REVIEW: Structure and Mechanism of Action of Type IA DNA Topoisomerases
D. V. Bugreev1 and G. A. Nevinsky1,2*
1Institute of Chemical Biology and Fundamental Medicine, Siberian Branch of Russian Academy of Sciences, pr. Lavrent’eva 8, 630090 Novosibirsk, Russia; fax: (3832) 33-3677; E-mail: Nevinsky@niboch.nsc.ru2Institute of Cytology and Genetics, Siberian Branch of Russian Academy of Sciences, Novosibirsk, Russia
* To whom correspondence should be addressed.
Received March 19, 2009; Revision received April 9, 2009
DNA topoisomerases are enzymes responsible for regulation of genomic DNA supercoiling. They participate in essential processes of cells such as replication, transcription, recombination, repair, etc., and they are necessary for normal functioning of the cells. Topoisomerases alter the topological state of DNA by either passing one strand of the helix through the other strand (type I) or by passing a region of duplex DNA through another region of duplex DNA (type II). Type I DNA topoisomerases are subdivided into enzymes that bind to the 5′- (type IA) or 3′-phosphate group (type IB) during relaxation of the cleavable DNA. This review summarizes the literature on type IA DNA topoisomerases. Special attention is given to particular properties of their structure and mechanisms of functioning of these enzymes.
KEY WORDS: type IA DNA topoisomerases, structure, reaction mechanismDOI: 10.1134/S0006297909130045
Abbreviations: scDNA, supercoiled DNA; ss and ds, single stranded and double stranded, respectively; topo I, DNA topoisomerase I.
Topological rearrangements of DNA play an important role in the
manifestation of its functional activities (replication, transcription,
recombination, etc.) as well as in the arrangement of higher order
structural organizations. Some of these problems have been analyzed in
monographs and reviews [1-9].
Topoisomerases play a major role in alteration of the topological state
of DNA. The reasons underlying multiple forms of topoisomerases in pro-
and eukaryotic cells have been considered in reviews [1, 8-12],
which also include the analysis of some problems on the mechanisms of
topoisomerase functioning [13-16]. The present review has been undertaken to
summarize the latest data on type IA DNA topoisomerases of higher and
lower organisms and also on the mechanisms of their functioning.
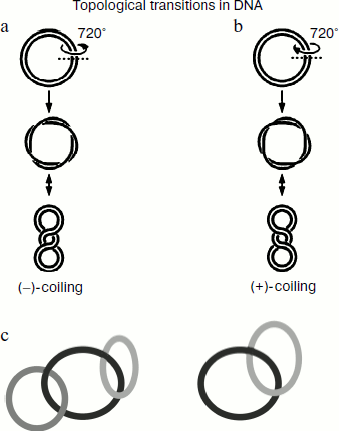
Modern concepts on the structure of genomic DNA suggest that individual transcriptionally active units behave as circular structural units that have lost free ends, and therefore any cell processes related to separation of complementary DNA strands (e.g. replication, transcription, recombination, and repair) induce various structural changes in DNA molecules including their supercoiling. In living cells, some DNA molecules (including the bacterial chromosome) exist in a supercoiled form. Figure 1 schematically shows examples of various topological structures of DNA. It is generally accepted that the negative supercoiling usually defined as “–” corresponds to anticlockwise DNA coiling, whereas the positive one (“+”) corresponds to the clockwise direction. The topological tension originating from genomic DNA supercoiling often represents an obstacle for numerous reactions catalyzed by various enzymes. However, a certain level of supercoiling is required for manifestation of specific functions of some cell proteins.
Supercoiling stimulates an initial stage of genetic recombination because it facilitates insertion of single-stranded fragments into DNA duplex [9]. Thus, the topological state of cell DNA can be used by living cells as a mechanism of regulation of total and local recombination. DNA supercoiling is required not only for genetic recombination but also for processes of normal cell division, replication of chromosomal and plasmid DNA, conjugative plasmid transfer between bacteria, and normal gene transcription [9]. DNA supercoiling also influences inducible mutagenesis. The processes of initiation of replication and transcription as well as differential regulation of transcription of various genes are especially sensitive to supercoiling processes because DNA supercoiling results in susceptibility of some sites for binding of certain proteins. During DNA supercoiling, its fragments (dC-dG) and (dT-dG) can undergo transition from right-handed into left-handed Z-DNA, which can appear in genomes of various organisms and have certain importance at certain stages of DNA functioning.Fig. 1. Schematic presentation of DNA supercoiling followed by formation of anticlockwise “–”-coiling (a) and clockwise “+”-coiling (b) of double-stranded DNA. The scheme shows two DNA catenane structures: two and three interlocked circular DNA molecules (c).
Enzymes that change and regulate the topological state of cell DNA are known as DNA topoisomerases. These are involved in basically all vital processes; they have been found in all pro- and eukaryotes and also in some viruses [1, 8]. Topoisomerases catalyze the reaction of DNA relaxation and insertion of negative and positive supercoils into DNA; these enzymes can also catalyze reactions of linkage and separation of DNA strands and promote renaturation of complementary single-stranded DNA circles.
All topoisomerases belong to the class of isomerases (subclass 99, sub-subclass 1); in accordance with their action mechanism, these enzymes are subdivided into two types (type I and type II). Type I enzymes (EC 5.99.1.2) temporarily cleave just one DNA strand and pass one strand through the break in the second DNA strand, and they do not require the presence of energy cofactors, whereas type II topoisomerases (EC 5.99.1.3) are ATP-dependent enzymes [17]. Type I enzymes are further subdivided into DNA topoisomerases IA and IB; during DNA hydrolysis, type IA enzymes covalently bind 5′-phosphate, whereas type IB enzymes form a bond with 3′-phosphate with the hydrolyzed DNA strand [17]. After DNA cleavage by type IB enzymes, strand transfer involves rotation around bonds that are distant to the break site. On the contrary, type IA enzymes cleave just one DNA strand and fix both ends at a certain distance from each other, whereas another strand (or in some cases double-stranded DNA) passes through this break.
It should be noted that in contrast to type I enzymes, type II topoisomerases insert temporal ATP-hydrolyzing double-stranded break, and they pass one site of double-stranded DNA through the other one. These enzymes can also be subdivided into relaxing topoisomerases and topoisomerases that can catalyze formation of topologically tensed DNA.
DNA gyrase (topoisomerase II) is a bacterial enzyme that introduces (in an ATP-dependent manner) negative supercoils into closed circular DNA molecules. Regulation of the topological state of chromosomal DNA in bacteria also involves DNA topoisomerases I, which relax negative supercoils and therefore exhibit the opposite effect (compared with DNA gyrase) on DNA. Combined effects of these two enzymes and their cell regulation maintain a certain genome level required for manifestation of activities of enzymes interacting with DNA.
In contrast to bacteria, cells of higher eukaryotes lack enzymes similar to DNA gyrase. However, the nuclear genome of Protozoa contains two gyrase genes; their protein products function in plastids [18]. It is possible that higher eukaryotes do not need such enzymes because alteration in topology of cell DNA is achieved during nucleosome formation. It is suggested that nucleosome structure of chromatin determines DNA supercoiling. Since winding of topologically closed DNA on histone globules causes formation of negative supercoils, this should be compensated by accumulation of the same number of positive supercoils between nucleosomes. This requires the presence of such relaxing enzymes as topoisomerase I. Eukaryotic topoisomerases I can relax both positive and negative supercoils, and together with topoisomerases II these enzymes are involved in regulation of supercoiling degree at the level of nucleosome DNA and also during processes influencing topology of DNA duplex.
CLASSIFICATION AND SOME PROPERTIES OF TOPOISOMERASES
The first DNA topoisomerase I (topo I) was isolated from E. coli in 1971 [19]; later this enzyme was characterized in more detail [20]. In 1972 eukaryotic topo I was found in mammalian cells [21], and then it was isolated from yeasts and from many mammals [21-29]. Subsequently, many topoisomerases I were isolated from numerous pro- and eukaryotic organisms: topo III from E. coli [31, 32] and yeasts [33], reverse gyrase from the thermophilic archaean Sulfolobus acidocaldarius [34], and quite recently topo V from the hyperthermophilic Methanopyrus kandleri [35, 36]. A new type IA topoisomerase, Dam topo III, was isolated from the thermophilic archaean Desulfurococcus amylolyticus [37].
