, T., Totsuka, R., Murata,
T., Takagi, T., Ohmachi, Y., Fujimura, H., and Takata, I. (1997)
Clin. Immunol. Immunopathol., 83, 173-178.
442.Firestein, G. S. (2004) Arthritis
Rheum., 50, 2381-2386.
443.Kisselev, A. F., and Goldberg, A. L. (2001)
Chem. Biol., 8, 739-758.
444.An, B., Goldfarb, R. H., Siman, R., and Dou, Q.
P. (1998) Cell Death Differ., 5, 1062-1075.
445.Drexler, H. C., Risau, W., and Konerding, M. A.
(2000) FASEB J., 14, 65-77.
446.Chen, L., Smith, L., Wang, Z., and Smith, J. B.
(2003) Mol. Pharmacol., 64, 334-345.
447.Fribley, A., and Wang, C. Y. (2006) Cancer
Biol. Ther., 5, 745-748.
448.Roccaro, A. M., Hideshima, T., Raje, N., Kumar,
S., Ishitsuka, K., Yasui, H., Shiraishi, N., Ribatti, D., Nico, B.,
Vacca, A., Dammacco, F., Richardson, P. G., and Anderson, K. C. (2006)
Cancer Res., 66, 184-191.
449.Adams, J., Palombella, V. J., Sausville, E. A.,
Johnson, J., Destree, A., Lazarus, D. D., Maas, J., Pien, C. S.,
Prakash, S., and Elliott, P. J. (1999) Cancer Res., 59,
2615-2622.
450.Teicher, B. A., Ara, G., Herbst, R.,
Palombella, V. J., and Adams, J. (1999) Clin. Cancer Res.,
5, 2638-2645.
451.Adams, J. (2002) Oncologist, 7,
9-16.
452.Maki, R. G., Kraft, A. S., Scheu, K., Yamada,
J., Wadler, S., Antonescu, C. R., Wright, J. J., and Schwartz, G. K.
(2005) Cancer, 103, 1431-1438.
453.Markovic, S. N., Geyer, S. M., Dawkins, F.,
Sharfman, W., Albertini, M., Maples, W., Fracasso, P. M., Fitch, T.,
Lorusso, P., Adjei, A. A., and Erlichman, C. (2005) Cancer,
103, 2584-2589.
454.Richardson, P. G., Barlogie, B., Berenson, J.,
Singhal, S., Jagannath, S., Irwin, D., Rajkumar, S. V., Srkalovic, G.,
Alsina, M., Alexanian, R., Siegel, D., Orlowski, R. Z., Kuter, D.,
Limentani, S. A., Lee, S., Hideshima, T., Esseltine, D. L., Kauffman,
M., Adams, J., Schenkein, D. P., and Anderson, K. C. (2003) N. Engl.
J. Med., 348, 2609-2617.
455.Ludwig, H., Khayat, D., Giaccone, G., and
Facon, T. (2005) Cancer, 104, 1794-1807.
456.Blade, J., Cibeira, M. T., and Rosinol, L.
(2005) Acta Oncol., 44, 440-448.
457.Feling, R. H., Buchanan, G. O., Mincer, T. J.,
Kauffman, C. A., Jensen, P. R., and Fenical, W. (2003) Angew. Chem.
Int. Ed. Engl., 42, 355-357.
458.Ahn, K. S., Sethi, G., Chao, T. H., Neuteboom,
S. T., Chaturvedi, M. M., Palladino, M. A., Younes, A., and Aggarwal,
B. B. (2007) Blood, 110, 2286-2295.
459.Ruiz, S., Krupnik, Y., Keating, M., Chandra,
J., Palladino, M., and McConkey, D. (2006) Mol. Cancer Ther.,
5, 1836-1843.
460.Chauhan, D., Catley, L., Li, G., Podar, K.,
Hideshima, T., Velankar, M., Mitsiades, C., Mitsiades, N., Yasui, H.,
Letai, A., Ovaa, H., Berkers, C., Nicholson, B., Chao, T. H.,
Neuteboom, S. T., Richardson, P., Palladino, M. A., and Anderson, K. C.
