REVIEW: From Structure and Functions of Steroidogenic Enzymes to New Technologies of Gene Engineering
L. A. Novikova1*, Ya. V. Faletrov2, I. E. Kovaleva1, S. Mauersberger3, V. N. Luzikov1#, and V. M. Shkumatov2
1Belozersky Institute of Physico-Chemical Biology, Lomonosov Moscow State University, 119991 Moscow, Russia; E-mail: novik@genebee.msu.su2Institute of Physico-Chemical Problems, Belorussian State University, Minsk, Belarus
3Institute of Microbiology, Dresden University of Technology, Dresden, Germany
* To whom correspondence should be addressed.
# Deceased.
Received December 25, 2008; Revision received February 10, 2009
This review summarizes data about structural and functional organization of steroidogenic P450-dependent enzymatic systems. Problems of catalysis of steroid substrate transformation, special features of mitochondrial type P450scc topogenesis, and abilities of some microbial electron transport proteins to support P450 activity in vitro and in vivo are considered. Principal steps in the creation and catalytic properties of transgenic strains of Escherichia coli, Saccharomyces cerevisiae, and Yarrowia lipolytica expressing both mammalian steroidogenic P450s and the corresponding electron transport proteins are also described. Achievements and prospects of using such transgenic strains for biotechnological synthesis and pharmacological screening are considered.
KEY WORDS: cytochrome P450, steroid hormone biosynthesis, transgenic microorganisms, topogenesis, electron transport proteins, cytochrome P450 inhibitorsDOI: 10.1134/S0006297909130057
Abbreviations: Ad, adrenodoxin, [2Fe-2S]‑ferredoxin from adrenal cortex; AdR, FAD‑containing NADPH‑adrenodoxin reductase; CPR, NADPH‑cytochrome P450‑reductase; ETS, (enzymatic) electron transport system; Fdc, bacterial [2Fe-2S]‑ferredoxin; Fld1 and Fld2, 1 and 2 type bacterial FMN-flavodoxins; FLDR, FAD-containing flavodoxin (ferredoxin) oxidoreductase; HSD, hydroxysteroid dehydrogenase; P450scc (CYP11A1), cytochrome P450 cholesterol hydroxylase/20,22‑lyase; P450c17 (CYP17), cytochrome P450 17α‑hydroxylase/17,20‑lyase; P450c11 (CYP11B1, P45011β), cytochrome P450 11β‑hydroxylase; P450c18 (CYP11B2), cytochrome P450 aldosterone synthase; P450c19 (CYP19, P450arom), cytochrome P450 aromatase; P450c21 (CYP21), cytochrome P450 21‑hydroxylase; SiR, NADPH-sulfite reductase.
Enzymatic systems containing cytochrome P450 (P450) as the terminal
oxidase responsible for regio- and stereoselective hydroxylation of
substrate molecules play the central role in biosynthesis of
physiologically active substances (steroids, D group vitamins, bile
acids, etc.) and oxidation of xenobiotics [1-3]. Due to their wide distribution, universal
structure, and the key role in biosynthesis and metabolism, these
systems can be used for studies on some fundamental problems of modern
biochemistry, such as (i) regulation of biosynthesis of physiologically
active substances on the tissue, subcellular, and molecular level; (ii)
principles of functioning of “multicenter” enzymes and
systems responsible for coordination of electron transfer, interaction
with substrates, and activation of molecular oxygen; (iii)
structural–functional aspects of protein–protein and
protein–ligand recognition; (iv) molecular mechanisms of
phylogenetic diversity of biosynthetic and metabolic pathways.
Findings in the field of monooxygenase catalysis accumulated during the last 30 years has led to the formulation of some applications, some realized and others in the stage of development. These applications include: (i) detection of genetic deficiencies which lead to disorders in biosynthesis of hormones and cause some severe diseases; (ii) development of economically efficient and ecologically pure methods of regio- and stereoselective enzymatic synthesis of hormones (chemical synthesis of which is inefficient, energy consuming, ecologically dangerous, or impossible); (iii) development of test systems based on substrate-specific P450s to be used in development and analysis of new antimicrobial agents and modifiers of steroid biosynthesis pathways.
This review summarizes data on steroid-transforming P450 monooxygenases in the field of structural–functional and molecular organization of the enzyme systems, features of protein topogenesis, substrate specificity, and interchangeability of mammalian and microbial electron transport proteins. These data form a basis for new approaches for reconstructing monooxygenase systems in microbial cells to create metabolic analogs of steroid-synthesizing organs of mammals.
BIOSYNTHESIS OF STEROIDS
Pathways of Steroid Biosynthesis
Biosynthesis of steroid hormones from cholesterol in mammals includes oxidative cleavage of the side chain of cholesterol with a subsequent regio- and stereoselective hydroxylations catalyzed by P450s: P450scc, P450c17, P450c21, P450c11, P450c18, and P450c19 [1, 3-5].
The compartmentalization principle is clearly manifested in steroid biosynthesis. P450scc, P450c18, and P450c11 function in mitochondria of adrenal cortex in cooperation with the FAD-containing flavoprotein adrenodoxin reductase (AdR) and [2Fe-2S]-ferredoxin (adrenodoxin (Ad)). P450c21 and P450c17 catalyze steroid hydroxylation in the endoplasmic reticulum in cooperation with the FAD/FMN-flavoprotein NADPH-cytochrome P450-reductase (CPR). Aromatase P450c19 functions additionally in the ovarian endoplasmic reticulum.
The rate of steroidogenesis is limited by delivery of cholesterol to the mitochondrial inner membrane. The delivery of cholesterol to mitochondria and factors that control this process are comprehensively reviewed in [6].
In adrenal cortex mitochondria, P450scc (scc is an abbreviation for “side chain cleavage”) catalyzes 22- and 20-hydroxylation and cleavage of the C20–C22 bond with removal of the cholesterol side chain. The resulting pregnenolone, which is a precursor of all steroid hormones, leaves mitochondria and enters the endoplasmic reticulum. Here the further stages of steroidogenesis are catalyzed by P450c17, which performs 17α‑hydroxylation and the C17–C20 lyase reaction, and by P450c21, which performs 21‑hydroxylation. In addition to these P450s, 3b-hydroxysteroid dehydrogenase/Δ4→Δ5 isomerase (3β‑HSD) is also involved in steroidogenesis in the endoplasmic reticulum. In the next stage, intermediate products of the synthesis 11-deoxycorticosterone and 11-deoxycortisol re-enter mitochondria or are secreted into the blood. From these intermediates the most important two corticosteroids, aldosterone and cortisol, are produced, respectively, in mitochondria of adrenal cortex cells under the influence of P450c18 (two-stage 18-oxidation) and P450c11 (11β‑hydroxylation).
Virtually all monooxygenase systems of mammalian adrenal cortex have a number of alternative activities catalyzed by the same form of the protein. This is represented by a dynamic model, which permits substrate sites of different P450s (determining regio- and stereospecificity of reactions) and the heme-bound oxidant to manifest multiple activities [3] and, consequently, the possibility of switching of steroid biosynthesis pathways in various molecular diseases associated with mutations in the steroidogenic enzymes. Qualitative differences in the composition of steroidogenic enzymes in different tissues and subcellular compartments and their substrate specificity determine the sequence of biosynthesis pathways of corticosteroids, progestins, and sex hormones. Quantitative differences in contents of P450c17, CPR, and cytochrome b5 determine the preferential realization of biosynthesis of either corticosteroids or androgens and estrogens [7, 8].
Features of enzymes involved in biosynthesis of steroid hormones are presented in some reviews. Thus, two classes of proteins responsible for steroidogenesis in human beings and mice, P450s and HSD, are reviewed in work [5] (nomenclature, encoding genes, chromosomal localization, tissues expressing these proteins, functional features). Molecular mechanisms and signaling pathways mediating steroidogenesis in adrenal cortex, sex glands, placenta, and also in nervous and cardiac tissues are considered in other works [4, 6, 9-13].
Reactions and Mechanisms of Catalysis by Cytochromes P450
It is obvious that creation of recombinant microorganisms synthesizing steroid-transforming P450s can be accompanied by changes in the reaction direction and in the spectra of products because activities of the microbial enzymes can be superposed on the substrate-specific activities of these P450s. To elucidate whether steroid metabolites are products of heterologously expressed P450s, it is necessary to consider the mechanism of catalysis by P450, especially with respect to “unusual” reactions.
P450s catalyze various redox reactions, such as C-hydroxylation, N-, S-, and O-dealkylation, oxidative deamination and desulfonation, N- and S-oxidation, oxidative dehalogenation, epoxidation, and production of peroxy compounds [14, 15]. Major reactions of steroid transformation by P450s include C-hydroxylation, oxidative cleavage of C–C bonds, and a mechanism-based inactivation by synthetic compounds containing ethynyl group. Some other transformations can be observed when steroid molecules contain some fragments uncommon for them (e.g. N- or S-atoms).
These enzymes can also catalyze such “unusual” reactions as cyclization, reduction and reducing dehalogenation, dehydration of oximes, aromatization and enlargement of cycles, ipso-substitution in para-substituted phenols [16, 17], and mechanism-based inhibition [18]. The term “unusual” does not denominate a new catalytic activity of an enzyme. The reaction type is determined by the nature of the P450 and structure of the substrate and also by environmental conditions during the reaction. All these processes can be explained by taking into account mechanisms of catalysis by P450 and general ideas about regularities of intramolecular rearrangements and reactivity of the resulting substrates (radicals, ions, ion-radicals). Some cases of “unusual” catalysis can be exemplified for a certain P450: (i) environment-caused changes in the stereo- and/or regiospecificity of reaction with a usual substrate; (ii) minor side reactions; (iii) transformation of a compound which is unusual as a physiological substrate of this P450; (iv) reactions conditioned by specific features of the substrate structure (different rearrangements of intermediates and mechanism-based inactivation; see also the section “Steroid-Transforming Transgenic Microorganisms for Studies in Pharmacology”).
Production of reactive oxygen species catalyzed by P450 is presented in Fig. 1. A reversible substrate attachment is believed to occur at the very beginning of the catalytic cycle (or at any other stage); therefore, it can be transformed under the influence of any active structure presented. Structures that directly realize oxidative transformation of a substrate are called “ultimate oxidants”. The most distributed transformation types are catalyzed by forms (4)-(7) [19]. The P450-catalyzed cleavage of an O–O bond (the change of (5) to (6)) is promoted by the mechanism of relay-race charge transfer. This so-called “push–pull” effect is as follows: the proximal ligand of the heme iron (cysteine thiolate for P450) and electrophilic fragments of the active site facilitate electron displacement in the O–O bond and production of new H–O and O–Fe bonds.
The scheme in Fig. 1 is usually considered together with another scheme that presents a pathway of hydrogen abstraction/oxygen rebound (Fig. 2). The considered mechanism helps us rationalize C-hydroxylation and, on taking into account transformations of radical intermediates, C–C bond cleavage [18, 20]. The cleavage of C–C bonds is catalyzed by P450scc, P450c17, P450c19, 14α‑demethylase, and some other P450s. Modern studies confirm that C–C cleavage in cases of deacylation is mediated by reactive oxygen species ((4) in the scheme in Fig. 1). This cleavage is also catalyzed by flavin monooxygenases by the BaeyerVilliger mechanism [21].Fig. 1. Scheme of the classical catalytic cycle of oxygen activation and oxidation of substrates by P450s [18]. I) Transmission of the first electron onto the heme iron from the protein partner of the ETS (electron transport system) resulting in transformation of the P450 heme iron ferri form (1) to ferro form (2); II) attachment of oxygen to ferro form with production of an intermediate (3); III) transmission of the second electron onto the heme iron from the protein partner of the ETS with production of a peroxo-iron intermediate (4); IV) attachment of a proton to the intermediate (4) resulting in its protonated form (5); V) attachment of another proton to the intermediate (5) with subsequent detachment of water and production of a perferryl intermediate (6); VI) isomerization of the perferryl intermediate (6) to a radical form (7); VII) oxidation of the substrate.
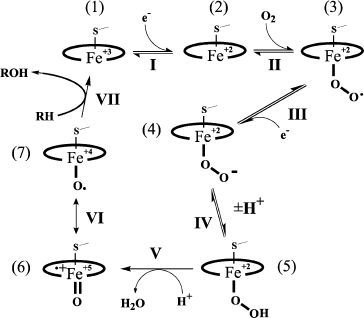
Moreover, various external and internal factors can induce the so-called “abortive detachment” of oxygen: upon transition to form (1), forms (3) and (4)-(5) (Fig. 1) produce, respectively, superoxide radicals and hydrogen peroxide. These particles can manifest their own oxidizing activity [22, 23]. They are relatively stable and can therefore oxidize both the active site fragments and any other component of the system.Fig. 2. Scheme of C-hydroxylation and cleavage of a C–C bond by P450. a) C-hydroxylation by the mechanism of hydrogen abstraction/oxygen rebound; b) cleavage of a C–C bond exemplified by the 17,20-lyase reaction of P450 by the BaeyerVilliger mechanism [18, 20]. a: 1) abstraction of a hydrogen atom from the substrate by a perferryl intermediate of the P450 heme; 2) rearrangements of the P450 heme and substrate intermediates resulting in reduction of the heme iron to ferri form and formation of a hydroxylated product (via attachment of a hydroxyl radical to the substrate intermediate). b: 1) attachment of the distal oxygen atom of the peroxide intermediate of the P450 heme to the substrate 20-keto-group carbon; 2) formation of a radical intermediate of the substrate as a result of homolytic cleavage of the O–O bond; 3) radical 2β‑cleavage of the substrate intermediate resulting in 17-keto-product and acetic acid.
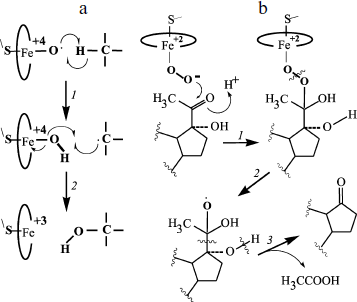
Peroxides, in turn, can attach to the P450 heme iron producing form (5) or its analog with subsequent formation of (6)-(7). Thus, P450s can manifest peroxidase activity [24]. However, such a “shunt” has minimal activity among P450s and is seldom realized, especially in vivo [25]. If iodosylbenzene is used as an artificial oxidant, P450 produces only forms (6)-(7), omitting forms (3)-(5) [26].
Although the above-presented mechanisms are rather reasonable, universal, and allow us to explain virtually all reactions catalyzed by P450, many studies suggest that the real chemistry of the processes can be more complicated and variable. The role of a ultimate oxidant can be played by HO+ and some other structures [27]. Note that these schemes do not take into account the significance for catalysis of the active site structure, especially that of amino acids that are proton donors or acceptors.
RECONSTRUCTION in vitro OF STEROID-TRANSFORMING SYSTEMS
FROM INDIVIDUAL PROTEINS
Preparation of Proteins
Monooxygenase systems were studied using high affinity interactions of immobilized proteins, modified substrates, and group-specific ligands, and this became a prerequisite to elaboration of enzymatic engineering approaches for steroid hydroxylases. More than 20 various homogenous proteins have been used by the authors of the present review for reconstruction of steroid-transforming systems and investigation of interchangeability of electron transport proteins. P450s with different substrate specificity and electron transport proteins of mitochondrial and microsomal systems of mammals and microorganisms have been prepared using original methods [3].