The table shows the existing classification of type I topoisomerases. According to their origin and due to differences in biochemical properties of eukaryotic and prokaryotic type I topoisomerases, these enzymes have been subdivided into two groups. There is a special group of viral topo enzymes. Prokaryotic and eukaryotic topoisomerases catalyze the relaxation of negatively supercoiled DNA (“–”, Fig. 1). In addition, eukaryotic topo I also catalyzes relaxation of positively supercoiled DNA (“+”, Fig. 1). Prokaryotic topo I can catalyze the knotting reaction of single-stranded circular DNA. The poxvirus enzyme is three times smaller than the eukaryotic topo and shares homology with its C-terminal site; it relaxes both “–” and “+”supercoiled (sc) DNA [38, 39].
Classification of type I topoisomerases
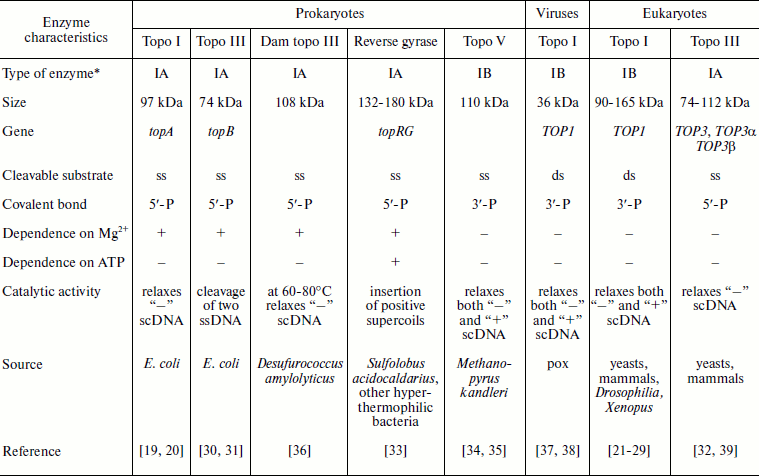
*All topoisomerases are class 5 enzymes (isomerases),
subclass 99, sub-subclass 1 (EC 5.99.1).
Discovery of new topo I enzymes and study of their properties have shown that their classification by origin does not adequately reflect the existing situation, and it has been proposed to subdivide type I topoisomerases by their primary structures [1].
For example, eukaryotic yeast topo III is very similar to E. coli topo I and III, and its specificity towards single-stranded DNA is similar to these enzymes [40]. Discovery of yeast topo III and its classification as a type IA topoisomerase has demonstrated that the topo classification into eukaryotic and prokaryotic enzymes is not entirely correct. Recently, it has been found that human cells also contain a TOP3 gene that is homologous to the yeast gene, but the protein product of the human gene contains an additional C-terminal site similar to the C-terminal site of the E. coli enzyme [41]. Discovery of the reverse gyrase originally isolated from S. acidocaldarius significantly extended the class of type IA topoisomerases. It was demonstrated that this enzyme catalyzes ATP-dependent introduction of positive supercoils into DNA [34, 42]. Catalytic activity similar to that of the S. acidocaldarius enzyme has also been found in many bacteria and archae [43-45]. The N-terminal part of the protein contains motifs found in DNA helicases, and a sequence of the C-terminal part shares similarity with the IA subclass of enzymes [46]. In addition, reverse gyrase from D. amylolyticus also exhibits the same sequence specificity towards cleavable DNA as bacterial topo I [47].
The existence of topoisomerases corresponding to the IB subclass was found in some prokaryotes after isolation and analysis of topo V from M. kandleri [35]. Biochemical properties of this enzyme are similar to the enzymes of IB subclass rather than IA subclass: it relaxes both “–” and “+” scDNA and during cleavage forms a covalent bond with the 3′-end of the cleavable DNA strand [35].
Properties of DNA topoisomerases I and IIIa from Bacillus cereus are similar to E. coli topo I and III [48]. Recently, an unusual topo IA was isolated from B. cereus and characterized [49]. This enzyme shares 64 and 33% homology with topo III from Bacillus subtilis and E. coli, respectively. Properties of this enzyme significantly differ from those of bacterial topo IA including the E. coli IA and IB enzymes. This enzyme caused only partial relaxation of “–” scDNA, and it was unable to totally relax DNA. In contrast to E. coli topo III, B. subtilis topo IIIa lacks decatenase activity, which cleaves DNA catenanes (Fig. 1 schematically shows DNA catenanes: two or more interlocked circular DNA molecules); this enzyme cannot compensate lack of E. coli topo III in vivo. Thus, topo IIIb from B. cereus was classed as a unique prokaryotic type IA topo.
SOME ASPECTS OF MECHANISMS OF TOPOISOMERASE ACTION
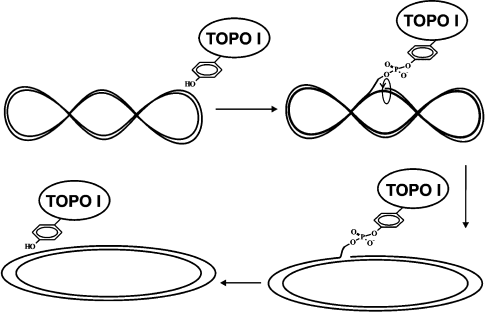
Convincing evidence now exists that the reaction catalyzed by all topoisomerases includes two sequential trans-etherification reactions. During the first stage the OH-group of the enzyme active site tyrosine residue attacks an internucleoside phosphate group, and this results in a covalent tyrosine-phosphate bond with one of the ends of cleaved DNA. The second reaction consists of religation of the DNA strand followed by release of the enzyme from its covalent complex with DNA (Fig. 2). Relaxation of scDNA occurs between these two reactions. The mechanism of DNA relaxation differs in pro- and eukaryotic enzymes, and this will be considered below [13-16].
In all known topoisomerases the active site tyrosine residue acts as a nucleophile in the reaction of DNA cleavage. By analogy with other phosphotransferase processes, the reaction catalyzed by topoisomerases should be classed as a reaction that follows the mechanism of acid–base catalysis. This viewpoint has been supported by results of the study of the kinetics of the reaction catalyzed by poxvirus topo I. The conclusion on base catalysis during tyrosine attack of a phosphodiester bond of DNA and acid catalysis providing protonation of the leaving 5′-hydroxyl group is based on the pH-dependence of the rate of cleavage and religation of the DNA strand [50, 51]. These data and also results of kinetic studies employing a substrate in which oxygen (that was not involved into formation of the phosphoester bond with DNA) was substituted for sulfur revealed that the cleavage reaction is accompanied by a conformational change of the topo I–DNA complex; this was not accompanied by cleavage of bonds formed by amino acid residues of the enzyme (involved into acid–base catalysis) with cleavable phosphate and released 5′-OH-group. In addition, the rate of the cleavage reaction is limited by effectiveness of covalent bond formation, whereas the religation rate is limited by reversible conformational changes of the enzyme [51].Fig. 2. Common mechanism of type I DNA topoisomerases: supercoiled DNA relaxation by the enzyme.
The phosphotyrosine intermediate formed between DNA and the topo enzyme during the reaction is obviously well shielded from solvent molecules at the active site of the enzyme because the religation reaction involving a hydroxyl group of the cleaved DNA strand is more probable than solvolysis of the phosphotyrosine bond. In case of incorrect spatial positioning of the deoxyribose hydroxyl group formed after DNA cleavage, hydrolysis or alcoholysis of the covalent DNA–topo I complex in E. coli and eucaryotes can occur [52-55]. These processes competing with the normal religation reaction can result in damage to the cell DNA. However, at neutral pH values the rate of solvolysis is rather low. It is possible that in contrast to an OH-group of DNA deoxyribose, an OH-group of a water molecule is not optimally oriented in the catalytic “pocket” of the enzyme and therefore cannot provide a high rate of phosphotyrosine bond hydrolysis.
Substitution of an oxygen atom on the phosphorus atom of the internucleoside phosphate group by a sulfur atom results creates a chiral center at the phosphorus atom and produces two chemical configurations known as the Rp and Sp diastereomers. Interaction of the cleavable internucleoside phosphate group with an amino acid residue of the E. coli topo I active site is stereospecific, because after substitution of the oxygen atom (that is not involved into formation of a phosphodiester bond) of the cleavable phosphate group by sulfur the enzyme could cleave only the Rp stereoisomers of the oligonucleotides [53]. Substitution of a phosphodiester bond for the 5′-phosphorothioate bond caused irreversible DNA cleavage by eukaryotic topo I because the 5′-sulfhydryl group formed after DNA cleavage could not serve as the nucleophile in the religation reaction [56].
