REVIEW: Nicking Endonucleases
L. A. Zheleznaya1*, G. S. Kachalova1,2, R. I. Artyukh1, A. K. Yunusova1, T. A. Perevyazova1, and N. I. Matvienko3#
1Institute of Theoretical and Experimental Biophysics, Russian Academy of Sciences, 142290 Pushchino, Moscow Region, Russia; E-mail: zheleznaya@iteb.ru2Max Plank Research Unit for Structural Molecular Biology, Hamburg, Germany
3Institute of Protein Research, Russian Academy of Sciences, 142290 Pushchino, Moscow Region, Russia
* To whom correspondence should be addressed.
# Deceased.
Received March 19, 2009; Revision received April 9, 2009
Nicking endonucleases are a new type of enzymes. Like restriction endonucleases, they recognize short specific DNA sequence and cleave DNA at a fixed position relatively to the recognition sequence. However, unlike restriction endonucleases, nicking endonucleases cleave only one predetermined DNA strand. Until recently, nicking endonucleases were suggested to be naturally mutated restriction endonucleases which had lost their ability to dimerize and as a result the ability to cleave the second strand. We have shown that nicking endonucleases are one of the subunits of heterodimeric restriction endonucleases. Mechanisms used by various restriction endonucleases for double-stranded cleavage, designing of artificial nicking endonucleases on the basis of restriction endonucleases, and application of nicking endonucleases in molecular biology are reviewed.
KEY WORDS: nicking endonuclease, restriction endonuclease, heterodimeric restriction endonucleaseDOI: 10.1134/S0006297909130033
Abbreviations: a.a., amino acid residue; bp, base pairs; dNTP, mixture of deoxynucleoside triphosphates; MTase, DNAmethyl transferase; NICKase, nicking endonuclease; ORF, open reading frame; PCR, polymerase chain reaction.
According to the nomenclature approved in 2003 [1],
nicking endonucleases include two groups of enzymes introducing a nick
in only one of two DNA strands. The first group includes endonucleases
associated inside cells only with m5C DNA methyltransferases. They
introduce a nick near unpaired bases of G-T pairs, emerging after
m5-cytosine deamination within a sequence, recognized by DNA
methyltransferase (MTase). Endonucleases of this type are designated by
the symbol “V” before the three-letter name of the
bacterial genus. For example, V.HpaII is associated with MTase HpaII in
Haemophilus parainfluenzae. Symbol “V”
originates from the best studied of this type of nicking endonuclease,
Vsr (very short patch repair).
The review deals with the second group of nicking endonucleases designated by the symbol “N” before abbreviated name of the bacterial genus. These endonucleases, like restriction endonucleases, recognize a short specific sequence in double-stranded DNA and cleave the DNA in a fixed position relative to the recognition sequence. However, unlike restriction endonucleases, nicking endonucleases cleave only one DNA strand. It is important that the nick is introduced into the predetermined DNA strand. Depending on the strand (top or bottom) nicked by the endonuclease, the latter is designated as “Nt” or “Nb”, respectively (only the single symbol “N” was used in early works). The discovery of this type of endonucleases served as the basis for introduction of a new subtype of endonucleases—nicking endonucleases.
The first nicking endonuclease N.BstSEI was isolated from strain SE of Bacillus stearothermophilus and described as “nickase” in 1996 by researchers of SibEnzyme (Russia) [2]. Below we shall also use the term “nickase”. After quite a long period during which nickase N.BstSEI remained the unique representative of a new type of endonucleases, several reports about different nicking endonucleases appeared almost simultaneously. In the year 2000, representatives of New England Biolabs reported on the isolation of the same enzyme, N.BstNBI, from strain NB of B. stearothermophilus [3]. In 2001 nickase N.BspD6I was isolated in Laboratory of N. I. Matvienko from strain D6 of Bacillus sp. [4]. In the same year, Dedkov et al. [5] described a nicking endonuclease N.Bst9I isolated from B. stearothermophilus strain 9. Two amazing facts are associated with these nicking endonucleases. First, although the abovementioned endonucleases were isolated from different sources, all of them exhibited identical specificity: they recognize in double-stranded DNA sequence (site) 5′-GAGTC-3′/5′-GACTC-3′ and cleave only the strand containing sequence 5′-GAGTC at a distance of four base pairs towards the 3′ end from the recognition sequence. However, the ability of these nickases to recognize identical sequences has a simple explanation. The point is that, unlike restriction endonucleases, it is rather difficult to detect nicking endonucleases in lysates of bacterial cells. The presence of restriction endonucleases is traditionally determined electrophoretically by formation of DNA fragments after incubation of substrate DNA with cell lysate. Plasmid and bacteriophage DNA (in particular, bacteriophage T7) with known base sequence are used as the substrate DNA. Nickases are able to fragment DNA only if two recognition sites in different orientation are close to each other. Such arrangement of recognition sites appears four times in DNA of bacteriophages T7. The high frequency of such combination of sites, in turn, is explained by the fact that the GAGTC sequence is incorporated in promoter sequences of T7 DNA. The second amazing fact is that sequencing of nickase N.BstNBI [6], N.BspD6I [7], and N.BstSEI [8] genes and adjacent regions has shown 100% coincidence between all these sequences. Such a coincidence can be due to the fact that all researchers were dealing with the same widespread strain (the enzymes were isolated in laboratories geographically remote from each other). However, it is possible that the strains were still different, and this would point to possible horizontal transfer of restriction–modification systems, and as it will be shown below, nicking endonucleases are a component of these systems.