Protein components of mitochondrial steroid-transforming systems were prepared using immobilized ferredoxins [28]. Adrenodoxin (Ad) was immobilized through α-NH2 groups of Ser1 and ε-amino groups of Lys5, Lys16, and Lys23 onto bromocyan-Sepharose. Based on the high affinity, chemically homogenous P450scc and P450c11 were selectively isolated in complex-producing pairs from crude extracts, and AdR was prepared by affinity chromatography on Ad-Sepharose. Ad-Sepharose with a spacer was synthesized by combining a pre-thiolated Ad with bromocyan-Sepharose. Using Ad-Sepharose with the spacer allowed us to significantly increase the activity of the immobilized Ad in the cholesterol side chain cleavage reaction due to more complete realization of its functions in electron transfer and complex-formation during catalysis by the monooxygenase.
Immobilized preparations of cytochrome c were synthesized in order to confirm the principle of complementary interaction of some electron transport proteins [29]. Modification of free carboxyl groups of cytochrome c by histamine enriched the protein with imidazole groups, which were more available for binding with bromoacetyl-Sepharose than His and Met residues in the native protein molecule. Using this scheme, cytochrome c was immobilized on bromoacetyl-Sepharose after insertion of three additional imidazole groups. Selectivity and efficiency of the synthesized sorbent were demonstrated on purification of Ad, hepatoredoxin, cytochrome b5, and isolation of a tryptic fragment of cytochrome b5.
Immobilization of P450scc and P450c11 through free SH-groups allowed us to study the distribution of available cysteine residues and assess their role for functioning of the hemoproteins. Immobilization of P450scc under denaturing conditions by coupling with thiopropyl-Sepharose was accompanied by release of 2.1-2.3 thiopyridone equivalents. Using the different availability of cysteine residues in the native and denatured protein and different distribution of cysteine residues in domains F1 and F2, a scheme was developed for selective separation of domains upon trypsinolysis of the native P450scc. In the crude chymotryptic hydrolysate of denatured P450scc immobilized on thiopropyl-Sepharose, two cysteine-containing peptides were found that contained residues Cys262 and Cys422. The latter residue was localized in a highly homologous conservative region and under native conditions acted as a ligand of the heme iron [30].
Reconstructions of Mitochondrial Type Monooxygenase Systems
Fundamental functional, physicochemical, and structural parameters that determine the functional analogy of reductase components in mitochondrial steroid-transforming systems involved in different pathways of cholesterol metabolism in mammals were evaluated: biosynthesis of steroid hormones in the adrenal cortex and bile acids the liver [31]. Kinetics of recombination of carbon monoxide with high- and low-spin forms of the hemoprotein were studied using pulse photolysis [32], the rate of elementary stages of electron transfer depending on stoichiometry of protein electron carriers was assessed by the stopped-flow method, and spectral changes generated by amino acid residues or the heme chromophore were evaluated by tandem differential spectroscopy of second derivatives in the mid UV and visible regions of the spectrum [33].
To compare the substrate specificity of P450scc and P45027A1, activities were determined of reconstructed steroid-hydroxylating systems with the reciprocal substitution of substrates (cholesterol and 3α,7α,12α-5β-cholestanetriol) and electron transport proteins [34]. The establishment of the ferredoxin active site structure was important from the viewpoint of interchangeability of electron transport proteins. Chemical modification of cysteine residues with 14C-labeled iodoacetamide and a subsequent establishment of the structure of cysteine-containing peptides and leveling of amino acid sequences of the polypeptide chains involved in the [2Fe-2S]-cluster formation resulted in suggestion of possible schemes of the spatial organization of the active site of ferredoxins expressed in adrenal cortex and liver [35].
Much data have been accumulated about kinetic, spectral, and thermodynamic parameters of protein–protein interactions between components of mitochondrial steroid-hydroxylating systems. Two hypothetical mechanisms are supposed for the interaction of adrenodoxin with its partners: a cluster mechanism that includes formation of an associated triple complex P450scc/Ad/AdR [36, 37] and a “shuttle” mechanism that includes the interaction of Ad in turn with P450scc and AdR [38]. Some findings also suggest production of Ad dimer [28] capable of interacting with AdR and P450scc, with a successive electron transfer from AdR to P450scc by the “bridge” formed by two Ads [39]. Based on reports about double complexes between Ad and AdR and Ad and P450 and about overlapping of the AdR and P450scc binding sites in the Ad molecule [40-43], some authors believe the “shuttle” mechanism to be the most likely [44], but the literature also presents data supporting the cluster mechanism [45-47].
Reciprocal Reconstruction of Microsomal and Mitochondrial Monooxygenase Systems and Interchangeability of Microbial and Mammalian Electron Transport Proteins
This reconstruction suggests a recombination of mitochondrial and microsomal proteins from mammals and microorganisms. Significant results were obtained showing an effective functional coupling of CPRs isolated from Candida maltosa and Yarrowia lipolytica with microsomal P450c21 and coumarin-7-hydroxylating P450 and also the possibility to use the plant sterol β-sitosterol as a substrate in the P450scc-system of cholesterol side chain cleavage. In contrast, use for reconstruction of microbial microsomal CPRs with the mammalian mitochondrial P450scc and P450c11 led to a 50-500-fold decrease in activity [48, 49].
Enzymatic electron transport systems (ETS) were detected later in microorganisms capable of transporting electrons onto the heme iron in a number of mammalian P450s under both in vitro and in vivo conditions. The presence of such proteins in a host microorganism during reconstruction of the steroid-transforming P450 monooxygenase systems partially or completely abolished the necessity for expressing proteins of the P450-dependent mammalian ETS.
Proteins of microbial electron transport systems capable of interacting with mammalian cytochromes P450. Ferredoxin from the bacterium E. coli (Fdc) is a small water-soluble anionic [2Fe-2S]‑protein [50] that is thought to be involved in a sophisticated system of iron-sulfur cluster formation in many proteins of E. coli [51, 52]. The primary structure and redox features of Fdc resemble those of ferredoxins from green plants and cyanobacteria and Ad and putidaredoxin of the camphor-hydrolyzing system from the bacterium Pseudomonas putida. Fdc can reduce type 1 and 2 flavodoxins (Fld). The half-reduction potential of Fdc is more negative (-380 mV) than that of the P450scc-cholesterol system (-290 mV) [53], and this suggests that Fdc can reduce P450scc and P450c11 heterologously expressed in E. coli [54].
Small anionic water-soluble FMN‑containing proteins Fld1 and Fld2 [55] are physiological partners of the FAD-containing flavodoxin (ferredoxin) oxidoreductase (FldR) [56]. Systems of FldRFld1 and FldRFld2 catalyze electron transfer from NAD(P)H onto some important elements of E. coli [57]. The back reaction of NADP+ reduction to NADPH is also catalyzed by the FldR–Fld systems and protects the bacteria against oxidative stress [58]. The system of NADPH/FldR/Fld contributes to the 17α‑hydroxylase and 17,20‑lyase activities of P450c17, but markedly weaker than CPR. Studies on the reduction kinetics have suggested that Fld can carry an electron from FldR onto the heme-containing domain of the flavocytochrome P450‑BM3 (a fatty acid hydroxylase from the bacterium Bacillus megaterium) and P450c17 by the “shuttle” mechanism, successively producing complexes with 1 : 1 stoichiometry [55, 59, 60]. Direct interaction with P450s is confirmed by the successful use of Fld-Sepharose for isolation and purification of P450c17 and P450c21 [61].
FldR and Fld from the cyanobacterium Anabaena can support 17α-hydroxylation of progesterone and reduction of cytochrome c by P450c17. The maximal rate of these reactions is two-threefold higher than for the system from E. coli but is significantly lower than the rate of the native system of CPR-P450c17 [62].
NADPH sulfite reductase (SiR) from E. coli is a multimeric hemoflavoprotein consisting of eight flavin α-subunits (SiR–FP) and four β-subunits of the heme. SiR catalyzes the six-electron reduction of sulfite to sulfide. Each α-subunit contains FAD and FMN as prosthetic groups. The primary structure of SiR–FP is similar to the CPR structure, and two C-terminal regions form a cationic FldR-like domain [63]. SiR–FP catalyzes the 17-hydroxylation of pregnenolone in the presence of P450c17 at 12-15-fold lower activity than the mammalian CPR. P450c17 interacts with the FMN-binding N-terminal region of SiR–FP [64, 65].
Yeast CPRs are functional with the host’s own P450s and can also support heterologously expressed microsomal P450c17, P450c21, and P450 1A [66-68]. In the yeast Saccharomyces cerevisiae, the genes YAH1 [69] and ARH1 [70] encode proteins that are highly homologous to mammalian Ad and AdR, respectively. In cooperation with Ad, Arh1p can catalyze the NADPH-dependent in vitro reduction of ferricyanide and cytochrome c and support the 11β‑hydroxylase activity of P450c11 [71]. However, Yah1p from S. cerevisiae could not act instead of Ad in the reconstructed systems of steroid hydroxylation. An Ad homolog from the fission yeast Schizosaccharomyces pombe etp1 containing a [2Fe-2S]‑cluster was shown to substitute for Ad in reconstructed systems and transfer electrons from AdR onto both the bovine P450scc and P450c11 and the human P450scc and P450c18 [72]. A homologous to AdR protein Arh1p from S. pombe displayed a weak ability for binding FAD, whereas modified Arh1 A18G had some spectral and functional features similar to those of AdR.
Covalent Sorptional Reconstruction of Steroid Hydroxylases and Preparation of Radiolabeled Steroids
Three approaches were used for covalent sorptional reconstruction: reaction in a volume, flow minicolumns, and direct spectrophotometric monitoring of functional states of immobilized proteins in flow microcuvettes. Multienzyme complexes of monooxygenase systems were established to be double or triple protein complexes possessing conformational mobility and ability for recombination. During separate elementary stages of the monooxygenase cycle (substrate binding, reduction, oxygenation), the monooxygenase system proteins are mainly in an associated state [73, 74].
The established features of the covalent sorptional reconstruction facilitated the development of some approaches for selective preparation of radiolabeled steroids from their available precursor cholesterol. Chemical methods for preparation of 3H- and 14C-labeled steroids are based on two fundamentally different approaches. 14C-Labeled steroids are prepared based on interaction of the labeled Grignard reagent with steroid enol-lactones. Tritium-labeled steroids can be prepared by hydration of unsaturated precursors with gaseous tritium. Enzymatic approaches allowed the same scheme to be used for preparation of a broad spectrum of the most important radiolabeled steroids from the same labeled precursor (14C- or 3H-labeled cholesterol) [75, 76].
SYSTEMS OF HETEROLOGOUS EXPRESSION FOR STUDIES ON TOPOGENESIS OF
CYTOCHROME P450scc
Data on proteins of steroid-transforming systems obtained in different host organisms are essential prerequisites for constructing transgenic microorganisms for biotechnological synthesis of steroid hormones from available precursors using the heterologous expression of P450s. Studies on P450scc are especially important for both theory and practice because expression of the P450scc monooxygenase system in microorganisms is promising for solution of the crucial problem of synthesis of C21-steroids, i.e. conversion of cholesterol (or its analogs) into pregnenolone.
P450scc is synthesized and provides for synthesis from cholesterol of many biologically active steroids in mammalian steroidogenic tissues, the central and peripheral nerve system cells [77] and cardiac tissue [78].
cDNAs of CYP11A1 have now been cloned from various sources [5]. The structure and localization of the human and mouse CYP11A1 gene are determined [79-82]. Amino acid sequences of P450scc from different mammals are highly homologous, e.g. for P450scc from bovine and human adrenals the homology is higher than 70% [5].
P450scc is a b-type cytochrome [83]; its mature form contains 1 mole heme per mole protein. Its prosthetic group, the heme, is ferroporphyrin IX, which is approximately parallel to the membrane surface and not submerged into the hydrophobic layer of the membrane [84].
Structural Organization of Cytochrome P450scc
The primary structure of the cholesterol-transforming P450scc was established by protein chemistry methods [85]. This problem was solved, first of all, due to development of a highly effective method for preparation of a homogenous P450scc by affinity chromatography on immobilized Ad, elucidation of the hemoprotein domain structure by limited proteolysis, and development of a method for preparing large fragments of the protein (F1 and F2) by thiol-disulfide exchange chromatography [30, 86]. The primary structure of the protein precursor of P450scc was established in work [87] based on DNA sequencing.
Attempts to prepare P450scc crystals suitable for X-ray crystallographic analysis have not been successful. The literature presents only computer-aided models of this protein on the base of experimental three-dimensional structures of P450cam (camphor hydroxylase from Pseudomonas putida), P450-BM3, P4502C5, and P4502B4 [88-91]. The alignment of amino acid sequences of P450scc from different mammals, site-directed mutagenesis, and analysis of the available models of spatial structure have provided information about organization of the functionally important regions of P450scc. A model of the P450scc active site presented in work [92] shows positions of the heme, substrate, and amino acid residues of the active site. The heme-binding loop, which includes the sequence Phe-X-X-Gly-Arg-X-Cys-X-Gly specific for all P450s and containing the absolutely conservative for P450s residue Cys (which is the fifth ligand of the heme iron), is localized in the C-terminal part of the polypeptide chain. In P450scc the site of interaction with Ad is immediately adjacent to the heme-binding domain [93], and negatively charged amino acid residues of Ad interact with positively charged residues of P450scc [89]. The region of the substrate primary binding is localized in the N-terminal part of the polypeptide chain (amino acids 8-28) [94, 95]. The substrate specificity and regioselectivity of hydroxylation in both bacterial and mammalian P450s are determined by specific residues in the loop B′-C [96, 97].
Features of Cytochrome P450scc Topogenesis
P450scc is synthesized in the cytosol as a precursor (preP450scc) with a detachable addressing presequence on the N-terminus. Then the precursor is translocated into mitochondria, undergoes proteolytic processing under the influence of a membrane-bound protease, and is inserted into the mitochondrial inner membrane [98, 99]. The precursor protein is capable of weak association with the mitochondrial inner membrane but can be inserted into the membrane only after the processing [100].
Import of cytochrome P450scc into heterologous systems. The protein possessing of a specific addressing sequence and ability for reversible unfolding are usually necessary and sufficient conditions for its import into mitochondria [101, 102]. Nevertheless, it was for a long time believed that preP450scc could be imported only into mitochondria of steroidogenic tissues. The idea of the tissue-specific import was a result of data on the absence of preP450scc import into cardiac muscle mitochondria [103, 104]. However, later the in vivo expression of active P450scc was shown in COS-1 cells [105], and a possibility of preP450scc import into plant mitochondria was indicated in an in vitro model system [106]. The idea of tissue-specific import of preP450scc also was not supported in the case of a modified precursor of P450scc that had in the addressing sequence on the N-terminus 13 additional amino acids, eight of them being positively charged (6His-preP450scc) [107]. Upon expression in bacterial cells and affinity purification, 6His-preP450scc was successfully translocated into mitochondria of rat liver and heart and into yeast mitochondria, and it was processed with production of the mature protein form (mP450scc). Together, these data demonstrated that P450scc can be studied using model systems of non-steroidogenic cells, and results of work [107] indicated that just the addressing presequence structure could be an important factor influencing the tissue specificity of the import.
Features of bovine P450scc were studied in detail in yeast mitochondria in both in vitro and in vivo systems [108-112]. An essential reason for choosing yeast systems for these studies was the availability of mutants in different components of the translocation complexes and mitochondrial proteases and also information about import signals used by yeast proteins for compartmentalization in mitochondria [113].