Escherichia coli TYPE I TOPOISOMERASE AS A TYPICAL MEMBER
OF THE IA SUBCLASS
Among type IA topoisomerases, topo I from E. coli (EcTopoI) is the best-studied enzyme [19]. It is a monomeric metal binding protein of 97 kDa (Fig. 3a). Escherichia coli topo I preferentially binds single-stranded (ss) DNA and can cleave ssDNA sites followed by subsequent ligation. The enzyme forms contacts with both 5′- and 3′-terminal regions of the DNA cleavage site [20]. The existence of a binding site for single-stranded DNA at the topo I catalytic center accounts for cleavage of even short oligonucleotides (seven nucleotides in length) by this enzyme. Cleavage occurs at the distance of three nucleotides from the 5′-end and four nucleotides from the 3′-end of the cleavable site [57].
After substrate cleavage, the enzyme still binds to the 5′-phosphate of DNA via the hydroxyl group of a tyrosine residue. In contrast to eukaryotic enzyme, E. coli topo I relaxes “–” scDNA. Such specificity of topo I is associated with its ability to recognize ss sites of DNA; the binding energy of topo I onto “–” scDNA is sufficient to overcome the energy barrier for strand separation at a short fragment of the duplex. In the case of relaxed or “+” scDNA this requires much more energy, thus explaining the inability of the topo to form a covalent intermediate with such DNA substrates [19, 20]. Sites of topo IA subclass enzyme that are located outside the catalytic “pocket” are also involved in DNA binding and the relaxation reaction of the DNA substrate. For example, E. coli topo I contains three zinc ions [58, 59] that bind in the C-terminal region of the protein (residues 598-737), where three tetracysteine domains involved in binding and manipulations with DNA strands are located [60, 61]. In the absence of all three Zn2+-binding sites, topo I is unable to relax DNA but can still bind and cleave DNA [59, 62, 63]. The first or first two of the three Zn2+-binding domains are required for manifestation of the relaxing activity: removal of the C-terminal part of the protein containing the first two tetracysteine sites resulted in enzyme inactivation [62]. Removal of zinc ions also eliminated the relaxing activity of topo I [64, 65]; however, the enzyme could bind and cleave single-stranded oligonucleotides [64]. The latest data on the mechanism of E. coli topo I suggest that the site of this enzyme that consists of C-terminal and Zn2+-binding domains (ZD-domain) is not involved in DNA recognition and catalytic cleavage/religation; it contacts with one of the duplex strands, which topo I passes through the break formed during relaxation of “–” scDNA [66]. A 30-kDa domain, which is resistant to proteolysis and located at the central part of the enzyme, contains one tyrosine residue that acts as a nucleophile in the reaction of DNA cleavage. This domain can cleave single-stranded oligonucleotides [10]. The topo I site (of 67 kDa), which includes the first 596 residues, shares sequence identity with all members of subclass IA topoisomerases [67]. This protein fragment cannot catalyze DNA relaxation but can bind and cleave ssDNA (as the full-length enzyme and the enzyme lacking zinc ions) [63]. These data suggest that the ZD-domain of this enzyme is responsible for manipulation with DNA strands during relaxation. The remaining part of topo is responsible for substrate binding, cleavage, and its subsequent relegation.
STRUCTURAL ANALYSIS OF ENZYMES
X-Ray analysis of enzymes and their complexes with ligands is one of the most informative methods used for elucidation of spatial structure of proteins and their contacts with DNA. The X-ray data represent a convincing basis for some suggestions on possible mechanisms of enzyme action. This is very important for studies of the main principles of protein–nucleic acid interactions. During recent years the structure of various fragments of type I DNA topoisomerases has been studied by X-ray analysis and nuclear magnetic resonance [67-72].
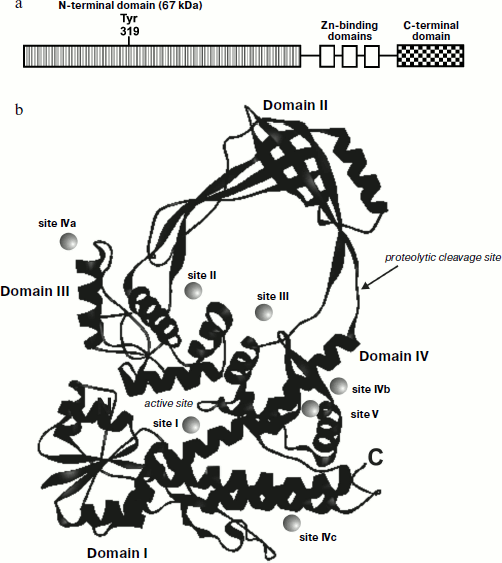
The structure of the N-terminal fragment (67 kDa) of E. coli 30 topo I was determined at 2.2 Å resolution [67]. This enzyme fragment consists of four domains, which form a toroid (Fig. 3b). The average distance between α-carbon atoms is 33.5 Å, and between polypeptide chains it is 27.5 Å. The first 160 residues form an α/β domain also known as a Rossman hairpin [73], which consists of four parallel β-chains fixed between four α-helices (domain I). This domain contains several highly conserved sites. The top of the toroid (domain II) is crescent-shaped due to intersection of several antiparallel β-sheets at an angle of 90°. The tyrosine residue involved in DNA cleavage is located in the third domain, which consists of several α-helices and contains many highly conserved residues. The remaining part of the 67 kDa fragment of the topo consists of several α-helices forming domain IV.
Recently the X-ray analysis of Thermotoga maritima topo I was performed with 1.7 Å resolution [74]. The full-length bacterial enzyme has a topo-like structure; it contains a conserved trans-esterification domain that includes domains I-V and a C-terminal sequence that binds zinc ion (domain V) and which is located just opposite domain IV.Fig. 3. Structure of E. coli type I DNA topoisomerase. a) Domain organization of topo I. b) Structure of N-terminal fragment (67 kDa) determined by X-ray analysis. Location of nucleotide binding sites of topo I was determined from X-ray data obtained for complexes of the enzyme with various mono- and trinucleotides [73].
Active site localization in E. coli topo is determined by the presence of catalytic Tyr319 (domain III) positioned between domains I and III (Fig. 3b). Tyr319 and also residues located in this region are involved in formation of contacts between these two domains; they form a site which is highly conserved for all proteins of this subclass [75]. Tyr319 interacts with several conserved residues (Glu9, Lys13, Asp111, Asp113, Glu115, Gln309, Glu313, Tyr312, Arg321), and this forms a wide network of hydrogen bonds that also includes water molecules and salt bridges (Fig. 4). Tyr319 forms a hydrogen bond with Asp111 via a water molecule and with Asp113 and Glu115, which involves two water molecules. The specific conformation of these three residues shares some structural resemblance with the exonuclease center of the Klenow fragment of E. coli DNA polymerase I [76] involved into metal ion binding. However, metal ion binding in this region was not detected during X-ray analysis of the N-terminal fragment of E. coli topo I; this is not surprising because these residues are involved in formation of a hydrogen bond network. Binding of magnesium ion by these residues might depend on conformational changes of the enzyme structure that occur during catalysis or when the protein adopts a conformation that allows interaction of these residues with the DNA substrate.
For identification of amino acid residues involved in the reaction between DNA and catalytically active Tyr319 of the topo active site, 12 highly conserved polar residues (Glu9, His33, Asp111, Glu115, Gln309, Glu313, Thr318, Arg321, Thr322, Asp323, His365, and Thr496) were substituted by alanine [77]. Results of that study showed that only mutations of Glu9 and Arg321 resulted in some decrease in relaxing activity of the enzyme. Topo with mutation R321A cleaved DNA with lower efficiency. Replacement of Arg321 by lysine had an insignificant influence on the enzyme activity, whereas substitution of Glu9 by glutamine significantly decreased relaxing capacity of topo I but had no influence on the reaction of DNA cleavage and religation. Thus, it appears that Glu9 plays a decisive role on the trans-esterification stage by interacting with the 3′-OH group. Positively charged Arg321 might also be involved in this process by interacting with cleavable DNA phosphate [77].Fig. 4. Spatial localization of various amino acid residues around catalytically active Tyr319 in E. coli DNA topoisomerase I. Dark dots indicate contacts between various residues.