The high similarity between nickases and restriction endonucleases resulted in appearance in the literature of the concept that nickases are a mutant form of restriction endonucleases that lost the ability for dimerization and, as a result, the ability to cleave DNA on both strands [1, 6]. We have shown for the first time that nicking endonucleases are a subunit of heterodimeric restriction endonucleases [9]. Before starting description of these data, we shall briefly consider general characteristics of type II restriction endonucleases.
STRATEGIES OF DNA CLEAVAGE ON TWO STRANDS BY TYPE II restriction
endonucleases
Restriction endonucleases are one of most numerous families, incorporating about 3900 biochemically characterized members and approximately the same number whose existence is predicted on the basis of bioinformatic analysis of DNA sequences found in GenBank. It is an extremely quickly evolving family. This follows from unusual diversity of strategies used by restriction endonucleases for DNA cleavage at two strands. Type II restriction endonucleases recognize specific 4-8 bp sequences, both symmetrical and non-symmetrical, and in the presence of Mg2+ they cleave DNA within the recognition sequence or near this sequence, and on one or both sides from it. Molecular mass values of endonucleases vary from ~30 to ~100 kDa. Protection of the host DNA against autohydrolysis is provided by the endonuclease-paired DNA methyltransferase (MTase) that recognizes the same sequence as the restriction endonuclease and methylates at a single base (adenine or cytosine) in each strand of the recognition sequence. The combination of two activities, endonuclease and MTase, was called the restriction–modification system, and it is considered as the last barrier on the way of penetration of foreign DNA into a bacterial cell.
Despite functional similarity, different restriction endonucleases use different strategies for DNA cleavage on two strands depending on the character of the recognition sequence and the presence in them of one or two catalytic centers in a monomeric molecule. Thus, cleavage of a palindromic sequence with identical top and bottom strands (like 5′-GAATTC-3′/5′-GAATTC-3′) is assured by dimerization of identical monomers, each of which contains a single catalytic center. Each center cleaves “its own” strand. According to X-ray data, the dimers not only interact with DNA, but they form numerous contacts with each other [10]. Establishing contacts between dimers serves as an additional control for recognition of a strictly defined sequence because recognition of a non-canonical and thus non-methylated sequence in host DNA will result in its hydrolysis. Probably, just emergence of these contacts activates the catalytic centers of the monomers. Replacement of an amino acid residue involved in specific base recognition in only a single subunit prevents cleavage of both strands [11].
When restriction endonucleases recognize an asymmetrical sequence (and, as a rule, cleave the DNA outside the recognition sequence) the problem of DNA cleavage on two strands is solved differently. Thus, some endonucleases require interaction with two copies of the recognition sequence. The best studied in this respect is restriction endonuclease FokI [12], for which it is shown that dimerization of monomers, each of which is bound to a separate copy of the recognition sequence, precedes DNA cleavage in both strands. In this case, catalytic domains of monomers are involved in dimerization, and DNA cleavage on both strands happens only near one recognition site. An example of a different type of DNA cleavage in both strands is restriction endonucleases containing two catalytic centers in a monomeric molecule [13-15], each of which cleaves “its own” DNA strand. Finally, endonuclease BflI exhibits unique activity. It interacts with DNA as a dimer and forms a single catalytic center upon dimerization. Besides, it not only does not require bivalent metal ions as a cofactor, it is even active in the presence of EDTA [16, 17].
HETERODIMERIC RESTRICTION ENDONUCLEASES
Endonucleases consisting of two different subunits got the name “heterodimeric”. We shall consider in detail this type of endonucleases because, as mentioned above, nicking endonucleases are a subunit of recently detected heterodimeric restriction endonucleases. The first such endonuclease Bpu10I was found in Bacillus pumilus strain 10 and described in 1998 [18].
Bpu10I recognizes in DNA an asymmetrical sequence but cleaves symmetrically within this sequence: 5′-CC↓TNAGC/5′-GC↑TNAGG (cleavage sites are shown by arrows). Endonuclease activity of Bpu10I was found only in cell extract, whereas after chromatography on the first column none of the fractions exhibited endonuclease activity. It became possible to understand the reason for such behavior of Bpu10I only after cloning the chromosomal DNA fragment containing the gene of the restriction endonuclease. This fragment contained two genes encoding DNA methyltransferases and two open reading frames (ORF). No endonuclease activity was detected in cell extracts containing a plasmid with either ORF alone. This activity was registered in extracts of cells containing a plasmid with both ORF as well as upon mixing cell extracts containing each plasmid separately. Proteins encoded by each ORF were purified, and only their mixture exhibited endonuclease activity. Thus, Bpu10I became the first restriction endonuclease functioning in the form of a heterodimer consisting of 34.5 and 34.0 kDa subunits. The amino acid sequences of the subunits reveal low homology (25% identical amino acids and 17% functionally similar ones). A two-subunit complex was not found, and because of this it was concluded that the interaction between the subunits is very weak.
Another heterodimeric endonuclease, BslI, was found in a strain of Bacillus sp. [19]. It recognizes symmetrical sequence CCNNNNN↓NNGG/CCNNNNN↑NNGG and cleaves the DNA symmetrically inside this sequence. Although BslI recognizes a symmetrical sequence and cleaves DNA symmetrically within the site, it consists of two subunits of 26 and 36 kDa, neither of which binds DNA or exhibits endonuclease activity. Only their mixture binds DNA and completely hydrolyzes it. Unlike Bpu10I, BslI subunits are able to form heterodimers (αβ), heterotetramers (α2β2), and possibly higher oligomers in the absence of DNA in solution. Based on the symmetry of the tetramer and recognition sequence (CCN7GG), it is supposed that the active form of BslI is the heterotetramer formed due to the interaction of two β subunits.