Studies on the import of in vitro synthesized preP450scc into isolated yeast mitochondria indicated that the apocytochrome was translocated at a high rate; however, the precursor was converted to the mature form with low efficiency [108]. As differentiated from the situation in adrenal cortex mitochondria, in the yeast the P450scc molecules were not inserted into the inner mitochondrial membrane but rather accumulated in the matrix. Here the heterologous protein was degraded under the influence of the endogenous soluble serine protease Pim1p with involvement of the chaperone Hsp70(Ssc1p) and associated co-chaperones (Mdj1p and Mge1p). In the absence of the proteolytically active protease Pim1p, the newly imported preP450scc aggregated [108]. These findings stimulated searches for addressing sequences of yeast proteins facilitating more effective insertion of P450scc into the yeast mitochondrial membrane. For this purpose, a protein Su9(112)-ΔP450 was constructed with the sequence of the 75 N-terminal amino acid residues of mP450scc substituted by the sequence of 112 amino acid residues of the subunit 9 of F1Fo-ATPase from Neurospora crassa. This sequence included the mitochondrial addressing sequence and the first transmembrane domain, which could ensure efficient insertion of reporter proteins into the inner membrane by the “re-export” mechanism [114, 115]. The hybrid protein Su9(112)-ΔP450 effectively bound with the inner membrane of yeast mitochondria, but the P450scc did not gain an active conformation and also underwent proteolysis, but in this case under the influence of the membrane-bound proteolytic complex Yta10p/Yta12p (m-AAA protease) [108]. This work was the first to show overlapping of the substrate specificities of Pim1p and Yta10p/Yta12p, and this depended on the conformational state and not the presence of specific sites in the targets.
P450scc with the presequence of subunit IV of yeast cytochrome c oxidase (preCoxIV-mP450scc) was imported and correctly processed in yeast mitochondria in vivo [109]. However, only a small share of the imported molecules was inserted into the inner membrane where the protein was capable of converting 22R-hydroxycholesterol to pregnenolone (in a reconstructed system which included isolated bovine Ad and AdR). Upon expression of P450scc with the presequence of subunit VI of yeast cytochrome c oxidase, the protein content in the inner membrane of yeast mitochondria was also low [116].
Topogenesis of preP450scc and Su9(1-112)-mP450scc with the full-length mP450scc was compared in vivo in yeast mitochondria [112]. Similarly to preCoxIV, the native sequence of preP450scc [109] ensured effective import of P450scc into the mitochondrial matrix, where the protein could be detected mainly as aggregates. Processing of the native presequence resulted in formation of mP450scc and an intermediate form, iP450scc. Protein Su9(1-112)-mP450scc was transferred into the mitochondrial matrix, processed, and inserted into the inner membrane directed by the “re-export” signal, and this was associated with aggregation of the protein molecules. It seemed that the complete transfer of the protein into the matrix resulted in its wrong folding, and its subsequent insertion into the membrane was not accompanied by recovery of the correct conformation. Measurements of the cholesterol hydroxylase/lyase activity indicated that retention in the inner membrane of yeast mitochondria of mP450scc and iP450scc molecules was associated with their complete transformation into the holo-form. Based on these findings, it was suggested for the P450scc topogenesis in yeast mitochondria its “retention” in the inner membrane on translocation from the cytosol should be crucial. This is illustrated by the scheme in Fig. 3.
This hypothesis was supported by data on the expression in yeast of hybrid constructions with other addressing signals [117]. Activities of the hybrid proteins (AAC-mP450scc, DLD(1-72)-mP450scc, Bcs1(1-83)-mP450scc) with a signal for translocation arrest in the membrane were significantly higher in mitochondrial fractions than the activity of the protein Su9(1-112)-mP450scc with the re-export signal; the hybrid Ad-mP450scc not inserting into the membrane lacked enzymatic activity.Fig. 3. Possible pathways of import of P450scc precursors into yeast mitochondria: 1) cotranslocational insertion into the membrane leading to production of an active protein (supposed pathway of preP450scc insertion into adrenal cortex mitochondria); 2) rapid translocation of the major share of preP450scc and CoxIV(1-25)-mP450scc to the yeast mitochondria matrix where they aggregate; 3) translocation of Su9(1-112)-mP450scc to the matrix and insertion into the inner membrane by the re-export mechanism. TIM23 is the translocase of inner membrane; PAM (presequence translocase-associated motor) is a complex of proteins that transfer precursors into the mitochondrial matrix.
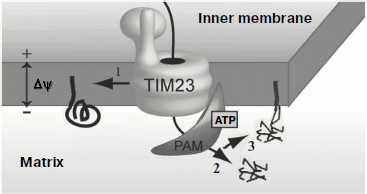
One should also know what amounts of imported heterologous proteins could compete for chaperones and proteases with natural targets of these proteases. The hybrid AdR-Ad imported in vivo into the yeast mitochondria matrix in amount up to 0.2% of the total mitochondrial protein failed to influence the development of the respiratory apparatus of the cells. However, AdR-Ad suppressed proteolysis and stimulated aggregation of P450scc inserted into the same mitochondria [110]. This finding indicated a limited size of the fund of mitochondrial chaperones and proteases (Pim1p, Yta10p/Yta12p, Yme1p) crucial for formation of respiratory complexes and determined certain limits for expression of heterologous proteins in yeast.
Changing the lipid composition of mitochondrial membranes was also a pathway for P450scc adaptation in yeast mitochondria. Aggregation of P450scc in yeast membranes could not be prevented by substitution of ergosterol by cholesterol in the mitochondrial membranes and also by chaperone Hsp78 superexpression [111].
The difference of P450scc topogenesis within yeast from its topogenesis within natural mitochondria (namely, the faster transfer to the matrix) can be due to different mechanisms of protein import in these systems. The yeast protein Tim44 (a component of the PAM-complex responsible for transfer of protein precursors to the matrix [118]) is tightly associated with the mitochondrial inner membrane, whereas the mammalian Tim44 is mainly localized in the soluble fraction of the matrix. Therefore, the more passive mtHsp70-dependent mechanism of precursor transfer to matrix is suggested to be mainly realized in mammalian mitochondria [119]. Recently a cell line of human adrenal carcinoma was obtained which retained the ability to react to adrenocorticotropic hormone [120]. Thus, both topogenesis and fine regulation of P450scc activity can be studied under conditions most similar to the natural ones.
Studies on Cytochrome P450scc Topogenesis Using a Model Prokaryotic System
It was shown in 1991 that expression of the mature P450scc (mP450scc) in E. coli cells most likely lacking proteins and lipids specific for steroidogenic mitochondria was associated with insertion of the newly synthesized protein into the cytoplasmic membrane and manifestation of the native P450scc features [121]. Consequently, bacterial cells synthesizing the mammalian P450scc can be used as an adequate model for studies on processes resulting in formation of the membrane holo-form of the protein (insertion into the lipid bilayer, heme attachment, the mP450scc polypeptide chain folding to a catalytically active conformation).
Cytochrome P450scc topology. P450scc is formally an integral membrane protein because it is not washed from the membrane upon the treatment of mitochondrial inner membrane preparations with sodium carbonate [100] and is solubilized with detergents [28]. However, analysis of the primary structure of mature P450scc did not reveal extended regions enriched with nonpolar amino acid residues that could act as transmembrane domains [122, 123] retaining the protein within the membrane. The N-terminal part of P450scc is the most tightly bound with the membrane. The C-terminal part (containing the heme and Ad binding sites) is weakly associated with the matrix surface of the mitochondrial inner membrane [100], and the substrate-binding site of P450scc is oriented to the mitochondrial matrix [124].
Based on experiments on binding of individual regions of the polypeptide with specific antibodies in mitoplast and submitochondrial particle preparations, a model was proposed of P450scc consisting of two domains crossing the mitochondrial inner membrane [125]. Later, studies on liposome preparations with inserted P450scc indicated that P450scc was tightly integrated with the membrane without traversing it [126, 127].
The hydrophobicity profile obtained for mitochondrial P450scc, P450c11, and P450 27A1 suggested the presence in their molecules of regions containing some hydrophobic amino acids that could be involved in binding with the membrane without penetrating it [128]. Both mitochondrial and microsomal P450s were supposed to associate with the membrane as a result of anchoring into the lipid bilayer of a large hydrophobic domain produced by a number of separate regions of the polypeptide chain [129]. Recent studies on topology of mitochondrial P450s revealed that an extended F-G loop in the central part of the polypeptide chain was responsible for interaction with the membrane [90]. Mutations in the region of the F-G loop of P450scc, P450 27A1, and P450 7A1 expressed in E. coli cells decreased the affinity of P450s for the membrane, which was manifested by changes in the subcellular distribution of the proteins [130-132]. In the case of P450scc, two amino acids from the F-G loop region (the sequence Val207-Arg226) were shown to be inserted into the lipid environment [132]. Moreover, the N-terminal A′-helix was also supposed to be involved in the binding of P450scc with the membrane; the removal of this helix also decreased the affinity of P450scc for the E. coli membrane [90].
Role of terminal protein sequence regions in protein structure formation. There are some works concerning the role of terminal sequences of P450s in the folding of the protein molecules. In some P450s, the conservative N-terminal proline-rich sequence (the PR-sequence) localized in the region connecting the addressing sequence and the catalytic domain plays an important role in formation of the protein structure [133, 134]. The PR-sequence of P450scc includes four proline residues (in positions 6, 8, 13, and 15), and proline in the position 13 is crucial for gaining the native conformation: substitution of this residue by alanine results in formation of a protein lacking the specific CO-differential spectrum [134]. But the data on the role of the PR-sequence are ambiguous. On one hand, this sequence is said to be necessary not only for correct folding of the protein, but also for stabilization of its final conformation through interaction with certain regions of the molecule [135, 136]. On the other hand, the PR-sequence of P450c17 was shown to be important only during the folding of the polypeptide chain, and not being required for supporting the structure of the folded protein [133]. The COOH-terminal sequence of P450 46A1 was also found to possess a proline-enriched region, but as differentiated from the PR-sequence, this region did not influence the polypeptide chain folding but ensured formation of protein dimers [137].
Properties of recombinant cytochrome P450scc synthesized in E. coli cells. Upon expression of cDNA of the mature form of P450scc from bovine adrenal cortex mitochondria (mP450scc) in E. coli cells, an active recombinant protein was detected in both the membrane and soluble fractions prepared by centrifugation of the cell homogenate under standard conditions [54]. It was shown by gel-permeation HPLC that the soluble fraction contained P450scc only inside particles with size above 400 kDa. These data confirmed by results of affinity chromatography and kinetic studies suggested that the particles containing P450scc should be complicated lipoprotein structures. No soluble P450scc in E. coli cells was found; thus, the insertion into the membrane was a necessary stage in formation of the holoenzyme. On insertion into the bacterial membrane, the protein had the same orientation as on insertion into the inner membrane of adrenal cortex mitochondria.
Upon addition of NADPH to a high-speed supernatant, the P450scc underwent one-electron reduction and catalyzed the conversion of 22R-hydroxycholesterol to pregnenolone. As a redox partner for P450scc in E. coli cells, bacterial ferredoxin [50] could be used, which resembled Ad in structure [51], and seemed to be reduced by the NADPH-dependent flavoprotein reductase structurally similar to AdR from adrenals [57]. Thus, a possibility of functional coupling of mammalian P450scc with the bacterial electron transport system was shown.
Comparative study on topogenesis of cytochrome P450scc and its hybrids with adrenodoxin expressed in E. coli cells. Data on the role of the N- and C-terminal sequences in folding of P450s were mainly obtained for proteins with deleted or mutation-carrying terminal regions [133-137]. Expression in E. coli cells of recombinant cDNAs encoding the hybrid proteins Ad–mP450scc and mP450scc–Ad was shown in work [138]. These proteins contain the amino acid sequence mP450scc, the N- or C-terminus of which is fused with a sequence of the mature form of a small (12 kDa) pronouncedly hydrophilic protein Ad with overall negative charge [44]. It was supposed that the presence of the Ad domain retaining features of the native protein [139] and, consequently, localized on the cytoplasmic membrane surface could produce a conformational tension in the structure of protein P450scc by fixing the ends of the molecule at the membrane surface. If the terminal regions of the P450scc chain were involved in its folding, the attachment of the Ad domain could influence features of P450scc.
The presence of the Ad sequence on the P450scc-domain termini did not prevent the binding of the heme with the protein and protein with the membrane. Similar results were obtained on the expression in E. coli cells of other model proteins with modified N-terminus: although the N-terminus of mP450scc was shielded by an extended sequence of 52 amino acids (His6preP450scc) or more than 50 amino acids (Δ1-53mP450scc) were removed, both model proteins as well as the unmodified mP450scc were detected in the cell membrane [140].
Fusion of the P450 N-terminus with Ad did not significantly affect the protein folding and induced no structural deformation of the P450 domain; modification of the C-terminus was associated with a significant increase in the protein fraction with incorrectly folded P450 (P450/P420 = 0.2). The specific activity (calculated per P450-form contents) of mP450scc-Ad was approximately equal to the activity of the unmodified mP450, and modification of the P450scc N-terminus in the protein Ad-mP450scc resulted in 30% decrease in the enzyme activity. Thus, the COOH-terminal region of the sequence is more important for the correct folding and stability of the protein than the NH2-terminal region, whereas the NH2-terminal region of the P450scc molecule seems to play a role in supporting the structure of the catalytically active enzyme.
Thus, studies on P450scc topogenesis using model systems based on yeast and bacterial cells revealed positive and negative sides of such heterologous models for expression of P450scc. Insertion of newly synthesized polypeptide chains of P450scc into the membrane was an obligatory stage in formation of the holo-form of the protein. Studies on mP450scc expression in E. coli indicated that characteristics of mP450scc synthesized in the bacteria corresponded to characteristics of the native protein and that mammalian P450scc could act as the enzyme using the bacterial cell proteins as redox partners. Studies on P450scc import into mitochondria of the yeast S. cerevisiae revealed the critical stage of this import and more or less confirmed the tissue specificity of P450scc, which manifested itself in the yeast at the stage of processing and intramitochondrial compartmentalization.
TRANSGENIC MICROORGANISMS FOR BIOTECHNOLOGICAL STEROID
PRODUCTION
Transgenic “substrate-specific P450–microorganism” systems must conform with the following principles of structural–functional organization of multienzymatic systems: (i) high affinity of proteinprotein interactions and possibility of substitution in the transgenic systems of mammalian ETS proteins by the native microbial proteins capable of more effective use of reducing equivalents on activation of molecular oxygen [3]; (ii) maintaining in the transgenic systems of the regulatory principle of balance between spin forms of P450 as a manifestation of preferential conformations and redox potentials in interactions of the expressed hemoproteins with physiological ligands [141]; (iii) maintaining the affinity in interactions with substrates and intermediates considering the necessity of directed transport and steric correspondence and in some cases manifestation of multiple activities by the same form of the enzyme [142, 143].
Commercial production of steroid hormones and steroid-derived drugs includes the microbiological transformation of a natural sterol (cholesterol, ergosterol, β-sitosterol) as an initial stage. This is associated with a complete degradation of the side chain and formation of androgens (androstenedione and androstenediendione) converted by subsequent chemical and enzymatic reactions to compounds starting the whole spectrum of steroid hormones (mineralocorticoids, glucocorticoids, sex hormones). Transgenic microorganisms producing proteins capable of synthesizing steroid hormones are promising for production of pregnanes by microbiological biotransformation combining several reactions to a single stage that fundamentally changes the technology of production of final steroid preparations. An important prerequisite for choosing an optimal recipient for molecular cloning is the determination of basal activity of microorganisms towards steroids and establishment of composition of microbial protein components promising as redox-partners of P450s. And it must be kept in mind that the presence of the microbe’s own enzymes capable of modifying steroid substrates can result in formation of both desired and undesired byproducts [143, 144].
Reconstruction of Steroid-Hydroxylating System of Mammalian Cytochrome P450scc in Microbial Cells
To create recombinant strains of microorganisms for metabolic transformation of steroids, the recipient cells need be inserted with some genes encoding proteins of one or several multicomponent systems. Different approaches for co-expression of proteins are described in the literature [145-151].
