REVIEW: Molecular Diversity of Spider Venom
A. A. Vassilevski*, S. A. Kozlov, and E. V. Grishin*
Shemyakin–Ovchinnikov Institute of Bioorganic Chemistry, Russian Academy of Sciences, ul. Miklukho-Maklaya 16/10, 117997 Moscow, Russia; E-mail: grev@ibch.ru; avas@ibch.ru* To whom correspondence should be addressed.
Received April 7, 2009; Revision received June 5, 2009
Spider venom, a factor that has played a decisive role in the evolution of one of the most successful groups of living organisms, is reviewed. Unique molecular diversity of venom components including substances of variable structure (from simple low molecular weight compounds to large multidomain proteins) with different functions is considered. Special attention is given to the structure, properties, and biosynthesis of toxins of polypeptide nature.
KEY WORDS: spider venom, toxin, neurotoxin, cytolytic peptide, acylpolyamine, latrotoxin, toxic enzymes, protein structure and function, biosynthesisDOI: 10.1134/S000297909130069
Abbreviations: a.a., amino acid residue; AMP, antimicrobial peptide; AP, acylpolyamine; CP, cytolytic peptide; CSαβ, cysteine-stabilized α-helix–β-sheet motif; DDH, disulfide-directed β-hairpin motif; ER, endoplasmic reticulum; ESM, extra structural motif; ICK, inhibitor cystine knot motif; LD50, median lethal dose; Mm, molecular mass; NMR, nuclear magnetic resonance; PSM, principal structural motif.
This review deals with spider venom, a factor that has played a decisive
role in evolution of one of the most successful groups of living
organisms (some facts of spider biology are given in a special appendix
at the end of this article). In our view, just during venom production
spiders have achieved unprecedented perfection, and in this case their
biological variability is especially pronounced. Spider venoms are
complex mixtures of biologically active compounds of different chemical
nature, from salts to large multidomain proteins. More than a hundred
different components can be found in the same venom, and in this
parameter spiders are leaders in living nature. During long-term
evolution, venom composition underwent continuous improvement and
adjustment for efficient functioning in the killing or paralyzing of
prey and/or as a repellent against aggressors. Different venom
components work synergistically, thus providing efficiency of action of
the mixture. Venom composition is highly species-specific and depends
on many factors including sex, nutrition, natural habitat, climate,
etc. [1-9]. Spiders carefully
spend their venom and learned to calculate accurately its dosage
depending on the victim’s size and resistance to the venom. If
the amount of venom necessary for a particular hunted object exceeds
its reserve, the spider wisely recedes [6, 10-12]. Insects are the main
natural targets for spider venoms. Specificity of action of some spider
toxins is unique: along with high toxicity for insects, they can be
absolutely harmless for members of other taxons, and this could be the
basis for creation of a new generation of insecticides [13-16]. Although the majority of
spiders are venomous, as a rule they are not dangerous to people or are
not more harmful than mosquitos or wasps; only a few spider species
present a real threat to humans (Table 1) [17]. Spider venoms are sources of highly specific
substances affecting different systems of membrane transport such as
ion channels, ionotropic receptors, etc. These substances are
indispensable instruments in studies of membrane systems and are used
widely in modern neurobiology. Because of the large number of spider
species and the complex composition of their venoms, specific
modulators of practically any membrane transport system can thus be
found for use not only in fundamental investigations but also in
treatment of diseases associated with compromised membrane transport
system functions [7, 18-29].
Table 1. Spiders exhibiting immediate threat
for human health
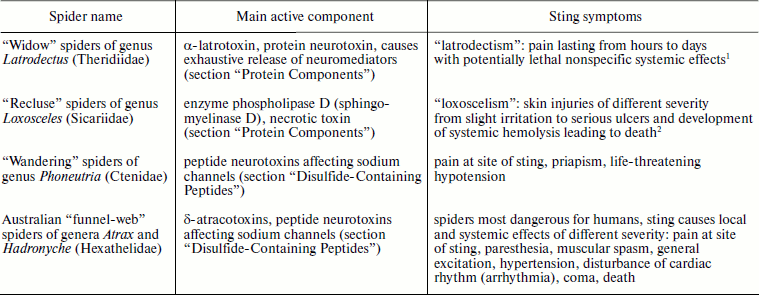
1Bites of spiders of genus Steatoda (“false
widows”) of the same family Theridiidae lead to
“steatodism” (a mild form of latrodectism).
2Spiders of genus Sicarius of the same family
Sicariidae produce even more powerful venom than members of genus
Loxosceles, but records concerning human victims are few because
these spiders live in deserts.
This review deals with results of investigations of spider venom composition and the structural and biological properties of their separate components. Special attention is paid to venom protein and peptide molecules and their biosynthesis, mechanisms of action, and application in pharmacology.
COMPOSITION OF SPIDER VENOM
Among the large number of spiders, the venoms of only about 100 species have been studied, and only a few of these have been thoroughly investigated. First of all, these are of course spiders of medical significance (Table 1) as well as some of those that have become standard objects of arachnologists or traditional for fauna of certain countries native to various research groups [6, 7, 16, 30-34].
Spider venoms are complex multicomponent mixtures of biologically active substances serving general purposes for both attacking (killing or paralyzing prey) and/or protecting (repellent) [1, 2]. In both cases, spider venoms are able to cause paralysis or severe pain. Two large venom groups are distinguished by the character of their action: neurotoxic and necrotic (cytolytic), although these effects can be exhibited simultaneously. Spider venoms include substances of different chemical nature that can be conventionally divided to three groups by molecular mass (Mm): (i) low molecular weight (<1 kDa) substances of various structure; (ii) peptides (1-10 kDa), with two main structure–functional subgroups being distinguished in this group—disulfide-containing neurotoxins and linear cytolytic peptides (CP); (iii) high molecular weight (>10 kDa) substances—different proteins including enzymes and neurotoxins.
The total number of individual components of spider venom studied using such methods of proteomics and peptidomics as two-dimensional electrophoresis, multimeric chromatography, and different types of mass spectrometry, etc., as well as genetic methods can exceed one thousand; therefore, their investigation is a complex problem [4, 18, 23, 28, 29, 35-38]. Despite extraordinary variety of chemical structure and biological function, often only a single type of venom components is favored. At present spiders are known which dominantly produce each of the above-mentioned groups of substances (Table 2). On average, spider venoms contain ~25% polypeptides by weight, and concentrations of major venomous components, acylpolyamines (AP) and polypeptides, reach tens of millimolar [7, 8, 37, 38].
Table 2. Main components of spider
venoms
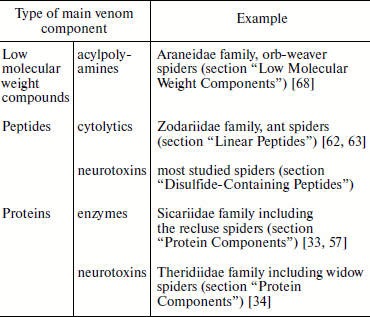
Most studied spiders produce venom with a predominance of disulfide-containing peptide neurotoxins considered in the section “Disulfide-Containing Peptides” of this review. Many of these substances, in turn, are characterized by a common cysteine motif of primary structure and belong to “knottins”, peptide molecules that form in space the “cystine knot” structure [39-43]. Up to several hundred of knottin molecules with similar spatial structure, stabilized by invariant cystine residues, can be present in the same venom. The specificity of action of each molecule is defined by its unique combination of variable amino acid residues located in the loop regions between the disulfide bridges (Tables 3 and 4). Such ensembles of the venom peptide molecules are usually called natural combinatorial libraries of biologically active substances [3, 16, 44-48]. Such “libraries” also appear in other animal venoms such as that of snakes, scorpions, cone snails, and sea anemones, but other motifs of amino acid sequences are specific in these cases and different types of spatial molecular structure are realized [49-54].
Table 3. Combinatorics of
disulfide-containing peptides in spider venom (example of
Chilobrachys jingzhao)
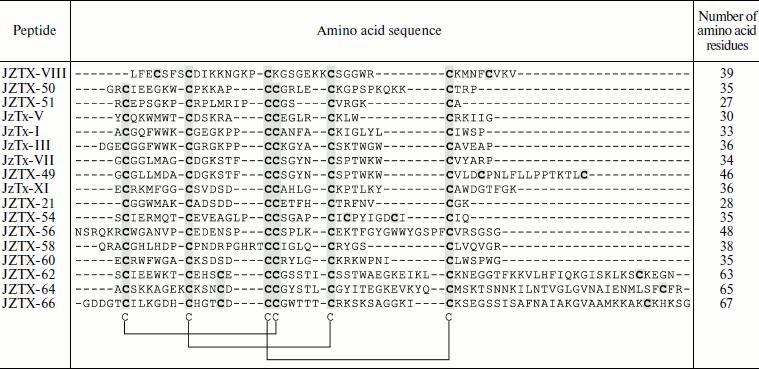
Note: An example of molecular diversity of spider venom components
(from [48]). Amino acid sequences of peptides
belonging to different families are shown. Cysteine residues are
darkened. Arrangement of disulfide bonds in the cystine knot motif is
shown below.
Table 4. Combinatorics of
disulfide-containing peptides in spider venom (example of Agelena
orientalis)
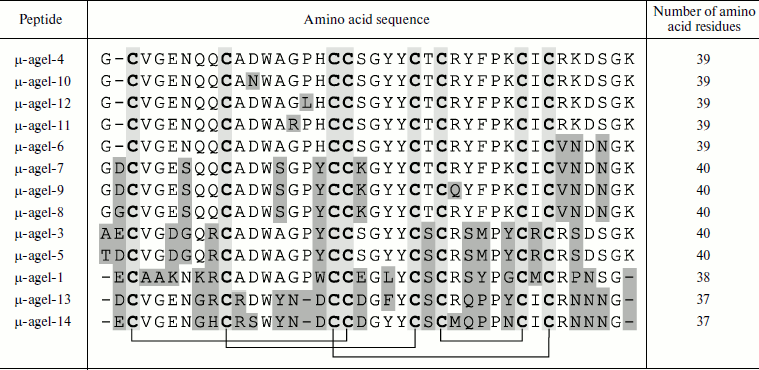
Note: An example of molecular diversity of spider venom components
(from [45]). Amino acid sequences of peptides
belonging to the same family are shown. Cysteine residues are shown by
slight darkening; differing residues are marked by heavier darkening.
Arrangement of disulfide bonds is shown below.
At least four exceptions from the above-described strategy of venom composition generation are known (Table 2). (a) “Widow” spiders of genus Latrodectus produce highly specialized protein neurotoxins called latrotoxins (section “Protein Components”) [34]. This property is also characteristic of other members of Theridiidae (tangle-web spiders) such as those of the Steatoda genus [55]. Note that another “classic” example of complex protein neurotoxins is tetanus and botulinum toxins of bacteria from the Clostridium genus [56]. (b) Different enzymes including phospholipase D are present in venom of spiders of the Sicariidae family (in particular, in venom of “recluse” spiders of the Loxosceles genus) (section “Protein Components”) [33, 57]. This property draws together the recluse spiders and pathogenic bacteria of genus Corynebacterium [58, 59]. Recall also that phospholipases A2 are an important component of bee and snake venoms [60, 61]. (c) “Ant spiders” of Central Asian species Lachesana tarabaevi (Zodariidae family) produce venom exhibiting pronounced cytolytic effect due to the presence of numerous various CP (section “Linear Peptides”) [62, 63]. This is also specific of different arthropods: bees, bumblebees, wasps, and ants [38, 60, 64-67]. (d) Finally, orb-weaver spiders of the Araneidae family produce various AP neurotoxins with intermediate action specificity (section “Low Molecular Weight Components”) [68]. AP are also found in wasp venoms [35, 68]. Thus, in cases (a) and (b) protein components are dominant, peptides are dominant in case (c), and polyamines in case (d). Any of these venom types can exhibit neurotoxic effect, while necrotic (cytolytic) venoms are characterized by the presence of polypeptide components. An “intermediate” type of venom composition or “repertoire” of produced toxins is characteristic of some spiders. For example, members of the Lycosoidea superfamily produce venoms with high CP content but a larger and/or functionally more important part of their venoms is represented by the knottin-type cysteine-containing neurotoxins, and in some cases the presence of significant amount of AP is found [18, 28, 38, 69-72]. Simultaneous presence in venoms of polyamine and peptide neurotoxins at high concentrations is characteristic of spiders of different taxonomic groups [7, 18, 28, 73]. Numerous protein components, mainly enzymes, are present in spider venoms, but in functional aspect these substances are considered as additional with exception of the already mentioned cases [4, 31, 32, 74].
Fine adjustment of the venom component composition, most efficiently providing for attack and/or protection function, was developed during evolution of venomous animals. Apparently two main strategies in formation of the venom composition emerged. One of them was elaborated by the honeybee and is based on utilization of a limited amount of active molecules. Main components of bee venom, the peptide melittin and enzyme phospholipase A2, are cytolytic substances of broad spectrum of action [60, 67]. The other strategy is characteristic of most studied venomous animals and suggests a wide “biomolecular” diversity of venom components [75].
It is supposed that at least four factors are used during evolutionary optimization of venoms of different chemical composition [8, 38, 62, 76]. (i) Functional diversity based on production of molecules affecting different targets. This provides for a sharp increase in the number of potential victims and lowers the probability of elaboration by them of resistance to the venom. For example, venom of North American “funnel-web” spider Agelenopsis aperta (Agelenidae family) contains α-agatoxins (AP inhibiting glutamate receptors and so preventing contraction of insect muscle cells), µ-agatoxins (peptide neurotoxins, modulators/activators of sodium channels in membranes of insect neuronal cells, which stimulate secretion of neuromediators), and a number of ω-agatoxins (peptide neurotoxins, inhibitors of calcium channels in neurons, prevent release of neuromediators), differently affecting various channels of insects and vertebrates [7]. (ii) Selectivity and efficiency, namely, selection of toxins as a high-precision weapon exhibiting the most specific and powerful effect. Due to such selection, molecules selectively recognizing certain targets appeared in hands of researchers. For example, ω-agatoxins IVA and IVB are usually used diagnostic ligands of P-type Ca2+-channels [24, 77-79]. (iii) Synergism: components of different structure and mechanism of action enhance functions of each other during combined use. In this case, the efficient active concentration of the separate components significantly decreases. Examples of functional synergism of different groups of venom components are known [6-8, 71, 76, 80, 81]. For example, synergism was found in the action of potassium salts, histamine, and cytolytic and neurotoxic peptide components of venom of Central American “wandering” spider Cupiennius salei (Ctenidae) [6, 8, 80]. In our view, the concept of “cabals”, elaborated for the cone snail toxins and suggesting coordination in action of several types of venom components for achievement of a certain physiological effect [82, 83], is also valid for spider venoms. For instance, α- and µ-agatoxins of the spider A. aperta venom form one such “cabal” causing rapid paralysis [76]. Another cabal is formed by slowly acting ω-agatoxins causing flaccid paralysis [7, 84]. (iv) Biomolecular diversity suggests production of great numbers of components with similar structure and function, but differing in specificity and mechanism of action [75]. As already mentioned above, according to present-day concepts, peptide venom components of many spiders, scorpions, anemones, snails, and snakes form the so-called natural combinatorial libraries of biologically active molecules selected during evolution [16, 44-54]. This factor is most specific for peptide neurotoxins, but it is also important for evolution of other venom components. The mechanism of formation of such biomolecular diversity is not yet known. It is probably universal for peptide neurotoxins and CP of spider venoms and is based on the existence of numerous gene families (multigene families) encoding these peptides. Biosynthesis of the venom peptide components follows a general scheme (section “Biosynthesis of Spider Venom Components”), most of them being characterized by “standard” organization of precursor molecules in the form of prepropeptides [85]. Preproregions of precursors are conservative, while mature chains are variable. The effect of mutation “concentration” in the gene regions corresponding to mature polypeptides has been called “accelerated” evolution specific of animals with biomolecular diversity of venom components [86-88].
The collected results suggest the concept of the existence of special functions for each venom component. However, much needs to be elucidated in this field. It should be kept in mind that secretory epithelium of spider venom glands is characterized by apocrine (with partial destruction of secretory cells) or even holocrine (with complete cell destruction) type of secretion, and usual cell components are inevitably present in the venom, and it is difficult to understand their specific functions [89-92].
Separate groups of the spider venom components will be considered below in accordance with the scheme of classification given at the beginning of this chapter – from structurally simpler low molecular weight substances to peptides and finally to proteins.
LOW MOLECULAR WEIGHT COMPONENTS
Low molecular weight compounds of different classes of inorganic and organic substances have been found in spider venoms, such as salts, carbohydrates, amino acids, biogenic amines, AP, etc. [1, 2, 29, 93].
Cation concentrations in the venom of wandering spider C. salei are ~10 mM Na+, ~200 mM K+, and ~1 mM Ca2+; different ratios are observed in the spider hemolymph: ~200 mM Na+, ~10 mM K+, and ~4 mM Ca2+ [6, 94]. High concentration of potassium ions in the venom attracts special attention. In such concentration they themselves are able to cause paralysis of the victim due to the potassium-induced depolarization of electro-excitable cell membranes. Also, synergism was revealed in action of the venom peptide neurotoxins and potassium salts, which is probably a widespread event noted not only for C. salei but for the venom of the scorpion Parabuthus transvaalicus as well [8, 95]. It is supposed that the synergism is based on the potassium-induced depolarization during which the voltage-gated ion channels, the targets of neurotoxins, are activated.
Biogenic amines like serotonin, histamine, noradrenalin, etc., as well as amino acids such as glutamate, taurine, or γ-aminobutyric acid are found in venoms of many spiders (often in high concentrations of the order of tens of millimolar and higher) [8, 74, 93, 94, 96-103]. Most of these compounds are known as neuromediators or neuromodulators in the insect nervous system; therefore, their function in the venom is obvious. In some cases synergism was found in action of neurotoxins and these compounds, the effect of which is probably similar to that of potassium salts: activation of neurotoxin receptor targets takes place [7, 8] (see below). Some amines are known to cause pain that can be the basis of the venom protective function and, in addition, they enhance the blood vessel permeability and increase local blood flow, thus contributing to spreading different toxic venom components.