A peculiarity of BslI is the presence in the amino acid sequence of its α subunit of two motifs characteristic of zinc fingers. Most zinc finger motifs are found in eucaryotic transcription factors. BslI is the first bacterial restriction endonuclease that contains motifs characteristic of zinc fingers. It is supposed that zinc is necessary for correct folding of the α subunit. BslI is distinguished among heterodimeric endonucleases by high thermal stability; it survives 30 PCR cycles.
The next heterodimeric endonuclease, BbvCI, was found in Bacillus brevis strain C [20, 21]. It recognizes a sequence differing from that recognized by Bpu10I by only the central base pair (shown in bold) CC↓TCAGC/GC↑TGAGG. DNA hydrolysis in both strands takes place only in the presence of both subunits. The central base pair in the sequence recognized by Bpu10I is N/N, but Bpu10I also recognizes the sequence recognized by BbvCI. However, molecular mass values of subunits forming BbvCI (31 and 32.6 kDa) are somewhat lower than those of subunits forming Bpu10I (34.5 and 34.0 kDa). This is probably due to the fact that, unlike Bpu10I, BbvCI recognizes a non-degenerate central base pair.
NICKING ENDONUCLEASES – SUBUNITS OF HETERODIMERIC
RESTRICTION ENDONUCLEASES
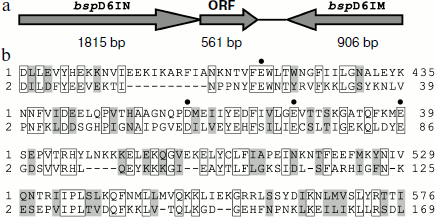
In genomic DNA of Bacillus sp. strain D6 an open reading frame encoding a protein of 186 a.a with a formerly unknown function is adjacent to the nickase gene (Fig. 1a).
The amino acid sequence encoded by the ORF has some homology to the C-terminal part of nickase: 29% identical and 20% homologous residues (Fig. 1b). When we determined the structure of nickase N.BspD6I [24] and revealed amino acid residues forming the catalytic center, it became clear that the ORF contains the same motif that forms the catalytic center of nickase. Therefore, it could be expected that the ORF product will have endonuclease activity.Fig. 1. a) Arrangement of gene complex containing gene of nicking endonuclease N.BspD6I. The size of each gene is shown in base pairs under the arrows. b) Comparison of amino acid sequences of proteins encoded by N.BspD6I (1) and ORF (2). Amino acid residues of nickase active center are shown by dots.
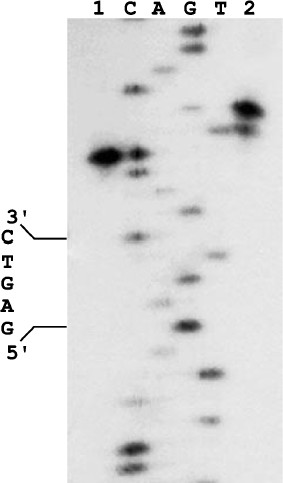
We cloned the ORF-containing DNA fragment and purified the corresponding protein, which did not bind DNA and did not form a complex with the nickase. However, the protein mixture with the nickase cleaved DNA in both strands (Fig. 2): the top strand was cleaved like by nickase, at the distance of four nucleotides from the recognition site, while the bottom strand was cleaved at the distance of six nucleotides from the site with some gap. Approximately 10% of molecules are cleaved at the distance of five nucleotides. The observed gap of the position in which the small subunit cleaves the bottom DNA strand is probably due to weak contact of the small subunit with the nickase and/or DNA.
Together these data indicate that the product of the ORF adjacent to the nickase gene in genomic DNA of Bacillus sp. strain D6 and the nickase are subunits of a heterodimeric restriction endonuclease. This endonuclease was named BspD6I. According to the nomenclature, subunits of this heterodimeric endonuclease should be designated as BspD6IA and BspD6IB. However, to make a simpler differentiation between these subunits we shall call the big subunit (70.8 kDa) “nicking endonuclease” or “nickase” as before, while the ORF product (21.6 kDa) will be called the “small subunit”.Fig. 2. Localization of phosphodiester bonds hydrolyzed by the nickase N.BspD6I mixture with small subunit. C, A, G, and T are products of M13mp19 phage DNA sequencing; 1) labeled M13mp19 phage DNA incubated with nickase mixture with small subunit; 2) labeled M13mp19 phage DNA incubated with nickase mixture with small subunit to which Klenow fragment and the dNTP mixture were added after enzyme inactivation. Electrophoresis in 6% polyacrylamide gel in the presence of 7 M urea at 50°C.
At the time of its detection by us in 2006, endonuclease BspD6I was a unique heterodimeric restriction endonuclease, one subunit of which outside the complex with the other exhibits specific nicking activity [9]. The peculiar properties of restriction endonuclease BspD6I (table) made it possible to classify it as a new type of heterodimeric restriction endonuclease not previously described in the literature.
Thus, we managed to show that nickase N.BspD6I and its homologs (N.BstSEI and N.BstNBI) function as independent nicking enzymes, and that they are large subunits of heterodimeric restriction endonucleases.