Construction and expression of fused proteins. Natural steroid-transforming enzymatic systems have relatively low functional activity: maximal values of the turnover number for P450scc, P450c11, or P450c17 do not exceed 100. The idea of creating fused monooxygenase systems combining within the same polypeptide chain all components necessary for catalysis appeared after natural “fused” monooxygenases were found [152, 153], in particular the bacterial P450-BM3 possessing a very high catalytic activity (more than 1000 turnovers). The polypeptide chain of this protein includes two catalytic domains (one of them contains a P450-like domain and the other contains binding sites of FAD and FMN similarly to CPR). The detection of the natural prototypes stimulated creation of two-component (microsomal) [154-157] and three-component (mitochondrial) [158-164] fused monooxygenases that were expected to display more effective electron transfer between partner proteins within the same molecule. Studies on two-component fused monooxygenases indicated that the components could interact more effectively in fused proteins than in systems with the activity determined by independent interaction of individual components [165-168]. Maximally active variants of fused proteins could be prepared by changing mutual positions of domains and interdomain linkers [155, 167]. However, in some cases activity of the fused protein increased upon addition of isolated CPR [169, 170]. Conclusions concerning correlation of activities of three-component systems and fused monooxygenases are also different. COS-1 cells expressing the fused proteins preP450scc-AdR-Ad [158, 159] and preP45027A1-AdR-Ad [160] displayed greater corresponding monooxygenase activity than cells co-expressing equimolar amounts of preP450scc, AdR, and Ad. However, on expression in the COS-1 cells of the fused deoxycortisol hydroxylase (preP450c11-Ad-AdR) the resulting system was less effective than the P450c11 functioning in combination with co-expressed and even with endogenous components of the monooxygenase [161].
Three-component monooxygenases containing prokaryotic P450cam [162] and microsomal P4501A1 [163] isolated after their expression in E. coli cells and characterized by routine enzymological approaches were shown to be self-sufficing catalytic units effectively functioning as a result of intramolecular electron transport. Activity of the isolated protein putidaredoxin reductase-putidaredoxin-P450cam was comparable with the activity of the system reconstructed of the individual proteins in 1 : 1 : 1 stoichiometry.
Interaction of catalytic domains in fused proteins composed of components of the cholesterol hydroxylase/lyase system. To increase the activity of the natural P450scc system, three- and two-component hybrid proteins were constructed composed of P450scc, Ad, and AdR from adrenal cortex. Then not only catalytic properties of the fused proteins were studied, but also folding of individual domains within the common polypeptide chain, formation of the domain active sites, and interaction between the system components within the fused protein [139, 164, 171].
Based on the artificial fused cDNA encoding preP450scc-AdR-Ad [158], cDNAs were constructed which encoded preP450scc(h)-AdR(h)-Ad(h) (a homologous variant of preF2(h), “h” signifying human) and pre-CoxIVP450scc(b)-AdR(h)-Ad(h) (a heterologous variant of preF2(b-h) with bovine preP450scc (“b” signifying bovine) with the leading sequence substituted by the presequence of the subunit IV of the yeast cytochrome c oxidase). Plasmids including these cDNAs provided for sufficiently effective expression of hybrid proteins in S. cerevisiae; however, upon the import into mitochondria the proteins F2(b-h) and F2(h) underwent intensive proteolysis [164]. The low level of active fused proteins in the yeast mitochondria prevented their more detailed characterization; therefore, the mature proteins F2(h) and F2(b-h) were expressed in E. coli cells [139]. Moreover, hybrid protein AdR(h)-Ad(h)-P450scc(h) (AAP) was constructed, which had a changed order of domain positions in the polypeptide chain as compared with F2(h) and the free COOH-terminus functionally important for the correct folding of P450scc [138]. Thus, replacement of the P450cam-fragment from the NH2-terminus onto the COOH-terminus of the hybrid protein increased threefold the activity of the fused monooxygenase [162]. Studies on triple hybrid proteins gave interesting findings: (i) a significant fraction of the recombinant proteins was inserted into the membrane of E. coli cells; (ii) spectra of the cell homogenate and membrane fraction were not characteristic for intact P450scc; (iii) the proteins underwent intracellular proteolysis, and sets of low molecular weight products immunogenic to antibodies against P450scc, AdR, and Ad were different for different hybrid proteins (the major product of F2(b-h) and F2(h) proteolysis was a 65-kDa protein, and the major product of AAP proteolysis was a 33-kDa protein). Therefore, the above-mentioned proteins were concluded to differ in structure because of different mutual localization of protein domains. The effective proteolysis of triple hybrids suggested that the folding of proteins into a compact globule was difficult, and they mainly took a conformation with protease-sensitive regions exposed into the environment. Attempts to increase the efficiency of protein compactization were unsuccessful either on the hybrid synthesis in Lon protease-deficient strains or on co-expression of the molecular chaperone GroESL.
Activities of fused three-component cholesterol hydroxylases were usually lower than the activity of the system reconstructed from isolated P450scc, AdR, and Ad (at the ratio of 1 : 1 : 1): specific activities of the proteins F2(b-h) and F2(h) determined in the cell homogenates were an order of magnitude lower, whereas the activity of the protein F2(b-h) isolated from lysates of E. coli cells by affinity chromatography was nearly two orders of magnitude lower than the activity of the reconstructed system. Activities of the proteins F2(b-h) and F2(h) significantly increased in both E. coli homogenates and lysates of S. cerevisiae mitochondria on addition of exogenous components of these proteins. Consequently, in fused proteins synthesized in the bacterial and yeast cells there were limitations on interactions of the catalytic domains of P450scc, Ad, and AdR and/or deformation of their active sites.
According to hypothetical mechanisms of interaction of Ad with AdR and P450scc (see section “The in vitro Reconstruction of Steroid-Transforming System from Individual Proteins”), to realize effective intramolecular transport of electrons in the fused cholesterol hydroxylase/lyase the positions of P450 and AdR domains should either provide formation of a transient triple complex with Ad or successively interact with it. Realization of the “shuttle” mechanism necessitates high mutual mobility of the domains: the fused molecule has to have a flexible mobile conformation, which immediately makes a potential target for intracellular proteases. A feedback was found between activities of the fused cholesterol hydroxylase/lyases and their resistance to proteolysis [139, 158]. And amounts of the most active polyenzymatic systems in the cells were insignificant, whereas contents of their fragments were high, which corresponded to the “shuttle” mechanism of functioning of the fused cholesterol hydroxylase/lyase system.
Folding of hybrid proteins was studied using simplified models, such as the two-component proteins AdR-Ad, Ad-P450scc, and P450scc-Ad [139]. As differentiated from triple hybrids, double hybrids were not proteolyzed on expression in E. coli cells. Determination of activities of hybrid molecules in the presence of exogenous individual proteins that were also their constituents revealed that each protein had a limiting component: in the proteins mP450scc-Ad and Ad-mP450scc it was P450scc and in the protein AdR-Ad it was Ad. Thus, upon addition of P450scc the activity of the proteins mP450scc-Ad and Ad-mP450scc increased, respectively, 20 and 10 times. The same domains could be formed differently within different hybrid proteins [139].
Thus, on folding of fused proteins constructed from the cholesterol hydroxylase/lyase system components, the formation of catalytic domains within the common polypeptide chain depended on the nature and mutual localization of amino acid sequences of the individual domains, and they were arranged as a result of a cooperative process that determined the nativity of these domains and the resulting activity of the fused monooxygenases.
Works in order to design a fused cholesterol hydroxylase/lyase system [139] revealed some general problems of formation and functioning of fused multi-enzymatic systems (deformation of active sites, structural limitations of their interactions, sensitivity to proteolysis) [139].
Co-expression of individual components of the cholesterol hydroxylase/lyase system in E. coli cells. Development of bi- and polycistronic systems for co-expression of proteins facilitated reconstruction of some active monooxygenase systems in bacterial cells [150, 172-174].
Based on the plasmid pTrc99A/P450scc [121], two genetic constructs were designed for co-expression of mature proteins of the cholesterol hydroxylase system. The first construct was pTrc99A/P450scc/AdR/Ad containing P450scc, AdR, and Ad cDNA within the same expression cassette. The other construct was pTrc/P450-AdR.Ad containing P450scc and AdR cDNA within the same expression cassette, and the gene of Ad was inserted into the same plasmid within a separate transcription unit (regulated by its own promoter and terminator) [175]. In E. coli cells these plasmids directed synthesis of all three proteins, and their intracellular localization was compatible with localization of the corresponding proteins of the bovine adrenal cortex. Homogenates of cells cultured under conditions of induced expression of heterologous cDNAs demonstrated in vitro the cholesterol hydroxylase/lyase activity due to the presence of catalytically active forms of all three proteins.
To ensure optimal in vivo functioning of the cholesterol hydroxylase/lyase system, Ad had to be in excess relative to the other proteins [176]. The plasmid pTrc/P450-AdR.Ad was expected to ensure the more effective translation of Ad from its own short mRNA molecule than the plasmid pTrc99A/P450scc/AdR/Ad. But determination of the stoichiometric correlation of the expressed proteins indicated that using the second construct (pTrc/P450-AdR.Ad) for co-expression of the proteins did not noticeably increase the Ad content relative to contents of the other components of the cholesterol hydroxylase/lyase system. The levels of heterologous protein expression in E. coli cells usually did not correspond to contents of functionally active proteins because some molecules of these proteins were present as inactive constituents of inclusion bodies. A significant share of the recombinant proteins of the P450scc, AdR, and Ad (~50% of the total contents) also formed inclusion bodies in bacterial cells [175].
Transgenic Microorganisms for Biotechnology
In 1989, P450c17 was heterologously expressed in S. cerevisiae under the control of the alcohol dehydrogenase promoter pADH1 [177]. Afterwards, the substrate specificity of the recombinant S. cerevisiae GRF18/YEp5117α containing the cDNA of P450c17 under the control of GAL10 promoter was studied in detail [178-180]. The constructed yeast catalyzed successive biotransformation of progesterone to 17α-hydroxyprogesterone and 17α,20α-dihydroxypregn-4-en-3-one. The structure of these compounds was proved by HPLC, TLC, and mass spectrometry. The reaction of 17a-hydroxylation was catalyzed as a result of functional coupling of the heterologously expressed P450c17 and the yeast CPR. The rate of this reaction decreased in the series of substrates: progesterone > 11β-hydroxyprogesterone > 11α-hydroxyprogesterone > 19-hydroxyprogesterone. The reaction of 20α-reduction recorded upon production of 17α-hydroxyprogesterone was catalyzed by the yeast orthologous protein of mammalian 20α-HSD. The rate of this reaction decreased in the series: 17α-hydroxyprogesterone > 21-hydroxyprogesterone > 19-hydroxyprogesterone [178-180].
Screening for functional regions of proteins and structural homology identified genes corresponding to the proteins responsible for side reactions: GCY1 (a galactose-induced crystalline-like yeast protein), YPR1 (yeast aldo-keto reductase), and ATF2 (O-acetyltransferase). Pregn-4-en-20(α,β)-ol-3-ones were converted by recombinant cells of S. cerevisiae GRF18/YEp5117α as follows: oxidation of 20(α,β)-dihydro-derivatives of progesterone to progesterone, 17α-hydroxylation, and 20(α,β)-reduction. 20-Oxidation was caused by the back reaction catalyzed by the yeast’s own enzyme and also by the P450c17 oxidase activity [180].
The last stage of glucocorticoid biosynthesis is catalyzed by P450c11. Successful expression of steroid 11β-hydroxylase in recombinant yeast is described in [181]. The authors succeeded in co-expression of mitochondrial Ad and modified P450c11 with the initial presequence substituted by the yeast presequence. It was shown that S. cerevisiae could synthesize its own mitochondrial protein resembling NADPH-dependent AdR. In S. cerevisiae with co-expressed P450c11 and P450scc, P450scc increased the hydroxylase activity of P450c11 under both in vivo and in vitro conditions [116].
These studies were continued by reconstructing in S. cerevisiae of two initial stages of mammalian steroidogenesis catalyzed by the system of cholesterol side chain cleavage that involved P450scc and 3β-HSD [148]. But using cholesterol as an initial substrate was associated with the problem of its penetration into the cell. To avoid this difficulty, the metabolism of S. cerevisiae grown on glucose- or ethanol-containing medium was changed to produce a new intermediate, a plant analog of cholesterol instead of the natural yeast sterol ergosterol. Ergosterol is distinct from cholesterol by the presence of double bonds at C-7 and C-22 and a methyl group at C-24, therefore, the solution of this problem was priced by disrupting one yeast gene (encoding Δ22-desaturase) and expressing of the Δ7-reductase gene from Arabidopsis thaliana. The resulting transgenic strain could synthesize a new intermediate, ergosta-5-enol instead of ergosterol. It was a great achievement because ergosta-5-enol could support the vital activity of the microorganisms by substituting for ergosterol in membranes, and at the same time it could serve as an initial substrate for the mammalian P450scc [148].
In [182], the synthesis of cortisol was completely reconstructed using recombinant S. cerevisiae. Cortisol was prepared from a simple source of carbon by an eight-stage process that included just the biosynthesis of yeast sterols, the stage of recombinant yeast (which used the plant enzyme), and five additional enzymatic reactions catalyzed by eight mammalian proteins. Expression of the plant enzyme Δ7-reductase resulted in a modified ergosterol biosynthesis, which led to production of the corresponding substrates of P450scc. Additionally, eight mammalian proteins (P450scc, P450c11, P450c17, P450c21, Ad, AdR, CPR, and 3β‑HSD) involved in steroid biosynthesis in the adrenal cortex of mammals were expressed. This work resulted in yeast strains that could synthesize cortisol as a major steroid upon growth on glucose or ethanol.
Thus, the most complicated metabolic pathway has been reconstructed in eukaryotic microorganisms. In other words, a biosynthetic analog of the mammalian adrenal cortex has been created. No doubt, this work is an outstanding achievement in metabolic proteomics [182]. Nevertheless, creation of new even more efficient transgenic microorganisms seems possible. Thus, the lines of searches for recipient microorganisms adapted for transformation of hydrophobic compounds and for simplification of gene engineering manipulations are still open.
Alkane-utilizing yeasts (Candida maltosa, C. tropicalis, or Yarrowia lipolytica) grow well on hydrophobic substrates (alkanes, alkenes, fatty acids, oils). These yeasts utilize hydrophobic substrates using some P450 systems of the CYP52 gene family that are characterized by very high enzymatic activities with turnover numbers of 1000-2000 min–1 under in vivo conditions. This is due to the presence of effective systems of substrate transport (inducible systems of absorption and intracellular transport of hydrophobic compounds), a high level of expression of the yeast’s own components responsible for electron transport, and a selective mechanism for excretion of oxidized products [183-185]. The mechanism of hydrocarbon utilization by Y. lipolytica cells has been intensively studied from the middle of the 1980s. The growth of cells on hydrocarbons is accompanied by induction of a special highly glycosylated extracellular protein, liposan, which is an emulsifier [186] facilitating entrance of hydrophobic substrates into the cell [187].
Expression of P450c17 in the yeast Y. lipolytica was associated with an active conversion of progesterone into 17α-hydroxyprogesterone [180, 188, 191, 194], which suggested an effective interaction between the expressed P450c17 and the yeast CPR. In addition to 17α-hydroxyprogesterone, the conversion of progesterone by the recombinant yeast Y. lipolytica E129A15 expressing P450c17 under the control of isocitrate lyase promoter pICL1 was accompanied by production of two steroid compounds – 17α,20β- and 17α,20α-dihydroxypregn-4-en-3-ones – the structures of which was proved by HPLC, TLC, and NMR spectroscopy [180]. The following sequence of transformations of 20α- or 20β-dihydroprogesterones is shown: initial 20(α,β)-oxidation, the main 17α-hydroxylation and minor 20(α,β)-reduction (Fig. 4). Oxidizing of steroid mixtures (17α-hydroxyprogesterone, 17α,20b- and 17α,20α-dihydroxypregn-4-en-3-one) by chromic acid was accompanied by cleavage of the C17–C20 bond in diols with production of androst-4-en-3,17-dione, which is another product of enzymatic reactions catalyzed by P450c17 [180, 188].