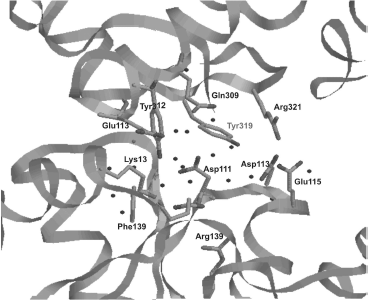
The X-ray analysis of the complex of the N-terminal fragment (67 kDa) of E. coli topo I with single-stranded DNA showed approaching of parallel DNA to the protein helix [78]. Results of the X-ray analysis of complexes of type IA topoisomerases with ssDNA suggest that Ser192, Arg195, and Gln197 are highly conserved residues that might be important for catalysis [79]. Site directed substitutions of these residues revealed that Arg195 and Gln197 are required for DNA cleavage and also for correct adaptation of the structure of the DNA G strand with the enzyme before its catalytic cleavage [80]. Mutation of Ser192 did not influence enzyme binding with scDNA, but it decreased the relaxation rate.
Recently, experimental theoretical analysis of the role of Ser10 and Lys13, which are highly conserved residues for all type IA topoisomerases, has been performed [81]. Substitution of these residues in E. coli topo I for Ala resulted in the decrease of hydrolytic and relaxing activities of this enzyme. Lys13 interacts with Glu9, which is believed to be involved into catalysis. Authors suggest that the decrease in the enzyme activity seen after Lys substitution may be attributed to the fact that this residue acts as a proton donor for Glu9 or cation facilitating the reaction of DNA hydrolysis. Ser10 forms hydrogen bond with internucleoside phosphate group and its substitution causes impairments in correct binding.
Site-directed mutagenesis of the conserved N-terminal Gly194 of the α-loop of E. coli topo I showed that increased mobility of amino acid residues around Gly194 is required for manifestations of the hydrolyzing and relaxing activities associated with opening and closing of the so-called “gate” of this enzyme during its binding with cleavable DNA G-strand and also for subsequent relaxation processes [82].
A 30-kDa fragment formed during proteolysis of E. coli topo I consists of domains II and III. The protease-sensitive site of this enzyme is located in the polypeptide chain connecting these domains with the other part of the protein globule (Fig. 3b). Interestingly, removal of these two domains insignificantly influenced the conformation of the other part of the protein molecule.
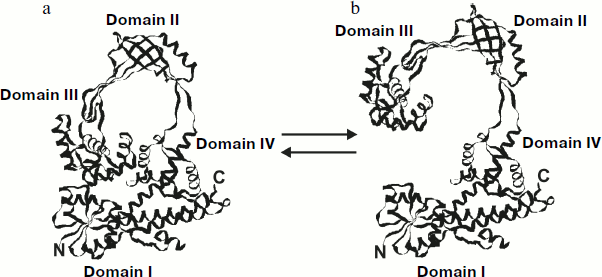
Topo I can adopt two possible conformations: in the “closed” conformation when domain III forms contacts domain I and in the “opened” conformation when domain III is separated from the main enzyme globule due to small rotation of a loop, formed by domains II and III, around two exposed proteolysis-sensitive polypeptides connecting these domains with the other part of the enzyme (Fig. 5).
The existence of more than one protein conformation is very important for DNA recognition by topo I and for manipulation with DNA strands. In the closed conformation the active site of topo I is inaccessible for interaction with DNA (Fig. 5a). In the opened conformation (Fig. 5b) the “catalytic” tyrosine and other residues of domain I involved in substrate recognition and catalysis can interact with DNA. In addition, ss- and dsDNA can easily enter (and leave) the inner cavity of the enzyme in the opened conformation; this occurs via the space opened between domains I and III. The existence of toroidal protein structure suggests that the inner cavity of the toroid might be a DNA binding site. This region contains a reasonable number of linearly located positively charged residues, and the size of this cavity suggests the possibility of dsDNA binding without any steric hindrance. The significant positive electrostatic potential inside the toroid also suggests a possible role of this site in DNA binding [67].Fig. 5. Conformational rearrangements in E. coli topo I: a) closed enzyme conformation; b) opened enzyme conformation.
One of the interesting aspects of the reaction catalyzed by this topo enzyme is the stage of scDNA relaxation. In the case of type IA enzyme, results of biochemical and structural studies support a reaction mechanism in which relaxation occurs via passage of the uncleaved DNA strand via the break formed in the second strand; this is accompanied by unwinding of just one supercoil.
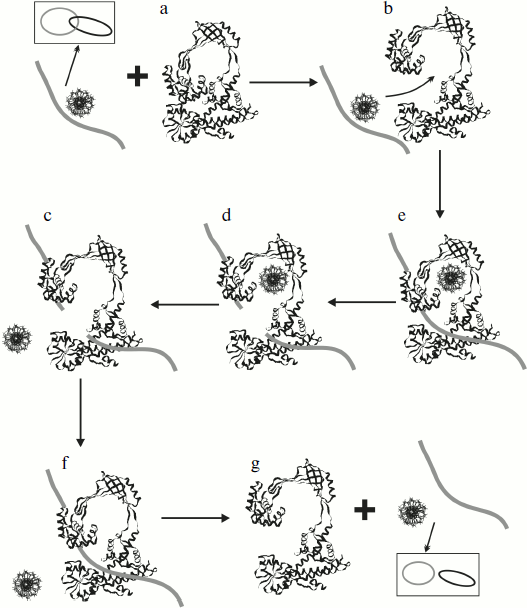
Results of the structural X-ray analysis of the topo I fragment (67 kDa) are also valuable for suggestion of a possible mechanism of passage of the DNA strand through the formed break (taking into consideration the existence of at least two conformational states of this enzyme, closed and opened). This mechanism can be well illustrated using the reaction of catenation/decatenation of dsDNA [67] (Fig. 6). Escherichia coli topo I can catalyze catenation/decatenation of two dsDNA provided that one of them contains a single stranded site (because this enzyme can cleave only ssDNA). This reaction has been well studied [83, 84]. Taking into consideration X-ray data, it is believed that initially the enzyme adopts the opened conformation (Fig. 6b) followed by subsequent binding of two DNA molecules (Fig. 6c): one of these molecules enters the inner cavity of topo I, whereas the other enters the active site. The enzyme then cleaves DNA at its single-stranded site followed by separation of the broken segment ends (Fig. 6d); after that the enzyme can pass the other DNA molecule through the transiently formed break (Fig. 6e), and then the cut ends are religated (Fig. 6f). Then the DNA molecule dissociates from the active site of the enzyme (Fig. 6g). This scheme implies significant conformational rearrangements of the enzyme required for separation of the broken DNA strand and entry of the second DNA molecule into the inner cavity of the enzyme. The unusual structure of topoisomerase meets all these criteria. One can see that active site susceptibility requires enzyme transition into the opened conformation. The cleavage reaction and separation of the cut strand occurs almost simultaneously. The enzyme holds DNA via covalent interaction between one half of the cleaved strand (plus end) with domain III and noncovalent interaction of the other half (minus end) with domain I. Analysis of the nucleotide sequence in the cleavage site region showed that noncovalent interactions of the enzyme with the minus end of the cleaved strand are specific: they involve preferential binding of four cytosines at the 5′-end of the cleavage site. The possibility of binding of the minus end of the cleaved strand by domain I is supported by the presence of the Rossman hairpin at this site; the Rossman hairpin was also found in nucleotide or dinucleotide binding sites of various proteins.
X-Ray data can be used to explain the mechanism of scDNA relaxation by E. coli topoisomerase I, the relaxation of only negative supercoils by this enzyme. Initially binding of dsDNA occurs in the inner cavity of topo I, and then the strands require decatenation for entry of one of them into the active site. Subsequent cleavage and passage of the second strand through the formed break unwinds one supercoil in the DNA. In the case of “+” scDNA, such decatenation of strands requires much energy, whereas in the case of “–” scDNA the process is easier. The amount of energy consumed for strand decatenation depends on degree of supercoiling, and so the rate of relaxation is higher when topological tension is higher in the DNA.Fig. 6. Mechanism of decatenation reaction of two circular DNA molecules (one of them contains a single stranded break) catalyzed by E. coli topo I (see explanations in the text) (adapted from [67]).