Comparison of properties of heterodimeric restriction endonucleases

Restriction endonuclease BspD6I did not remain a unique enzyme for a long time. Soon two other heterodimeric endonucleases, BsrDI and BtsI, were found [22], and one of their subunits exhibits nicking activity. BsrDI was isolated from B. stearothermophilus D70. It recognizes the GCAATG/CATTGC sequence and cleaves the top strand two nucleotides from the site and the bottom strand immediately after the site. BtsI was isolated from Bacillus thermoglucosidasius. It recognizes the GCAGTG/CACTGC sequence and cleaves DNA like BsrDI. Both endonucleases consist of two subunits: BsrDI of 56 and 25 kDa, and BtsI of 38 and 18 kDa, and each contain one catalytic center. In the absence of the small subunits, the large subunits of both endonucleases act as nickases. The comparison of endonucleases BspD6I, BsrDI, and BtsI clearly shows the diversity of heterodimeric restriction endonucleases. Despite “external” similarity revealed in nicking activity of one subunit, molecular mass values of the large subunits are much different: 70 (BspD6I), 56 (BsrDI), and 38 kDa (BtsI). In this case, molecular mass values of the small subunits are close to each other (about 20 kDa). Thus, the large subunits have changed during evolution, probably towards compaction and lowering molecular mass.
SPATIAL STRUCTURES OF SUBUNITS OF HETERODIMERIC RESTRICTION
ENDONUCLEASE BspD6I
We were able to obtain a crystal of nickase N.BspD6I and to determine the enzyme spatial structure at high resolution (1.8 Å) [23, 24]. The full-size molecule (604 a.a.) is a compact elongated globule of 50 × 60 × 80 Å. The structure consists of three domains (Fig. 3): N-terminal (302 a.a.), linker (303-380 a.a.), and C-terminal (381-604 a.a.). The N-terminal domain consists of two subdomains. Stacking of the two subdomains is a much distorted version of a helix–turn–helix motif. The C-terminal domain has α/β stacking similar to the general core motif of type II restriction endonucleases. The linker domain joining the N- and C-terminal domains is an α-helical bundle of three helices. The structure is in many respects similar to the structure of restriction endonuclease FokI that, like nickase N.BspD6I, cleaves DNA aside from the recognition site. The N-terminal part in FokI is responsible for DNA recognition and binding, while the C-terminal part carries the catalytic function [25].
The structure of FokI was determined in its complex with DNA (PDB code: 1fok), in which subdomains D1 and D2 interact with the recognition sequence. The high structural similarity between D1 and D2 nickase subdomains with similar domains of FokI indicated that the N-terminal domain of nickase N.BspD6I is also the recognition domain.Fig. 3. Structure of nicking endonuclease N.BspD6I (PDB code: 2ewf). Subdomain D1 is shown in blue, subdomain D2 of the N-terminal domain is shown in violet, the catalytic domain in red, and the linker domain in green.

For type II restriction endonucleases a characteristic sequence of amino acid residues forming the catalytic center, motif PD…(D/E)XK, was identified [26, 27]. This motif includes amino acid residues of a β-hairpin and adjacent α-helices. Highly conservative aspartic acid in the first part of this sequence PD...(D/E)XK is incorporated into the N-terminus of the first short β-strand that together with the underlying loop makes a bend. This bending (about 90°) is provided by a proline residue preceding the aspartic acid residue in the polypeptide chain. The second acidic residue in the PD...(D/E)XK motif is located in a neighboring long β-strand. The rest of the motif sequence consists of XK amino acid residues, where X is a hydrophobic residue, and other positively charged residues (glutamine, arginine) sometimes play the role of the last element of the catalytic motif instead of lysine. Such specific arrangement of the catalytic motif residues provides for cleavage of a phosphodiester bond in DNA. A similar sequence of P455, D456, E469, V470, and E482 residues was also revealed in the amino acid sequence of the C-terminal domain in N.BspD6I. These residues were assigned to the catalytic center on the basis of their mutual spatial arrangement and according to data of mutagenesis [6].
We have determined the structure of the small subunit at 1.5 Å resolution [23, 24]. Despite low homology between amino acid sequences of the small subunit and the catalytic C-domain of nicking endonucleases, their structures are very similar (Fig. 4). Structural similarity even in the absence of homology in amino acid sequence is typical of restriction endonucleases. The structure of the small subunit, like that of the N.BspD6I C-domain, comprises a β-sheet consisting of five strands surrounded by α-helices. Remarkably, there is a much extended loop of 16 a.a. in the small subunit structure, while the C-domain of nickase has no such extended loop. Extended loops in homodimeric endonuclease structures are usually involved in interactions between monomers. The loop can also be involved in formation of contacts with the N.BspD6I C-domain.
Amino acid residues D60, E73, and E86 are located in the active center of the small subunit analogously to those of corresponding amino acids in the active center of the nicking endonuclease (Fig. 5). The peculiarity of the small subunit and nickase active centers is that all three amino acid residues are negatively charged, whereas one positively charged residue (usually lysine) is always present in known restriction endonucleases. Up to the present time only one endonuclease, BamHI, is known that contains three negatively charged residues in the active center [28].Fig. 4. Spatial structures of C-terminal domain of nicking endonuclease N.BspD6I (a) and of small subunit (PDB code: 2P14) (b). L, the loop in small subunit.
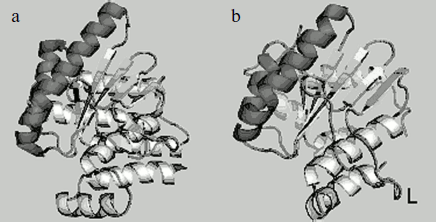
Fig. 5. Structure of the small subunit catalytic center and its environment. Hydrogen bonds are designated by dotted lines.