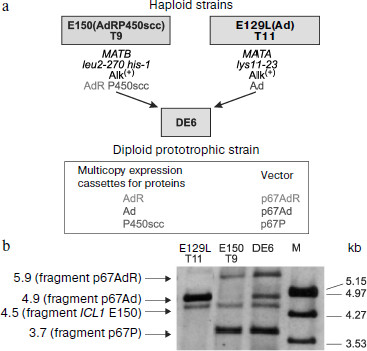
More recently, recombinant strains of Y. lipolytica were prepared that could express the cholesterol hydroxylase/lyase system proteins (P450scc, AdR, Ad) (Fig. 5) and also co-express the P450scc-system and P450c17 [189], which promoted coupling of the cholesterol conversion to pregnenolone (progesterone) with synthesis of 17α-hydroxylated derivatives. On constructing the above-mentioned recombinant strains of Y. lipolytica, we used a new approach for inserting some heterologous cDNAs into the yeast genome. This approach included (i) the co-integration of multiple copies of various expression cassettes in the region of repeated elements of the yeast genome (rDNA regions or LTR zeta fragments of the retrotransposon Ylt1 [190]) and (ii) the construction of diploid strains using haploid multicopy transformants of different mating types that allowed us to increase the quantity and obtain new combinations of expression cassettes with different heterologous cDNAs within the same diploid strain (Fig. 5a).Fig. 4. Enzymatic conversions of pregn-4-en-20β-ol-3-one (I) and pregn-4-en-20α-ol-3-one (II) into progesterone (III) with its subsequent transformation to 17α-hydroxyprogesterone (IV), and 17α,20α-dihydroxypregn-4-en-3-one (V) and 17,20β-dihydroxypregn-4-en-3-one (VI) by the transgenic yeast Y. lipolytica E129A15 expressing P450c17 [188].
Integrative vectors containing cDNAs of mature proteins of the cholesterol-transforming P450scc-system and P450c17 under control of the strong and regulated promoter and terminator of the isocitrate lyase gene ICL1 (induced by ethanol and alkanes, repressed by glucose [191-194]) were constructed based on the multicopy integrative plasmids p64PT and p67PT (integration provided by sequences of rDNA or LTR zeta of retrotransposon Ylt1, multicopy selection marker ura3d4, and pICL1-SphI-ICL1t) [189, 194a]. Southern hybridization indicated that up to three vectors were integrated into the genome along with concurrent transformation of haploid recipient strains of Y. lipolytica by a mixture of similar plasmids, which included pICL1-controlled expressing cassettes with different heterologous cDNAs. Subsequently, diploid strains were obtained that contained three to five expression cassettes for proteins of the cholesterol hydroxylase/lyase P450scc-system and P450c17. The expression of proteins was proved by Western blotting. Functional expression of two systems of mammalian P450 in Y. lipolytica was confirmed by the ability of the yeast cells to convert cholesterol into pregnenolone and further into 17α-hydroxypregnenolone and also of progesterone into 17α-hydroxyprogesterone ([189], unpublished results). These results indicated the suitability of the above-described method for constructing transgenic Y. lipolytica strains capable of expressing up to five or six heterologous proteins that, in particular, is promising for creation of new strains co-expressing even more proteins involved in steroidogenesis.Fig. 5. Construction of the recombinant diploid strain DE6 of the yeast Y. lipolytica co-expressing components of the mammalian cholesterol hydroxylase/lyase system (P450scc, AdR, and Ad) (a); Southern hybridization of strain DE6 and the parental haploid multicopy transformants E150 T9 and E129L T11 of Y. lipolytica (b). Crossing was used to combine three expression cassettes with different heterologous cDNAs into the same diploid strain. The parental strains E150(AdRP450scc) T9 (E150 Т9) and E129L(Ad) Т11 (E129L Т11) used for the crossing were obtained, respectively, as a result of strain E129L transformation by the multicopy plasmid p67Ad (expression cassette for adrenodoxin) and of simultaneous transformation of E150 by the multicopy plasmids p67AdR (for adrenodoxin reductase) and p67P (for P450scc bovine). The yeast chromosomal DNA (restricted by endonuclease NcoI) was analyzed using as a probe the SalI-fragment of the vector p67AdR (pICL1-AdR), and this allowed detection of fragments of the gene ICL1 and expression cassettes with heterologous cDNAs (due to the presence of pICL1) contained in the integrative vectors. Preparations of λ-DNA restricted by EcoRI/HindIII were used as molecular weight markers (M).
The advantage of Y. lipolytica for steroid biotransformation as compared to S. cerevisiae can be exemplified by the recombinant strains Y. lipolytica E129A15 (expression of P450c17), Y. lipolytica DC3, Y. lipolytica DC5, and Y. lipolytica DE (co-expression of P450c17 and the yeast CPR) [195]. Similar values of catalytic parameters Vmax and Km determined for different recombinant strains of Y. lipolytica suggested that in various recombinant microorganisms P450c17 was synthesized in its native state. Additional expression of CPR was accompanied by a decrease in synthesis of the heterologous P450c17, but by an increase in the yield of 17α-hydroxyprogesterone.
In the case of Y. lipolytica DC5, 98% of progesterone was converted into 17α-hydroxyprogesterone as a result of adapted cultivation and biotransformation conditions using pICL1-controlled expression of P450c17 and, in contrast to the transgenic S. cerevisiae [178, 182], this abolished the necessity to destroy genes encoding the orthologous proteins 20α- and 20β-HSD. This highly selective production of 17α-hydroxyprogesterone was achieved by the selected conditions for P450c17-expression by the isocitrate lyase promoter and for biotransformation (absence of galactose-induced protein Gcy1p, which in case of the pGAL10-controlled P450c17 expression in S. cerevisiae on galactose led to formation of 17α,20α-dihydroxypregn-4-en-3-one) and by a rapid excretion of the desired product 17α-hydroxyprogesterone by the yeast cells [195].
STEROID-TRANSFORMING TRANSGENIC MICROORGANISMS FOR STUDIES IN
PHARMACOLOGY
The development of new drugs is now often initiated by identification of a protein of known structure that is responsible for the onset or progress of a disease. The next stage is the search for a ligand (inhibitor, activator, or modifier), that has the maximally strong and specific affinity for this protein. The optimal ligand is then directly synthesized based on parameters of the mutual conformational correlation of functional sites of the protein and compounds to be synthesized. According to the general tendency for reducing the number of animals used in preliminary stages of biomedical testing, the development of systems for heterologous expression of mammalian enzymes or enzymatic systems in microbial cells can be considered as a promising new line that allows metabolic proteomics approaches to be used in pharmacological studies. Note that design of selective inhibitors for P450s is still difficult because no three-dimensional structures of membrane-bound steroid-synthesizing P450s have been experimentally prepared to date.
Molecular targets for selective inhibitors that are being developed and/or used as drugs include the following steroid-transforming P450s: P450c17 in the treatment malignant and benign forms of prostate hyperplasia [196-203]; P450c19 in the treatment of breast and ovary carcinomas [202, 204-206]; P450c18 in the treatment of hyperaldosteronism and hyperaldosteronism-mediated hypertension, myocardial ischemia, and fibrosis [196, 207-210].
Various inhibitors of P450c17 were proposed for use as drugs [197-201]. Many of them are characterized by selectivity and high affinity for P450c17 (their affinity for P450c17 can be 10 times or more higher than for other P450s [199, 200]). The high homology of the pairs P450c17 and P450c21, and P450c18 and P450c11 and also the lyase and hydroxylase reactions catalyzed in the same active site of P450c17 cause difficulties associated with concurrent inhibition of pathological and normal physiological reactions. Nevertheless, in some cases the concurrent inhibition of P450c17 and other P450s can be favorable for pharmacology, e.g. inhibition of P450c19 in the treatment of estrogen-derived diseases [202], of P450c24 (24-hydroxylase) upon prescription of vitamin D preparations in the treatment of prostate tumor [203], and of liver P450 minimizing biodegradation of prescribed drugs [203]. Some of these compounds (inhibitors of P450c17) can also inhibit the 5α-steroid reductase (another key enzyme of androgen biosynthesis) [200] and act as antagonists of androgen receptors [198].
Inhibitors of P450 involved in biosynthesis of steroid hormones are considered in several recent reviews [200, 214-217].
The major structural fragments of most of these inhibitors are (i) a substrate-derived steroid nucleus and (ii) a heterocycle capable of acting as the sixth ligand of the P450 heme iron. These two fragments are crucial in structures of the three main classes of P450 steroidogenesis inhibitors: steroidal, heterocyclic non-steroidal, and heterocyclic steroidal. Heterocyclic inhibitors are promising for decreasing activities of some other P450s, whereas steroidal inhibitors can decrease the activity of other steroidogenic enzymes, in particular those not belonging to P450s, and also act as agonists or antagonists of steroid receptors. Note that a heterocyclic ligand and pharmacophore responsible for mechanism-dependent inactivation (see further) is localized in the inhibitor structure similarly to the site of oxidation of the steroid substrate, i.e. close to the heme [200, 214, 216].
Among steroidal inhibitors of P450c17 of the 17-heteroaryl androstane class and of some other groups, 3β‑hydroxy-5-en-derivatives exhibit higher inhibitory ability than 3‑keto‑Δ4, similarly to P450c17 substrates progesterone and pregnenolone [200]. Steroidal inhibitors of P450c19 formestane and exemestane are also derivatives of the thermodynamically more favorable substrate androstendione (17-ketone) and not of testosterone (17β-ol) [215, 216]. An additional insertion of the Δ16-bond increases the inhibitory activity of some steroidal inhibitors of P450c17 [200]. Steroidal inhibitors of P450c17 from 3β‑hydroxy-5-en-derivatives of androstanes containing 17β-(methyl sulfoxide)-, vinyl fluoride 20-F-Δ17,20-, and 21,21-F2-Δ20,21-20-CH3-groups, which imitate, respectively, 20-keto-group-, 17,20-, and 20,21-enol tautomers of pregnenolone are also of interest [200, 201].
In addition to reversible competition with the substrate, steroidal inhibitors of P450 can cause direct or mechanism-dependent inactivation of P450. Direct inactivation occurs due to the presence of sterically available electrophilic fragments that can covalently modify nucleophilic groups of amino acids and N-atoms of the heme in the active site of a P450 target. This is exemplified by 3,4-diketone (formestane), coupled 1-en-3-one and 6-methylen-4-en-3-one (exemestane, 6-methyleneprogesterone), and 17-azridin-2-yl and 20-carbaldehyde structural fragments of P450c17 and P450c19 inhibitors. Mechanism-dependent inactivation occurs in the presence of fragments whose oxidation by P450s yields intermediates causing inactivation, e.g. 17-O- or 17-N-cyclopropyl (P450c17) or ethynyl or vinyl fragments in positions 17, 18, and 19 (P450c18 and P450c19, respectively) [18, 196, 200, 216].
Different azolyl, pyridinyl, and pyridine-2,6-dione fragments are more often used as heterocyclic ligands of the P450 heme iron. Coordination features of the N-atom within the same heterocycle can be significantly changed depending on position and type of substituents as a result of displacement of electron density and of steric obstacles. And similarly, in the same type heterocycles (in particular, triazoles) this feature additionally depends on the mutual position and nature of heteroatoms. Thus, the ability of biphenyl derivatives to inhibit P450c17 was the highest in the case of imidazole, 1,2,4-triazol-1-yl derivatives were less active, whereas 1,2,4-triazol-4-yl virtually did not inhibit P450c17 [218].
Heterocycles combine functions of inhibitor delivery to the active center of P450 and its inhibition preventing electron transfer from CPR and attachment of O2 to the heme iron. A steroidal or non-steroidal residue of a heterocyclic inhibitor of P450 participating in steroid hormone biosynthesis prevents substrate binding and is responsible for additional interactions in the active site of a target enzyme determining the selectivity and increasing the stability of the P450inhibitor complex. Non-steroidal residues of heterocyclic inhibitors of P450 in most cases are combinations of planar aromatic structures resembling cycles of the steroid core. Thus, a biphenyl fragment imitates the system of AC rings; a naphthalene fragment imitates CD or AB (P450c17); 1-methylene-1,2,3,4-tetrahydronaphthalene and 1-methylene-2,3-dihydro-1H-indene fragments imitate AB (P450c18), a 2-phenylnaphthalene fragment imitates ABD (P450c17) [219-221]. In the structure of virtually all inhibitors of P450, the heterocycle is bound with the aromatic fragment through the carbon atom (a methylene group). The presence of polar substituents imitating the 3β‑hydroxyl or 3-keto-group, and less frequently the 17α-hydroxyl of steroidal substrates, is also fundamentally important [200, 220]. Additional fragments can be inserted into inhibitor molecules considering data on inhibitor docking into active sites of P450 targets in computer-aided models.
At present, the most promising parameters for clinical practice belong to such inhibitors of P450c17 as abiraterone, which is now under stage II of clinical testing [222], and to the steroidal inhibitor 17-1H-benzo[d]imidazole VN/124-1, which acts also as an antagonist of androgen receptors [198]. Inhibitors of P450c19 anastrozole (Arimidex (AstraZeneca)), letrozole (Femara (Novartis)) (non-steroidal triazoles), and the steroidal inactivator exemestane (Aromasin (Pfizer)) are approved drugs [215, 216] (Fig. 6).
Transgenic bacteria and yeast expressing enzymes of steroid biosynthesis as test systems for primary screening of potential drugs. Recombinant microorganisms expressing in vivo a functionally active P450-target can be used as test systems for screening of potential inhibitors of the corresponding P450s [209, 210]. Activity of P450s within recombinant microorganisms is provided by co-expression of proteins of the natural ETS of these P450s or by the presence of the microbe’s own ETS proteins capable of supporting activities of heterologous P450s [56, 72, 223]. Some microorganisms are shown to possess functional analogs of mammalian HSDs [178, 224], which are also considered as pharmacological targets for treatment of similar diseases [225].Fig. 6. Structures of P450c17 and P450c19 inhibitors that are used as approved drugs (lower row) and of some inhibitors that are most promising for medicine [198, 215, 216, 222].
Using recombinant microorganisms for screening of P450 inhibitors presents an opportunity for evaluation of general toxic effects of substances under study and also of the contribution of the substance transport, binding with proteins, and induction of P450 and other proteins to inhibition of the P450-target activity [226, 227].
Transgenic E. coli bacteria co-expressing P450c17 and CPR used as a test system demonstrated both 17α-hydroxylase and 17,20-lyase activities of P450c17 on addition of the corresponding steroid substrates to the cell culture [228].
Recombinant S. cerevisiae yeast expressing P450c17 was also proposed as a test system (Fig. 7). The yeast displayed progesterone 17-hydroxylase activity of the heterologous P450c17, which functioned in cooperation with the yeast CPR capable of supporting the activity of this P450. The yeast was assessed as a possible test system using five steroidal and two non-steroidal compounds, including ketoconazole, and also in parallel experiments with microsomes of the yeast [195, 228-230]. At first, to justify the use of the recombinant S. cerevisiae GRF18/YEp5117α as a pharmacological model, known drugs modifying biosynthesis of steroids were used. The effects of some drugs (ketoconazole, metyrapone, dexamethasone, mifepristone, danazol, levonorgestrelum) on the ratio of biosynthetic and inactivating functions of the recombinant microorganisms were studied (Fig. 7). Values of Km/Vmax, inhibition constants, and the type of inhibition of these drugs in reactions of progesterone 17α-hydroxylation were determined.