In 1994, an unusual glutamate receptor blocker, sulfated fucopyranosyl guanosine, was found in the venom of North American funnel-web spider Hololena curta (Agelenidae family) [104, 105]. Later it was shown by NMR of whole venoms of many spider species that sulfated nucleosides can be their main components [93, 106]. These are mono- and disulfates of ribonucleosides (guanosine and xanthosine), and some of them are additionally glycosylated (contain one or two fucose residues). Different type nucleosides, whose function is probably potentiation of the toxicity of the polypeptide components, were also found in spider venoms [103, 107, 108].
Investigations of the first half of the 1980s showed that venoms of the Araneidae and Nephilidae family spiders such as Argiope lobata, Nephila clavata, and Araneus gemma are able to disturb nerve–muscle transmission in arthropods by blocking glutamatergic synapses [109-111]. In 1986, the main component of A. lobata (widespread in the Old World) venom, the 636-Da compound argiopin, was isolated and its structure was determined using NMR and mass spectrometry [112]. Four different fragments were found in argiopin: residues of asparagine and arginine, 2,4-dioxyphenylacetic acid, and polyamine (Fig. 1).
Argiopin was the first member of a broad class of toxins from spider venoms that got the common name of “acylpolyamine” or “polyamine” toxins. It was shown in experiments on neuromuscular preparation of flesh fly larvae and on isolated frog spinal cord that argiopin at concentrations ~10–8-10–6 M blocks ion channels activated by glutamate [113-115]. In addition to argiopin, eight different antagonists of glutamate receptors were found in A. lobata venom such as argiopinines and pseudoargiopinines having similar mechanism of action but different affinity to receptors (Fig. 1). All identified toxins selectively blocked ion currents that are activated upon application to membrane of isolated rat hippocampal neurons of glutamate or an agonist of one of the glutamate receptor types – kainic acid [116]. All inhibitors except pseudoargiopinine III (a shortened form of pseudoargiopinines) exhibited pronounced structural homology, contained an arginine residue, aliphatic polyamines, and chromophoric aromatic groups modified by asparagine or lysine [117-119]. The structures of other inhibitors of glutamate receptors from venoms of Asian spiders Nephila pilipes (NSTX-3) and N. clavata (JSTX-3) were determined at approximately the same time [120, 121].Fig. 1. Combinatorics of acylpolyamine toxins in spider venom (example of Argiope lobata). An example of molecular diversity of spider venom components (according to [117, 118]). Structural formulas of substances are shown with corresponding names given below. The following fragments are designated in the structure of argiopin: 1) acyl radical; 2) intermediate group (amino acid residue); 3) polyamine chain; 4) terminal group (arginine residue).
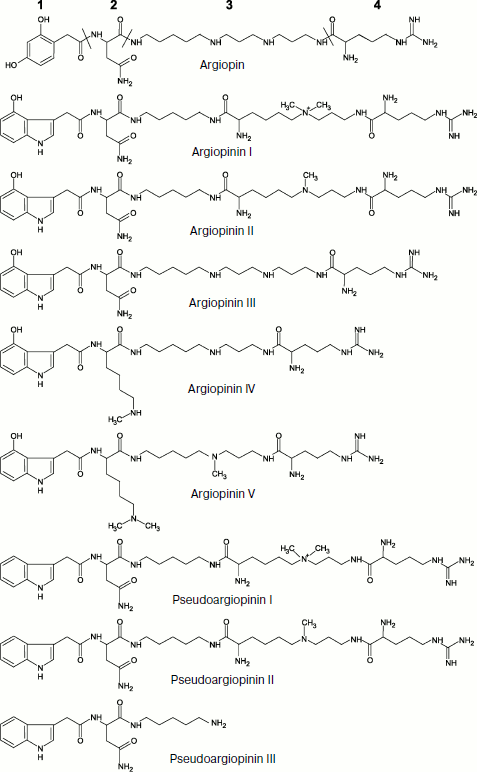
The structures of more than a hundred polyamine toxins from venoms of tens of spider species of different taxonomic groups are now known. All these toxins exhibit notable structural homology, i.e. based on a polyamine chain having primary, quaternary amino-, or guanidine group or an arginine residue at one end of the molecule and in most cases an aromatic group (of variable type) – acyl radical – at the other end. This group joins the polyamine either via an amino acid residue or directly by an amide bond; other modifications can be also introduced (Fig. 1) [7, 18, 35, 68, 122-125]. The polyamine chain resembles structurally well-known polyamines such as spermine, spermidine, putrescine, and cadaverine, which are also found in some spider venoms [101, 126]. As already noted, several different AP modifications can be simultaneously present in the same spider venom. For instance, venom of A. aperta contains over 30 different AP (α-agatoxins) whose structures were determined by mass spectrometry [127]. This is an example of biomolecular diversity (section “Composition of Spider Venom”), as though spiders follow principles of combinatorial chemistry. Toxin molecules exhibit different efficiency and specificity and their resulting mixture is active against a broad range of targets. Note that some insects synthesize combinatorial libraries of macrocyclic polyamines for protection [128]. The discovery of philantotoxins, AP, in the beewolf Philanthus triangulum venom probably illustrates convergent evolution of the wasp and spider venom components [104, 129].
AP are non-competitive inhibitors of activated states of glutamate receptors. In other words, polyamine toxins do not interact with the agonist-binding center, and the receptor should be activated before beginning of their action [68, 130]. In this connection, simultaneous presence of AP and glutamate in venoms of the orb-weaver spiders should be noted. Receptor activation by glutamate of the venom immediately results in their efficient blocking by polyamines, and so there is functional synergism between different venom components [81]. In venoms of funnel-web spiders (Agelenidae family), the functional role of glutamate is played by µ-agatoxins, which results in activation of glutamate receptors due to stimulation of neuromediator release; venom efficiency (the development of paralysis) is much higher when a mixture of α- and µ-agatoxins is used instead of the separate components [7, 76].
Although polyamine toxins are produced by spiders mainly to paralyze invertebrate victims by blocking their glutamate receptors, they are also efficient against glutamate and acetylcholine receptors of the vertebrate nervous system. Moreover, they influence other ionotropic receptors and ion channels, and in most cases they are pore blockers that are efficient in micro- and submicromolar concentrations (10–8-10–6 M). Other mechanisms of action are also possible [68, 122, 123, 131-136]. Comparison of efficiency of natural and synthetic analogs made it possible to identify functional significance of different regions in AP molecules such as the polyamine chain length and modification extent and the size and type of acyl radical and linker region. In some investigations, it was possible to achieve increased selectivity for different receptor types (up to ~103-fold) and efficiency (Kd of complex with receptor ~10–9 M) of polyamine toxins by directed modification of their molecules and by chemical synthesis of the corresponding analogs [18, 116, 137, 138]. Polyamine toxins are indispensable tools in investigations of glutamate receptors. For example, JSTX-3 is used for identification of glutamate receptor subunits (a variable amino acid residue is responsible for high sensitivity to the toxin) [139]. Computer modeling revealed important information concerning structure of the pore region and principles of functioning of different types of receptors as well as enabled the design of selective ligands [139-143].
The function of many known components of spider venoms is still not clear. For example, citrate was found in venom of some spiders [100, 144]. The function of this compound in bee and snake venoms is inhibition of Ca2+-dependent phospholipases, i.e. protection against their own toxins. After venom is injected into an aggressor or victim organism, the mixture is diluted and the inhibitory effect is eliminated [145]. Keeping in mind the development of analytical methods, one should expect that the chemical assortment of known low molecular weight substances of spider venoms will be soon significantly expanded.
LINEAR PEPTIDES
Linear peptides, i.e. those free of disulfide bonds, are rather frequent in spider venoms [18, 28, 29, 38]. They mainly exhibit cytolytic effect not characteristic of disulfide-containing venom components. For this reason, it is convenient to distinguish linear peptides as a special group of spider venom constituents. However, as investigations are developed, isolation of this group will probably seem more and more arbitrary. For instance, short linear kinin-like, bradykinin-potentiating peptides as well as inhibitors of angiotensin-transforming enzyme have been isolated from some spider venoms, which is specific of venoms of different animals [146-149]. These compounds interfere with the victim’s physiological status, and probably in this way they contribute to the overall toxicity of the venom.
Necrotic activity of many spider venoms was established long ago [1]. According to current thinking, it can be due to the presence of two types of components—linear peptides and large proteins with phospholipase activity [33, 38]. Prototypes of both substance groups are presently considered as “classic” main components of the honeybee Apis mellifera venom CP melittin and phospholipase A2 protein, the presence of which is responsible for high and nonspecific cytotoxicity of bee venom [60, 67, 150]. As for spiders, cytolytic activity of venoms of 26 spider species was screened in 1998 [151], and then amino acid sequence was determined for the first time for two CP from venom of North American tarantula Hogna carolinensis (Lycosa carolinensis; “wolf” spiders of the Lycosidae family) [69]. Due to later works in this field, a concept arose concerning CP as an important component of arthropod venom [38, 152].
About 30 CP assigned to 12 groups have been isolated from venoms of five spider species (families Ctenidae, Lycosidae, “lynx” spiders of Oxyopidae, Zodariidae) and characterized (Table 5; amino acid sequences are given in Swiss-Prot section of UniProt database [153]) [62, 63, 69-72, 154]. In addition to linearity, they are characterized by the following features: they are short (contain <50 a.a. with exception of cyto-insectotoxins that will be discussed below), cationic (relatively high positive charge of molecules at neutral pH, pI >10), and amphiphilic (see below) polypeptides prone to formation of α-helices and exhibiting affinity to lipid bilayers [38]. All these features are characteristic of peptides from very different organisms that carry out protective function against pathogens. They form the biggest group of antimicrobial peptides (AMP) [155], so-called α-helical AMP: in aqueous media these peptides are disordered, and they acquire α-helical conformation upon contact with membranes [156]. Cytoplasmic membrane serves as target for CP action – their positive charge provides for electrostatic binding to its surface, amphiphilicity allows the peptides to incorporate into the bilayer, and just disturbance of the bilayer integrity is responsible for cell death [157-159].
Table 5. Cytolytic peptides of spider
venom
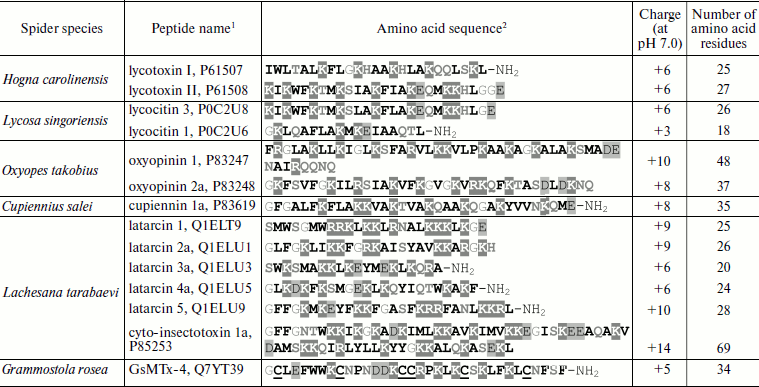
1The UniProt database codes are shown [153].
2Hydrophobic residues are marked in bold, positively
charged residues are shaded dark grey, negatively charged residues
– light grey, glycine residues are in grey print, hydrophilic
uncharged residues – in bold grey. Half-cystine residues are
underlined. C-Terminal amidation is marked as NH2.
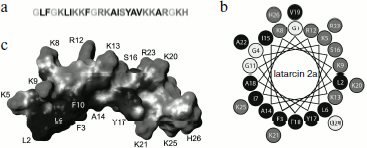
The amphiphilic (amphipathic) property is expressed in spatial separation of hydrophobic and hydrophilic (including charged) amino acid residues, forming clusters or patches on the surface of molecules. Amphiphilicity can be revealed at the level of primary structure; for example, in melittin positively charged amino acid residues are clustered in the C-terminal region [160]. For most CP from spider venoms and for α-helical AMP, amphiphilicity is revealed at the level of secondary structure and does not result in any particular amino acid residues distribution along the amino acid sequence. It is convenient to visualize amphipathic α-helices using projection on a plane such as “helical wheel” (Schiffer–Edmundson projection) (Fig. 2) [161]. In some cases, amphiphilicity becomes obvious when spatial structure is considered, as in the case of cupiennins [72]. Spatial structure is now known for three CP from spider venoms (they were studied by NMR and are presented in the PDB database [162]): cupiennin 1a (from C. salei venom) in aqueous trifluoroethanol imitating a membrane environment (code in PDB database is 2K38) [163], and two latarcins, 1 and 2a (from L. tarabaevi venom) in complex with sodium dodecyl sulfate micelles (2PCO, 2G9P) [164, 165]. All three structures are similar: the peptides form amphipathic α-helices. Owing to the amphipathic properties, CP are characterized by a peculiar intramolecular gradient of hydrophobic potential (Fig. 2) [166, 167] and can efficiently incorporate into membranes. In this case, side chains of hydrophilic amino acid residues are oriented to the aqueous phase (and/or to membrane “interface” where they interact with lipid “heads”), while side chains of hydrophobic amino acid residues are immersed in the nonpolar phase of lipid “tails”.
CP from spider venoms exhibit activity against a broad spectrum of target cells of pro- and eukaryotic origin in the micromolar range of concentrations. Compared to neurotoxins, they seem less specific and efficient. Most of these substances are characterized by low selectivity and correspondingly by receptor-independent mechanism of action. It will not be an exaggeration to say that CP from venoms of spiders and other arthropods are typical membranolytics, but nevertheless efficiency of their action depends on the membrane lipid composition and membrane potential [38, 62, 63, 67, 71, 152, 154, 156-158, 168-171]. Several models have been proposed to explain the increase in permeability of target cell membranes in response to CP. Three of them appear most often in the literature. According to the “barrel-stave” model, CP molecules form oligomers piercing the membrane with channel or pore formation. Experimental confirmation of this model was obtained for the peptide antibiotic (peptaibol) alamethicin [172]. Another model suggests formation of a “carpet” of numerous incorporated peptide molecules on the membrane that is destroyed after achievement of a threshold concentration. This model was proposed for AMP from amphibian skin (dermaseptins) and insect hemolymph (cecropins) [173-175] and is suggested for many latarcins [164, 165, 168]. The model of “toroidal” pores formed both by peptide molecules and membrane lipids obtained experimental grounds for the majority of CP (such as magainin from frog skin and melittin) [176, 177]. It is supposed that most CP from spider venoms act just in this way [163, 165, 168, 170, 171]. The possibility of peptide action using several mechanisms simultaneously should be noted along with the absence of really clear borders between the above-described models; there have been attempts to join all models into one [159, 168, 178]. Besides, we believe that in biological sense there is no principal significance of detailed mechanism of CP action, and only the final result is important – realization of venomous function.Fig. 2. Cytolytic peptide latarcin 2a from the spider Lachesana tarabaevi venom. a) Amino acid sequence (code in the UniProt database Q1ELU1 [153]). b) “Helical wheel” projection. Hydrophobic, hydrophilic, and glycine residues are marked in different shades of gray. c) Spatial structure in complex with detergent micelles (code in PDB database 2G9P [162]). The virtual surface of the peptide molecule is darkened in accordance with hydrophobic potential [166, 167] using the MOLMOL program (http://hugin.ethz.ch/wuthrich/software/molmol/).
The following additional characteristics of α-helical CP from spider venoms should be mentioned. (i) Obvious prevalence of lysine residues over arginines. Reasons for this prevalence are not quite clear; more often replacement of some positively charged amino acid residues by others does not influence functional properties of CP [179]. Arginine residues are incorporated in processing motifs of the venom peptide components and their elimination from the mature peptide sequences can be associated with assurance of correct maturation (section “Biosynthesis of Spider Venom Components”). (ii) Frequent posttranslational modification – C-terminal amidation. This modification is due to the presence of an additional C-terminal glycine residue in precursor molecules (section “Biosynthesis of Spider Venom Components”) [180]. (iii) Often glycine or serine appears as the N-terminal amino acid residue, which is probably due to peculiarities of limited proteolysis of propeptides, namely, to the specificity of processing enzymes. (iv) Despite general preference for amino acid residues with high potential to form helical conformation, glycine and/or proline residues are frequent in the middle of CP sequences. As a result, peptides are characterized by helix–loop–helix type of structure. The appearance of local regions with disordered structure or with quite pronounced conformational lability influences functional features of molecules and their specificity toward different types of membranes [163-165, 171, 181, 182].
For comparison, Table 5 gives the sequence of the disulfide-containing peptide GsMTx-4 (code in UniProt database is Q7YT39 [153]) from the venom of the Chilean rose bird-eating spider Grammostola rosea (Grammostola spatulata; Theraphosidae family). This peptide is an inhibitor of mammalian mechanosensitive receptors and also exhibits cytolytic activity [183, 184]. Structurally it is a typical neurotoxin of spider venom, and it belongs to the class of molecules forming the so-called “cystine knot” in space (section “Disulfide-Containing Peptides”) [185]. A great number of AMP from plants and hemolymph of arthropods belong to the same class [186, 187]. So, evolutionary parallelism in the CP molecule structure is revealed in this way. It was found that the classic blocker of potassium channels charybdotoxin from the venom of the scorpion Leiurus quinquestriatus hebraeus also exhibits antimicrobial activity, and in spatial structure (motif of “cysteine-stabilized α-helix–β-sheet”, CSαβ) it resembles AMP from other sources [188, 189]. The existence of molecules simultaneously exhibiting neuro- and cytotoxic activities can be explained by (a) random emergence of the less specific and less powerful cytolytic activity due to the necessary structural requirements of amphiphilicity, the presence of a charge, etc. [190]; (b) directed selection of such molecules that are either a peculiar point of neurotoxin and CP divergence or successfully combine both necessary functions.