ARTIFICIAL NICKING ENDONUCLEASES
The practical need for nicking endonucleases and the limited set of natural enzymes has stimulated intensive work on constructing artificial nickases. The first artificial nickase, N.MlyI, was obtained by site-directed mutagenesis of restriction endonuclease MlyI causing it to lose the ability for dimerization [6]. The second artificial nickase, N.AlwI, was obtained by replacement of the C-terminal domain of restriction endonuclease AlwI by the C-terminal domain of nickase N.BstNBI [30].
A real breakthrough in nicking endonuclease construction on the basis of restriction endonucleases began after the discovery of heterodimeric restriction endonucleases and endonucleases containing two active centers in a monomeric molecule. Inactivation of the catalytic center of one subunit of heterodimeric endonucleases Bpu10I and BbvCI produced nicking endonucleases hydrolyzing only the top or bottom DNA strand depending on which subunit was inactivated [18, 20]. A similar way of inactivation of one catalytic center was used to obtain nicking endonucleases based on restriction endonucleases with two catalytic centers in a monomeric molecule, such as SapI [13], BsaI, BsmIA, and BsmIB [14]. The list of nicking endonucleases given in the REBASE database (http://rebase.neb.com) consists mainly of artificial enzymes.
An original technique for introducing a nick into DNA was proposed by Frank-Kamenetskii et al. [31, 32]. They suggested the use of a peptide oligonucleotide (PNA) complementary to the sequence of the DNA strands in the region of site recognized by a restriction endonuclease (they used PleI). The peptide oligonucleotide is hybridized to a corresponding DNA strand by displacement of the other strand, and the endonuclease cuts the hybrid site on both strands. After removal of the peptide oligonucleotide, the DNA cleaved on only one strand is obtained.
PRACTICAL APPLICATION OF NICKING ENDONUCLEASES
As shown in the previous section, the number of nicking endonucleases of different specificity is constantly increasing. The development of these enzymes is motivated by their usefulness as new tools in molecular biology. They have opened the possibility of improving or simplifying some already existing methods such as site-directed mutagenesis, DNA labeling, and isothermal DNA amplification with strand replacement.
Methods for isothermal amplification of DNA with strand replacement are presently widely used both in fundamental science and in applied investigations like library screening, detection of point mutations in DNA, estimation of mRNA expression level, SNP-analysis of genomic DNA, and even in immunohistochemistry for detection of surface and intracellular antigens. An advantage of this method of DNA amplification is that the reaction is carried out at constant temperature, and therefore instruments for cyclic regulation of the reaction mixture temperature are not necessary. In many cases, circular DNA is used as the template, which provides for so-called rolling circle amplification (RCA) [33-35]. In this case numerous copies of this DNA combined in concatemers are synthesized.
Nicking endonucleases can be used to overcome some methodical difficulties. To start synthesis, DNA polymerases require a free 3′-OH end (primer). In some cases an oligonucleotide complementary to the region of template sequence is used as the primer. However, preliminary denaturing of the template DNA (if it is double-stranded) is necessary for primer annealing, which results in partial degradation of the DNA. Introducing a nick into one DNA strand obviates the need for the stage of DNA denaturing.
Nicking endonucleases can also significantly simplify methods of isothermal amplification of DNA with strand displacement, in particular, the SDA (strand displacement amplification) technique in which thiophosphate derivatives of DNA are used for nicking one DNA strand [36]. Restriction endonucleases do not cleave fully modified DNA, but some of them, such as HincII, are able to introduce a nick into semi-modified sites where only one strand is modified. Correspondingly, the unmodified strand is hydrolyzed by the endonuclease. However, this method is not always applicable (for example, in the case of total amplification of genomic DNA), and it is also rather expensive.
Another field where nicking endonucleases can substitute for traditional methods is cloning of DNA such as PCR fragments. When cloning PCR fragments, sites for restriction endonucleases are usually introduced into the primer sequences. After amplification, the PCR product and vector are treated by the corresponding endonucleases. In this case protruding ends of 1-4 bp are formed. However, a site for nicking endonuclease, cleaving aside from the recognition site, can be introduced into the primer sequence instead of sites for restriction endonucleases. In this case, sticky ends of any length can be planned. This will obviate the need for ligation and allow a single annealing procedure for the vector to insert sticky ends.
The number of works on practical use of nicking endonucleases is gradually growing. In particular, nickase Nt.BbvCI was used in a work studying recombination and repair processes in E. coli for obtaining nicked DNA and DNA with single-stranded regions [37]. Nickases Nt.BbvCI and Nb.BbvCI were used to obtain nicked plasmids in a work investigating the seven-subunit protein complex Ctf18-RFC of Saccharomyces cerevisiae [38].
Novel methods have also been proposed based on nicking endonucleases. The principle of one of them is very simple [39]. An oligonucleotide duplex containing a sequence recognized by nickase Nt.BstNBI is incubated with the nickase and DNA polymerase. After single strand cleavage, the formed single-stranded fragment (12 nucleotides) is released into solution because its melting temperature is below the reaction temperature (55°C). DNA polymerase uses the free 3′OH end that appears after cleavage and begins synthesis. However, as soon as DNA polymerase completes the duplex, the nickase again introduces a nick into the completed strand. This process is repeated many times, and thus a great amount of 12 bp long oligonucleotide is synthesized. Since the method is rather sensitive (it is possible to achieve over 106 times signal amplification), it is suggested for use in detection of small amounts of genomic DNA and, in general, for use instead of real time PCR.