In the initial stages of biotransformation, the drugs danazol, mifepristone, and ketoconazole decreased the production of 17α-hydroxyprogesterone and consumption of progesterone. The decreased conversion of progesterone was a result of two consecutive processes: 17α-hydroxylation and 20α-reduction. At inhibitor/P450c17 ratio 1000 : 1 and inhibitor/progesterone ratio 1 : 1, no irreversible inhibition of P450c17 was recorded under in vivo conditions due to covalent binding of danazol or mifepristone in the active site of the hemoprotein. Similarly to the product, danazol and mifepristone has a coupled Δ4-3-keto-structure and 17-OH group and also failed to modify the yeast analog 20α-HSD by the structure-dependent mechanism because in the initial stages the ratio of 17α-hydroxyprogesterone and 17α,20α-dihydroxypregn-4-en-3-one was the same in the control experiment and in the presence of the modifiers. The decreased production of 17α-hydroxyprogesterone under the influence of ketoconazole in the initial stage of progesterone biotransformation reflected substitution by the substrate of the imidazole-containing inhibitor ketoconazole bound with the P450c17 heme iron, but without its influence on functional features of the 20α-HSD analog [195, 228-230].Fig. 7. Enzymatic conversions of progesterone (I) to 17α-hydroxyprogesterone (II) and 17α,20α-dihydroxypregn-4-en-3-one (III) in the recombinant yeast S. cerevisiae GRF18/YEp5117α and structures of some modifiers of steroid hormone biosynthesis: 1) danazol; 2) levonorgestrelum; 3) mifepristone; 4) ketoconazole (2R,4R-isomer) [228].
Recombinant S. pombe yeast expressing P450c18 or P450c11 (90% homology) was also proposed as a corresponding test system. This yeast displayed activities of aldosterone synthase and 11β-hydroxylase of the corresponding P450s owing to proteins of its own ETS (see section “The in vitro Reconstruction of Steroid-Transforming System from Individual Proteins”). The test system was assessed using nine non-steroidal azoles and one steroidal compound, and also by parallel experiments with cultures of mammalian cells (V73MZ (Chinese hamster) and H295R (human adrenal cortex tumor)) capable of expressing the two P450s [209, 210, 231].
A recombinant S. pombe yeast expressing highly homologous P450c21 and P450c17 was used for comparative assessment of the influence of P450c17 inhibitors on P450c21 [232].
To study metabolism and toxicity of drugs and to analyze the effect of mutations on functional properties of the enzymes, a new approach is being developed using transgenic bacterial cells of E. coli and Salmonella typhimurium co-expressing P450 and CPR [233]. A computerized model of the three-dimensional structure of the target protein, data on docking of potential ligands, a directed synthesis, and using the transgenic yeast S. cerevisiae GRF18/YEp5117α allowed us to establish that 1-(3-benzyloxyphenyl)-1H-tetrazole could be an inhibitor of P450c17, which is important in the development of human prostate tumors [195, 227].
CONCLUSIONS
Intensive long-term studies on steroidogenic enzymes have significantly enlarged concepts about the structure and functions of monooxygenase systems and steroid hydroxylases involved in the synthesis of steroid hormones. Successful cloning of cDNA of these enzymes opened the way for modern molecular-biological studies.
Studies on structural–functional relationships of steroid-synthesizing P450s and mechanisms of regulation of steroid hormone biosynthesis are a prerequisite for, on one hand, elucidation of biochemical bases of different disorders in steroidogenesis and development of approaches for correcting adrenal cortex dysfunction, and, on the other hand, for constructing biotechnological systems for directed synthesis of physiologically active steroids.
Conventional methods of steroid hormone production (extraction from plants and animal tissues, complete chemical synthesis, chemoenzymatic synthesis) have made available for medicine a wide spectrum of drugs, but at present the potential of these methods is virtually exhausted from the standpoint of commercial technology. Application of transgenic microorganisms obtained by recombinant DNA techniques is a fundamentally new technology in the synthesis of drug substances. This approach combines biotechnological advantages of enzymatic transformations and the possibility of creation of analogs imitating pathways of the hormone biosynthesis in steroidogenic tissues of mammals, including humans. Constructing such recombinant microorganisms has to be preceded by studies on expression and features of steroid-transforming system proteins in different host organisms in order to assess the ability of the protein to gain native features in a heterologous medium and to choose the best recipient microorganism. For this purpose, systems were used based on bacterial and yeast cells capable of heterologous expression of proteins involved in steroidogenesis in mammals.
The alkane-utilizing “non-conventional” yeast Y. lipolytica has an obvious advantage as a system for biotransformation of hydrophobic steroids and seems promising for application in biotechnology. The constructed “humanized” microorganisms can be also successfully used for screening and directed synthesis of compounds that have substrate-specific P450s as molecular targets. It seems that three-dimensional structures of membrane-bound steroid-transforming P450s will be established very soon, and this will facilitate works for creation of effective new drugs for treatment of diseases associated with disorders in functioning of steroidogenic enzymes.
This work was supported by the Russian Foundation for Basic Research (projects Nos. 05-04-48049a and 08-04-00842a), the Belorussian Foundation for Basic Research (project No. BO4MC-030), and the INTAS project CA 03-51-4366.
REFERENCES
1.Nelson, D. R., Koymans, L., Kamataki, T., Stegeman,
J. J., Feyereisen, R., Waxman, D., Waterman, M. R., Gotoh, O., Coon, M.
J., Estabrook, R. W., Gunsulus, I. C., and Nebert, D. W. (1996)
Pharmacogenetics, 6, 1-42.
2.Werck-Reichardt, D., and Feyereisen, R. (2000)
Genome Biol., 1, 3003.1-3003.9.
3.Shkumatov, V. M., Usanov, S. A., Chashchin, V. L.,
and Akhrem, A. A. (1985) Pharmazie, 10, 757-773.
4.Miller, W. L. (1988) Endocr. Rev., 9,
295-318.
5.Payne, A. H., and Hales, D. B. (2004) Endocr.
Rev., 25, 947-970.
6.Sewer, M. B., Dammer, E. B., and Jagarlapudi, S.
(2007) Drug Metab. Rev., 39, 371-388.
7.Miller, W. L., Auchus, R. J., and Geller, D. H.
(1997) Steroids, 62, 133-142.
8.Usanov, S. A., Chashchin, V. L., and Akhrem, A. A.
(1990) in Molecular Mechanisms of Adrenal Steroidogenesis and
Aspects of Regulation and Application (Ruckpaul, K., and Rein, H.,
eds.) Frontiers in Biotransformation, Vol. 3, Akademie-Verlag,
Berlin, pp. 1-57.
9.Miller, W. L. (2005) Endocrinology,
146, 2544-2550.
10.Bose, H., Lingappa, V. R., and Miller, W. L.
(2002) Nature, 417, 87-91.
11.Lala, D. S., Rice, D. A., and Parker, K. L.
(1992) Mol. Endocrinol., 6, 1249-1258.
12.Morohashi, K., Honda, S., Inomata, Y., Handa, H.,
and Omura, T. (1992) J. Biol. Chem., 267,
17913-17919.
13.Mensah-Nyagan, A. G., Do-Rego, J. L., Beaujean,
D., Loo-The, V., Pelletier, G., and Vaudry, H. (1999) Pharmacol.
Rev., 51, 63-81.
14.Katzung, B. G. (1998) Fundamental and Clinical
Pharmacology [in Russian], Binom-Nevskii Dialekt, Moscow-St.
Petersburg, Vol. 1.
15.Metelitsa, D. I. (1984) Modeling Redox
Enzymes [in Russian], Nauka i Tekhnika, Minsk.
16.Ohe, T., Ashino, T., and Akihirobe, A. (1997)
Drug Metab. Dispos., 25, 116-122.
17.Coon, M. J., Vaz, A. D. N., McGinnity, F., and
Peng, H.-M. (1998) Drug Metab. Dispos., 26,
1190-1193.
18.Guengerich, F. P. (2001) Curr. Drug
Metab., 2, 93-115.
19.Lin, H. L., Kent, U. M., and Hollenberg, P. F.
(2002) J. Pharmacol. Exp. Ther., 301, 160-167.
20.Gupta, M. K., Guryev, O. L., and Auchus, R. J.
(2003) Arch. Biochem. Biophys., 418, 151-160.
21.Kamerbeek, N. M., Janssen, D. B., van Berkel, W.
J. H., and Fraaije, M. W. (2003) Adv. Synth. Catal., 6/7,
106-120.
22.Kashem, M. A., and Dunford, H. B. (1987)
Biochem. Cell. Biol., 65, 486-492.
23.Guengerich, F. P. (1992) FASEB J.,
6, 669-673.
24.Mansuy, D. (1994) Pure Appl. Chem.,
66, 737-744.
25.Cirino, P. C., Tang, Y., Takahashi, K., Tirrell,
D. A., and Arnold, F. H. (2003) Biotechnol. Bioeng., 83,
729-734.
26.Guengerich, F. P., Vaz, A. D., Raner, G. N.,
Pernecky, S. J., and Coon, M. J. (1997) Mol. Pharmacol.,
51, 147-151.
27.Hlavica, P. (2004) Eur. J. Biochem.,
271, 4335-4360.
28.Akhrem, A. A., Lapko, V. N., Lapko, A. G.,
Shkumatov, V. M., and Chashchin, V. L. (1979) Acta Biol. Med.
Germ., 38, 257-273.
29.Akhrem, A. A., Gilevich, S. N., Shkumatov, V. M.,
and Chashchin, V. L. (1984) Biomed. Biochem. Acta, 43,
165-177.
30.Chashchin, V. L., Vasilevsky, V. I., Lapko, V.
N., Adamovich, T. B., Berikbaeva, T. M., Shkumatov, V. M., and Akhrem,
A. A. (1984) Biochim. Biophys. Acta, 791, 375-383.
31.Chashchin, V. L., Usanov, S. A., Lapko, V. N.,
Adamovich, T. B., Shkumatov, V. M., and Akhrem, A. A. (1986) in
Chemistry of Peptides and Proteins (Voelter, W., Bayer, E.,
Ovchinnikov, Yu., and Ivanov, V., eds.) Walter de Greyter and Co,
Berlin-New York, pp. 177-189.
32.Akhrem, A. A., Dzhagarov, B. M., Rumas, B. K.,
Timinsky, Yu. V., Shkumatov, V. M., and Chashchin, V. L. (1979)
Dokl. Akad. Nauk SSSR, 5, 1256-1259.
33.Smettan, G., Shkumatov, V. M., Ristau, O., Rein,
H., and Ruckpaul, K. (1989) in Cytochrome P450: Biochemistry and
Biophysics (Schuster, I., ed.) Taylor and Francis, London-New
York-Philadelphia, pp. 435-438.
34.Gilevich, S. N., Guryev, O. L., Shkumatov, V. M.,
Chashchin, V. L., and Akhrem, A. A. (1987) Biokhimiya,
52, 198-213.
35.Chashchin, V. L., Shkumatov, V. M., Lapko, A. G.,
Degtyareva, L. N., and Sherman, S. N. (1989) Biokhimiya,
54, 559-563.
36.Turko, I. V., Usanov, S. A., Akhrem, A. A., and
Chashchin, V. L. (1988) Biokhimiya, 53, 1352-1356.
37.Hara, T., and Kimura, T. (1989) J.
Biochem., 105, 601-605.
38.Lambeth, J. D., Seybert, D. W., and Kamin, H.
(1979) J. Biol. Chem., 254, 7255-7264.
39.Pikuleva, I. A., Tesh, K., Waterman, M. R., and
Kim, Y. (2000) Arch. Biochem. Biophys., 373, 44-55.
40.Tuls, J., Geren, L., Lambeth, J. D., and Millett,
F. (1987) J. Biol. Chem., 262, 10020-10025.
41.Lambeth, J. D., Geren, L. M., and Millett, F.
(1984) J. Biol. Chem., 259, 10025-10029.
42.Vickery, L. E. (1997) Steroids, 62,
124-127.
43.Muller, E.-C., Lapko, A., Otto, A., Muller, J.,
Ruckpaul, R., and Heinemann, U. (2001) Eur. J. Biochem.,
268, 1837-1843.
44.Grinberg, A. V., Hannemann, F., Schiffler, B.,
Muller, J., Heinemann, U., and Bernhardt, R. (2000) Proteins,
40, 590-612.
45.Kido, T., and Kimura, T. (1979) J. Biol.
Chem., 254, 11806-11815.
46.Hara, T., Koba, C., Takeshima, M., and Sagara, Y.
(2000) Biochem. Biophys. Res. Commun., 276, 210-215.
47.Ivanov, Y. D., Kanaeva, I. P., Karuzina, I. I.,
Usanov, S. A., Hui Bon Hoa, G., Sligar, S. G., and Archakov, A. I.
(2001) J. Inorg. Biochem., 87, 175-184.
48.Juvonen, R. O., Shkumatov, V. M., and Lang, M. A.
(1988) Eur. J. Biochem., 171, 205-211.
49.Smettan, G., Shkumatov, V. M., Pommerening, K.,
and Ruckpaul, K. (1985) in Cytochrome P-450. Biochemistry,
Biophysics and Induction of Cytochrome P-450 (Vereczkey, L., and
Mayar, K., eds.) Akademie Kiado, Budapest, pp. 207-210.
50.Ta, D. T., and Vickery, L. E. (1992) J. Biol.
Chem., 267, 11120-11125.
51.Kakuta, Y., Horio, T., Takahashi, Y., and
Fukuyama, K. (2001) Biochemistry, 40, 11007-11012.
52.Knoell, H. E., and Knappe, J. (1974) Eur. J.
Biochem., 50, 245-252.
53.Pember, S. O., Powell, G. L., and Lambeth, J. D.
(1983) J. Biol. Chem., 258, 3198-3206.
54.Shkumatov, V. M., Radyuk, V. G., Faletrov, Ya.
V., Vinogradova, A. A., Luzikov, V. N., and Novikova, L. A. (2006)
Biochemistry (Moscow), 71, 884-892.
55.Hall, D. A., Vander Kooi, C. W., Stasik, C. N.,
Stevens, C. Y., Zuiderweg, E. R., and Matthews, R. G. (2001) Proc.
Natl. Acad. Sci. USA, 98, 9521-9526.
56.Ingelman, M., Bianchi, V., and Eklund, H. (1997)
J. Mol. Biol., 268, 147-157.
57.Wan, J. T., and Jarrett, J. T. (2002) Arch.
Biochem. Biophys., 406, 116-126.
58.Leadbeater, C., McIver, L., Campopiano, D. J.,
Webster, S. P., Baxter, R. L., Kelly, S. M., Price, N. C., Lysek, D.
A., Noble, M. A., Chapmann, S. K., and Munro, A. W. (2000) Biochem.
J., 352, 257-266.
59.McIver, L., Leadbeater, C., Campopiano, D. J.,
Baxter, R. L., Daff, S. N., Chapman, S. K., and Munro, A. W. (1998)
Eur. J. Biochem., 257, 577-585.
60.Jenkins, C. M., and Waterman, M. R. (1994) J.
Biol. Chem., 269, 27401-27408.
61.Jenkins, C. M., Pikuleva, I., Kagava, N., and
Waterman, M. R. (1997) Arch. Biochem. Biophys., 347,
93-102.
62.Jenkins, C. M., Genzor, C. G., Fillat, M. F.,
Waterman, M. R., and Gymez-Moreno, C. (1997) J. Biol. Chem.,
272, 22509-22513.