(2005) Cancer Cell, 8, 407-419.
461.Yaron, A., Gonen, H., Alkalay, I., Hatzubai,
A., Jung, S., Beyth, S., Mercurio, F., Manning, A. M., Ciechanover, A.,
and Ben-Neriah, Y. (1997) EMBO J., 16, 6486-6494.
462.Yang, Y., Ludwig, R. L., Jensen, J. P., Pierre,
S. A., Medaglia, M. V., Davydov, I. V., Safiran, Y. J., Oberoi, P.,
Kenten, J. H., Phillips, A. C., Weissman, A. M., and Vousden, K. H.
(2005) Cancer Cell, 7, 547-559.
463.Vassilev, L. T., Vu, B. T., Graves, B.,
Carvajal, D., Podlaski, F., Filipovic, Z., Kong, N., Kammlott, U.,
Lukacs, C., Klein, C., Fotouhi, N., and Liu, E. A. (2004)
Science, 303, 844-848.
464.LaRusch, G. A., Jackson, M. W., Dunbar, J. D.,
Warren, R. S., Donner, D. B., and Mayo, L. D. (2007) Cancer
Res., 67, 450-454.
465.Ambrosini, G., Sambol, E. B., Carvajal, D.,
Vassilev, L. T., Singer, S., and Schwartz, G. K. (2007)
Oncogene, 26, 3473-3481.
466.Issaeva, N., Bozko, P., Enge, M., Protopopova,
M., Verhoef, L. G., Masucci, M., Pramanik, A., and Selivanova, G.
(2004) Nat. Med., 10, 1321-1328.
467.Ding, K., Lu, Y., Nikolovska-Coleska, Z., Wang,
G., Qiu, S., Shangary, S., Gao, W., Qin, D., Stuckey, J., Krajewski,
K., Roller, P. P., and Wang, S. (2006) J. Med. Chem., 49,
3432-3435.
468.Jones, R. J., Chen, Q., Voorhees, P. M., Young,
K. H., Bruey-Sedano, N., Yang, D., and Orlowski, R. Z. (2008) Clin.
Cancer Res., 14, 5416-5425.
469.Butz, K., Denk, C., Ullmann, A., Scheffner, M.,
and Hoppe-Seyler, F. (2000) Proc. Natl. Acad. Sci. USA,
97, 6693-6697.
470.Chen, Q., Xie, W., Kuhn, D. J., Voorhees, P.
M., Lopez-Girona, A., Mendy, D., Corral, L. G., Krenitsky, V. P., Xu,
W., Moutouh-de Parseval, L., Webb, D. R., Mercurio, F., Nakayama, K.
I., Nakayama, K., and Orlowski, R. Z. (2008) Blood, 111,
4690-4699.
471.Harris, J. R. (1968) Biochim. Biophys.
Acta, 150, 534-537.
472.DeMartino, G. N., and Goldberg, A. L. (1979)
J. Biol. Chem., 254, 3712-3715.
473.Wilk, S., and Orlowski, M. (1980) J.
Neurochem., 35, 1172-1182.
474.Hase, J., Kobashi, K., Nakai, N., Mitsui, K.,
Iwata, K., and Takadera, T. (1980) Biochim. Biophys. Acta,
611, 205-213.
475.Kleinschmidt, J. A., Hugle, B., Grund, C., and
Franke, W. W. (1983) Eur. J. Cell Biol., 32, 143-156.
476.Schmid, H. P., Akhayat, O., Martins De Sa, C.,
Puvion, F., Koehler, K., and Scherrer, K. (1984) EMBO J.,
3, 29-34.
477.Kloetzel, P. M., Falkenburg, P. E., Hossl, P.,
and Glatzer, K. H. (1987) Exp. Cell Res., 170,
204-213.
478.Arrigo, A. P., Tanaka, K., Goldberg, A. L., and
Welch, W. J. (1988) Nature, 331, 192-194.