In addition to short latarcins, long linear cyto-insectotoxins exhibiting equally pronounced antimicrobial, cytolytic, and insecticidal activities were found in L. tarabaevi venom [63, 154]. These peptides twice exceed “average” CP both by length and charge (Table 5), but on the basis of other characteristics they belong to α-helical AMP. They are now perhaps the longest known peptides of this class. Biological activity draws them together with “usual” peptide toxins, but the absence in their structure of disulfide bonds made it possible to identify cyto-insectotoxins as an independent group of spider venom components, as well as CP and AMP. Cyto-insectotoxins can be considered as composed of two short peptides (about 30 a.a. in length) joined by a linker resembling the processing motif of precursors from spider venom glands (section “Biosynthesis of Spider Venom Components”). It is supposed that these compounds emerged in evolution from “binary” precursors due to mutation of the key arginine residue in the processing site, and therefore they are unusual “modular” molecules of the CP–CP type. The mutation was fixed because it had a positive effect: cyto-insectotoxins, unlike short CP, exhibited high insecticidal activity. Another example of modular peptide toxins is also known: modules of linear CP and CSαβ are combined in the structure of some polypeptides from scorpion venoms – the CP–CSαβ type [191, 192]. We believe that the variability of modular toxins can be wider, and their emergence correlates with the general evolutionary vector “from simple to complex”.
Active function of AMP as effector molecules of innate immunity in plants and animals is generally accepted [193-195]. Bacteria and fungi also produce CP to displace competitors from ecological niches [196]. A number of possible functions of spider venom CP have been proposed [38]. (i) Direct toxic effect is most often considered as the main function of CP in venoms [63, 67, 69]. However, it is necessary to note pronounced differences both in cyto- and total toxicity of different CP, and some specificity of action is observed. Besides, this function is obvious only if corresponding peptides are the major venom component; otherwise, their contribution to toxicity is insignificant [62, 152]. (ii) Synergism with neurotoxins in action. This effect was shown experimentally for oxyopinins and cupiennins [8, 71]. The molecular basis of synergism is not clear; it is supposed that CP serve as an original “guide”, “spreading factor” for neurotoxins, due to their cytolytic properties CP “clear” the way to neurons through protective cellular barriers. (iii) Direct antimicrobial effect. Many arthropods live in large aggregates or colonies where decrease in risk of epidemic is important. It is possible that the CP-containing venom is used as an antiseptic [66, 197]. Besides, CP concentration in a stung victim is usually sufficient to clear all microorganisms, and these compounds can be used as conservants. Finally, the CP function might also be direct protection of venom glands against infection [191, 198, 199]. It is still not clear why authors of works on the venom cytolytic components do not consider their function in external digestion common among spiders. In our view, the ability of CP to make food digestion easier via destruction of the victim’s cell and tissue structures is obvious, and such CP function is probably a leading one.
DISULFIDE-CONTAINING PEPTIDES
At the present time approximately 500 peptides with Mm <10 kDa (amino acid sequences are shown in the Swiss-Prot section of the UniProt database [153]) are characterized for 60 spider species of 20 families; most of these peptides contain disulfide bonds and exhibit neurotoxic properties. However, it is only a small part of the actual natural diversity. Peptidomic and genetic investigations point to simultaneous presence in the same venom of up to several hundred and more peptides, so with account for the remarkable spider species variability [200, 201], it can be admitted that researchers deal with a huge natural library having tens of million or more molecules [3, 4, 31, 32, 45, 46, 48, 202, 203]. Neurotoxins are as a rule efficient against corresponding receptors already at nanomolar concentrations, a complex with numerous contacts being formed (Kd of toxin complex with receptor ~10–9 M), i.e. there is specific target recognition. Compared to nonselective substances such as CP, neurotoxins exhibit their effect in significantly lower (at least by one order of magnitude) doses. In this case, the so-called median lethal dose (LD50), causing 50% lethality, depends on the target organism and the route of toxin introduction (it can vary in a broad range from fractions of µg/kg to tens of mg/kg and up to the absence of significant toxicity), which is first of all due just to selectivity towards certain receptors.
In addition to peptides from spider venoms, there are also many well characterized families of polypeptide toxins affecting different targets and isolated from animals of various taxonomic groups like snakes, scorpions, cone snails, and sea anemones; the number of known substances is more than one thousand. For successful orientation in this diversity, adequate toxin classification and nomenclature are necessary. The recent tendency in toxin denomination using Greek symbols and names of producing animals is clear. The conus toxin nomenclature, elaborated in detail, suggests that a Greek letter in front of polypeptide name points to its main target [83]. However, this principle does not always agree with accepted norms in nomenclature of other animal toxins. Moreover, division only on the basis of main targets is not enough due to numerous toxins with no detected pharmacological effect; therefore, other criteria are used additionally for toxin classification. For example, considering cysteine residue distribution in amino acid sequence, the same conotoxins are subdivided into several superfamilies, and within each of them separate groups are formed according to the mechanism of action [83, 204]. Classification based on primary structure homology and position of structurally important amino acid residues was established for scorpion toxins affecting potassium channels [205, 206]. For spider peptide toxins, a principle of classification following the pattern of the primary structure motifs was proposed based on analysis of amino acid sequences using a special algorithm and not considering pharmacological properties [45]. The next step was a recently proposed “global” classification of animal toxins, which combines pharmacological toxin parameters and data on taxonomic position of the producing animal [207]. The most frequent symbols in names of spider toxins should be noted: α – the targets are chemo-excitable ionotropic receptors of postsynaptic membrane (nicotine acetylcholine and glutamate), κ – K+ channels, µ – Na+ channels (in the novel nomenclature the symbol µ is used only for pore blockers, while the symbol δ is used for modulators of these channels), ω – Ca2+ channels.
Unlike conotoxins [208], spider peptide toxins seldom contain modified amino acid residues. Widespread posttranslational modifications of these molecules include C-terminal amidation and splitting off C-terminal positively charged amino acid residues, “standard” transformations in biosynthesis of secreted polypeptides in eukaryotes (section “Biosynthesis of Spider Venom Components”). Unique exceptions are PLTX-II from the venom of Plectreurys tristis (Plectreuridae family; P34079) and ω-agatoxin IVB from the venom of A. aperta (P37045). In the first case the C-terminal amino acid residue is modified by palmitic acid [209], and in the second case chirality of one amino acid residue is altered, which results in significant increase in toxin activity (by one-two orders of magnitude) and stability [210, 211]. ω-Agatoxins IA and IB (P15969, P15970) are two-chain polypeptides (Mm ~7.5 kDa) whose chains are fixed by a disulfide bond. Both toxin chains are encoded by the same gene, and their separation is the result of propeptide cleavage in accordance with characteristic motifs of limited proteolysis (section “Biosynthesis of Spider Venom Components”) [85, 212]. Another example of two-chain peptide is CSTX-13 (Mm ~7.5 kDa, P83919), so-called neurotoxic enhancer from the venom of C. salei [80], probably synthesized in a similar way.
Independently of their biological activity, peptide spider toxins are characterized by a number of features of amino acid sequence that determine the polypeptide chain folding in space. Due to their small size, the three-dimensional structure of these molecules is usually studied by NMR. Now it is possible to speak definitely about three types of fold characteristic of spatial structure of disulfide-containing peptides of spider venoms (presented in the PDB database [162]). All three types of fold are not unique for spiders, but are extremely widespread in nature. Functional variability of peptides with identical type of spatial structure has caused great interest of scientists in these folds, which represent peculiar structural “scaffolds” for creation of compounds with desired properties. After the immunoglobulin domains, the fold types shown below are favorite objects of structural biology, rational design, and structural–functional investigations of biologically active compounds [213, 214].
The polypeptide chain fold described by a structural motif of the so-called “inhibitor cystine knot” (ICK) (Tables 3 and 4) is characteristic of the overwhelming majority of known spider peptide toxins. The same motif appears in numerous peptides with various functions isolated from very diverse sources, i.e. animals, plants, fungi, viruses: C1 X2-7 C2 X3-11 C3 X0-7 C4 X1-17 C5 X1-19 C6, where X is any amino acid residue. The following arrangement of disulfide bonds is observed in all molecules of this type: C1–C4, C2–C5, C3–C6. Spatial structure of peptides with ICK motif is characterized by the presence of a β-hairpin and a peculiar “knot” (origin of its name): the third disulfide bond (C3–C6) pierces the ring formed by the other two disulfides and the main chain atoms joining these bonds. Polypeptides with the ICK fold are also often called knottins; now over a thousand knottins with different biological functions are known such as protease inhibitors, AMP, insecticidal and antihelminthic peptides, neurotoxins, humoral regulators, etc. [39, 41-43, 186, 187]. It should be noted that some molecules posses the conditions of the primary structure motif and are characterized by the same type of disulfide arrangement, but the knot structure is not formed in space; therefore, these molecules do not belong to the ICK class [215]. Thus, the primary structure motifs and even arrangement of disulfide bonds not always unambiguously define the molecular fold.
Three main fragments can be distinguished in the “canonical” structure of the knottin type spider toxins containing six half-cystine residues (Fig. 3): N-terminal region of about 8-10 amino acid residues (to the second half-cystine residue); central fragment richest in half-cystine residues and containing specific “doubled” CC motif, and finally, C-terminal fragment after the fifth half-cystine residue characterized by the highest variability both in size and amino acid composition. Strict regularity was noted in the distribution of cysteine residues in the N-terminal and central parts of the toxin amino acid sequence, which resulted in formulation for them of a special primary structure motif. For the overwhelming majority of toxins the distance between the first and second cysteine residues corresponds to 6 a.a., while the third and fourth cysteine residues usually occupy adjacent positions. These observations made it possible to identify the so-called principal structural motif, PSM: C1 X6 C2………C3 C4, where X is any amino acid residue. In practical aspect, the presence of the PSM motif in any studied spider venom peptide structure is probably a sufficient condition to suppose reliably the neurotoxic function. This is usually important when amino acid sequence is deduced from the nucleotide sequence. Analysis of sequences containing more than six cysteine residues revealed a different regularity: in more than 70% of analyzed structures the fifth and sixth, as well as the seventh and eighth cysteine residues are arranged at interval of one amino acid residue (Table 4). The extra structural motif (ESM) was proposed: C5 X C6………C7 X C8, where X is any amino acid residue. The presence in the studied structure of an ESM motif without PSM does not point reliably to neurotoxic function of the peptide. Examples of PSM and ESM motifs revealed in spider toxins are shown in Fig. 3, and several rarer varieties of these motifs were also found [45].
The ICK type of fold abundance in spider peptide neurotoxins is confirmed by investigation of over 30 spatial structures, but while the general fold is retained there is high variability in detail (examples are shown in Figs. 4 and 5). For numerous toxins this type of fold is implied on the basis of the arrangement of cysteine residues in the primary structure and (when known) the general scheme of disulfide bond formation. Peptides form in space tightly packed coils with extremely stable structures. In terms of PSM and ESM motifs, both N- and C-terminal regions of the molecule are fixed at the central core. In this case, prevalent elements of secondary structure are β-sheets and β-turns, and α-helical conformation is rare. The size of the β-hairpin depends on the distance between the fifth and sixth cysteine residues of the “canonical” cystine knot, and an additional third strand of antiparallel β-structure, formed by the N-terminal region of the molecule, is present in many toxin structures.Fig. 3. Amino acid sequences of spider peptide toxins with indication of PSM and ESM motifs. Examples are agelenin from Allagelena opulenta (code in UniProt database P31328 [153]), heteropodatoxin-1 from Heteropoda venatoria (P58425), hainantoxin-IV from Haplopelma hainanum (P83471), ω-Lsp-IA from Geolycosa sp. (P85079), SFI 1 from Segestria florentina (P61095), δ-palutoxin IT1 from Paracoelotes luctuosus (P83256). Breaks are introduced for optimization of sequence comparison. Numbering of the first five cysteine residues is shown above.
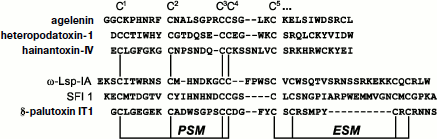
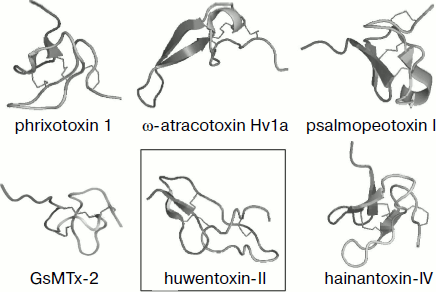
Fig. 4. Spatial structure models for spider venom peptide toxins with six cysteine residues. Ribbon models were drawn using the PyMOL program (http://www.pymol.org/). Examples are phrixotoxin 1 from Paraphysa scrofa (code in PDB database 1V7F [162]), GsMTx-2 from Grammostola rosea (1LUP), ω-atracotoxin Hv1a from Hadronyche versuta (1AXH), psalmopeotoxin I from Psalmopoeus cambridgei (1X5V), hainantoxin-IV from Haplopelma hainanum (1NIY) (ICK motif), and huwentoxin-II from Haplopelma schmidti (1I25; DDH motif, shown in frame).
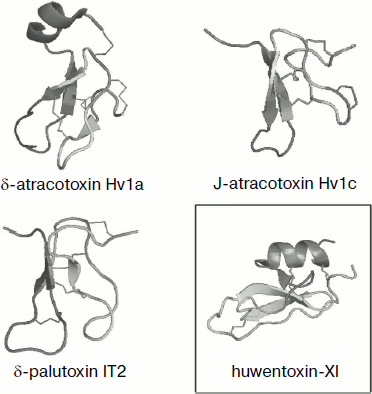
Over six half-cystine residues can be present in molecules with cystine knot fold, and in this case disulfide bonds are arranged so that the ICK motif is not altered (Fig. 5). Moreover, approximately equal numbers of spider toxins with three and four disulfide bonds are known. The latter are usually characterized by the presence of an ESM motif (Fig. 3), and the scheme of S–S bridge arrangement is as follows: C1–C4, C2–C5, C3–C8, C6–C7; the fourth “additional” disulfide bond is introduced into the β-hairpin structure that is usually elongated in this case (δ-palutoxin IT2 structure, 1V91, is shown as an example in Fig. 5). An additional structural element of δ-atracotoxins from venom of Australian funnel-web spiders (Atrax and Hadronyche genera, Hexathelidae family) is fixation by the fourth disulfide bond of the C-terminal region of the molecule; in this case a specific CCC triplet is present in the primary structure [216, 217]. A rare vicinal disulfide bond between cysteine residues in adjacent positions was revealed for the same spider venom insectotoxins called κ- or J-atratoxins (J-ACTXs). This bond is not involved in stabilization of polypeptide chain fold, but it is critical for biological activity, probably due to participation in direct contact with the receptor [218].Fig. 5. Spatial structure models for spider venom peptide toxins with eight cysteine residues. Ribbon models were drawn using the PyMOL program. Examples are δ-atracotoxin-Hv1a (1VTX) and J-atracotoxin-Hv1c (1DL0) from Hadronyche versuta, δ-palutoxin IT2 (1V91) from Paracoelotes luctuosus (ICK motif), and huwentoxin-XI from Haplopelma schmidti (2JOT; Kunitz motif, shown in frame).
It was found during analysis of J-ACTX spatial structures that they exhibit the highest similarity with peptides free of cystine knot but characterized by the “disulfide-directed β-hairpin” (DDH) structural motif C1 X5-19 C2 X2 G/P X2 C3 X6-19 C4, where X is any amino acid residue, and arrangement of disulfide bonds is C1–C3, C2–C4 [218]. This motif implies formation in space of β-hairpins stabilized by two invariant disulfide bonds; in this case the presence of additional secondary structure elements and disulfides is not limited. It is supposed that the DDH motif came earlier in evolution and the ICK type of fold was based on it [219, 220]. For example, the DDH motif is specific of huwentoxin-II of Chinese bird-eating spider Haplopelma schmidti (syn. Haplopelma huwenum, Ornithoctonus huwena, Selenocosmia huwena, Theraphosidae family; 1I25) with disulfide arrangement C1–C3, C2–C5, C4–C6 (Fig. 4), and is also possible for a number of other peptides [219].
Huwentoxin-XI (2JOT), the spatial structure of which is described by the Kunitz motif [221], was also found in H. schmidti spider venom. This motif is widespread in nature and is specific of a number of protease inhibitors (such as the best known bovine pancreatic trypsin inhibitor or aprotinin), K+-channel blockers (from sea anemones and snakes), and other peptides [222-226]. The arrangement of disulfide bonds observed in these molecules is C1–C6, C2–C4, C3–C5, and the spatial structure includes a short N-terminal 310-helix, C-terminal α-helix, and three strands of antiparallel β-structure (Fig. 5).
Note that specific motifs corresponding to the above-mentioned folds are absent from amino acid sequences of some spider venom peptide components. Thus, the diversity of the molecular folds can be significantly wider than that shown here.
Disulfide-containing peptides from spider venoms are characterized by high variability of functional features (Table 6). Many of the first studied spider peptide toxins, like ω-agatoxins from A. aperta venom affecting calcium channels (1990), and hanatoxins from G. rosea venom affecting potassium channels (1995), have become indispensable tools for investigation of their targets [7, 21, 24, 79, 227, 228]. In biological aspect, the main targets of spider toxins should be looked for in insects. The effect on mammals can be: (i) also biologically justified and carry out function of protection from attack; (ii) the result of homology of target molecules among members of different taxons; (iii) accidental performing necessary structural requirements to ligands of corresponding receptors. Main targets (receptors) of presently characterized disulfide-containing peptides from spider venoms are, in accordance with their neurotoxic effect, protein components of electro-excitable cell membranes (of neurons and myocytes). First of all these are different voltage-gated ion channels (potassium, sodium, calcium) but also chemo-, thermo-, and mechanosensitive ionotropic receptors (Table 6), which is the main subject of many reviews [18, 23, 28, 29, 35, 36, 44].
Table 6. Some targets of
disulfide-containing spider toxins
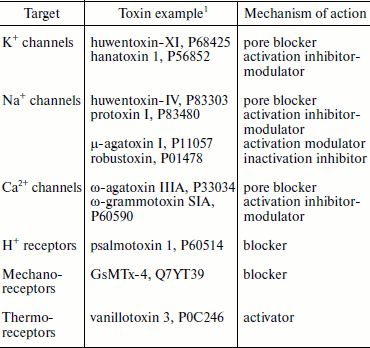
1The UniProt database codes are shown [153].