63.Gruez, A., Zeghouf, M., Bertrand, J.,
Eschenbrenner, M., Coves, J., Fontecave, M., Pignol, D., and
Fontecilla-Camps, J.-C. (1998) Acta Crystallogr., 54,
135-136.
64.Zeghouf, M., Fontecave, M., Macherel, D.,
and Coves, J. T. (1998) Biochemistry, 37, 6114-6123.
65.Zeghouf, M., Defaye, G., Fontecave, M., and
Coves, J. (1998) Biochem. Biophys. Res. Commun., 246,
602-605.
66.Sakaki, T., Akiyoshi-Shibata, M., Yabusaki, Y.,
Manabe, K., Murakami, H., and Ohkawa, H. (1991)
Pharmacogenetics, 1, 86-93.
67.Wu, D. A., Hu, M. C., and Chung, B. C. (1991)
DNA Cell Biol., 10, 201-209.
68.Sakaki, T., Shinkyo, R., Takita, T., Ohta, M.,
and Inouye, K. (2002) Arch. Biochem. Biophys., 401,
91-98.
69.Barros, M. H., and Nobrega, F. G. (1999)
Gene, 233, 197-203.
70.Manzella, L., Barros, M. H., and Nobrega, F. G.
(1998) Yeast, 14, 839-846.
71.Lacour, T., Achstetter, T., and Dumas, B. (1998)
J. Biol. Chem., 273, 23984-23992.
72.Schiffler, B., Bureik, M., Reinle, W.,
Müller, E. C., Hannemann, F., and Bernhardt, R. (2004) J.
Inorg. Biochem., 98, 1229-1237.
73.Radyuk, V. G., Shkumatov, V. M., Chashchin, V.
L., and Akhrem, A. A. (1983) Biokhimiya, 48, 454-463.
74.Shkumatov, V. M., Radyuk, V. G., Gaponova, G. I.,
Chashchin, V. L., Akhrem, A. A., Smettan, G., and Rukpaul, K. (1988)
Biokhimiya, 53, 1962-1971.
75.Shkumatov, V. M., Radyuk, V. G., Gilevich, S. N.,
and Chashchin, V. L. (1987) USSR Patent No. 365709.
76.Shkumatov, V. M., Radyuk, V. G., and Chashchin,
V. L. (1988) USSR Patent No. 1461000.
77.Compagnone, N. A., Bulfone, A., Rubenstein, J.
L., and Mellon, S. H. (1995) Endocrinology, 136,
2689-2696.
78.Kayes-Wandover, K. M., and White, P. C. (2000)
J. Clin. Endocrinol. Metab., 85, 2519-2525.
79.Morohashi, K., Sogawa, K., Omura, T., and
Fujii-Kuriyama, Y. (1987) J. Biochem. (Tokyo), 101,
879-887.
80.Oonk, R. B., Parker, K. L., Gibson, J. L., and
Richards, J. S. (1990) J. Biol. Chem., 265,
22392-22401.
81.Chung, B. C., Matteson, K. J., Voutilainen, R.,
Mohandas, T. K., and Miller, W. L. (1986) Proc. Natl. Acad. Sci.
USA, 83, 8962-8966.
82.Youngblood, G. L., Nesbitt, M. N., and Payne, A.
H. (1989) Endocrinology, 125, 2784-2786.
83.Nelson, D. R., Kamataki, T., Waxman, D. J.,
Guengerich, F. P., Estabrook, R. W., Feyereisen, R., Gonzalez, F. J.,
Coon, M. J., Gunsalus, I. C., and Gotoh, O. (1993) DNA Cell
Biol., 12, 1-51.
84.Blum, H., Leigh, J. S., Salerno, J. C., and
Ohnishi, T. (1978) Arch. Biochem. Biophys., 187,
153-157.
85.Chashchin, V. L., Lapko, V. N., Adamovich, T. B.,
Lapko, A. G., Kuprina, N. S., and Akhrem, A. A. (1986) Biochim.
Biophys. Acta, 871, 217-223.
86.Chashchin, V. L., Vasilevsky, V. I., Lapko, V.
N., Adamovich, T. B., Berikbaeva, T. M., Shkumatov, V. M., and Akhrem,
A. A. (1984) Biochim. Biophys. Acta, 791, 375-383.
87.Morohashi, K., Fujii-Kuriyama, Y., Okada, Y.,
Sogawa, K., Hirose, T., Inayama, S., and Omura, T. (1984) Proc.
Natl. Acad. Sci. USA, 81, 4647-4651.
88.Vijayakumar, S., and Salerno, J. C. (1992)
Biochim. Biophys. Acta, 1160, 281-286.
89.Usanov, S. A., Graham, S. E., Lepesheva, G. I.,
Azeva, T. N., Strushkevich, N. V., Gilep, A. A., Estabrook, R. W., and
Peterson, J. A. (2002) Biochemistry, 41, 8310-8320.
90.Headlam, M. J., Wilce, M. C., and Tuckey, R. C.
(2003) Biochim. Biophys. Acta, 1617, 96-108.
91.Sivozhelezov, V., and Nicolini, C. (2005) J.
Theor. Biol., 234, 479-485.
92.Storbeck, K. H., Swart, P., and Swart, A. C.
(2007) Mol. Cell. Endocrinol., 265/266, 65-70.
93.Chashchin, V. L., Turko, I. V., Akhrem, A. A.,
and Usanov, S. A. (1985) Biochim. Biophys. Acta, 828,
313-324.
94.Tsujita, M., and Ichikawa, Y. (1993) Biochim.
Biophys. Acta, 1161, 124-130.
95.Lewis, D. F., and Lee-Robichaud, P. (1998) J.
Steroid Biochem. Mol. Biol., 66, 217-233.
96.Graham-Lorence, S., and Peterson, J. A. (1996)
FASEB J., 10, 206-214.
97.Gotoh, O. (1992) J. Biol. Chem.,
267, 83-90.
98.Nabi, N., Kominami, S., Takemori, S., and Omura,
T. (1983) J. Biochem., 94, 1517-1527.
99.Omura, T., and Ito, A. (1991) Meth.
Enzymol., 206, 75-81.
100.Ou, W. J., Ito, A., Morohashi, K.,
Fujii-Kuriyama, Y., and Omura, T. (1986) J. Biochem. (Tokyo),
100, 1287-1296.
101.Pfanner, N., Rassow, J., van der Klei, I. J.,
and Neupert, W. (1992) Cell, 68, 999-1002.
102.Nguen, V., Argan, C., Lusty, C. J., and Shore,
G. C. (1986) J. Biol. Chem., 261, 800-805.
103.Matocha, M. F., and Waterman, M. R. (1984)
J. Biol. Chem., 259, 8672-8678.
104.Ogishima, T., Okada, Y., and Omura, T. (1985)
J. Biochem. (Tokyo), 98, 781-790.
105.Zuber, M. X., Mason, J. I., Simpson, E. R., and
Waterman, M. R. (1988) Proc. Natl. Acad. Sci. USA, 85,
699-703.
106.Luzikov, V. N., Novikova, L. A., Whelan, J.,
Hugosson, M., and Glaser, E. (1994) Biochem. Biophys. Res.
Commun., 199, 33-36.
107.Novikova, L. A., Savel’ev, A. S.,
Zvyagil’skaya, R. A., and Luzikov, V. N. (1996) FEBS
Lett., 378, 182-184.
108.Savel’ev, A. S., Novikova, L. A.,
Kovaleva, I. E., Luzikov, V. N., Neupert, W., and Langer, T. (1998)
J. Biol. Chem., 273, 20596-20602.
109.Savel’ev, A. S., Novikova, L. A., Drutsa,
V. L., Isaeva, L. V., Chernogolov, A. A., Usanov, S. A., and Luzikov,
V. N. (1997) Biochemistry (Moscow), 62, 779-786.
110.Saveliev, A. S., Kovaleva, I. E., Novikova, L.
A., Isaeva, L. V., and Luzikov, V. N. (1999) Arch. Biochem.
Biophys., 363, 373-376.
111.Kovaleva, I. E., Grivennikov, S. I., and
luzikov, V. N. (2000) Biochemistry (Moscow), 65,
1206-1211.
112.Kovaleva, I. E., Novikova, L. A., Nazarov, P.
A., Grivennikov, S. I., and Luzikov, V. N. (2003) Eur. J.
Biochem., 270, 222-229.
113.Stuart, R. A., Ono, H., Langer, T., and
Neupert, W. (1996) Cell Struct. Funct., 21, 403-406.
114.Rojo, E. E., Guiard, B., Neupert, W., and
Stuart, R. A. (1999) J. Biol. Chem., 274,
19617-19722.
115.Rojo, E. E., Guiard, B., Neupert, W., and
Stuart, R. A. (1998) J. Biol. Chem., 273, 8040-8047.
116.Cauet, G., Balbuena, D., Achstetter, T., and
Dumas, B. (2001) Eur. J. Biochem., 268, 4054-4062.
117.Minenko, A. N., Novikova, L. A., Luzikov, V.
N., and Kovaleva, I. E. (2008) Biochim. Biophys. Acta,
1780, 1121-1130.
118.Hoogenraad, N. J., Ward, L. A., and Ryan, M. T.
(2002) Biochim. Biophys. Acta, 1592, 97-105.
119.Bohnert, M., Pfanner, N., and van der Laan, M.
(2007) FEBS Lett., 581, 2802-2810.
120.Parmar, J., Key, R. E., and Rainey, W. E.
(2008) J. Clin. Endocrinol. Metab., 93, 4542-4546.
121.Wada, A., Mathew, P. A., Barnes, H. J.,
Sanders, D., Estabrook, R. W., and Waterman, M. R. (1991) Arch.
Biochem. Biophys., 290, 376-380.
122.Morohashi, K., Fujii-Kuriyama, Y., Okada, Y.,
Sogawa, K., Hirose, T., Inayama, S., and Omura, T. (1984) Proc.
Natl. Acad. Sci. USA, 81, 4647-4651.
123.Von Heijne, G. (1988) Biochim. Biophys.
Acta, 947, 307-333.
124.Churchill, P. F., and Kimura, T. (1979) J.
Biol. Chem., 254, 10443-10448.
125.Usanov, S. A., Chernogolov, A. A., and
Chashchin, V. L. (1990) FEBS Lett., 275, 33-35.
126.Schwarz, D., Kruger, V., Chernogolov, A. A.,
Usanov, S. A., and Stier, A. (1993) Biochem. Biophys. Res.
Commun., 195, 889-896.
127.Schwarz, D., Richter, W., Kruger, V.,
Chernogolov, A., Usanov, S., and Stier, A. (1994) J. Struct.
Biol., 113, 207-215.
128.Wimley, W. C., and White, S. H. (1996) Nat.
Struct. Biol., 3, 842-848.
129.Williams, P. A., Cosme, J., Sridhar, V.,
Johnson, E. F., and McRee, D. E. (2000) Mol. Cell, 5,
121-131.
130.Nakayama, K., Puchkaev, A., and Pikuleva, I. A.
(2001) J. Biol. Chem., 276, 31459-31465.
131.Pikuleva, I. A., Puchkaev, A., and Bjorkhem, I.
(2001) Biochemistry, 40, 7621-7629.
132.Pikuleva, I. (2004) Mol. Cell.
Endocrinol., 215, 161-164.
133.Kusano, K., Kagawa, N., Sakaguchi, M.,
Waterman, M. R., and Omura, T. (2001) J. Biochem. (Tokyo),
129, 259-269.
134.Kusano, K., Kagawa, N., Sakaguchi, M., Omura,
T., and Waterman, M. R. (2001) J. Biochem. (Tokyo), 129,
271-277.
135.Chen, C. D., Doray, B., and Kemper, B. (1998)
Arch. Biochem. Biophys., 350, 233-238.
136.Kemper, B. (2004) Toxicol. Appl.
Pharmacol., 199, 305-319.
137.Mast, N., Andersson, U., Nakayama, K., Bjorhem,
I., and Pikuleva, I. (2004) Arch. Biochem. Biophys., 428,
99-108.
138.Vinogradova, A. A., Luzikov, V. N., and
Novikova, L. A. (2007) Biochemistry (Moscow), 72,
208-214.
139.Nazarov, P. A., Drutsa, V. L., Miller, W. L.,
Shkumatov, V. M., Luzikov, V. N., and Novikova, L. A. (2003) DNA
Cell. Biol., 22, 243-252.
140.Vinogradova, A. A., Luzikov, V. N., and
Novikova, L. A. (2004) in Reports of Young Scientists [in
Russian], MGU Publishers, Moscow, Vol. 1, pp. 31-41.
141.Shkumatov, V. M., Smettan, G., Ristau, O.,
Rein, H., Ruckpaul, K., Chaschin, V. L., and Akhrem, A. A. (1988)
Chem.-Biol. Interact., 68, 71-83.
142.Shkumatov, V. M. (1993) 2nd Int. Symp. on
Cytochrome P450 of Microorganisms and Plants, Hachioji-Tokyo, June
13-17, L‑28.
143.Shkumatov, V. M. (1991) Cytochrome
P450-Containing Enzymatic Systems of Biosynthesis of Biologically
Active Substances: Author’s abstract of Doctoral dissertation
[in Russian], Bach Institute of Biochemistry, Moscow.
144.Shkumatov, V. M. (1992) Proc. 7th Int. Conf.
“Biochemistry, Biophysics of Cytochrome P-450”,
Joint Stock Company, Moscow, p. 481.
145.Uchida, E., Kagawa, N., Sakaki, T., Urushino,
N., Sawada, N., Kamakura, M., Ohta, M., Kato, S., and Inouye, K. (2004)
Biochem. Biophys. Res. Commun., 323, 505-511.
146.Aoyama, T., Nagata, K., Yamazoe, Y., Kato, R.,
Matsunaga, E., Gelboin, H. V., and Gonzalez, F. J. (1990) Proc.
Natl. Acad. Sci. USA, 87, 5425-5429.
147.Benson, R. E., Gottlin, E. B., Christensen, D.
J., and Hamilton, P. T. (2003) Antimicrob. Agents Chemother.,
47, 2875-2881.
148.Duport, C., Spagnoli, R., Degryse, E., and
Pompon, D. (1998) Nat. Biotechnol., 16, 186-189.
149.Shimada, T., Tsumura, F., Gillam, E. M. J.,
Guengerich, F. P., and Inoue, K. (2000) Protein Expres. Purif.,
20, 73-80.
150.Ehmer, P. B., Jose, J., and Hartmann, R. W.
(2000) J. Steroid Biochem. Mol. Biol., 75, 57-63.
151.Steinborn, G., Gellissen, G., and Kunze, G.
(2007) FEMS Yeast Res., 7, 1197-1205.
152.Narhi, L. O., and Fulco, A. J. (1986) J.
Biol. Chem., 261, 7160-7169.
153.McMillan, K., Bredt, D. S., Hirsch, D. J.,
Snyder, S. H., Clark, J. E., and Masters, B. S. (1992) Proc. Natl.
Acad. Sci. USA, 89, 11141-11145.
154.Murakami, H., Yabusaki, Y., Sakaki, T.,
Shibata, M., and Ohkawa, H. (1987) DNA, 6, 189-197.
155.Yabusaki, Y., Murakami, H., Sakaki, T.,
Shibata, M., and Ohkawa, H. (1988) DNA, 7, 701-711.
156.Chun, Y. J., Shimada, T., and Guengerich, F. P.
(1996) Arch. Biochem. Biophys., 330, 48-58.
157.Fisher, C. W., Shet, M. S., Claude, D. L.,
Martin-Wixtrom, C. A., and Estabrook, R. W. (1992) Proc. Natl. Acad.