INTERACTION OF E. coli TOPOISOMERASE I WITH DNA
Recent X-ray data obtained for various complexes of E. coli topo I with mono- and trinucleotides indicate that there are at least five potential DNA binding sites on the protein globule of this enzyme [85]. Figure 2 shows the localization of these binding sites. The data suggest that during the recognition process topo I forms contacts the sugar-phosphate backbone of nucleotide ligands, and it obviously does not interact with the bases [85].
The first DNA binding site is located outside the active site of the topoisomerase, in the cavity between domains I, III, and IV; the mononucleotide phosphate group interacts with Arg114 and Arg161. Two other arginines (Arg136 and Arg493) located near these residues provide high positive potential at this particular site, thus optimizing conditions for DNA binding [85]. The second site is located in the large central cavity of topo I. It appears that this site is involved in binding of one of DNA strands that enters the inner enzyme cavity after decatenation of duplex strands. Recognition can involve Arg296 and Arg396, which form bonds with phosphate groups. The third site is located in the central cavity of topo I in close proximity to the positively charged protein cluster, and this makes it the high-affinity DNA recognition site. Direct contact with ligands is determined by Arg516, Gln291, and Arg515 [85]. The fourth site specifically binds 3′,5′ADP (5′-pAp-3′), which does not interact with any other detected sites. During ligand binding, 3′-phosphate contacts with Glu520; this is a rather unusual case of interaction between a protein carboxyl group and an internucleoside phosphate group of DNA. In the forming structure, 3′,5′ADP forms a bridge between adjacent protein molecules; this suggests that nucleotide binding at this site of the enzyme might represent a feature of the crystal package. However, 3′,5′ADP stimulated conformational changes including the active site of the enzyme; this suggests that this site can contribute to DNA binding at the first recognition site [85]. The fifth site was classified as a phosphate-binding site formed by Arg535 and Arg202 of domain IV.
Minimal substrates for E. coli topo I are either oligoadenylates seven nucleotides in length or oligothymidylates eight nucleotides in length [57]. DNA cleavage requires enzyme contacts with three internucleoside phosphate groups at the 3′-end and two phosphate groups at the 5′-end (versus the cleavage site). It should be noted that the interaction of topo I with cleavable and (–1)-phosphate groups is not stereospecific; however, the enzyme was 10 times less effective in cleavage of a substrate containing a (Rp)-thio-isomer at the (+1) position compared with cleavage of its (Sp)-isomer [86]. The reverse dependence was observed for phosphate groups at the (+3) and (–2)-positions, where the (Rp)-conformation was preferential to the (Sp)-isomer. The difference in the cleavage rate of various thio-isomers was significantly higher than differences in enzyme affinity to modified ligands [86]. This suggests that the cleavage can occur only in the case of correct substrate localization at the active site, when conformational changes in DNA required for the catalytic stage occur most effectively.
Escherichia coli DNA TOPOISOMERASE III
Escherichia coli DNA topoisomerase III, a polypeptide that consists of 653 residues [87], was originally isolated from cells carrying a deletion in the gene encoding topo I (topA) and characterized as a protein that could relax scDNA [32]. Later it was found that this enzyme also demonstrated highly effective catalytic strand decatenation during replication [88]. It was shown that in E. coli cells this enzyme functions as a decatenase [89].
In contrast to topo I, topo III cannot relax negatively scDNA under standard reaction conditions (10 mM Mg2+, 50 mM Na+ or K+, 37°C), this catalytic reaction requiring higher temperature (52°C) and lower salt concentration (<20 mM) [32, 88]. However, both decatenation of plasmid DNA dimers and unlinking of DNA strands during replication catalyzed by topo III do not require such strict limitations and can be performed under standard conditions [88]. in vitro topo I cannot catalyze the reaction of strand decatenation during replication [90], and this discriminates the role of topoisomerases in the cells: topo III decatenates newly synthesized DNA molecules during replication, whereas topo I is involved in maintenance of a certain level of supercoiling of chromosomal DNA [88].
Recently it has been demonstrated that the biological role of topo III in recombination is similar to that of RuvABC protein [91]. Since E. coli cells are nonviable only when they are simultaneously deficient in two type IA enzymes, topo I and III, it is believed that these enzymes share the main topoisomerase functions in the cells [92].
Topo III is a sequence-specific protein. This enzyme exhibits higher catalytic efficiency with respect to oligonucleotides containing the following sequence.
![]()
The minimal DNA fragment required for manifestation of catalytic activity of topo III consists of seven nucleotides in length: six nucleotides 5′ to the cleavage site and just one nucleotide 3′ to the cleavage site [93]. Topo also asymmetrically interacts with the DNA sequence and protects against nuclease hydrolysis of a fragment of about 14 nucleotides: two nucleotides 3′ to the cleavage site and 12 nucleotides 5′ to the cleavage site of the DNA sequence [93]. Thus, during catalysis the enzyme forms tight noncovalent bonds with the 5′-site and only a minimal number of contacts with the 3′-site of DNA. Such asymmetric interaction is reasonable, because during catalysis the enzyme is covalently bound to the 3′-fragment and, consequently, it does not require tight fixation of this part of the DNA by noncovalent bonds. In contrast to topo I, this enzyme can cleave not only DNA but also RNA molecules [94].
Topo III as well as topo I exhibits highly effective interaction with ssDNA and rather weak interaction with duplexes [95]. In the case of scDNA relaxation by this enzyme, its binding with single-stranded sites can occur right after decatenation of duplex strands, which is facilitated by the presence of negative supercoiling. During decatenation of DNA strands formed in the process of replication, topo III can bind to short DNA fragments formed in the replicating DNA after removal of RNA primers [93]. This suggestion is supported by the fact that the reaction of decatenation of DNA dimers catalyzed by topo III is significantly facilitated by the presence of even small single-stranded sites in the DNA substrate [88].
Analysis of the gene encoding topo III (topB) showed that this enzyme shares significant homology with topo I [86]. However, in spite of significant homology, these enzymes catalyze different reactions. It should be noted that homology between these proteins covers the first 600 residues. Although C-terminal sites of topo I and topo III are involved in DNA binding, they demonstrate significant difference [95, 96]. The C-terminal site of topo I contains three “zinc fingers” and a large number of Lys and Arg [58]. In topo III, this site lacks any known motifs, but it is also rich in positively charged residues [95]. Sequential removal of C-terminal residues of this protein is accompanied by a gradual decrease in the affinity of the enzyme to DNA. Removal of all these residues decreased the enzyme processivity in relaxation of “–” scDNA, but did not influence selection of the cleavage site [95]. Removal of 49 C-terminal residues of topo III resulted in a two orders of magnitude decrease in the enzyme affinity to the single-stranded substrate and a similar decrease in catalytic activity of this enzyme [93]. Replacement of these residues in topo III for the C-terminal fragment of topo I caused almost total recovery of relaxing activity, but decatenase activity towards DNA strands improved insignificantly [93]. Thus, structural differences of the C-terminal domains of these enzymes are mainly responsible for the differences in catalytic activity between these enzymes.
X-Ray analysis of E. coli topo III showed that the structural organization of this enzyme is similar to that of type IA topoisomerases [97]. The protein globule is composed of four domains that form a toroid structure similar to that of topo I; however, relative positioning of the four domains significantly differs. The main difference between these proteins consists of the presence of an additional 17 residues in topo III; these residues form a positively charged loop protruding from the central cavity of the enzyme. This fragment might be involved in the reaction of catenation/decatenation of DNA [85]. Indeed, removal of this domain resulted in a sharp (four orders of magnitude) decrease in topo III activity in the catalytic decatenation of DNA strands, whereas DNA relaxing activity demonstrated just a 20-fold decrease [98]. The topo III site responsible for binding of single-stranded DNA is a groove located on the protein globule and directed towards the active site [97].