Spider venom neurotoxins studied to date can be divided to two large groups by their mechanism of action. (1) Pore blockers (direct inhibition of ion conductivity due to interaction with the receptor pore region). For example, huwentoxin-IV (P83303) from the venom of H. schmidti and Tx1 (P17727) from the venom of Brazilian wandering spider Phoneutria nigriventer (Ctenidae family) are pore blockers of Na+ channels and interact with the so-called receptor site 1 where classic inhibitors tetrodotoxin and saxitoxin as well as µ-conotoxins bind [229-233]. Similarly, huwentoxin-XI (P68425) from H. schmidti spider venom is also a typical pore inhibitor of K+ channels; its structure contains a specific “functional dyad” of amino acid residues found in potassium channel blockers from different sources and with various 3D structure [221, 234]. ω-Agatoxin IIIA (P33034) supposedly is a pore blocker of calcium channels [235]. (2) Different types of modulators influencing activation and inactivation of appropriate targets. For instance, hanatoxins 1 and 2 (HaTx; P56852, P56853) from G. rosea venom interact with the voltage-sensitive domain of K+ channels and interfere with their activation [20, 21, 228, 236-238]. The hanatoxin-homologous inhibitor of Ca2+ channels ω-grammotoxin SIA (ω-GrTx SIA; P60590) from the same spider venom acts similarly [239, 240]. Protoxins I and II (ProTx; P83476, P83480) from the venom of Chilean spider Thrixopelma pruriens (Theraphosidae family) have similar effect on Na+ channels [241, 242]. Moreover, these toxins are characterized by cross-activity and interact with the conservative region of the ion channel voltage-sensitive domains, which is called the “paddle” motif [20, 21, 243-248]. Numerous peptides of spider venoms are modulators of Na+ channels; they retard inactivation or cause a shift in activation potential and interact with the so-called receptor sites 3 and 4, the binding sites of α- and β-toxins from scorpion venoms, respectively [233, 249, 250]. For example, the main toxic venom components of dangerous for humans spider genera Atrax and Hadronyche (Table 1) are δ-atracotoxins that similarly to the scorpion α-toxins retard inactivation of Na+ channels and bind to site 3 [249, 251, 252]. The best studied are δ-atracotoxins Ar1a (robustoxin; P01478) and Hv1a (versutoxin; P13494), major components of Atrax robustus and Hadronyche versuta venoms, respectively (LD50 ~0.2 mg/kg in the case of subcutaneous and ~50-100 ng/kg in the case of intracerebral introduction into mice) [253-256]. In the same way components of P. nigriventer venom influencing sodium channels similarly to δ-atracotoxins are responsible for toxicity in mammals: Tx2-1 (P29423), Tx2-5 (P29424), Tx2-6 (δ-CNTX-Pn2a; P29425), and Tx2-9 (P29426) [257, 258], and peptide Tx4(6-1) (δ-CNTX-Pn1a; P59368) selectively influences Na+ channels in insects, also by binding to site 3 [259, 260]. It should be noted that now available efficient antidotes ameliorate bite symptoms of dangerous Australian funnel-web and South American wandering spiders [17, 261]. Like scorpion β-toxins, insectotoxins of funnel-web spiders (µ-agatoxins I-VI from A. aperta venom, P11057-P11062, P60177, and curtatoxins I-III from H. curta venom, P15967, P15968, P60177) cause a shift in the Na+ channel activation potentials and probably interact with site 4 [76, 262]. However, homologous to the latter δ-palutoxins IT1-IT4 (P83256-P83259) from the venom of Asian spider Pireneitega luctuosa (Amaurobiidae family) also bind to site 4, but they inhibit channel inactivation similarly to the scorpion α-toxins [263, 264].
The widespread mechanism of action of peptide neurotoxins (characteristic, for example, of classic acetylcholine receptor inhibitors from snakes and cone snails), namely competitive inhibition of natural agonist interaction with its binding site [265], is not characteristic of spider toxins, but this might be due to insufficiency of our knowledge.
Recent studies suggest two main types of spider toxin interactions with receptors: (i) membrane-independent, direct association with receptor from aqueous solution in the absence of toxin molecule contact with membrane; (ii) membrane-mediated; during interaction with the receptor, the toxin establishes contacts with membrane lipids. The second type of action is still poorly studied, but it is specific of some classic non-peptide toxins and is probably widespread [20, 184, 245, 266-271]. As already mentioned, the main structural requirement to peptides interacting with membranes is their amphiphilicity. This draws neurotoxins acting via membrane-mediated mechanism together with CP and AMP, and they can exhibit cytolytic properties (section “Linear Peptides”).
In addition to target type diversity, many spider toxins are characterized by selective action towards particular receptor groups. Perhaps the best example is toxins influencing calcium channels [272, 273]. Earlier (section “Composition of Spider Venom”) it has been already said that the A. aperta spider venom contains various inhibitors of Ca2+ channels – ω-agatoxins. These peptides form four families with different specificity towards various types of mammalian calcium channels, and in this case the usual biological target for their action is insect channels [7, 227]. ω-Agatoxins IA and IB (P15969, P15970) are two-chain (66 and 3 a.a.) peptides with unknown type of fold (cystine knot fold is specific of members of other families), inhibitors of L-type channels [212, 274-276]. ω-Agatoxins IIA and IIB (90 and 95 a.a., P15971) influence N-type channels [276], whereas ω-agatoxins IIIA-IIID (76 a.a.; P33034, P81744-P81746), despite homology to family II members, exhibit a broad spectrum of activity towards L-, N-, P/Q-, and R-type channels, the only exception being T-type channels [235, 277]. Finally, ω-agatoxins IVA and IVB (48 a.a.; P30288, P37045) are highly specific inhibitors of P/Q type channels [24, 77-79, 278, 279]. The venom of another well-studied spider, P. nigriventer, also contains various peptides influencing Ca2+ channels [30]. For example, neurotoxin Tx3-2 (O76201) is active against L-type channels [280], Tx3-4 (Pn3-4a, ω-PTx-IIA; P81790) is structurally similar to ω-agatoxins III and irreversibly inhibits P/Q- and N-type channels, whereas its action against R-type channels is incomplete and reversible [281]; Tx3-3 (ω-PnTx3-3; P81789) and Tx3-6 (P81792) exhibit a broad spectrum of specificity [282, 283]. Highly selective inhibitors of calcium channels in the insect central neurons were isolated from venoms of Atrax and Hadronyche spider genera; they are the shorter atracotoxins of the ω-ACTX-1 family (36-37 a.a.) and the longer members of the ω-ACTX-2 family (42-44 a.a.) [15, 16]. Activity of ω-atracotoxins Hv1a (P56207) and Hv2a (P82852) from H. versuta venom was studied in detail, and in this case the ratio of toxin concentrations efficient towards insect and mammalian channels, i.e. specificity towards certain taxonomic animal groups, reaches 104 [284-287]. In all the above-mentioned cases both functional diversity and synergism in action against various targets in the victim organism provide for highest efficiency of the mixture.
In our view, with regard of extreme variability of spiders themselves and peptides in their venom compositions, the probability of detection of a ligand to practically any pre-assigned receptor is rather high, and this has been repeatedly confirmed experimentally. For instance, screening of a large species collection resulted in detection of psalmotoxin 1 (PcTx1; P60514) in the venom of the bird-eating Trinidad chevron tarantula Psalmopoeus cambridgei (Theraphosidae) that blocked with high affinity and selectivity subtype 1a of acid-sensing ion channels (ASICs) [19, 288]. A number of mechanosensitive channel ligands were obtained from G. rosea venom such as α-mechanotoxin (GsAF II, P61409), GsMTx-2 (P60273), and GsMTx-4 (Q7YT39) [27, 183, 185]. Finally, agonists of vanilloid receptor TRPV1 called vanillotoxins (VaTx 1-3; P0C244-P0C246) were also found in the venom of P. cambridgei [26, 289].
The variability of targets of disulfide-containing peptides from spider venoms is not restricted to ion transport systems. For example, the lectin-like peptide huwenlectin-1 (SHLP-I; Q86C51) causing agglutination of erythrocytes was isolated from the venom of H. schmidti [290, 291]. A report concerning isolation from P. cambridgei venom of the two peptides psalmopeotoxins I and II (PcFK; P0C201, P0C202) exhibiting antimalarial properties also deserves attention [292, 293]. These peptides in vitro inhibit development of malarial Plasmodium falciparum within infected erythrocytes and do not exhibit hemolytic, cytolytic, and antimicrobial activity; therefore, the molecular target of their action remains unknown. It is also reported that peptide Tx3-4 from P. nigriventer venom, in addition to effect on calcium channels, is also able to block reverse capture of glutamate in synaptosomes, probably by influencing the glutamate transporter [294, 295]. The function of some peptides, as in the case of CSTX-13 from C. salei venom, might be just potentiation of activities of other components [80].
PROTEIN COMPONENTS
Independently of chosen strategy of component composition formation (section “Composition of Spider Venom”), various proteins are usually found in spider venoms. However, the diversity of the protein component has been thoroughly investigated using methods of proteomics and genomics for venoms of only five spider species [4, 31, 32, 296-299]. For instance, proteomic investigations of venoms of Chinese tarantulas Chilobrachys jingzhao (Chilobrachys guangxiensis) and H. schmidti (Theraphosidae family), mainly rich with peptide components, revealed the presence of at least 90 and 300 proteins, respectively, with Mm >10 kDa. Among proteins for which homologies were found in the UniProt database [153], there were various enzymes and transport, regulatory, and structural proteins, and most of them are characterized by intracellular localization and fulfill certain intracellular functions. The reasons for appearance of these proteins in venoms are most likely associated with apocrine or holocrine type of venom gland secretion; they also might get there accidentally together with secreted toxins, or due to contamination of venom samples, or they may carry out definite functions in venoms proper. Most proteins could not be assigned to a certain function or no known homologous sequences were found for them in general, which however could be due to inadequacy of the identification techniques [4, 31, 32].
Many publications deal with characteristics of different enzymes in spider venoms (proteases, nucleases, lipases, glycosidases, etc.), but results of investigations sometimes contradict each other, which might be the result of venom sample contaminations, for example, by digestive juices. First of all, this is associated with proteolytic enzymes, because proteolytic activity is usually absent from pure venom preparations or is very low, except for rare cases [29, 74, 300-310]. Nevertheless, as already noted in the case of CP, one of functions of spider venom components might be the involvement in external digestion, and a central role in this process could be played by enzymes. Hyaluronidase is a frequent component in spider venoms [94, 103, 306, 308, 311, 312]. Taking into account the ability of this enzyme to destroy structure of extracellular matrix, hyaluronidase is usually considered as a “spreading factor” of venoms; the same function can be also carried out by proteases [33, 38, 306, 309, 311, 313]. It appears that spiders, unlike bees and snakes [60, 61], rarely produce type A phospholipases, though such cases have been described [312, 314].
In 1994, a work appeared that reported the presence of enzyme peptidyl-isomerase in A. aperta venom (code in UniProt database AAB34913, AAB34914 [153]) [210, 315]. This enzyme catalyzes the final step of ω-agatoxin IVB biosynthesis by changing chirality of Ser46 (section “Disulfide-Containing Peptides”). A similar situation with the final stage biosynthesis taking place in the extracellular space with involvement of peptidyl-isomerases occurs in other venomous animals as well [316-318].
Auxiliary functions are more often attributed to protein components of most spider venoms. However, the situation is quite different in recluse spiders of genus Loxosceles (Sicariidae family) that are widespread mainly in the Western hemisphere. Their venom contains a great number of various enzymes—phosphatases, hyaluronidases, phospholipases, and different proteases [33, 301, 304-309, 319-321]. Homologous protein necrotoxins (Mm 30-35 kDa) responsible for toxic activity are main components of their venom. In biochemical aspect, these proteins are type D phospholipases active against sphingomyelin and are therefore also called “sphingomyelinases D” [308, 320-324]. Amino acid sequences of a number of necrotoxins have been determined, and for one of them (from Loxosceles laeta) the crystal structure was obtained (code in PDB database 1XX1 [162]) revealing the TIM-barrel type fold, which is very widespread in various enzymes [325]. About ten different isoforms of sphingomyelinase D were identified in venoms of three Loxosceles species using proteomic investigations [326]. Genes of a number of toxins have been cloned, and their recombinant analogs causing “loxoscelism” symptoms (Table 1) and efficient immune sera preventing development of pathology have been produced [299, 308, 327-335]. It should be stressed that similar enzymes do not appear in any other animals but are found in some bacteria, which suggests quite ancient horizontal gene transfer, because introns are present within the spider genes [58, 336]. Despite the success achieved, the molecular mechanism of action of necrotoxins and the basis of their specificity are still not clear. For example, rabbits and guinea pigs exhibit sensitivity to Loxosceles venoms similar to that in humans, whereas no skin ulcers are formed in mice and rats [337]. The involvement of certain blood plasma and immune system components of the victim in development of necrosis was shown [320, 338-341]. An interesting discovery, widening the already broad pharmacology of sphingomyelinase D, is detection of K+ channel activation in response to enzyme action on cells [342].
Spiders of the genus Latrodectus (Theridiidae family) known in Russia and USA as karakurt and “black widow” are among arachnid species most dangerous for man because antidotes are still not widely available [17, 261]. A family of high molecular weight neurotoxins, latrotoxins (Mm >100 kDa) exhibiting strong toxic effect against various animals was found in their venoms [343, 344]. Similar proteins were found in spider venoms of the close genus Steatoda [55, 345]. Over 400 publications deal with investigation of latrotoxins and their receptors. The best studied is α-latrotoxin (α-LTX; P23631), which is effective against vertebrates. It was possible to show using electrophysiological techniques that this toxin causes exhaustive release of neuromediators from the vertebrate nerve terminals resulting in blockade of signal transmission [346]. α-LTX stimulated exocytosis and caused secretion of all known types of neuromediators. Venomous effect was accompanied by membrane depolarization and calcium ion entry into neurons, but the presence of Ca2+ was not an obligatory condition for its action (Ca2+ could be replaced by Mg2+) [347, 348].
Fractionation of the Latrodectus mactans spider venom revealed a number of protein components active towards vertebrates, insects, and crustaceans [349]. At least seven different karakurt toxins are now identified; this serves as an example of biomolecular diversity of spider venom components (section “Composition of Spider Venom”). These are α-latrotoxin for vertebrates, five toxins for insects – latroinsectotoxins α-LIT (Q02989), β-LIT, γ-LIT, δ-LIT (Q25338), and ε-LIT, as well as toxin for crustaceans – α-latrocrustotoxin (α-LCT; Q9XZC0) [350]. This nomenclature is generally accepted for toxins of karakurt and related spiders; it should not be confused with nomenclature for peptide toxins (section “Disulfide-Containing Peptides”). Latrotoxins are high molecular weight proteins with Mm >100 kDa (Table 7). In electrophysiological investigations, all toxins of this family stimulated release of neuromediators from nerve terminals in appropriate animals. Latroinsectotoxins had no visible effect on vertebrates and crustaceans (Table 8), and latrocrustotoxin did not influence insects and mice [351-353]. These results directly correlated with data on measuring the radiolabeled α-LTX and α-LIT binding to preparations of vertebrate and insect neuronal membranes [354]. Apparently, karakurt secretes toxins that are the most perfect concerning their specificity towards animals of certain taxonomic groups. In this case, combination of different toxin activities results in broad specificity of the whole venom. One of the main features of latrotoxin action is formation in artificial membranes of pores permeable for cations [355, 356]. However, specific toxin effect on cell membranes is mediated by specialized receptors.
Table 7. Latrotoxin characteristics
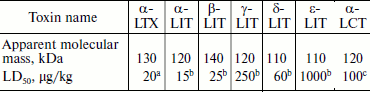
Note: Testing was carried out on micea, larvae of the
greater wax moth Galleria mellonellab, and on Cuban
crayfish Procambarus cubensisc.
Table 8. Toxic activity of
α-latroinsectotoxin
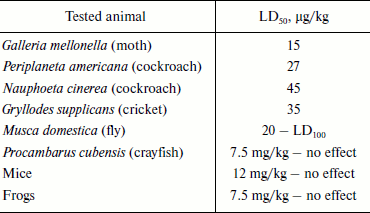
Note: LD50 and LD100 are doses causing death of 50
and 100% of experimental animals, respectively.
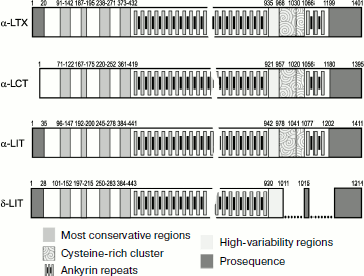
Structures of four latrotoxins were determined in the 1990s [34, 357-360]. Three of them (α-LTX, α-LIT, and α-LCT) consist of ~1100-1200 a.a. and δ-LIT of ~1000 a.a.: probably all latrotoxins consist of several domains (Fig. 6). Comparison of toxin structures revealed rather high level of their homology (on average over 30% identical amino acid residues). The N-terminal part of latrotoxins does not exhibit noticeable similarity with other known proteins, while the central part contains tandems of ankyrin repeats of 30-35 a.a. each. Similar repeats exist in hundreds of proteins exhibiting very different functions and are believed to be important for protein–protein interactions [361, 362]. The number of ankyrin repeats in latrotoxins varies from 13 for δ-LIT to 20 for α-LTX. Repeats 15-17 in α-LTX, α-LIT, and α-LCT contain unusual clusters of six cysteine residues that are absent from δ-LIT. An interesting feature of these toxins is the presence in their N-terminus of two conservative hydrophobic regions of ~30 a.a. Thus, two main parts can be distinguished in latrotoxin structures: N-terminal with two conservative hydrophobic regions and central with ankyrin repeats [34].