Sci. USA, 89, 10817-10821.
158.Harikrishna, J. A., Black, S. M., Szklarz, G.
D., and Miller, W. L. (1993) DNA Cell Biol., 12,
371-379.
159.Black, S. M., Harikrishna, J. A., Szklarz, G.
D., and Miller, W. L. (1994) Proc. Natl. Acad. Sci. USA,
91, 7247-7251.
160.Dilworth, F. J., Black, S. M., Guo, Y. D., and
Miller, W. L. (1996) Biochem. J., 320, 267-271.
161.Cao, P. R., Bulow, H., Dumas, B., and
Bernhardt, R. (2000) Biochim. Biophys. Acta, 1476,
253-264.
162.Sibbesen, O., de Voss, J. J., and Montellano,
P. R. (1996) J. Biol. Chem., 271, 22462-22469.
163.Lacour, T., and Ohkawa, H. (1999) Biochim.
Biophys. Acta, 1433, 87-102.
164.Novikova, L. A., Nazarov, P. A.,
Savel’ev, A. S., Drutsa, V. L., Sergeev, V. N., Miller, V. L.,
and Luzikov, V. N. (2000) Biochemistry (Moscow), 65,
1362-1366.
165.Shet, M. S., Fisher, C. W., Holmans, P. L., and
Estabrook, R. W. (1996) Arch. Biochem. Biophys.,
330, 199-208.
166.Wilks, A., Black, S. M., Miller, W. L., and
Ortiz de Montellano, P. R. (1995) Biochemistry, 34,
4421-4427.
167.Sakaki, T., Shibata, M., Yabusaki, Y.,
Murakami, H., and Ohkawa, H. (1990) DNA Cell Biol., 9,
603-614.
168.Sakaki, T., Kominami, S., Takemori, S., Ohkawa,
H., Akiyoshi-Shibata, M., and Yabusaki, M. J. (1994)
Biochemistry, 33, 4933-4939.
169.Chaurasia, C. S., Alterman, M. A., Lu, P., and
Hanzlik, R. P. (1995) Arch. Biochem. Biophys., 317,
161-169.
170.Helvig, C., and Capdevila, J. H. (2000)
Biochemistry, 39, 5196-5205.
171.Isaeva, L. V., Spiridonova, V. A., Lyumovich,
D., Kovaleva, I. E., Chernogolov, A. A., Usanov, A. S., and Luzikov, V.
N. (1996) Biochemistry (Moscow), 61, 1026-1033.
172.Gillam, E. M. J., Aguinaldo, A. M. A., Notley,
L. M., Kim, D., Mundkowski, R. G., Volkov, A. A., Arnold, F. H.,
Soucek, P., deVoss, J. J., and Guengerich, F. P. (1999) Biochem.
Biophys. Res. Commun., 265, 469-472.
173.Sawada, N., Sakaki, T., Kitanaka, S., Takeyama,
K., Kato, S., and Inouye, K. (1999) Eur. J. Biochem.,
265, 950-956.
174.Sakaki, T., Sawada, N., Komai, K., Shiozawa,
S., Yamada, S., Yamamoto, K., Ohyama, Y., and Inouye, K. (2000) Eur.
J. Biochem., 267, 6158-6165.
175.Shashkova, T. V., Luzikov, V. N., and Novikova,
L. A. (2006) Biochemistry (Moscow), 71, 810-814.
176.Usanov, S. A., Chernogolov, A. A., Petrashin,
A. I., Akhrem, A. A., and Chashchin, V. L. (1987) Biol. Membr.
(Moscow), 4, 1102-1115.
177.Sakaki, T., Shibata, M., Yabusaki, Y.,
Murakami, H., and Ohkawa, H. (1989) DNA, 8, 409-418.
178.Shkumatov, V. M., Usova, E. V., Polyako, Yu.
S., Frolova, N. S., Radyuk, V. G., Mauersberger, S., Chernogolov, A.
A., Honeck, H., and Schunck, W.-H. (2002) Biochemistry
(Moscow), 67, 456-467.
179.Shkumatov, V. M., Usova, E. V., Radyuk, V. G.,
Frolova, N. S., Raikov, A. V., Novikova, L. A., Nazarov, P. A., Drutsa,
V. L., and Luzikov, V. N. (2001) in Selected Works of Belorussian
State University [in Russian], Khimiya, Minsk, Vol. V, pp.
446-450.
180.Shkumatov, V. M., Usova, E. V., Radyuk, V. G.,
Kashkan, Zh. N., Kovganko, N. V., Juretzek, T., and Mauersberger, S.
(2003) Bioorg. Khim., 29, 640-647.
181.Dumas, B., Cauet, G., Lacour, T., Degryse, E.,
Laruelle, L., Ledoux, C., Spagnoli, R., and Achstetter, T. (1996)
Eur. J. Biochem., 238, 495-504.
182.Szczebara, F. M., Chandelier, C., Villeret, C.,
Masurel, A., Bourot, S., Duport, C., Blanchard, S., Groisillier, A.,
Testet, E., Costaglioli, P., Cauet, G., Degryse, E., Balbuena, D.,
Winter, J., Achstetter, T., Spagnoli, R., Pompon, D., and Dumas, B.
(2003) Nat. Biotechnol., 21, 143-149.
183.Mauersberger, S., Ohkuma, M., Schunck, W.-H.,
and Takagi, M. (1996) Candida maltosa, in Nonconventional
Yeasts in Biotechnology (Wolf, K., ed.) Springer-Verlag,
Berlin-Heidelberg-New York, pp. 411-580.
184.Barth, G., and Gaillardin, C. (1996)
Yarrowia lipolytica, in Nonconventional Yeasts in
Biotechnology (Wolf, K., ed.) Springer-Verlag,
Berlin-Heidelberg-New York, pp. 313-388.
185.Fickers, P., Benetti, P. H., Wache, Y., Marty,
A., Mauersberger, S., Smit, M. S., and Nicaud, J.-M. (2005) FEMS
Yeast Res., 5, 527-543.
186.Cirigliano, M. C., and Carman, G. M. (1984)
Appl. Environ. Microbiol., 48, 747-750.
187.Mauersberger, S., Wang, H. J., Gaillardin, C.,
Barth, G., and Nicaud, J.-M. (2001) J. Bacteriol., 183,
5102-5109.
188.Shkumatov, V. M., Frolova, N. S., Rudaya, E.
V., Faletrov, Ya. V., Mauersberger, S., and Barth, G. (2006) Prikl.
Biokhim. Mikrobiol., 42, 539-546.
189.Novikova, L. A., Yovkova, V., Faletrov, Ya.,
Agalarov, Ya., Luzikov, V. N., Shkumatov, V. M., Barth, G., and
Mauersberger, S. (2008) in Proc. of IV Congr. Russian Soc. of
Biochemists and Molecular Biologists [in Russian], Arta,
Novosibirsk, p. 353.
190.Schmid-Berger, N., Schmid, B., and Barth, G.
(1994) J. Bacteriol., 176, 2477-2482.
191.Juretzek, T., Mauersberger, S., Schunck, W.-H.,
Nicaud, J.-M., and Barth, G. (1999) Curr. Genet., 35,
307.
192.Juretzek, T., Wang, H.-J., Nicaud, J.-M.,
Mauersberger, S., and Barth, G. (2000) Biotechnol. Bioprocess.
Eng., 5, 320-326.
193.Juretzek, T., Le Dall, M.-T., Mauersberger, S.,
Gaillardin, C., Barth, G., and Nicaud, J.-M. (2001) Yeast,
18, 97-113.
194.Juretzek, T., Mauersberger, S., and Barth, G.
(2000) Patents DE19932811, WO0003008.
194a.Holz, M., Förster, A., Mauersberger, S., and Barth, G. (2009)
Appl. Microbiol. Biotechnol., 81, 1087-1096.
195.Shkumatov, V. M. (2007) EMBO Workshop: The
Chemistry and Biochemistry of Catalysis by Biological Systems,
20-22 June, EMBL, Hamburg, p. 20.
196.Delorome, C., Piffeteau, A., Sobrio, F., and
Marquet, A. (1997) Eur. J. Biochem., 254, 252-260.
197.Zhu, N., Ling, Y., Lei, X., Handratta, V., and
Brodie, A. M. (2003) Steroids, 68, 603-611.
198.Handratta, V. D., Vasaitis, T. S., Njar, V. C.,
Gediya, L. K., Kataria, R., Chopra, P., Newman, D., Jr., Farquhar, R.,
Guo, Z., Qiu, Y., and Brodie, A. M. (2005) J. Med. Chem.,
48, 2972-2984.
199.Matsunaga, N., Kaku, T., Itoh, F., Tanaka, T.,
Hara, T., Miki, H., Iwasaki, M., Aono, T., Yamaoka, M., Kusaka, M., and
Tasaka, A. (2004) Bioorg. Med. Chem., 12, 2251-2273.
200.Jarman, M., Smith, H. J., Nicols, P. J., and
Simons, C. (1998) Natural Prod. Rep., 15, 495-512.
201.Weintraub, P. M., Holland, A. K., Gates, C. A.,
Moore, W. R., Resvick, R. J., Bey, P., and Peet, N. P. (2003)
Bioorg. Med. Chem., 11, 427-431.
202.Hartmann, R. W., Palusczak, A., Lacan, F.,
Ricci, G., and Ruzziconi, R. (2004) J. Enzyme Inhib. Med. Chem.,
19, 145-155.
203.Peeh, D. M., Seto, E., Hsu, J. Y., and Feldman,
D. (2002) Urology, 168, 1583-1588.
204.Saberi, M. R., Shah, K., and Simons, C. (2005)
J. Enzyme Inhib. Med. Chem., 20, 135-141.
205.Gasi, K. M., Stojanovic, S. Z., Sakac, M. N.,
Popsavin, M., Santa, S. J., Stankovic, S. M., Klisuric, O. R., Andric,
N., and Kovacevic, R. (2005) Steroids, 70, 47-53.
206.Pouget, C., Yahiaoui, S., Fagnere, C.,
Habrioux, G., and Chulia, A. J. (2004) Bioorg. Chem., 32,
494-503.
207.Ulmschneider, S., Muller-Vieira, U., Klein, C.
D., Antes, I., Lengauer, T., and Hartmann, R. W. (2005) J. Med.
Chem., 48, 1563-1575.
208.Ulmschneider, S., Muller-Vieira, U., Mitrenga,
M., Hartmann, R. W., Oberwinkler-Marchais, S., Klein, C. D., Bureik,
M., Bernhardt, R., Antes, I., and Lengauer, T. (2005) J. Med.
Chem., 48, 1796-1805.
209.Ehmer, P. B., Bureik, M., Bernhardt, R.,
Muller, U., and Hartmann, R. W. (2002) J. Steroid Biochem. Mol.
Biol., 81, 173-179.
210.Bureik, M., Hubel, K., Dragan, C. A., Scher,
J., Becker, H., Lenz, N., and Bernhardt, R. (2004) Mol. Cell.
Endocrinol., 217, 249-254.
211.Yee, S. W., and Simons, C. (2004) Bioorg.
Med. Chem. Lett., 14, 5651-5654.
212.Posner, G. H., Crawford, K. R., Yang, H. W.,
Kahramana, M., Jeon, H. B., Li, H., Lee, J. K., Suha, B. C., Hatcher,
M. A., Labonte, T., Usera, A., Dolan, P. M., Kensler, T. W., Peleg, S.,
Jones, G., Zhang, A., Korczak, B., Saha, U., and Chuang, S. S. (2004)
J. Steroid Biochem. Mol. Biol., 89/90, 5-12.
213.Parise, R. A., Egorin, M. J., Kanterewicz, B.,
Taimi, M., Petkovich, M., Lew, A. M., Chuang, S. S., Nichols, M.,
El-Hefnawy, T., and Hershberger, P. A. (2006) Int. J. Cancer,
119, 1819-1828.
214.Hakki, T., and Bernhardt, R. (2006)
Pharmacol. Therap., 111, 27-52.
215.Smith, I. E., and Dowsett, M. (2003) New
Engl. J. Med., 348, 2431-2442.
216.Brueggemeier, R. W., Hackett, J. C., and
Diaz-Cruz, E. S. (2005) Endocr. Rev., 26, 331-345.
217.Brunoa, R. D., and Njar, V. C. O.
(2007) Bioorg. Med. Chem., 15, 5047-5060.
218.Zhuang, Y., Wachall, B. G., and Hartmann, R. W.
(2000) Bioorg. Med. Chem., 8, 1245-1252.
219.Handratta, V. D., Jelovac, D., Long, B. J.,
Kataria, R., Nnane, I. P., Njar, V. C. O., and Brodie, A. M. H. (2004)
J. Steroid Biochem. Mol. Biol., 92, 155-165.
220.Jagusch, C., Negri, M., Hille, U. E., Hu, Q.,
Bartels, M., Jahn-Hoffmann, K., Pinto-Bazurco Mendieta, M. A.,
Rodenwaldt, B., Muller-Vieira, U., Schmidt, D., Lauterbach, T.,
Recanatini, M., Cavalli, A., and Hartmann, R. W. (2008) Bioorg. Med.
Chem., 16, 1992-2010.
221.Pinto-Bazurco Mendieta, M. A. E., Negri, M.,
Jagusch, C., Hille, U. E., Muller-Vieira, U., Schmidt, D.,
Hansenc, K., and Hartmann, R. W. (2008) Bioorg. Med. Chem.
Lett., 18, 267-273.
222.O’Donnell, A., Judson, I., Dowsett, M.,
Raynaud, F., Dearnaley, D., Mason, M., Harland, S., Robbins, A.,
Halbert, G., Nutley, B., and Jarman, M. (2004) Br. J.
Cancer, 90, 2317-2325.
223.Aoyama, Y., Yoshida, Y., Kubota, S., Kumaoka,
H., and Furumichi, A. (1978) Arch. Biochem. Biophys.,
185, 362-369.
224.Vico, P., Cauet, G., Rose, K., Lathe, R., and
Degryse, E. (2002) Yeast, 19, 873-886.
225.Ngatcha, B. T., Van, Luu-The, and Poirier, D.
(2000) Bioorg. Med. Chem. Lett., 10, 2533-2536.
226.Gomez-Lechon, M. J., Donato, M. T., Castell, J.
V., and Jover, R. (2004) Curr. Drug Metab., 5,
443-462.
227.Shkumatov, V. M., Faletrov, Y. V., Frolenkov,
K. A., Mauersberger, S., Barth, G., Novikova, L. A., and Luzikov, V. N.
(2008) 9th Int. Symp. Cytochrome P450 Biodiversity and
Biotechnology, Nice, France, June 8-12, p. 67.
228.Shkumatov, V. M., Usova, E. V., Frolova, N. S.,
Barth, G., and Mauersberger, S. (2006) Biomed. Khim., 52,
298-308.
229.Shkumatov, V. M., Faletrov, Ya. V.,
Mauersberger, S., Barth, G., Novikova, L. A., and Luzikov, V. N. (2007)
XVIII Mendeleev Congr. General and Applied Chemistry [in
Russian], Moscow, Vol. 4, p. 492.
230.Shkumatov, V. M. (2007) The 1st Congr. of
Scientists of Belarus Republic [in Russian], Minsk, November 1-2,
2007, pp. 413-420.
231.Muller-Vieira, U., Angotti, M., and Hartmann,
R. W. (2005) J. Steroid Biochem. Mol. Biol., 96,
259-270.
232.Dragan, C. A., Hartmann, R. W., and Bureik, M.
(2006) J. Enzyme Inhib. Med. Chem., 21, 547-556.
233.Fujita, K., and Kamataki, T. (2002) Drug
Metab. Pharmacokinet., 17, 1-22.