REVERSE GYRASE
Reverse gyrase is a protein with unusual functions that is produced by hyperthermophilic organisms. In contrast to other topoisomerases, this enzyme performs ATP (dATP)-dependent insertion of positive supercoils into DNA [99]. In the presence of other triphosphates (UTP, GTP, or CTP), reverse gyrase catalyzes only relaxation of “–” scDNA [100]. The enzyme more effectively hydrolyzes ATP in the presence of ssDNA rather than dsDNA; this suggests preferential binding of the protein to single-stranded sites of DNA [100]. Reverse gyrase was originally isolated from S. acidocaldarius [34]. Study of the action mechanism of the enzyme has shown that during reaction reverse gyrase forms a covalent bond with the 5′-phosphate of cleavable DNA [47] and, consequently, it can be classified as a type IA topoisomerase. Subsequently it was demonstrated that such type of activity is present in all hyperthermophilic bacteria [101], and “+” supercoiling provided by this enzyme is required for stabilization of the duplex structure of chromatin at high temperatures; this prevents local melting of DNA and also facilitates renaturation of the double-stranded structure after passage of these DNA sites by the transcription complex [89].
Reverse gyrase is a large monomeric protein [99]. Analysis of the amino acid sequence of the enzyme from S. acidocaldarius showed that the C-terminal domain shares rather high structural resemblance with corresponding domains of E. coli topo I and yeast topo III [46]. The N-terminal domain of this enzyme lacks any homology with known topoisomerases, but it contains several helicase motifs including the ATP-binding site [46]. The topoisomerase domain of this enzyme can exhibit DNA relaxing activity, whereas the helicase fragment cannot catalyze melting of the duplex chains; it appears that within the full-length enzyme the latter acts as an ATPase responsible for conformational changes that are required for manifestation of catalytic activity of the enzyme [101]. In contrast to reverse gyrases obtained from other sources, the enzyme isolated from M. kandleri is a heterodimer that consists of 43 kDa (RgyA) and 138 kDa (RgyB) subunits, which (by analogy with monomeric enzymes) exhibits topoisomerase and ATPase activities, respectively [99].
Reverse gyrase as well as bacterial topo I exhibits highly effective binding to single-stranded sites of DNA [45, 100]; this is not surprising because of the existence of certain homology between these enzymes. Such preference can be attributed to direct functions of these enzymes. In the case of topo I, effective formation of single-stranded sites is associated with excess of negative supercoils, whereas in the case of reverse gyrase the single-stranded binding sites are formed due to denaturation of dsDNA occurring at high temperatures.
The study of specificity of reverse gyrase from Desulfurococcus amylolyticus showed that its higher efficiency is observed in the cleavage of the sequence:
![]()
DNA TOPOISOMERASE III FROM THERMOPHILIC ARCHAE (Dam topo
III)
Dam topo III is a new type IA topoisomerase isolated from the thermophilic archaean D. amylolyticus [37]. It is a monomeric protein of 108 kDa. Dam topo III is specific for a single-stranded site of DNA, and the catalytic effect of the enzyme consists of alteration of the number of catenanes in the spiral structure of circular DNA [37]. The enzyme exhibits various activities that depend on temperature and reaction conditions: at 60-80°C it relaxes only “–” scDNA (increases catenane number), but at 82-99°C it can relax both “+” (decrease catenanes number) and “–” scDNA. Interestingly, the ability of the enzyme to relax “+” scDNA depends on the growth phase of D. amylolyticus cells: Dam topo III activity is higher in the exponential phase compared with the stationary phase [37]. Catalytic activity requires Mg2+, whereas addition of ATP does not influence catalytic activity [37]. This type of activity is typical not only for D. amylolyticus, but also for other thermophilic archaea [37].
It appears that the ability of Dam topo III to relax “+” scDNA does not have physiological importance and represents a result of the effect of high temperatures on DNA. Indeed, melting of “+” scDNA strands at high temperatures should result in appearance of single-stranded sites. Dam topo III exhibits specificity to such regions and can relax DNA. At lower temperatures “+” scDNA (in contrast to “–” scDNA) is more resistant to unlinking of DNA complementary strands, and consequently it is not a substrate.
Thus, at permissive (for D. amylolyticus) temperature up to 97°C, Dam topo III will promote denaturation of cell DNA rather than stabilization of its duplex structure. Consequently, it seems unlikely that this enzyme is involved in regulation of the topological state of cell DNA, particularly in relaxation of “+” scDNA, because high affinity of reverse gyrase to single-stranded sites formed in cell DNA would prevent Dam topo III binding to these regions [37]. Besides maintenance of DNA in a certain topological state, the role of bacterial topoisomerases can also consist of unlinking of newly formed DNA molecules during replication [37]. Dam topo I (reverse gyrase) obviously cannot play this role due to its inability to reduce the number of catenanes in DNA and therefore to unlink two DNA molecules [37]. In the case of Dam topo III, total unlinking of complementary strands should be the final product of sequential unwinding of scDNA at high temperatures [37]. Consequently, Dam topo III is an analog of topo I of mesophilic bacteria, and it is obviously necessary for unlinking of structures formed during replication [37].
EUKARYOTIC DNA TOPOISOMERASE III
Eukaryotic topo III was originally found in 1989 [33]. A gene encoding yeast topo III was identified by its ability to suppress mitotic recombination between repeating sequences. The protein product of this gene shared homology with E. coli topo I and III and did not exhibit any similarity with known eukaryotic topoisomerases [33]. The biochemical properties of this enzyme were similar to those of E. coli topo III. The protein was highly effective in binding of single-stranded DNA, catalyzed catenane unlinking, and was able to relax only “–” scDNA by forming a covalent bond at the 5′-end of DNA of the cleavage site [102]. However, in contrast to other eukaryotic topo I it exhibited rather weak relaxing activity [33]. It appears that lack of such enzyme with low topoisomerase activity would insignificantly influence development of cells containing topo I and II. However, in reality yeast cells lacking topo III exhibited slower growth, had increased ability for recombination, and were unable to form spores [33]. Thus, it is believed that in vivo the major role of yeast topo III consists of decatenation of complementary duplex strands after their separation by helicase rather than regulation of the topological state of cell DNA [103].
Topo III can function together with cell telomerase and influence telomere stability [103]. Telomere stabilization in yeasts mediated by topo III represents one example illustrating the fact that various DNA topoisomerases, besides their canonic function (alteration of DNA topology), play an important role in stabilization of the cell genome. Mechanisms by which topoisomerases provide genome stability by preventing mitotic recombination still remain unknown. It is believed that these enzymes are required for unlinking of recombination intermediates when DNA strands are supercoiled [103]. The recombination process can be thus interrupted by helicase catalyzing separation of two incorrectly paired DNA strands [103]. Involvement of helicases in the decrease in recombination frequency is also confirmed in [104, 105]. Yeast SGS1 helicase interacts with topo III [106, 107]. Mutation of the sgs1 gene results in a decrease in life expectancy phenotypically similar to gene mutation seen in humans with Werner’s syndrome [106]. Thus, topo III functions in a complex with proteins of the SGS1 family and thus might be necessary for maintenance of genome stability and regulation of cell aging [106].
In prokaryotes and lower eukaryotes only one form of topo III has been found [106], whereas in mammalian cells two isoforms of topo III are known [106, 108]. The gene encoding human topo IIIα (hTOP3α) homologous to yeast topo III is located within the 17p11.2-12 chromosome [41]. The murine analog is important for the stage of early embryogenesis [109]. The second gene encoding human topo III (hTOP3β) is located within the 22q11 chromosome [110]. Three alternative transcripts of this gene encode proteins differing in the C-terminal site that is involved in DNA binding [106]. A similar situation was observed during expression of the hTOP3α gene [106]. Different mode of expression of genes encoding topo III resulting in synthesis of various isoforms of the protein suggests different physiological functions of these topoisomerases, especially if we take into consideration the fact that the action of topo IIIα in the cell cannot be replaced by topo IIIβ or other topoisomerases [106]. Various forms of topo III can demonstrate different catalytic activity with respect to various DNA substrates. Such conclusion can be made taking into consideration that enzyme differences involve the C-terminal domain. Indeed, even small differences in the structure of this site in E. coli topo I and topo III result in different catalytic activity of these significantly homologous enzymes [106].
Mouse topo IIIβ shares 36% homology with topo IIIα. This enzyme relaxes “–” scDNA and its catalytic activity increases with increase in supercoiling degree [108]. Topo IIIβ only partially relaxes scDNA at 37°C, while full DNA conversion occurs at higher temperatures [108]. This suggests that topo IIIβ is specific to single-stranded sites that are easily formed in “–” scDNA at a high level of supercoiling. During partial relaxation, formation of single-stranded DNA regions is hampered but can occur at high temperatures due to local melting of DNA duplex.