Even high-purity α-LTX preparations contain two components with Mm ~130 and 8 kDa. The low molecular weight component (latrodectin, LMWP; P49125) consists of 70 a.a. with three intramolecular disulfide bonds [363-365]. LMWP is devoid of its own toxicity, has no pore-forming activity in bilayer lipid membranes, and structurally resembles crustacean hyperglycemic hormone [366]. It is supposed that its functional role is stabilization of α-LTX structure, and as a result potentiation of its biological activity; in addition, this polypeptide probably decreases selectivity of action of latrotoxins, which is also biologically justified [367].Fig. 6. Organization of latrotoxin amino acid sequences. Established sequences of protoxins α‑LTX (P23631), α-LCT (Q9XZC0), α-LIT (Q02989), and δ-LIT (Q25338) are shown schematically according to annotation in UniProt database [153]. Profragments are shown in dark gray. In mature chains positions of ankyrin repeats, cysteine-rich cluster (ornament), as well as the most conservative (medium gray) and conversely the most variable (light gray) regions are shown. Breaks are introduced into the δ-LIT sequence to optimize comparison with other latrotoxins.
Attempts to obtain α-LTX crystals have not been successful. However, rather interesting data on its spatial structure were obtained using electron microscopy. The first works showed that α-LTX exists in the form of oligomers [368]. Later its spatial structure with resolution of about 15 Å was determined using cryoelectron microscopy. It was shown that the toxin exists in the form of tetramer [369], and symmetrical tetramer is formed only in the presence of calcium or magnesium ions, while without them α-LTX exists as a stable asymmetrical homodimer. Three domains are distinguished in the monomer structure, while the tetrameric form has in its center a channel of ~10 Å in the narrowest place. It is supposed that just the toxin tetramer is able to incorporate into the membrane, forming the ion pore. Actually, the amount of tetramer form of α-LTX evidently correlates with its ability to stimulate exocytosis of neuromediators [370]. Probably, Ca2+-dependent effect of latrotoxins is caused by their tetramerization and incorporation into the presynaptic membrane with pore formation, which results in calcium ion entry into the nerve terminals.
As already mentioned, the α-LTX binding to the cell membrane is mediated by specific receptors. Labeled toxin was used as a research instrument to localize the receptors. In addition, affinity sorbents with immobilized α-LTX were obtained [371-373]. Three different receptors/acceptors of this toxin have been identified: neurexin Iα, latrophilin, and tyrosine phosphatase σ, among which latrophilin is a G-protein-coupled receptor [374-378]. It was found that in the case of α-LTX the presence of membrane receptor is enough for pore-forming activity, and no interaction with other presynaptic proteins is required. Neurexin binding takes place only in the presence of calcium ions, whereas interaction with other receptors is Ca2+-independent. Far from all α-LTX effects can be explained by its channel-forming properties. Besides incorporation into neuronal membrane and pore formation, α-LTX is able, after binding to latrophilin or tyrosine phosphatase, to modulate activity of phospholipase C and cause release of calcium ions from intracellular stores [379, 380]. Latroinsectotoxin receptors are not known now despite the presence in insect genomes of homologs of all three types of α-latrotoxin receptors [14, 381]. Latrophilin homolog was found in the genome of nematode Caenorhabditis elegans; it was found that this protein is responsible for the strong toxic effect of latrotoxins (apparently ε-LIT: LD50 ~1-2 µg/kg) in these animals [382].
It should be noted that α-LTX is one of the most efficient instruments of investigation of molecular processes of neuromediator exocytosis now known. Undoubtedly, further investigation of latrotoxins and their receptors will be of great importance for recognition of molecular mechanisms of neurosecretion [14, 380, 383].
To conclude, protein components of spider venoms carry out various functions: they exert direct toxic effect (necrotoxins or neurotoxins), appear as venom “spreading factors” (enzymes destroying tissue structures), and take part in final stages of toxin maturation.
BIOSYNTHESIS OF SPIDER VENOM COMPONENTS
Mechanisms of biosynthesis of various spider venom components have been studied to different extents, but on the whole this field is not sufficiently studied. For example, there are no data in the literature concerning biosynthesis of low molecular weight components including well-known AP. As for polypeptides, information about the mechanism of their production is rather diverse, although it is possible to speak about understanding the general scheme of the process [85].
At the present time the structure of genes encoding a very limited set of spider venom polypeptides is established. Among them are insectotoxins of the spider Diguetia canities [384], a number of H. schmidti peptide neurotoxins [385, 386], sphingomyelinase D of several species of Loxosceles and Sicarius [330, 335], and some latrotoxins [360, 387]. An interesting peculiarity of genes of H. schmidti peptides and latrotoxins is the absence of introns, which is unique compared to other animal toxin genes and is more characteristic of bacterial proteins. The largest amount of information about maturation of the venom polypeptide components was obtained during analysis of mRNA (more accurately, cDNA) from venom glands. cDNA libraries containing more than one hundred sequences have been obtained for several spider species [4, 45, 46, 48, 330].
Apparently all latrotoxins are synthesized as inactive precursors (Fig. 6). The protoxin molecule undergoes processing during which the kexin/furin type protease (also see below) cleaves off the N-terminal (~20-30 a.a.) and C-terminal (up to ~200 a.a.) fragments [359, 360, 367, 387]. Thus, the latrotoxin precursor molecules can be divided into three regions: cleaved N- and C-terminal fragments and the formed mature chain.
Peptide components of spider venoms are usually synthesized as precursors within which three main elements can be distinguished. (i) Since all molecules under consideration are secreted, precursors include N-terminal, so-called signal or leader (pre-)peptides causing cotranslational delivery of the growing polypeptide chain into the endoplasmic reticulum (ER). (ii) The presence of prosequences (profragments), eliminated posttranslationally due to so-called limited proteolysis, is characteristic of the overwhelming majority of known toxin precursors. The function of prosequences is very poorly studied, but it apparently consists in assurance of correct peptide maturation, folding, and sorting. In the case of CP, the function of these elements is blocking of mature chain activities and, as a result, protection of producing cells against cytolysis [154, 388, 389]. (iii) Sequences corresponding to mature peptides are localized in the C-terminal regions of the precursors. Thus, structural organization of precursors is described by the term prepropeptides. Similar structural organization is observed in the case of a great number of secreted molecules including conotoxins [86].
Figure 7 shows a scheme of processing of peptide precursors from spider venoms. Initial cleavage of prepropeptide molecules, namely, separation of leader peptides by signal peptidases takes place on entrance into ER [390]. The next step of propeptide limited proteolysis by specific enzymes results in prosequence removal. An important discovery was detection of motifs that define proteolysis of toxin and CP precursors from spider venoms. These motifs were called PQM (processing quadruplet motif) (shown by arrow in Fig. 7) and iPQM (inverted PQM) [45, 85]. The PQM motif can be described by X1X2X3R expression, where any Xn = E. In other words, according to the generally accepted Schechter–Berger nomenclature [391], the P1 residue forming the hydrolyzed bond P1–P1′ is an arginine residue, while in positions P2–P4 (sometimes up to P6) there are glutamic acid residues. The iPQM motif is a symmetrical copy of PQM: the P1 position is also occupied by an arginine residue, while glutamic acid residues are in the P1′–P5′ region. Mnemonic description of these processing motifs EtoR/EafterR, showing mutual arrangement of key amino acid residues, is proposed as well.
Motifs PQM and iPQM differ from classic “dibasic” R(K)toR motifs found in numerous peptide precursors in various animals and recognized by subtilisin-like processing enzymes of kexin/furin type [85, 392]. Note that no limited proteolysis enzymes have been found in the venom glands of spiders, and their localization is also not known along with the place of the secretory pathway from ER to extracellular space, where maturation proceeds. In this connection, peptidyl isomerase (section “Protein Components”) should be mentioned once again along with the presence of immature toxin precursors in some spider venoms. On one hand, this might be an accidental result of the mechanism of venom secretion or milking, and on the other, it might point to extracellular processing of these peptides [31, 393, 394]. Besides, proteases found in venoms usually exhibit low activity (section “Protein Components”) and can be enzymes of limited proteolysis. The following analysis of protein precursors from various animals has shown that the EtoR/EafterR motif seems to be more frequent and specific not only of toxins [85]. Analysis of all known toxin precursors produced by animals of different taxonomic groups has shown that maturation following the PQM motif is characteristic of practically all known spider toxins (Fig. 8), with rare exceptions such as magi-4 (P83560) from the venom of spider Macrothele gigas [395]. Toxins of scorpions, sea anemones, snakes, and insects probably undergo maturation following the R(K)toR motif. Most cone snail toxins also belong to the group with the R(K)toR motif processed by kexin/furin type proteases, although there is a great number of conotoxins maturating with involvement of different types of proteolytic enzymes [396]. Thus, maturation of the spider venom peptide components differs from that known for all other venomous animals. Note that detection of EtoR/EafterR motifs predicts well the structure of mature molecules, which is especially important in analysis of translated nucleotide sequences. For example, ω-agatoxins IA and IB (P15969, P15970) are cut into two chains precisely marked by symmetrical PQM and iPQM.Fig. 7. Processing of the spider venom toxin precursors. Main stages of maturation as well as key regions of the precursor amino acid sequence are shown on the scheme. Signal peptide, profragments, and the mature chain sequence are designated with different hues of gray. The PQM (processing quadruplet motif) is shown by arrows.
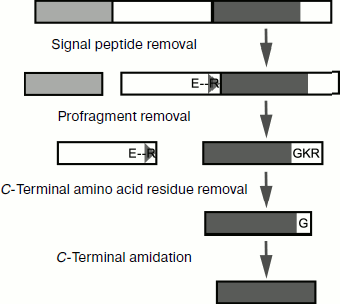
Often, in addition to prosequence separation, precursors undergo other posttranslational transformations. C-Terminal amidation includes splitting off the C-terminal glycine residue with simultaneous amide formation at the preceding amino acid residue. The so-called amidating enzyme or peptidylglycine α-amidating monooxygenase catalyzes this process. It appeared that in fact this is a complex consisting of two enzymes: peptidylglycine-α-hydroxylating monooxygenase, converting glycine residue to an α-hydroxylated intermediate, and peptidyl-α-hydroxyglycine α-amidating lyase that catalyzes hydroxylate cleavage with final amide formation [397]. C-Terminal positively charged amino acid residues are cleaved off by type D or E carboxypeptidases [398, 399]. The change in chirality of one amino acid residue in ω-agatoxin IVB (P37045) has been discussed above (section “Protein Components”), while the mechanism of PLTX-II (P34079) modification by palmitic acid is still unknown.Fig. 8. Diagram of the limited proteolysis motif distribution in toxin precursor proteins from different animal groups. The bar height corresponds to the number of known toxin precursor proteins containing a given motif type.
CP precursors are apparently characterized by great diversity, but now only cDNA sequences for peptides from L. tarabaevi venom are known [62, 63]. The above-described precursor structure with a single PQM motif is characteristic of most CP. Simultaneously two (“binary” precursors) and more (“complex” precursors) PQM as well as iPQM motifs were found within other CP precursors, due to which these molecules are processed with formation of several mature peptides. We believe that structural variability of spider venom polypeptide precursors is still not completely identified.
CONCLUSION
The study of spider venom, first initiated by the goal of elucidating the nature of the “active principle”, to understand the mechanism of action of toxins dangerous for humans, and to design appropriate antidotes, is now much more diverse: the discovered molecular diversity of venoms increased the breadth of their applicability.
It appears that spider venom includes very different components having various targets and modulating numerous physiological processes in certain directions. Thus, these unique mixtures can be considered as natural pharmacopoeias, and the main practical interest in them is now due to the possibility of obtaining new pharmacological preparations. Fundamental interest in venom components is associated with obtaining highly selective tools for investigation of their targets, as was mentioned repeatedly above. As for practical interest, it seems that among each group of compounds from spider venoms (in accordance with the sections of this review) there are candidates for drug design.
For instance, poorly studied sulfated nucleotides are probably able to interfere in different processes with involvement of usual nucleotides [106]. It was proposed to use AP for various purposes, and we consider the most interesting the directed effect on the activity of glutamate receptors and thus prevention of epileptic events [400, 401]. In the case of bites of venomous animals, the use of purified polypeptide toxin preparations or their recombinant analogs for obtaining efficient sera seems obvious [261, 334]. Interest in CP is due to their antimicrobial activity; it is supposed that CP (AMP) will become a new generation of antibiotic drugs that will be introduced into clinical practice against the background of lowering potential of usual antibiotics due to development of resistance to them in pathogenic microorganisms. Nevertheless, first of all it is necessary to solve two main problems: the increase in CP efficiency and specificity towards pathogenic cells and creation of technology for their production in industrial amounts for broad practical application [158, 159, 388, 389, 402-404]. It seems that disulfide-containing peptides are the most promising. Relatively simple molecular structure, stability, specificity, and efficiency of action, diversity of recognizable receptors as well as combinatorics of their amino acid sequences are distinctive parameters of this group of compounds, which facilitate rational drug design on their basis. An example is blocker of proton-activated channels (ASICs) playing a crucial role in generation of pain stimuli. This peptide is the prototype of an analgesic drug of a new generation [19, 22, 25, 288]. An important problem of contemporary medicine is channelopathy – dysfunction of ion channels. Selective ligands binding to these proteins may return their activities to physiological norm and thus eliminate pathology. In this case, not blockers but rather “milder” modulators changing the level of target activity but not completely inhibiting it may be most useful [405-408].
Another aspect of practical interest in spider venoms is following biological logic: since insects comprise the main prey of spiders, then insectotoxins of their venoms can be used as bioinsecticides in “green” biotechnology to decrease agricultural losses. Many ways of realizing this idea have been proposed including creation of toxin-producing transgenic plants [409-411]. Creation of viral vectors affecting insect organisms (for example, on the basis of baculoviruses) containing toxin genes is of particular interest because in this case the problem of toxin delivery to its target in the victim’s organism is solved automatically [13-16]. CP can also be used for obtaining transgenic plants resistant to diseases or as biopesticides [412-415].
In summary, current problems in the study of spider venoms are mainly particularly practical and concern technology of searching for new components with desired properties for creation on their basis of drug prototypes or insecticides. However, there are more distant problems and tasks that require a new level of thinking. We shall mention some of these.
(i) How does the molecular diversity of venom form? What is the mechanism of emergence of natural combinatorial libraries of compounds? Sources of variability can be studied at different levels: gene duplication, alternative splicing, RNA-editing, precursor protein processing, and posttranslational modifications. Now, in the absence of information about genomes of spiders and other venomous animals with biomolecular diversity of venom components, it is difficult to answer these questions. Moreover, even confirmation of hypothesis concerning existence of multigene toxin families does not give an exhaustive answer yet. Actually, analysis of available data shows that distinctions between toxin genes are distributed unevenly: sequences of signal peptides are the most conserved, whereas those of mature chains, on the contrary, are hypervariable, while cysteine residues retain invariance (Tables 3 and 4) [49, 203]. The enhanced variability of toxin genes is described by the term “accelerated” evolution specific of genes regulating interactions with variable factors [75]. Understanding the mechanism of selective mutation introduction into the gene region corresponding to the mature chain and of cysteine codon protection still lies ahead.
(ii) What are real scales of natural variability? As already noted, the venoms of only about one hundred spider species, i.e. ~0.25% of known species have been studied. Note that many spiders are characterized by small size and only fractions of microliters of their venoms can be obtained. This is one of the reasons responsible for extreme sparseness or complete absence of information about venoms of members of many families. For broad investigations of these objects, standard methods of fractionation and structural analysis of active components are not optimal, and new technologies are necessary. The approach that combines genetic (obtaining cDNA from venom glands) and proteomic or peptidomic (based on analytical chromatography and mass spectrometry) methods is promising in this respect [3, 4, 46, 152, 416]. Besides, due to broad scales of expected molecular diversity (several tens of million or more individual compounds), efficient analysis of newly discovered components as well as searching compounds with desired properties will require new methods for variability analysis and screening.
(iii) Finally, in conclusion we should pose the following question: what will modern researchers learn from spiders and will they be able to excel the latter in their art? Actually, spiders have to their credit quite a number of achievements in biological chemistry. Among them are an apparatus of combinatorial chemistry of organic compounds, combinatorial libraries of polypeptide molecules with common type of fold, complex multidomain proteins with the highest action selectivity, and naturally, the spider’s web. One should hope that all these achievements will be taken in and used by humankind.
APPENDIX: SOME FACTS OF SPIDER BIOLOGY
Spiders (order Araneae, class Arachnida, subphylum Chelicerata, phylum Arthropoda) comprise one of the most evolutionarily successful groups of living organisms. They appeared on the Earth more than 300 million years ago, and from that time on they prosper. The insects that are largely the basis of the spider’s ration are followed by the latter as the most widespread and variable organisms. In fact, members of over 40,000 presently described spider species [200, 201] live practically everywhere on the Earth and occupy different ecological niches from deserts to water reservoirs, from the subterranean caves to high mountains. Such wide spreading of spiders is the result of specific for these organisms remarkable biological diversity, already noted species, morphological, and behavioral, as well as chemical and molecular diversity. Dwarfs (body length less than 0.5 mm) and giants (leg span over 25 cm) appear among spiders (sex dimorphism is also characteristic of spiders, and females can exceed males more than a hundred-fold by weight), short-lived and long-lived (lifetime from several months to decades), lone and social (with common spider’s web, in this case individuals transmit vibration signals to each other). Unlike insects, spiders have four pairs of legs (as well as chelicerae and pedipalps) and simple eyes. Spider pigmentation is very different, mainly being colors making them unnoticeable, but bright colors are also possible, especially in tropical species. Almost all, without exception, spiders are predators and live on living catch, but they are able to survive for several weeks without food [417-421].