The use of nicking endonuclease showed the possibility of transferring “cargo” representing an oligonucleotide [40]. The transferred oligonucleotide includes the site sequence of the strand not cleaved by the nickase. An oligonucleotide together with three others, hybridized at short intervals to the first, constitute the “rails” for transporting the “cargo”. As a result, oligonucleotides form a comb-like construct. Half of each of three oligonucleotides in the comb is complementary to the “rail” oligonucleotide, while the other is not complementary and incorporates the sequence of the site of the nickase-cleavable strand. In the case of cargo-oligonucleotide binding to the comb tooth, nickase cleaves the oligonucleotide-tooth, its cleaved-off part leaves into solution, while the cargo-oligonucleotide hybridizes to the adjacent oligonucleotide-tooth. To start the movement from the first tooth, the sequence of the latter is chosen so that the probability of hybridization to it exceeded that to other teeth. In this work, the cargo-oligonucleotide contained at its end a fluorescence quencher, while the second and third teeth contained different fluorophores. Subsequent fluorescence quenching was confirmed by cargo-oligonucleotide transfer. The rate of cargo transfer was 1 Å/sec. The authors supposed that such a linear motor can be used to solve some problems in nanotechnologies.
We have used the ability of nicking endonucleases to cut only one DNA strand in elaboration of a new method of DNA target detection in a reaction proceeding at constant temperature (55°C) [41, 42]. A molecular beacon and a nickase (Nt.BspD6I isolated by us was used in this work) are involved in the reaction. The molecular beacon is an oligonucleotide that in the free state has a hairpin structure with a loop. The loop sequence is complementary to that of target DNA region. A fluorophore is located at one end of the oligonucleotide, while a fluorescence quencher is at the other. Owing to the beacon hairpin structure, fluorophore and fluorescence quencher are in the immediate vicinity of each other; therefore, the fluorophore fluorescence is quenched by the fluorescence quencher. In the case of hybridization of the beacon with single-stranded DNA target, spatial uncoupling of the fluorophore and quencher takes place, generating a fluorescence signal [43].
The principle of our method is as follows. The loop of the molecular beacon is complementary to the DNA target and contains the sequence of nickase recognition site GAGTC (the strand cleaved by nickase), while the DNA target should contain the GACTC sequence (not cleaved strand). This is not a strong limitation because such sequence is rather frequent, on the average of one per thousand base pairs in a random sequence. Hybridization of the molecular beacon loop with the target results in formation of double-stranded DNA, nickase cleaves the molecular beacon, and two single-stranded fragments are formed. Both fragments separate from the DNA target if the melting temperature of either of them is below that of the reaction (55°C). This means that irreversible enhancement of fluorescence will take place in the reaction of beacon cleavage. The target freed of fragments is able to accept the next molecular beacon. Thus, the hybridization–cleavage process can be repeated many times on the same target, providing for signal accumulation. Unlike traditional methods of DNA target detection, where the increase in fluorescence signal during reaction is due to increase in the amount of target DNA, in our method the signal is enhanced due to the increase in the amount of cleaved molecular beacon.
Recently a method based on the use of nicking endonucleases was proposed for labeling unique sequences in double-stranded DNA [44]. The method is based on introduction of two nicks in the same DNA strand at a distance of 15-25 nucleotides from each other. The fragment flanked by nicks is replaced by an oligonucleotide in the strand displacement reaction and is covalently “ligated” to the DNA by ligase. The long 3′-end of the oligonucleotide is not complementary to the DNA target and represents a branched DNA (3′-flap). This end is used as the primer in the rolling circle amplification, and the produced DNA is registered using the fluorescent probe. The proposed method can be used for detection of double-stranded viral DNA.
It is also supposed that nicking enzymes can be used to solve some problems in DNA nanotechnologies and DNA computers [45, 46].
So far, nicking endonucleases are not very widely used in research and medical diagnosis. This is explained by insignificant variety of these enzymes and by the fact that they have become available only rather recently. However, interest in them is continuously growing. It can be expected that in several years nicking endonucleases will become as widespread as polymerases, restriction endonucleases, ligases, and other enzymes used in molecular biology are now.
CONCLUSION
Our finding of a new type of heterodimeric endonuclease has shed light on the origin of nicking endonucleases, formerly considered as natural mutants of restriction endonucleases that had lost their ability for dimerization and, as a result, for DNA cleavage on both strands. Detection of heterodimeric endonucleases BtsI and BsrDI [22] whose properties are similar to those of BspD6I, found by us, shows that heterodimeric endonucleases, one subunit of which in the absence of the other exhibits activity of nicking endonuclease, are probably widespread in nature. However, it is rather difficult to register the presence of heterodimeric endonucleases in bacterial cell lysates.
The presence of restriction endonucleases is traditionally registered by formation of DNA fragments after incubation of substrate DNA with bacterial cell lysate. The low number of detected heterodimeric restriction endonucleases (six) might be due to peculiarities of their behavior during isolation. Heterodimeric endonucleases can be registered only at the stage of substrate incubation of DNA with cell lysate. However, they are lost during subsequent purification. Owing to this, researchers stopped further work with such endonucleases [4, 18, 20]. Thus, at initial stages of nickase isolation directly from the strain Bacillus sp. D6, we came into collision with the fact that restriction endonuclease called by us BspD6III was eluted from the first column in addition to nickase [4]. However, during further purification of this endonuclease only nickase was eluted from the column instead of endonuclease. At the present time, when it has already been shown that the subunits do not interact with each other in the absence of DNA, it became clear why these endonucleases “are lost” during isolation. During chromatography, subunits are released in different peaks, and, as a result, endonuclease activity (substrate DNA fragmentation) cannot be registered in any peak.