CONCLUSIONS
Since the 1970s studies of topoisomerases have revealed wide diversity of these enzymes in pro- and eukaryotic cells. During this period, biological functions of the enzymes of various types and also analogies and differences in their structure and mechanisms of actions have been recognized.
DNA topoisomerases type IA break one of the strands, pass the other strand through this break, and thus remove topological stress of supercoiled DNA. Topoisomerases type II form a double-strand break using ATP or other nucleotides as the energy source and pass DNA fragments through the formed break.
Analysis of topoisomerases using various methods, including X-ray analysis, steady state and rapid kinetics, site-directed mutagenesis, etc., have revealed particular roles of certain amino acids in both catalysis, and cooperative interactions between various domains.
Thus, in pro- and eukaryotic cells there are several topoisomerases differing in their biological functions. The existence of various topoisomerases provides equilibrium between relaxed and weakly and strongly helical forms of DNA.
This work was supported by a grant from the Program for Basic Research of the Presidium of the Russian Academy of Sciences “Molecular and Cell Biology” (No. 10.5) and an Integration grant from the Presidium of the Siberian Branch of the Russian Academy of Sciences.
REFERENCES
1.Wang, J. C. (1996) Annu. Rev. Biochem.,
65, 635-692.
2.Merino, A., Madden, K. R., Lane, W. S., Champoux,
J. J., and Reinberg, D. (1993) Nature, 365, 227-232.
3.Trowbridge, P. W., Roy, R., and Simmons, D. T.
(1999) Mol. Cell Biol., 19, 1686-1694.
4.Zhu, J., and Schiestl, R. H. (1996) Mol. Cell
Biol., 16, 1805-1812.
5.Shuman, S. (1989) Proc. Natl. Acad. Sci.
USA, 86, 3489-3493.
6.Pommier, Y., Pourquier, P., Fan, Y., and Strumberg,
D. (1998) Biochim. Biophys. Acta, 1400, 83-105.
7.Kato, S., and Kikuchi, A. (1998) Nagoya J. Med.
Sci., 61, 11-26.
8.Champoux, J. J. (2001) Annu. Rev. Biochem.,
70, 369-413.
9.Wang, J. C. (2002) Nat. Rev. Mol. Cell
Biol., 3, 430-440.
10.Sharma, A., and Mondragon, A. (1995) Curr.
Opin. Struct. Biol., 5, 39-47.
11.Gupta, M., Fujimori, A., and Pommier, Y. (1995)
Biochim. Biophys. Acta, 1262, 1-14.
12.Berger, J. M. (1998) Biochim. Biophys.
Acta, 1400, 3-18.
13.Viard, T., and de la Tour, C. B. (2007)
Biochimie, 89, 456-467.
14.Champoux, J. J. (2002) Trends Phamacol.
Sci., 23, 199-201.
15.Corbet, K. D., and Berger, J. M. (2004) Annu.
Rev. Biophys. Struct., 33, 95-118.
16.Leppard, J. B., and Champoux, J. J. (2005)
Chromosoma, 114, 75-85.
17.Champoux, J. J. (2002) Proc. Natl. Acad. Sci.
USA, 99, 11998-12000.
18.Wilson, R. J. (2002) J. Mol. Biol.,
319, 257-274.
19.Wang, J. C. (1971) J. Mol. Biol.,
55, 523-533.
20.Kirkegaard, K., and Wang, J. C. (1985) J. Mol.
Biol., 185, 625-637.
21.Champoux, J. J., and Dulbecco, R. (1972) Proc.
Natl. Acad. Sci. USA, 69, 143-146.
22.Durnfoed, J. M., and Champoux, J. J. (1978) J.
Biol. Chem., 253, 1086-1089.
23.Riou, G. F., Gabillot, M., Douc-Rasy, S., Kayser,
A., and Barrois, M. (1983) Eur. J. Biochem., 253,
479-484.
24.Pylleyblank, D. E., and Morgan, A. R. (1975)
Biochemistry, 14, 5206-5209.
25.Hsieh, T., and Brutlag, D. (1980) Cell,
21, 115-125.
26.Liu, L. F. (1983) Meth. Enzymol.,
100, 133-137.
27.Tanizawa, A., Tabuchi, A., Bertrand, R., and
Pommier, Y. (1993) J. Biol. Chem., 268, 25463.
28.Brun, G., Vannier, P., Scovassi, I., and Callen,
J.-C. (1981) Eur. J. Biochem., 268, 407-415.
29.Castora, F. J., and Kelly, W. G. (1986) Proc.
Natl. Acad. Sci. USA, 83, 1680-1684.
30.Lazarus, G. M., Henrich, J. P., Kelly, W. G.,
Schmitz, S. A., and Castora, F. J. (1987) Biochemistry,
26, 6195-6203.
31.Dean, F., Krasnow, M. A., Otter, R., Matzuk, M.
M., Spengler, S. J., and Cozzarelli, N. R. (1983) Cold Spring Harb.
Symp. Quant Biol., 47, 769-777.
32.Srivenugopal, K. S., Lockshon, D., and Morris, D.
R. (1984) Biochemistry, 23, 1899-1906.
33.Wallis, J. W., Chrebet, G., Brodsky, G., Rolfe,
M., and Rothstein, R. (1989) Cell, 58, 409-419.
34.Kikuchi, A., and Asai, K. (1984) Nature,
309, 677-681.
35.Slesarev, A. I., Stetter, K. O., Lake, J. A.,
Gellert, M., Krah, R., and Kozyavkin, S. A. (1993) Nature,
364, 735-737.
36.Slesarev, A. I., Lake, J. A., Stetter, K. O.,
Gellert, M., and Kozyavkin, S. A. (1994) J. Biol. Chem.,
269, 3295-3303.
37.Slesarev, A. I., Zaitzev, D. A., Kopylov, V. M.,
Stetter, K. O., and Kozyavkin, S. A. (1991) J. Biol. Chem.,
266, 12321-12328.
38.Sekiguchi, J., and Shuman, S. (1994) J. Biol.
Chem., 269, 29760-29764.
39.Shuman, S., and Prescott, J. (1990) J. Biol.
Chem., 265, 17826-17836.
40.Kim, R. A., and Wang, J. C. (1992) J. Biol.
Chem., 267, 17178-17185.
41.Hanai, R., Caron, P. R., and Wang, J. C. (1996)
Proc. Natl. Acad. Sci. USA, 93, 3653-3657.
42.Nadal, M., Jaxel, C., Portemer, C., Forterre, P.,
Mirambeau, G., and Duguet, M. (1988) Biochemistry, 27,
9102-9108.
43.Bouthier de la Tour, C., Portemer, C., Nadal, M.,
Stetter, K. O., Forterre, P., and Duguet, M. (1990) J.
Bacteriol., 172, 6803-6808.
44.Bouthier de la Tour, C., Portemer, C., Huber, R.,
Forterre, P., and Duguet, M. (1991) J. Bacteriol., 173,
3921-3923.
45.Slesarev, A. I., and Kozyavkin, S. A. (1990)
J. Biomol. Struct. Dyn., 7, 935-942.
46.Confalonieri, F., Elie, C., Nadal, M., de La
Tour, C., Forterre, P., and Duguet, M. (1993) Proc. Natl. Acad. Sci.
USA, 90, 4753-4757.
47.Kovalsky, O. I., Kozyavkin, S. A., and Slesarev,
A. I. (1990) Nucleic Acids Res., 18, 2801-2805.
48.Li, Z., Hiasa, H., and DiGate, R. (2005)
Nucleic Acids Res., 33, 5415-5425.
49.Li, Z., Hiasa, H., and DiGate, R. (2006) Mol.
Microbiol., 60, 140-151.
50.Stivers, J. T., Shuman, S., and Mildvan, A. S.
(1994) Biochemistry, 33, 327-339.
51.Stivers, J. T., Shuman, S., and Mildvan, A. S.
(1994) Biochemistry, 33, 15449-15458.
52.Tse-Dinh, Y. C., and Wang, J. C. (1986) J.
Mol. Biol., 191, 321-331.
53.Domanico, P. L., and Tse-Dinh, Y. C. (1988)
Biochemistry, 27, 6365-6371.
54.Domanico, P. L., and Tse-Dinh, Y. C. (1991) J.
Inorg. Biochem., 42, 87-96.