Mimicry is characteristic of spiders. Some spiders are practically able to fuse with flowers or resemble a shoot, but the ability to resemble their victims such as ants, bees, and flies seems the most surprising. For example, some jumping spiders of the Salticidae family became adapted to life among ants and hunting for them. In this case, mimicry is carried out at morphological (the body shape acquires features characteristic of these insects) and behavioral (spiders use the fore pair of legs like ants use antennas) levels as well as at chemical level (pheromones characteristic of ants are produced) [422-425]. Spiders are congenital hunters—quickness of their reaction practically has no analogs in terrestrial animals. Apparently, among all Arthropoda, just spiders have the most centralized nervous system that perhaps gives in by this parameter only to Cephalopoda molluscs among invertebrates. Hunting methods in spiders are extremely diverse. Some of them use complex spider’s webs and are sitting there expecting an entangled victim, others cast the spider’s web as a seine; still others (American bolas spiders of Mastophora genus) produce chemical substances, pheromones of butterflies, thus attracting the latter over long distances; another group in general does not use spider’s web and represents “active” hunters [417-419, 426-429]. In the case of damage by poisonous victim, some spiders perform autotomy of an injured leg [430]. Many spiders have an astonishing ability to walk along smooth vertical surfaces and ceilings, which is explained by the presence on legs of thousands of fine hairs forming contacts with the surface at microscopic distances, or by use of a special type of silk [431-433].
One can say with pronounced simplification that two factors that are distinguishing features of these organisms, i.e. invention of spider’s web and venom apparatus, played the decisive role in spider evolution. The first factor – the spider’s web – is a unique material of protein nature, technical characteristics of which much exceed presently used synthetic fibers. Many spiders are skilful weavers, and the property of diversity is revealed here as well: there are several types of thread depending on function, and in addition spiders of different families spin structurally different webs. The fact that thread up to several tens and even hundreds of meters can be obtained from a common European garden spider seems surprising [434-438]. This review deals with the other factor, spider venoms. For hunting and protection, most spiders use strong chelicerae, on the fangs of which ducts of venom glands open.
Due to their distinctive behavioral and morphological peculiarities, spiders from ancient times appear in art, religion, and philosophy of different cultures, symbolizing patience and invention as well as evil and insidiousness. Despite an inclination to arachnophobia, rather widespread among contemporary people, one should recognize that these astonishing animals possess unique natural information, and modern science helps to expose its virtually inexhaustible treasure trove.
This work was supported by the Russian Foundation for Basic Research (grant Nos. 08-04-00454 and 08-04-90444), the Federal Agency for Science and Innovation (grant No. NSh‑639.2008.4; State contract No. 02.512.12.2020 of 27.06.2008), the Federal Agency for Education (State contract No. P1388 of 02.09.2009) of the Russian Federation, and the Program of the Russian Academy of Sciences “Molecular and Cell Biology”.
REFERENCES
1.Bettini, S., and Alsop, D. W. (1978)
Arthropod Venoms, Springer-Verlag, Berlin-New York,
977 p.
2.Mebs, D. (2002) Venomous and Poisonous
Animals: a Handbook for Biologists
Toxicologists, and Toxinologists,
Physicians and Pharmacists, CRC Press, Medpharm,
Stuttgart-Boca Raton, 339 p.
3.Escoubas, P., Quinton, L., and Nicholson, G. M.
(2008) J. Mass Spectrom., 43, 279-295.
4.Liang, S. (2008) Expert Rev.
Proteomics, 5, 731-746.
5.Vapenik, Z., and Nentwig, W. (2000)
Toxicon, 38, 293-298.
6.Kuhn-Nentwig, L., Schaller, J., and Nentwig, W.
(2004) Toxicon, 43, 543-553.
7.Adams, M. E. (2004) Toxicon, 43,
509-525.
8.Wullschleger, B., Nentwig, W., and Kuhn-Nentwig, L.
(2005) J. Exp. Biol., 208, 2115-2121.
9.Herzig, V., Khalife, A. A., Chong, Y., Isbister, G.
K., Currie, B. J., Churchill, T. B., Horner, S., Escoubas, P.,
Nicholson, G. M., and Hodgson, W. C. (2008) Toxicon,
51, 1167-1177.
10.Wigger, E., Kuhn-Nentwig, L., and Nentwig, W.
(2002) Toxicon, 40, 749-752.
11.Malli, H., Kuhn-Nentwig, L., Imboden, H.,
and Nentwig, W. (1999) J. Exp. Biol., 202, 2083-2089.
12.Boeve, J. L., Kuhn-Nentwig, L., Keller, S., and
Nentwig, W. (1995) Toxicon, 33, 1347-1357.
13.Nicholson, G. M. (2007) Toxicon,
49, 490-512.
14.Rohou, A., Nield, J., and Ushkaryov, Y. A. (2007)
Toxicon, 49, 531-549.
15.King, G. F. (2007) Toxicon, 49,
513-530.
16.Tedford, H. W., Sollod, B. L., Maggio, F., and
King, G. F. (2004) Toxicon, 43, 601-618.
17.Vetter, R. S., and Isbister, G. K. (2008)
Annu. Rev. Entomol., 53, 409-429.
18.Estrada, G., Villegas, E., and Corzo, G. (2007)
Nat. Prod. Rep., 24, 145-161.
19.Diochot, S., Salinas, M., Baron, A., Escoubas,
P., and Lazdunski, M. (2007) Toxicon, 49, 271-284.
20.Huang, P. T., Shiau, Y. S., and Lou, K. L. (2007)
Toxicon, 49, 285-292.
21.Swartz, K. J. (2007) Toxicon, 49,
213-230.
22.Rajendra, W., Armugam, A., and Jeyaseelan,
K. (2004) Toxicon, 44, 1-17.
23.Grishin, E. (1999) Eur. J. Biochem.,
264, 276-280.
24.Olivera, B. M., Miljanich, G. P.,
Ramachandran, J., and Adams, M. E. (1994) Annu. Rev.
Biochem., 63, 823-867.
25.Lewis, R. J., and Garcia, M. L. (2003) Nat.
Rev. Drug. Discov., 2, 790-802.
26.Cromer, B. A., and McIntyre, P. (2008)
Toxicon, 51, 163-173.
27.Bowman, C. L., Gottlieb, P. A.,
Suchyna, T. M., Murphy, Y. K., and Sachs, F. (2007)
Toxicon, 49, 249-270.
28.Corzo, G., and Escoubas, P. (2003) Cell. Mol.
Life Sci., 60, 2409-2426.
29.Rash, L. D., and Hodgson, W. C. (2002)
Toxicon, 40, 225-254.
30.Gomez, M. V., Kalapothakis, E., Guatimosim,
C., and Prado, M. A. (2002) Cell. Mol. Neurobiol., 22,
579-588.
31.Liao, Z., Cao, J., Li, S., Yan, X., Hu, W., He,
Q., Chen, J., Tang, J., Xie, J., and Liang, S. (2007)
Proteomics, 7, 1892-1907.
32.Yuan, C., Jin, Q., Tang, X., Hu, W., Cao, R.,
Yang, S., Xiong, J., Xie, C., Xie, J., and Liang, S. (2007) J.
Proteome Res., 6, 2792-2801.
33.Da Silva, P. H., da Silveira, R. B., Appel, M.
H., Mangili, O. C., Gremski, W., and Veiga, S. S. (2004)
Toxicon, 44, 693-709.
34.Grishin, E. V. (1998) Toxicon, 36,
1693-1701.
35.De Beleboni, O. R., Pizzo, A. B., Fontana,
A. C., de Coutinho-Netto, O. G. C. R., and dos Santos, J. W. F. (2004)
Eur. J. Pharmacol., 493, 1-17.
36.Escoubas, P., Diochot, S., and Corzo, G. (2000)
Biochimie, 82, 893-907.
37.Jackson, H., and Parks, T. N. (1989)
Annu. Rev. Neurosci., 12, 405-414.
38.Kuhn-Nentwig, L. (2003) Cell. Mol. Life
Sci., 60, 2651-2668.
39.The KNOTTIN database (http://knottin.cbs.cnrs.fr).
40.Mouhat, S., Jouirou, B., Mosbah, A., de Waard,
M., and Sabatier, J. M. (2004) Biochem. J., 378,
717-726.
41.Norton, R. S., and Pallaghy, P. K. (1998)
Toxicon, 36, 1573-1583.
42.Craik, D. J., Daly, N. L., and Waine, C. (2001)
Toxicon, 39, 43-60.
43.Gracy, J., Le-Nguyen, D., Gelly, J. C., Kaas, Q.,
Heitz, A., and Chiche, L. (2008) Nucleic Acids Res., 36,
D314-D319.
44.Escoubas, P., and Rash, L. (2004)
Toxicon, 43, 555-574.
45.Kozlov, S., Malyavka, A., McCutchen, B., Lu, A.,
Schepers, E., Herrmann, R., and Grishin, E. (2005) Proteins,
59, 131-140.
46.Escoubas, P., Sollod, B., and King, G. F. (2006)
Toxicon, 47, 650-663.
47.Escoubas, P. (2006) Mol. Divers.,
10, 545-554.
48.Chen, J., Deng, M., He, Q., Meng, E., Jiang, L.,
Liao, Z., Rong, M., and Liang, S. (2008) Cell. Mol. Life Sci.,
65, 2431-2444.
49.Sollod, B. L., Wilson, D., Zhaxybayeva, O.,
Gogarten, J. P., Drinkwater, R., and King, G. F. (2005)
Peptides, 26, 131-139.
50.Olivera, B. M., Hillyard, D. R., Marsh, M., and
Yoshikami, D. (1995) Trends Biotechnol., 13,
422-426.
51.Conticello, S. G., Gilad, Y., Avidan, N.,
Ben-Asher, E., Levy, Z., and Fainzilber, M. (2001) Mol. Biol.
Evol., 18, 120-131.
52.Fry, B. G., Scheib, H., van der Weerd, L., Young,
B., McNaughtan, J., Ramjan, S. F., Vidal, N., Poelmann, R. E., and
Norman, J. A. (2008) Mol. Cell. Proteomics, 7,
215-246.
53.Fry, B. G., and Wuster, W. (2004) Mol. Biol.
Evol., 21, 870-883.
54.Wang, Y., Yap, L. L., Chua, K. L., and Khoo, H.
E. (2008) Toxicon, 51, 1374-1382.
55.Cavalieri, M., D’Urso, D., Lassa, A.,
Pierdominici, E., Robello, M., and Grasso, A. (1987)
Toxicon, 25, 965-974.
56.Schiavo, G., Matteoli, M., and Montecucco,
C. (2000) Physiol. Rev., 80, 717-766.
57.Binford, G. J., and Wells, M. A. (2003) Comp.
Biochem. Physiol. B: Biochem. Mol. Biol., 135,
25-33.
58.Bernheimer, A. W., Campbell, B. J., and
Forrester, L. J. (1985) Science, 228, 590-591.
59.Bernheimer, A. W. (1996) Med.
Microbiol. Immunol., 185, 59-63.
60.Habermann, E. (1972) Science, 177,
314-322.
61.Koh, D. C., Armugam, A., and Jeyaseelan, K.
(2006) Cell. Mol. Life Sci., 63, 3030-3041.
62.Kozlov, S. A., Vassilevski, A. A., Feofanov,
A. V., Surovoy, A. Y., Karpunin, D. V., and Grishin, E. V. (2006) J.
Biol. Chem., 281, 20983-20992.
63.Vassilevski, A. A., Kozlov, S. A.,
Samsonova, O. V., Egorova, N. S., Karpunin, D. V.,
Pluzhnikov, K. A., Feofanov, A. V., and Grishin, E. V. (2008)
Biochem. J., 411, 687-696.
64.Argiolas, A., and Pisano, J. J. (1984) J.
Biol. Chem., 259, 10106-10111.
65.Argiolas, A., and Pisano, J. J. (1985) J.
Biol. Chem., 260, 1437-1444.
66.Orivel, J., Redeker, V., le Caer, J. P., Krier,
F., Revol-Junelles, A. M., Longeon, A., Chaffotte, A., Dejean, A.,
and Rossier, J. (2001) J. Biol. Chem., 276,
17823-17829.
67.Raghuraman, H., and Chattopadhyay, A. (2007)
Biosci. Rep., 27, 189-223.
68.Mellor, I. R., and Usherwood, P. N. (2004)
Toxicon, 43, 493-508.
69.Yan, L., and Adams, M. E. (1998) J. Biol.
Chem., 273, 2059-2066.
70.Budnik, B. A., Olsen, J. V., Egorov, T. A.,
Anisimova, V. E., Galkina, T. G., Musolyamov, A. K., Grishin, E.
V., and Zubarev, R. A. (2004) J. Mass Spectrom.,
39, 193-201.
71.Corzo, G., Villegas, E., Gomez-Lagunas, F.,
Possani, L. D., Belokoneva, O. S., and Nakajima, T. (2002) J. Biol.
Chem., 277, 23627-23637.
72.Kuhn-Nentwig, L., Muller, J., Schaller, J.,
Walz, A., Dathe, M., and Nentwig, W. (2002) J. Biol. Chem.,
277, 11208-11216.
73.Quistad, G. B., Reuter, C. C., Skinner, W. S.,
Dennis, P. A., Suwanrumpha, S., and Fu, E. W. (1991) Toxicon,
29, 329-336.
74.Schanbacher, F. L., Lee, C. K., Hall, J. E.,
Wilson, I. B., Howell, D. E., and Odell, G. V. (1973) Toxicon,
11, 21-29.
75.Mebs, D. (2001) Toxicon, 39,
87-96.
76.Adams, M. E., Herold, E. E., and Venema, V. J.
(1989) J. Comp. Physiol. A, 164, 333-342.
77.Mintz, I. M., Venema, V. J., Swiderek, K. M.,
Lee, T. D., Bean, B. P., and Adams, M. E. (1992) Nature,
355, 827-829.
78.Adams, M. E., Mintz, I. M., Reily, M. D.,
Thanabal, V., and Bean, B. P. (1993) Mol. Pharmacol., 44,
681-688.
79.Meir, A., Ginsburg, S., Butkevich, A., Kachalsky,
S. G., Kaiserman, I., Ahdut, R., Demirgoren, S., and
Rahamimoff, R. (1999) Physiol. Rev., 79,
1019-1088.
80.Wullschleger, B., Kuhn-Nentwig, L., Tromp, J.,
Kampfer, U., Schaller, J., Schurch, S., and Nentwig, W. (2004) Proc.
Natl. Acad. Sci. USA, 101, 11251-11256.
81.Early, S. L., and Michaelis, E. K. (1987)
Toxicon, 25, 433-442.
82.Terlau, H., Shon, K. J., Grilley, M., Stocker,
M., Stuhmer, W., and Olivera, B. M. (1996) Nature, 381,
148-151.
83.Terlau, H., and Olivera, B. M. (2004) Physiol.
Rev., 84, 41-68.
84.Bindokas, V. P., Venema, V. J., and Adams, M. E.
(1991) J. Neurophysiol., 66, 590-601.
85.Kozlov, S. A., and Grishin, E. V. (2007)
Toxicon, 49, 721-726.
86.Duda, T. F., Jr., and Palumbi, S. R. (1999)
Proc. Natl. Acad. Sci. USA, 96, 6820-6823.
87.Nakashima, K., Nobuhisa, I., Deshimaru, M.,
Nakai, M., Ogawa, T., Shimohigashi, Y., Fukumaki, Y., Hattori, M.,
Sakaki, Y., Hattori, S., and Ohno, M. (1995) Proc. Natl. Acad. Sci.
USA, 92, 5605-5609.
88.Zhijian, C., Feng, L., Yingliang, W., Xin, M.,
and Wenxin, L. (2006) Toxicon, 47, 348-355.
89.Malli, H., Kuhn-Nentwig, L., Imboden, H.,
Moon, M. J., and Wyler, T. (2000) Cell Tissue Res., 299,
417-426.
90.Dos Santos, V. L., Franco, C. R., Viggiano,
R. L., da Silveira, R. B., Cantao, M. P., Mangili, O. C., Veiga,
S. S., and Gremski, W. (2000) Toxicon, 38, 265-285.
91.Yigit, N., and Guven, T. (2006) Toxicon,
47, 58-67.
92.Silva, L. M., Botelho, A. C., Nacif-Pimenta, R.,
Martins, G. F., Alves, L. C., Brayner, F. A., Fortes-Dias, C. L., and
Pimenta, P. F. (2008) Toxicon, 51, 693-706.
93.Schroeder, F. C., Taggi, A. E., Gronquist,
M., Malik, R. U., Grant, J. B., Eisner, T., and Meinwald, J. (2008)
Proc. Natl. Acad. Sci. USA, 105, 14283-14287.
94.Kuhn-Nentwig, L., Schaller, J., and Nentwig, W.
(1994) Toxicon, 32, 287-302.
95.Inceoglu, B., Lango, J., Jing, J., Chen, L.,
Doymaz, F., Pessah, I. N., and Hammock, B. D. (2003) Proc. Natl.
Acad. Sci. USA, 100, 922-927.
96.Welsh, J. H., and Batty, C. S. (1963)
Toxicon, 1, 165-170.
97.Rash, L. D., King, R. G., and Hodgson, W. C.
(1998) Toxicon, 36, 367-375.
98.Frew, R., Hamilton, M. G., and Lundy, P. M.
(1994) Toxicon, 32, 511-515.
99.Rash, L. D., King, R. G., and Hodgson, W. C.
(2000) Toxicon, 38, 1111-1127.
100.Duffield, P. H., Duffield, A. M., Carroll,
P. R., and Morgans, D. (1979) Biomed. Mass Spectrom.,
6, 105-108.
101.Lange, C., Paris, C., and Celerier, M. L.
(1992) Rapid Commun. Mass Spectrom., 6, 289-292.
102.Lange, C., Paris, C., and Celerier, M. L.
(1992) Rapid Commun. Mass Spectrom., 6, 517-519.
103.Savel-Niemann, A. (1989) Biol. Chem.
Hoppe-Seyler, 370, 485-498.
104.Meinwald, J., and Eisner, T. (1995) Proc.
Natl. Acad. Sci. USA, 92, 14-18.
105.McCormick, J., Li, Y., McCormick, K., Duynstee,
H. I., van Engen, A. K., van der Marel, G. A., Ganem, B., van Boom, J.
H., and Meinwald, J. (1999) J. Am. Chem. Soc., 121,
5661-5665.