In conclusion, we shall consider arrangement of genes of the restriction–modification systems containing heterodimeric restriction endonucleases (Fig. 6). The arrangement is similar in most systems. First, all of them, except for the BslI system, contain two genes of DNA methyltransferase. The presence of two MTase genes is characteristic of systems that recognize an asymmetrical sequence. This is due to differences in the top and bottom strand sequences; therefore, each strand is recognized and methylated by its “own” MTase. The BslI system includes one MTase because the system recognizes a symmetrical sequence. However, in systems containing two MTases the MTase genes are arranged differently relative to genes encoding subunits, this suggesting different mechanisms of regulation of gene expression in different systems.
The system similarity is also found in arrangement of genes encoding individual subunits: these genes are oriented unidirectionally and are almost immediately adjacent to each other. Thus, genes of Bpu10I endonuclease subunits are separated from each other by only three base pairs, in the BspD6I system by one nucleotide, while in the other systems they are even overlapped. In the BslI and BsrDI systems, the last adenine of the TAA stop-codon of one subunit is incorporated in the initiating codon of the second subunit. In the BbvCI system, all genes including those of MTases are overlapped by approximately six nucleotides. Such situation in the subunit gene arrangement suggests that this is a stage of evolution which might be followed by complete fusion of genes encoding individual subunits of heterodimeric endonucleases, i.e. emergence of restriction endonucleases with two catalytic centers.Fig. 6. Arrangement of restriction–modification systems containing genes encoding subunits of heterodimeric restriction endonucleases. Dark gray arrows with subscripts above them R1 or R2 point to genes encoding individual subunits, light gray arrows with subscripts above them M1 and M2 point to genes encoding DNA methyltransferases.
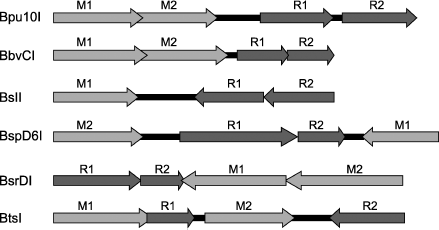
However, the evolution vector might be quite different. Thus, in the BtsI system, genes encoding individual subunits are not simply far apart from each other but they are separated by the MTase gene. Then the situation of subunit gene arrangement can be interpreted quite differently; namely, this stage of evolution does not precede fusion of subunit genes, but on the contrary, this stage precedes complete separation of subunits from each other. This variant of evolution of heterodimeric restriction endonucleases seems preferable for the following reasons. Introduction of a nick into one DNA strand is involved in such important cell processes as replication, recombination, and repair. Therefore, it can be supposed that systems containing only nicking endonuclease are formed in bacterial cells on the basis of restriction–modification systems.
Finally, another fact should be noted: up to the present time, all natural nicking endonucleases have been isolated from bacilli. The only exceptions are two endonucleases, Nt.CviPII and Nt.CvQXI, isolated from the NYs-1 chlorella virus and recognizing a degenerate trinucleotide site [46, 47]. There is still no answer to the question whether the presence of nicking endonucleases is a peculiarity of bacilli.
This work was supported by the Russian Foundation for Basic Research (grants 06-04-48947 and 05-04-48901) and the Naukograd Foundation (grant 04-04-97313).
REFERENCES
1.Roberts, R. J., Belfort, M., Bestor, T., et al.
(2003) Nucleic Acids Res., 31, 1805-1812.
2.Abdurashitov, M. A., Belichenko, O. A., Shevchenko,
A. V., and Degtyarev, S. Kh. (1996) Mol. Biol. (Moscow),
30, 1261-1267.
3.Morgan, R. D., Calvet, C., Demeter, M., Agra, R.,
and Kong, H. (2000) Biol. Chem., 381, 1123-1125.
4.Zheleznaya, L. A., Perevyazova, T. A., Alzhanova,
D. V., and Matvienko, N. I. (2001) Biochemistry (Moscow),
66, 989-993.
5.Dedkov, V. S., Abdurashitov, M. A., Yankovsky, N.
K., Kileva, E. V., Myakisheva, T. V., Popinchenko, D. V., Belichenko,
O. A., and Degtyarev, S. Kh. (2001) Biotekhnologiya, 4,
3-8.
6.Higgins, L. S., Besnier, C., and Kong, H. (2001)
Nucleic Acids Res., 29, 2492-2501.
7.Perevyazova, T. A., Rogulin, E. A., Zheleznaya, L.
A., and Matvienko, N. I. (2003) Biochemistry (Moscow),
68, 984-987.
8.Gololobova, N. S., Okhapkina, S. S., Abdurashitov,
M. A., and Degtyarev, S. Kh. (2005) Mol. Biol. (Moscow),
39, 960-964.
9.Yunusova, A. L., Rogulin, E. A., Artyukh, R. I.,
Zheleznaya, L. A., and Matvienko, N. I. (2006) Biochemistry
(Moscow), 71, 815-818.
10.Stahl, F., Wende, W., Wenz, C., Jeltsch, A., and
Pingoud, A. (1998) Biochemistry, 37, 5682-5688.
11.Pingoud, A., and Jeltsch, A. (2001) Nucleic
Acids Res., 29, 3705-3727.
12.Vanamee, E. S., Santagata, S., and Aggarwal, A.
K. (2001) J. Mol. Biol., 309, 69-78.
13.Samuelson, J. C., Zhu, Z., and Xu, S. (2004)
Nucleic Acids Res., 32, 3661-3671.
14.Zhu, Z., Samuelson, J. C., Zhou, J., Dore, A.,
and Xu, S.-Y. (2004) J. Mol. Biol., 237, 573-583.