55.Christiansen, K., Knudsen, B. R., and
Westergaard, O. (1994) J. Biol. Chem., 269,
11367-11373.
56.Burgin, A. B., Huizenga, B. N., and Nash, H. A.
(1995) Nucleic Acids Res., 23, 2973-2979.
57.Tse-Dinh, Y. C., McCarron, B. G., Arentzen, R.,
and Chowdhry, V. (1983) Nucleic Acids Res., 11,
8691-8701.
58Tse-Dinh, Y. C., and Beran-Steed, R. K. (1988) J. Biol. Chem.,
263, 15857-15859.
59.Tse-Dinh, Y. C. (1991) J. Biol. Chem.,
266, 14317-14320.
60.Tse-Dinh, Y. C., and Beran, R. K. (1988) J.
Mol. Biol., 202, 735-742.
61.Zhu, C. X., and Tse-Dinh, Y. C. (1994)
Biochem. Mol. Biol. Int., 33, 195-204.
62.Zumstein, L., and Wang, J. C. (1986) J. Mol.
Biol., 191, 333-340.
63.Lima, C. D., Wang, J. C., and Mondragon, A.
(1993) J. Mol. Biol., 232, 1213-1216.
64.Zhu, C. X., Samuel, M., Pound, A., Ahumada, A.,
and Tse-Dinh, Y. C. (1995) Biochem. Mol. Biol. Int., 35,
375-385.
65.Samuel, M., Zhu, C. X., Villanueva, G. B., and
Tse-Dinh, Y. C. (1993) Arch. Biochem. Biophys., 300,
302-308.
66.Ahumada, A., and Tse-Dinh, Y. C. (2002) BMC
Biochem., 3, 13-14.
67.Lima, C. D., Wang, J. C., and Mondragon, A.
(1994) Nature, 367, 138-146.
68.Sharma, A., Hanai, R., and Mondragon, A. (1994)
Structure, 2, 767-777.
69.Lue, N., Sharma, A., Mondragon, A., and Wang, J.
C. (1995) Structure, 3, 1315-1322.
70.Yu, L., Zhu, C. X., Tse-Dinh, Y. C., and Fesik,
S. W. (1995) Biochemistry, 34, 7622-7628.
71.Redinbo, M. R., Stewart, L., Kuhn, P., Champoux,
J. J., and Hol, W. G. (1998) Science, 279, 1504-1513.
72.Stewart, L., Redinbo, M. R., Qiu, X., Hol, W. G.,
and Champoux, J. J. (1998) Science, 279, 1534-1541.
73.Brandeen, C. I. (1980) Q. Rev. Biophys.,
13, 317-338.
74.Hansen, G., Harrenga, A., Wieland, B., Schomburg,
D., and Reinemer, P. (2006)
[javascript:AL_get(this,%20'jour',%20'J%20Mol%20Biol.'); J. Mol. Biol.
], 358, 1328-1340.
75.Caron, P. R., and Wang, J. C. (1994) Adv.
Pharmacol., 29B, 271-297.
76.Beese, L. S., and Steitz, T. A. (1991) EMBO
J., 10, 25-33.
77.Chen, S. J., and Wang, J. C. (1998) J. Biol.
Chem., 273, 6050-6056.
78.Perry, K., and Mondragon, A. (2004) Structure,
12, 7-9.
79.Changela, A., DiGate, R. J., and Mondragon, A.
(2001) Nature, 411, 1077-1081.
80.Cheng, B., Feng, J., Mulay, V., Gadgil, S.,
and Tse-Dinh, Y. C. (2004)
[javascript:AL_get(this,%20'jour',%20'J%20Biol%20Chem.'); J. Biol.
Chem.], 279, 39207-39213.
81.Strahs, D., Zhu, C. X., Cheng, B., Chen, J., and
Tse-Dinh, Y. C. (2006)
[javascript:AL_get(this,%20'jour',%20'Nucleic%20Acids%20Res.'); Nucleic
Acids Res.], 34, 1785-1797.
82.Cheng, B., Feng, J., Gadgil, S., and Tse-Dinh, Y.
C. (2004) J. Biol. Chem., 279, 8648-8654.
83.Brown, P. O., and Cozzarelli, N. R. (1981)
Proc. Natl. Acad. Sci. USA, 78, 843-847.
84.Tse, Y., and Wang, J. C. (1980) Cell,
22, 269-276.
85.Feinberg, H., Changela, A., and Mondragon, A.
(1999) Nat. Struct. Biol., 6, 961-968.
86.Roche, C. J., and Tse-Dinh, Y. C. (2001) Int.
J. Biol. Macromol., 29, 175-180.
87.DiGate, R. J., and Marians, K. J. (1989) J.
Biol. Chem., 264, 17924-17930.
88.DiGate, R. J., and Marians, K. J. (1988) J.
Biol. Chem., 263, 13366-13373.
89.Nurse, P., Levine, C., Hassing, H., and Marians,
K. J. (2003) [javascript:AL_get(this,%20'jour',%20'J%20Biol%20Chem.');
J. Biol. Chem.], 278, 8653-8660.
90.Hiasa, H., DiGate, R. J., and Marians, K. J.
(1994) J. Biol. Chem., 269, 2093-2099.
91.Lopez, C. R., Yang, S., Deibler, R. W., Ray, S.
A., Pennington, J. M., Digate, R. J., Hastings, P. J., Rosenberg,
S. M., and Zechiedrich, E. L. (2005)
[javascript:AL_get(this,%20'jour',%20'Mol%20Microbiol.'); Mol.
Microbiol.], 58, 80-101.
92.Stupina, V. A., and Wang, J. C. (2005)
[javascript:AL_get(this,%20'jour',%20'J%20Biol%20Chem.'); J. Biol.
Chem.], 280, 355-360.
93.Zhang, H. L., Malpure, S., and DiGate, R. J.
(1995) J. Biol. Chem., 270, 23700-23705.
94.DiGate, R. J., and Marians, K. J. (1992) J.
Biol. Chem., 267, 20532-20535.
95.Zhang, H. L., and DiGate, R. J. (1994) J.
Biol. Chem., 269, 9052-9059.
96.Beran-Steed, R. K., and Tse-Dinh, Y. C. (1989)
Proteins, 6, 249-258.
97.Mondragon, A., and DiGate, R. (1999) Structure
Fold Des., 7, 1373-1383.
98.Li, Z., Mondragon, A., Hiasa, H., Marians, K. J.,
and DiGate, R. J. (2000) Mol. Microbiol., 35,
888-895.
99.Krah, R., O’Dea, M. H., and Gellert, M.
(1997) J. Biol. Chem., 272, 13986-13990.
100.Shibata, T., Nakasu, S., Yasui, K., and
Kikuchi, A. (1987) J. Biol. Chem., 262, 10419-10421.
101.Declais, A. C., Marsault, J., Confalonieri, F.,
de La Tour, C. B., and Duguet, M. (2000) J. Biol. Chem.,
275, 19498-19504.
102.Gangloff, S., de Massy, B., Arthur, L.,
Rothstein, R., and Fabre, F. (1999) EMBO J., 18,
1701-1711.
103.Kim, R. A., Caron, P. R., and Wang, J. C.
(1995) Proc. Natl. Acad. Sci. USA, 92, 2667-2671.
104.Kodadek, T. (1991) J. Biol. Chem.,
266, 9712-9718.
105.Morel, P., Hejna, J. A., Ehrlich, S. D., and
Cassuto, E. (1993) Nucleic Acids Res., 21, 3205-3209.
106.Ng, S. W., Liu, Y., Hasselblatt, K. T., Mok, S.
C., and Berkowitz, R. S. (1999) Nucleic Acids Res., 27,
993-1000.
107.Bennett, R. J., Noirot-Gros, M. F., and Wang,
J. C. (2000) J. Biol. Chem., 275, 26898-26905.
108.Seki, T., Seki, M., Onodera, R., Katada, T.,
and Enomoto, T. (1998) J. Biol. Chem., 273,
28553-28556.
109.Li, W., and Wang, J. C. (1998) Proc. Natl.
Acad. Sci. USA, 95, 1010-1013.
110.Kawasaki, K., Minoshima, S., Nakato, E.,
Shibuya, K., Shintani, A., Schmeits, J. L., Wang, J., and Shimizu, N.
(1997) Genome Res., 7, 250-261.