106.Taggi, A. E., Meinwald, J., and Schroeder, F.
C. (2004) J. Am. Chem. Soc., 126, 10364-10369.
107.Horni, A., Weickmann, D., and Hesse, M. (2001)
Toxicon, 39, 425-428.
108.Chan, T. K., Geren, C. R., Howell, D. E., and
Odell, G. V. (1975) Toxicon, 13, 61-66.
109.Kawai, N., Niwa, A., and Abe, T. (1982)
Brain Res., 247, 169-171.
110.Usmanov, P. B., Kalikulov, D., Shadyeva, N.,
and Tashmukhamedov, B. A. (1983) Dokl. Akad. Nauk SSSR,
273, 1017-1018.
111.Usherwood, P. N., Duce, I. R., and Boden, P.
(1984) J. Physiol. (Paris), 79, 241-245.
112.Grishin, E. V., Volkova, T. M., Arseniev, A.
S., Reshetova, O. S., Onoprienko, V. V., Magasanik, L. G., Antonov, S.
M., and Fedorova, I. M. (1986) Bioorg. Khim., 12,
1121-1124.
113.Magasanik, L. G., Antonov, S. M., Fedorova, I.
M., Volkova, T. M., and Grishin, E. V. (1986) Biol. Membr.
(Moscow), 3, 1204-1219.
114.Antonov, S. M., Grishin, E. V., Magazanik,
L. G., Shupliakov, O. V., Vesselkin, N. P., and Volkova, T.
M. (1987) Neurosci. Lett., 83, 179-184.
115.Antonov, S. M., Shupliakov, O. V.,
Magazanik, L. G., Vesselkin, N. P., Volkova, T. M., and
Grishin, E. V. (1988) Dokl. Akad. Nauk SSSR, 298,
505-508.
116.Kiskin, N. I., Krystal’, O. A.,
Tsyndrenko, A. Ya., Volkova, T. M., and Grishin, E. V. (1989)
Neirofiziologiya, 21, 748-756.
117.Grishin, E. V., Volkova, T. M., and Arseniev,
A. S. (1988) Bioorg. Khim., 14, 883-892.
118.Grishin, E. V., Volkova, T. M., and
Arseniev, A. S. (1989) Toxicon, 27, 541-549.
119.Grishin, E. V., Volkova, T. M., and
Arseniev, A. S. (1989) J. Protein. Chem., 8,
320-322.
120.Aramaki, Y., Yasuhara, T., Higashijima,
T., Yoshioka, M., Miwa, A., Kawai, N., and Nakajima, T. (1986) Proc.
Japan Acad., 62B, 359-362.
121.Aramaki, Y., Yasuhara, T., Higashijima,
T., Miwa, A., Kawai, N., and Nakajima, T. (1989) Biomed.
Res., 8, 167-173.
122.Rogoza, L. N., Salakhutdinov, N. F., and
Tolstikov, G. A. (2006) Bioorg. Khim., 32,
27-41.
123.Norris, T. M., Moya, E., Blagbrough, I. S., and
Adams, M. E. (1996) Mol. Pharmacol., 50,
939-946.
124.Tzouros, M., Chesnov, S., Bienz, S., Hesse, M.,
and Bigler, L. (2005) Toxicon, 46, 350-354.
125.Hisada, M., Fujita, T., Naoki, H., Itagaki, Y.,
Irie, H., Miyashita, M., and Nakajima, T. (1998) Toxicon,
36, 1115-1125.
126.Cabbiness, S. G., Gehrke, C. W., Kuo, K. C.,
Chan, T. K., Hall, J. E., Hudiburg, S. A., and Odell, G. V. (1980)
Toxicon, 18, 681-683.
127.Chesnov, S., Bigler, L., and Hesse, M. (2001)
Helv. Chim. Acta, 84, 2178-2197.
128.Schroder, F. C., Farmer, J. J.,
Attygalle, A. B., Smedley, S. R., Eisner, T., and Meinwald,
J. (1998) Science, 281, 428-431.
129.Eldefrawi, A. T., Eldefrawi, M. E., Konno, K.,
Mansour, N. A., Nakanishi, K., Oltz, E., and Usherwood, P. N. (1988)
Proc. Natl. Acad. Sci. USA, 85, 4910-4913.
130.Antonov, S. M., Dudel, J., Franke, C., and
Hatt, H. (1989) J. Physiol., 419, 569-587.
131.Williams, K. (1997) Biochem. J.,
325 (Pt. 2), 289-297.
132.Tikhonov, D. B., Mellor, I. R., and
Usherwood, P. N. (2004) Biophys. J., 87,
159-170.
133.Kitaguchi, T., and Swartz, K. J. (2005)
Biochemistry, 44, 15544-15549.
134.Yakehiro, M., Furukawa, Y., Koike, T., Kimura,
E., Nakajima, T., Yamaoka, K., and Seyama, I. (2001) Br. J.
Pharmacol., 132, 63-72.
135.Lee, J. K., John, S. A., and Weiss, J. N.
(1999) J. Gen. Physiol., 113, 555-564.
136.Sutton, K. G., Stapleton, S. R., and Scott, R.
H. (1998) Neurosci. Lett., 251, 117-120.
137.Nakanishi, K., Choi, S.-K., Hwang, D., Lerro,
K., Orlando, M., Kalivretenos, A. G., Eldefrawi, A., Eldefrawi,
M., and Usherwood, P. N. R. (1994) Pure Appl. Chem., 66,
671-678.
138.Stromgaard, K., Jensen, L. S., and
Vogensen, S. B. (2005) Toxicon, 45, 249-254.
139.Blaschke, M., Keller, B. U., Rivosecchi,
R., Hollmann, M., Heinemann, S., and Konnerth, A. (1993) Proc. Natl.
Acad. Sci. USA, 90, 6528-6532.
140.Gu, J. G., Albuquerque, C., Lee, C. J., and
MacDermott, A. B. (1996) Nature, 381, 793-796.
141.Tikhonov, D. B., Mellor, J. R., Usherwood,
P. N., and Magazanik, L. G. (2002) Biophys. J., 82,
1884-1893.
142.Andersen, T. F., Tikhonov, D. B., Bolcho,
U., Bolshakov, K., Nelson, J. K., Pluteanu, F., Mellor, I. R.,
Egebjerg, J., and Stromgaard, K. (2006) J. Med. Chem.,
49, 5414-5423.
143.Tikhonov, D. B. (2007) Mol. Membr.
Biol., 24, 135-147.
144.Fenton, A. W., West, P. R., Odell, G. V.,
Hudiburg, S. M., Ownby, C. L., Mills, J. N., Scroggins, B. T., and
Shannon, S. B. (1995) Toxicon, 33, 763-770.
145.Odell, G. V., Fenton, A. W., Ownby, C. L.,
Doss, M. P., and Schmidt, J. O. (1999) Toxicon, 37,
407-409.
146.Sosnina, N. A., Golubenko, Z., Akhunov, A. A.,
Kugaevskaya, E. V., Eliseeva, Yu. E., and Orekhovich, V. N. (1990)
Dokl. Akad. Nauk. SSSR, 315, 236-239.
147.Ferreira, L. A., Alves, W. E., Lucas, M. S.,
and Habermehl, G. G. (1996) Toxicon, 34,
599-603.
148.Ferreira, L. A., Lucas, S. M., Alves, E. W.,
Hermann, V. V., Reichl, A. P., Habermehl, G., and Zingali, R. B. (1998)
Toxicon, 36, 31-39.
149.Akchunov, A. A., Golubenko, Z., and
Sosnina, N. (1992) Agents Actions Suppl., 38 (Pt.
1), 469-474.
150.Schmidt, J. O. (1995) Toxicon,
33, 917-927.
151.Cohen, E., and Quistad, G. B. (1998)
Toxicon, 36, 353-358.
152.Kozlov, S. A., Vassilevski, A. A., and
Grishin, E. V. (2008) in Peptidomics: Methods and Applications
(Soloviev, M., Shaw, C., and Andren, P., eds.) John Wiley
& Sons, Hoboken, pp. 55-70.
153.UniProt database of polypeptide sequences
(Universal Protein Resource) (http://www.uniprot.org).
154.Vassilevski, A. A. (2008) Cytolitic and
Antimicrobial Peptides from Lachesana tarabaevi Spider
Venom: Candidate’s dissertation [in Russian],
Shemyakin–Ovchinnikov Institute of Bioorganic Chemistry, Moscow,
101 p.
155.Antimicrobial Peptide Database (http://aps.unmc.edu/AP/main.php).
156.Tossi, A., Sandri, L., and Giangaspero, A.
(2000) Biopolymers, 55, 4-30.
157.Sitaram, N., and Nagaraj, R. (2002) Curr.
Pharm. Des., 8, 727-742.
158.Tossi, A., and Sandri, L. (2002) Curr.
Pharm. Des., 8, 743-761.
159.Yeaman, M. R., and Yount, N. Y. (2003)
Pharmacol. Rev., 55, 27-55.
160.Habermann, E., and Jentsch, J. (1967)
Hoppe-Seyler’s Z. Physiol. Chem., 348, 37-50.
161.Schiffer, M., and Edmundson, A. B. (1967)
Biophys. J., 7, 121-135.
162.Protein Data Bank of Spatial Polypeptide
Structures (http://www.rcsb.org/pdb/home/home.do).
163.Pukala, T. L., Boland, M. P., Gehman, J. D.,
Kuhn-Nentwig, L., Separovic, F., and Bowie, J. H. (2007)
Biochemistry, 46, 3576-3585.
164.Dubovskii, P. V., Volynsky, P. E.,
Polyansky, A. A., Chupin, V. V., Efremov, R. G., and
Arseniev, A. S. (2006) Biochemistry, 45,
10759-10767.
165.Dubovskii, P. V., Volynsky, P. E.,
Polyansky, A. A., Karpunin, D. V., Chupin, V. V., Efremov, R. G.,
and Arseniev, A. S. (2008) Biochemistry, 47,
3525-3533.
166.Efremov, R. G., Gulyaev, D. I., Vergoten,
G., and Modyanov, N. N. (1992) J. Protein. Chem., 11,
665-675.
167.Efremov, R. G., and Alix, A. J. (1993) J.
Biomol. Struct. Dyn., 11, 483-507.
168.Vassilevski, A. A., Kozlov, S. A., Zhmak, M.
N., Kudelina, I. A., Dubovsky, P. V., Shatursky, O. Ya., Arseniev, A.
S., and Grishin, E. V. (2007) Bioorg. Khim., 33,
405-412.
169.Belokoneva, O. S., Villegas, E., Corzo, G.,
Dai, L., and Nakajima, T. (2003) Biochim. Biophys. Acta,
1617, 22-30.
170.Belokoneva, O. S., Satake, H.,
Mal’tseva, E. L., Pal’mina, N. P., Villegas, E.,
Nakajima, T., and Corzo, G. (2004) Biochim. Biophys. Acta,
1664, 182-188.
171.Nomura, K., and Corzo, G. (2006) Biochim.
Biophys. Acta, 1758, 1475-1482.
172.He, K., Ludtke, S. J., Worcester, D. L., and
Huang, H. W. (1996) Biophys. J., 70, 2659-2666.
173.Shai, Y. (1999) Biochim. Biophys. Acta,
1462, 55-70.
174.Oren, Z., and Shai, Y. (1998)
Biopolymers, 47, 451-463.
175.Pouny, Y., Rapaport, D., Mor, A., Nicolas, P.,
and Shai, Y. (1992) Biochemistry, 31,
12416-12423.
176.Ludtke, S. J., He, K., Heller, W. T., Harroun,
T. A., Yang, L., and Huang, H. W. (1996) Biochemistry,
35, 13723-13728.
177.Yang, L., Harroun, T. A., Weiss, T. M., Ding,
L., and Huang, H. W. (2001) Biophys. J., 81,
1475-1485.
178.Bechinger, B., and Lohner, K. (2006)
Biochim. Biophys. Acta, 1758, 1529-1539.
179.Giangaspero, A., Sandri, L., and Tossi, A.
(2001) Eur. J. Biochem., 268, 5589-5600.
180.Bleakman, A., and Smyth, D. G. (1987) Eur.
J. Biochem., 167, 161-165.
181.Pukala, T. L., Brinkworth, C. S., Carver,
J. A., and Bowie, J. H. (2004) Biochemistry, 43,
937-944.
182.Kuhn-Nentwig, L., Dathe, M., Walz, A.,
Schaller, J., and Nentwig, W. (2002) FEBS Lett., 527,
193-198.
183.Suchyna, T. M., Johnson, J. H., Hamer, K.,
Leykam, J. F., Gage, D. A., Clemo, H. F., Baumgarten, C. M., and Sachs,
F. (2000) J. Gen. Physiol., 115, 583-598.
184.Jung, H. J., Kim, P. I., Lee, S. K., Lee, C.
W., Eu, Y. J., Lee, D. G., Earm, Y. E., and Kim, J. I. (2006)
Biochem. Biophys. Res. Commun., 340, 633-638.
185.Oswald, R. E., Suchyna, T. M., McFeeters,
R., Gottlieb, P., and Sachs, F. (2002) J. Biol. Chem.,
277, 34443-34450.
186.Craik, D. J., Daly, N. L., Mulvenna, J., Plan,
M. R., and Trabi, M. (2004) Curr. Protein Pept. Sci.,
5, 297-315.
187.Bulet, P., and Stocklin, R. (2005) Protein
Pept. Lett., 12, 3-11.
188.Gimenez-Gallego, G., Navia, M. A., Reuben, J.
P., Katz, G. M., Kaczorowski, G. J., and Garcia, M. L. (1988)
Proc. Natl. Acad. Sci. USA, 85, 3329-3333.
189.Yount, N. Y., and Yeaman, M. R. (2004) Proc.
Natl. Acad. Sci. USA, 101, 7363-7368.
190.Lee, S. Y., and MacKinnon, R. (2004)
Nature, 430, 232-235.
191.Conde, R., Zamudio, F. Z., Rodriguez, M. H.,
and Possani, L. D. (2000) FEBS Lett., 471, 165-168.
192.Diego-Garcia, E., Schwartz, E. F.,
D’Suze, G., Gonzalez, S. A., Batista, C. V., Garcia, B. I., de la
Vega, R. C., and Possani, L. D. (2007) Peptides, 28,
31-37.
193.Brogden, K. A. (2005) Nat. Rev.
Microbiol., 3, 238-250.
194.Brown, K. L., and Hancock, R. E. (2006)
Curr. Opin. Immunol., 18, 24-30.
195.Zasloff, M. (2002) Nature, 415,
389-395.
196.Cotter, P. D., Hill, C., and Ross, R. P. (2005)
Nat. Rev. Microbiol., 3, 777-788.
197.Torres-Larios, A., Gurrola, G. B., Zamudio, F.
Z., and Possani, L. D. (2000) Eur. J. Biochem., 267,
5023-5031.
198.Moerman, L., Bosteels, S., Noppe, W.,
Willems, J., Clynen, E., Schoofs, L., Thevissen, K., Tytgat, J., van
Eldere, J., van der Walt, J., and Verdonck, F. (2002) Eur. J.
Biochem., 269, 4799-4810.
199.Dai, L., Corzo, G., Naoki, H.,
Andriantsiferana, M., and Nakajima, T. (2002)
Biochem. Biophys. Res. Commun., 293,
1514-1522.
200.The World Spider Catalog, version 9.5 by Norman
I. Platnick, American Museum of Natural History (http://research.amnh.org/entomology/spiders/catalog/index.html).
201.Database on Spider Taxonomics and Geographic
Distribution in Russia and Former USSR countries, Design of K. G.
Mikhailov (Zoological Museum, Moscow State University) on the basis of
Catalog of Spiders (Arachnida, Aranei) of Former USSR
Territories (1997) (http://www.zin.ru/BioDiv/aranei.asp).
202.Pan, Z., Barry, R., Lipkin, A., and
Soloviev, M. (2007) BMC Mol. Biol., 8, 32.
203.Jiang, L., Peng, L., Chen, J., Zhang, Y.,
Xiong, X., and Liang, S. (2008) Toxicon, 51,
1479-1489.
204.Olivera, B. M., and Cruz, L. J. (2001)
Toxicon, 39, 7-14.
205.Miller, C. (1995) Neuron, 15,
5-10.
206.Tytgat, J., Chandy, K. G., Garcia, M. L.,
Gutman, G. A., Martin-Eauclaire, M. F., van der Walt, J. J., and
Possani, L. D. (1999) Trends Pharmacol. Sci.,
20, 444-447.
207.King, G. F., Gentz, M. C., Escoubas, P., and
Nicholson, G. M. (2008) Toxicon, 52, 264-276.
208.Buczek, O., Bulaj, G., and Olivera, B. M.
(2005) Cell. Mol. Life Sci., 62, 3067-3079.
209.Branton, W. D., Rudnick, M. S., Zhou, Y.,
Eccleston, E. D., Fields, G. B., and Bowers, L. D. (1993)
Nature, 365, 496-497.
210.Heck, S. D., Siok, C. J., Krapcho, K. J.,
Kelbaugh, P. R., Thadeio, P. F., Welch, M. J., Williams, R. D., Ganong,
A. H., Kelly, M. E., Lanzetti, A. J., Gray, W. R., Phillips, D., Parks,
T. N., Jackson, H., Ahlijanian, M. K., Saccomano, N. A., and Volkmann,
R. A. (1994) Science, 266, 1065-1068.
211.Kuwada, M., Teramoto, T., Kumagaye, K. Y.,
Nakajima, K., Watanabe, T., Kawai, T., Kawakami, Y., Niidome, T.,
Sawada, K., Nishizawa, Y., and Kouichi, K. (1994) Mol.
Pharmacol., 46, 587-593.
212.Santos, A. D., Imperial, J. S., Chaudhary,
T., Beavis, R. C., Chait, B. T., Hunsperger, J. P., Olivera, B. M.,
Adams, M. E., and Hillyard, D. R. (1992) J. Biol. Chem.,
267, 20701-20705.
213.Skerra, A. (2000) J. Mol. Recognit.,
13, 167-187.
214.Hosse, R. J., Rothe, A., and Power, B. E.