15.Armalyte, E., Bujnicki, J. M., Gledriene, J.,
Gasiunas, J., and Lubys, A. (2005) J. Biol. Chem., 280,
41584-41594.
16.Sapranauskas, R., Sasnauskas, G., Lagunavicius,
A., Vilkaitis, G., Lubys, A., and Siksnys, V. (2000) J. Biol.
Chem., 275, 30878-30885.
17.Sasnauskas, G., Halford, S. E., and Siksnys, V.
(2003) Proc. Natl. Acad. Sci. USA, 100, 6410-6415.
18.Stankevicius, K., Lubys, A., Timinskas, A.,
Vaitkeicius, D., and Janulaitis, A. (1998) Nucleic Acids Res.,
26, 1084-1091.
19.Hsieh, P.-C., Xiao, J.-P., O’Loane, D., and
Xu, S.-Y. (2000) J. Bacteriol., 182, 945-955.
20.Heiter, D. F., Lunnen, K. D., and Wilson, G. G.
(2005) J. Mol. Biol., 348, 631-640.
21.Bellamy, S. R. W., Milsom, S. E., Scott, D. J.,
Daniels, L. E., Wilson, G. G., and Halford, S. E. (2005) J. Mol.
Biol., 348, 641-653.
22.Xu, S. Y., Zhu, Z., Zhang, P., Chan, S. H.,
Samuelson, J. C., Xiao, J., Ingalls, D., and Wilson, G. G. (2007)
Nucleic Acids Res., 35, 4608-4618.
23.Kachalova, G. S., Rogulin, E. A., Artyukh, R. I.,
Perevyazova, T. A., Zheleznaya, L. A., Matvienko, N. I., and Bartunik,
H. D. (2005) Acta Crystal., F61, 332-334.
24.Kachalova, G. S., Rogulin, E. A., Yunusova, A.
K., Artyukh, R. I., Perevyazova, T. A., Matvienko, N. I., Zheleznaya,
L. A., and Bartunik, H. D. (2008) J. Mol. Biol., 348,
489-502.
25.Wah, D. A., Hirsch, J. A., Dorner, L. F.,
Schildkraut, I., and Aggarwal, A. K. (1997) Nature,
388, 97-100.
26.Pingoud, A., and Jeltsch, A. (2001)
Nucleic Acids Res., 29, 3705-3727.
27.Pingoud, A., Fuxreiter, M., Pingoud, V., and
Wende, W. (2005) Cell. Mol. Life Sci., 62, 685-707.
28.Viadiu, H., and Aggarwal, A. K. (1998) Nat.
Struct. Biol., 5, 910-916.
29.Kachalova, G. S., Yunusova, A. K., Artyukh, R.
I., Rogulin, E. A., Perevyazova, T. A., Zheleznaya, L. A., Matvienko,
N. I., and Bartunik, H. D. (2007) Acta Crystal., F63,
795-797.
30.Xu, Y., Lunnen, D., and Kong, H. (2001) Proc.
Natl. Acad. Sci. USA, 96, 12990-12995.
31.Demidov, V. V., Protozanova, E., Izvolsky, K. I.,
Price, C., Nielsen, P. E., and Frank-Kamenetskii, M. D. (2002) Proc.
Natl. Acad. Sci. USA, 99, 15953-15958.
32.Protozanova, E., Demidov, V. V., Soldatenkov, V.,
Chasovskikh, S., and Frank-Kamenetskii, M. D. (2002) EMBO Rep.,
3, 956-961.
33.Nuovo, G. J. (2000) Diagn. Mol. Pathol.,
9, 195-202.
34.Andras, S. C., Power, J. B., Cocking, E. C., and
Davey, M. R. (2001) Mol. Biotechnol., 9, 29-44.
35.Schweitzer, B., and Kingsmore, S. (2001) Curr.
Opin. Biotechnol., 12, 21-27.
36.Walker, G. T., Little, M. C., Nadeau, J. G., and
Shank, D. D. (1992) Proc. Natl. Acad. Sci. USA, 89,
392-396.
37.Hebert, M. L., and Wells, R. D. (2005) J. Mol.
Biol., 353, 961-979.
38.Bylund, G. O., and Burgers, P. M. (2005) Mol.
Cell Biol., 25, 5445-5455.
39.Van Ness, J., van Ness, L. K., and Galas, D. J.
(2003) Proc. Natl. Acad. Sci. USA, 100, 4504-4509.
40.Bath, J., Green, S. J., and Turberfield, A. J.
(2005) Angew. Chem. Int. Ed., 44, 4358-4361.
41.Tyagi, S., and Kramer, F. R. (1996) Nat.
Biotechnol., 14, 303-308.
42.Zheleznaya, L. A., Perevyazova, T. A.,
Zheleznyakova, E. N., and Matvienko, N. I. (2002) Biochemistry
(Moscow), 67, 498-502.
43.Zheleznaya, L. A., Kopein, D. S., Rogulin, E. A.,
Gubanov, S. I., and Matvienko, N. I. (2006) Anal. Biochem.,
248, 123-126.
44.Kuhn, H., and Frank-Kamenetskii, M. D. (2008)
Nucleic Acids Res., 36, e40.
45.Wang, L., Hall, J. G., Lu, M., Liu, Q., and
Smith, L. M. A. (2001) Nat. Biotechnol., 19,
1053-1059.
46.Zhang, X., Yan, H., Shen, Z., and Seeman, N. C.
(2002) J. Am. Chem. Soc., 124, 12940-12941.
47.Chan, S.-H., Zhu, Z., van Etten, J. L., and Xu,
S.-Y. (2004) Nucleic Acids Res., 32, 6187-6199.