(2006) Protein Sci., 15, 14-27.
215.Jimenez-Barbero, J., Javier Canada, F.,
Asensio, J. L., Aboitiz, N., Vidal, P., Canales, A., Groves, P.,
Gabius, H. J., and Siebert, H. C. (2006) Adv. Carbohydr. Chem.
Biochem., 60, 303-354.
216.Fletcher, J. I., Chapman, B. E., Mackay,
J. P., Howden, M. E., and King, G. F. (1997) Structure,
5, 1525-1535.
217.Pallaghy, P. K., Alewood, D., Alewood, P.
F., and Norton, R. S. (1997) FEBS Lett., 419,
191-196.
218.Wang, X., Connor, M., Smith, R., Maciejewski,
M. W., Howden, M. E., Nicholson, G. M., Christie, M. J., and King, G.
F. (2000) Nat. Struct. Biol., 7, 505-513.
219.Shu, Q., Lu, S. Y., Gu, X. C., and Liang, S. P.
(2002) Protein Sci., 11, 245-252.
220.Wen, S., Wilson, D. T., Kuruppu, S.,
Korsinczky, M. L., Hedrick, J., Pang, L., Szeto, T., Hodgson, W. C.,
Alewood, P. F., and Nicholson, G. M. (2005) Peptides,
26, 2412-2426.
221.Peng, K., Lin, Y., and Liang, S. P. (2006)
Acta Biochim. Biophys. Sin. (Shanghai), 38,
457-466.
222.Berndt, K. D., Guntert, P., Orbons, L. P., and
Wuthrich, K. (1992) J. Mol. Biol., 227, 757-775.
223.Antuch, W., Berndt, K. D., Chavez, M. A.,
Delfin, J., and Wuthrich, K. (1993) Eur. J. Biochem.,
212, 675-684.
224.Harvey, A. L. (2001) Toxicon, 39,
15-26.
225.Schweitz, H., Bruhn, T., Guillemare, E.,
Moinier, D., Lancelin, J. M., Beress, L., and Lazdunski, M. (1995)
J. Biol. Chem., 270, 25121-25126.
226.Andreev, Y. A., Kozlov, S. A., Koshelev,
S. G., Ivanova, E. A., Monastyrnaya, M. M., Kozlovskaya, E. P., and
Grishin, E. V. (2008) J. Biol. Chem., 283,
23914-23921.
227.Adams, M. E., Bindokas, V. P., Hasegawa,
L., and Venema, V. J. (1990) J. Biol. Chem., 265,
861-867.
228.Swartz, K. J., and MacKinnon, R. (1995)
Neuron, 15, 941-949.
229.Peng, K., Shu, Q., Liu, Z., and Liang, S.
(2002) J. Biol. Chem., 277, 47564-47571.
230.Martin-Moutot, N., Mansuelle, P., Alcaraz,
G., dos Santos, R. G., Cordeiro, M. N., de Lima, M. E.,
Seagar, M., and van Renterghem, C. (2006) Mol. Pharmacol.,
69, 1931-1937.
231.Li, R. A., and Tomaselli, G. F. (2004)
Toxicon, 44, 117-122.
232.French, R. J., and Terlau, H. (2004) Curr.
Med. Chem., 11, 3053-3064.
233.Billen, B., Bosmans, F., and Tytgat, J. (2008)
Curr. Pharm. Des., 14, 2492-2502.
234.Mouhat, S., de Waard, M., and Sabatier, J. M.
(2005) J. Pept. Sci., 11, 65-68.
235.Mintz, I. M. (1994) J. Neurosci.,
14, 2844-2853.
236.Swartz, K. J., and MacKinnon, R. (1997)
Neuron, 18, 665-673.
237.Swartz, K. J., and MacKinnon, R. (1997)
Neuron, 18, 675-682.
238.Li-Smerin, Y., and Swartz, K. J. (2000) J.
Gen. Physiol., 115, 673-684.
239.Lampe, R. A., Defeo, P. A., Davison, M. D.,
Young, J., Herman, J. L., Spreen, R. C., Horn, M. B., Mangano, T.
J., and Keith, R. A. (1993) Mol. Pharmacol., 44,
451-460.
240.McDonough, S. I., Lampe, R. A., Keith, R. A.,
and Bean, B. P. (1997) Mol. Pharmacol., 52,
1095-1104.
241.Middleton, R. E., Warren, V. A., Kraus, R. L.,
Hwang, J. C., Liu, C. J., Dai, G., Brochu, R. M., Kohler, M. G., Gao,
Y. D., Garsky, V. M., Bogusky, M. J., Mehl, J. T., Cohen, C. J., and
Smith, M. M. (2002) Biochemistry, 41, 14734-14747.
242.Priest, B. T., Blumenthal, K. M., Smith, J. J.,
Warren, V. A., and Smith, M. M. (2007) Toxicon, 49,
194-201.
243.Li-Smerin, Y., and Swartz, K. J. (1998)
Proc. Natl. Acad. Sci. USA, 95, 8585-8589.
244.Smith, J. J., Cummins, T. R., Alphy, S., and
Blumenthal, K. M. (2007) J. Biol. Chem., 282,
12687-12697.
245.Milescu, M., Vobecky, J., Roh, S. H., Kim, S.
H., Jung, H. J., Kim, J. I., and Swartz, K. J. (2007) J. Gen.
Physiol., 130, 497-511.
246.Alabi, A. A., Bahamonde, M. I., Jung, H. J.,
Kim, J. I., and Swartz, K. J. (2007) Nature, 450,
370-375.
247.Bosmans, F., Martin-Eauclaire, M. F., and
Swartz, K. J. (2008) Nature, 456, 202-208.
248.Swartz, K. J. (2008) Nature, 456,
891-897.
249.Catterall, W. A., Cestele, S., Yarov-Yarovoy,
V., Yu, F. H., Konoki, K., and Scheuer, T. (2007) Toxicon,
49, 124-141.
250.Possani, L. D., Becerril, B., Delepierre,
M., and Tytgat, J. (1999) Eur. J. Biochem., 264,
287-300.
251.Little, M. J., Wilson, H., Zappia, C., Cestele,
S., Tyler, M. I., Martin-Eauclaire, M. F., Gordon, D., and
Nicholson, G. M. (1998) FEBS Lett., 439,
246-252.
252.Nicholson, G. M., Little, M. J., and
Birinyi-Strachan, L. C. (2004) Toxicon, 43, 587-599.
253.Sheumack, D. D., Claassens, R., Whiteley, N.
M., and Howden, M. E. (1985) FEBS Lett., 181,
154-156.
254.Brown, M. R., Sheumack, D. D., Tyler, M.
I., and Howden, M. E. (1988) Biochem. J., 250,
401-405.
255.Nicholson, G. M., Willow, M., Howden, M.
E., and Narahashi, T. (1994) Pflugers Arch., 428,
400-409.
256.Nicholson, G. M., Walsh, R., Little, M. J., and
Tyler, M. I. (1998) Pflugers Arch., 436, 117-126.
257.Cordeiro Mdo, N., Diniz, C. R.,
Valentim Ado, C., von Eickstedt, V. R., Gilroy, J., and
Richardson, M. (1992) FEBS Lett., 310, 153-156.
258.Matavel, A., Cruz, J. S., Penaforte, C. L.,
Araujo, D. A., Kalapothakis, E., Prado, V. F., Diniz, C. R., Cordeiro,
M. N., and Beirao, P. S. (2002) FEBS Lett., 523,
219-223.
259.Figueiredo, S. G., Garcia, M. E., Valentim, A.
C., Cordeiro, M. N., Diniz, C. R., and Richardson, M. (1995)
Toxicon, 33, 83-93.
260.De Lima, M. E., Stankiewicz, M., Hamon, A., de
Figueiredo, S. G., Cordeiro, M. N., Diniz, C. R., Martin-Eauclaire, M.,
and Pelhate, M. (2002) J. Insect Physiol., 48, 53-61.
261.Isbister, G. K., Graudins, A., White, J., and
Warrell, D. (2003) J. Toxicol. Clin. Toxicol., 41,
291-300.
262.Skinner, W. S., Adams, M. E., Quistad, G. B.,
Kataoka, H., Cesarin, B. J., Enderlin, F. E., and Schooley, D. A.
(1989) J. Biol. Chem., 264, 2150-2155.
263.Corzo, G., Escoubas, P., Stankiewicz, M.,
Pelhate, M., Kristensen, C. P., and Nakajima, T. (2000) Eur. J.
Biochem., 267, 5783-5795.
264.Corzo, G., Escoubas, P., Villegas, E., Karbat,
I., Gordon, D., Gurevitz, M., Nakajima, T., and Gilles, N. (2005)
Biochemistry, 44, 1542-1549.
265.Tsetlin, V. I., and Hucho, F. (2004) FEBS
Lett., 557, 9-13.
266.Suchyna, T. M., Tape, S. E., Koeppe, R. E.,
2nd, Andersen, O. S., Sachs, F., and Gottlieb, P. A. (2004)
Nature, 430, 235-240.
267.Smith, J. J., Alphy, S., Seibert, A. L., and
Blumenthal, K. M. (2005) J. Biol. Chem., 280,
11127-11133.
268.Jung, H. J., Lee, J. Y., Kim, S. H., Eu, Y. J.,
Shin, S. Y., Milescu, M., Swartz, K. J., and Kim, J. I. (2005)
Biochemistry, 44, 6015-6023.
269.Posokhov, Y. O., Gottlieb, P. A.,
Morales, M. J., Sachs, F., and Ladokhin, A. S. (2007)
Biophys. J., 93, L20-L22.
270.Wee, C. L., Bemporad, D., Sands, Z. A.,
Gavaghan, D., and Sansom, M. S. (2007) Biophys. J., 92,
L07-L09.
271.Wang, S. Y., and Wang, G. K. (2003) Cell.
Signal., 15, 151-159.
272.Norton, R. S., and McDonough, S. I. (2008)
Curr. Pharm. Des., 14, 2480-2491.
273.McDonough, S. I. (2007) Toxicon,
49, 202-212.
274.Bindokas, V. P., and Adams, M. E. (1989) J.
Neurobiol., 20, 171-188.
275.Scott, R. H., Dolphin, A. C., Bindokas, V.
P., and Adams, M. E. (1990) Mol. Pharmacol., 38,
711-718.
276.Venema, V. J., Swiderek, K. M., Lee, T. D.,
Hathaway, G. M., and Adams, M. E. (1992) J. Biol. Chem.,
267, 2610-2615.
277.Yan, L., and Adams, M. E. (2000) J. Biol.
Chem., 275, 21309-21316.
278.Sather, W. A., Tanabe, T., Zhang, J. F., and
Tsien, R. W. (1994) Ann. N. Y. Acad. Sci., 747,
294-301.
279.Sidach, S. S., and Mintz, I. M. (2000) J.
Neurosci., 20, 7174-7182.
280.Kalapothakis, E., Penaforte, C. L., Leao, R.
M., Cruz, J. S., Prado, V. F., Cordeiro, M. N., Diniz, C. R.,
Romano-Silva, M. A., Prado, M. A., Gomez, M. V., and Beirao, P. S.
(1998) Toxicon, 36, 1971-1980.
281.Dos Santos, R. G., van Renterghem, C.,
Martin-Moutot, N., Mansuelle, P., Cordeiro, M. N., Diniz, C. R., Mori,
Y., de Lima, M. E., and Seagar, M. (2002) J. Biol. Chem.,
277, 13856-13862.
282.Leao, R. M., Cruz, J. S., Diniz, C. R.,
Cordeiro, M. N., and Beirao, P. S. (2000)
Neuropharmacology, 39, 1756-1767.
283.Vieira, L. B., Kushmerick, C.,
Hildebrand, M. E., Garcia, E., Stea, A., Cordeiro, M.
N., Richardson, M., Gomez, M. V., and Snutch, T. P. (2005) J.
Pharmacol. Exp. Ther., 314, 1370-1377.
284.Fletcher, J. I., Smith, R., O’Donoghue,
S. I., Nilges, M., Connor, M., Howden, M. E., Christie, M. J., and
King, G. F. (1997) Nat. Struct. Biol., 4, 559-566.
285.Wang, X. H., Connor, M., Wilson, D., Wilson, H.
I., Nicholson, G. M., Smith, R., Shaw, D., Mackay, J. P., Alewood, P.
F., Christie, M. J., and King, G. F. (2001) J. Biol. Chem.,
276, 40306-40312.
286.Bloomquist, J. R. (2003) Invert.
Neurosci., 5, 45-50.
287.Chong, Y., Hayes, J. L., Sollod, B., Wen, S.,
Wilson, D. T., Hains, P. G., Hodgson, W. C., Broady, K. W., King,
G. F., and Nicholson, G. M. (2007) Biochem. Pharmacol.,
74, 623-638.
288.Escoubas, P., de Weille, J. R., Lecoq, A.,
Diochot, S., Waldmann, R., Champigny, G., Moinier, D., Menez, A., and
Lazdunski, M. (2000) J. Biol. Chem., 275,
25116-25121.
289.Siemens, J., Zhou, S., Piskorowski, R., Nikai,
T., Lumpkin, E. A., Basbaum, A. I., King, D., and Julius, D.
(2006) Nature, 444, 208-212.
290.Liang, S. P., and Pan, X. (1995)
Toxicon, 33, 875-882.
291.Lu, S., Liang, S., and Gu, X. (1999) J.
Protein. Chem., 18, 609-617.
292.Choi, S. J., Parent, R., Guillaume, C.,
Deregnaucourt, C., Delarbre, C., Ojcius, D. M., Montagne, J. J.,
Celerier, M. L., Phelipot, A., Amiche, M., Molgo, J.,
Camadro, J. M., and Guette, C. (2004) FEBS Lett.,
572, 109-117.
293.Pimentel, C., Choi, S. J., Chagot, B., Guette,
C., Camadro, J. M., and Darbon, H. (2006) Protein Sci.,
15, 628-634.
294.Reis, H. J., Prado, M. A., Kalapothakis,
E., Cordeiro, M. N., Diniz, C. R., de Marco, L. A., Gomez, M. V., and
Romano-Silva, M. A. (1999) Biochem. J., 343 (Pt.
2), 413-418.
295.Reis, H. J., Gomez, M. V., Kalapothakis,
E., Diniz, C. R., Cordeiro, M. N., Prado, M. A., and Romano-Silva, M.
A. (2000) Neuroreport, 11, 2191-2194.
296.Duan, Z. G., Yan, X. J., He, X. Z., Zhou, H.,
Chen, P., Cao, R., Xiong, J. X., Hu, W. J., Wang, X. C., and Liang, S.
P. (2006) Comp. Biochem. Physiol. B: Biochem. Mol. Biol.,
145, 350-357.
297.Richardson, M., Pimenta, A. M., Bemquerer, M.
P., Santoro, M. M., Beirao, P. S., Lima, M. E., Figueiredo, S. G.,
Bloch, C., Jr., Vasconcelos, E. A., Campos, F. A., Gomes, P. C., and
Cordeiro, M. N. (2006) Comp. Biochem. Physiol. C: Toxicol.
Pharmacol., 142, 173-187.
298.Chen, J., Zhao, L., Jiang, L., Meng, E., Zhang,
Y., Xiong, X., and Liang, S. (2008) Toxicon, 52,
794-806.
299.Fernandes-Pedrosa, M. F.,
Junqueira-de-Azevedo, I. L., Goncalves-de-Andrade, R. M., Kobashi,
L. S., Almeida, D. D., Ho, P. L., and Tambourgi, D. V. (2008) BMC
Genomics, 9, 279.
300.Perret, B. A. (1977) Toxicon, 15,
505-510.
301.Norment, B. R., Jong, Y. S., and Heitz, J. R.
(1979) Toxicon, 17, 539-548.
302.Atkinson, R. K., and Wright, L. G. (1992)
Comp. Biochem. Physiol. C: Pharmacol. Toxicol.
Endocrinol., 102, 125-128.
303.Akhunov, A. A., Makevnina, L. G.,
Golubenko, Z., and Paskhina, T. S. (1996)
Immunopharmacology, 32, 160-162.
304.Feitosa, L., Gremski, W., Veiga, S. S., Elias,
M. C., Graner, E., Mangili, O. C., and Brentani, R. R. (1998)
Toxicon, 36, 1039-1051.
305.Veiga, S. S., da Silveira, R. B., Dreyfus,
J. L., Haoach, J., Pereira, A. M., Mangili, O. C., and Gremski, W.
(2000) Toxicon, 38, 825-839.
306.Young, A. R., and Pincus, S. J. (2001)
Toxicon, 39, 391-400.
307.Da Silveira, R. B., dos Santos Filho, J. F.,
Mangili, O. C., Veiga, S. S., Gremski, W., Nader, H. B., and von
Dietrich, C. P. (2002) Toxicon, 40, 815-822.
308.Barbaro, K. C., Knysak, I., Martins, R., Hogan,
C., and Winkel, K. (2005) Toxicon, 45, 489-499.
309.Da Silveira, R. B., Wille, A. C., Chaim, O. M.,
Appel, M. H., Silva, D. T., Franco, C. R., Toma, L., Mangili, O.
C., Gremski, W., Dietrich, C. P., Nader, H. B., and Veiga, S. S. (2007)
Biochem. J., 406, 355-363.
310.Devaraja, S., Nagaraju, S.,
Mahadeswaraswamy, Y. H., Girish, K. S., and Kemparaju, K. (2008)
Toxicon, 52, 130-138.
311.Schanbacher, F. L., Lee, C. K., Wilson, I.
B., Howell, D. E., and Odell, G. V. (1973) Comp. Biochem. Physiol.
B: Biochem. Mol. Biol., 44, 389-396.
312.Nagaraju, S., Mahadeswaraswamy, Y. H., Girish,
K. S., and Kemparaju, K. (2006) Comp. Biochem. Physiol. C: Toxicol.
Pharmacol., 144, 1-9.
313.Kreil, G. (1995) Protein Sci., 4,
1666-1669.
314.Usmanov, P. B., and Nuritova, F. A. (1994)
Toxicon, 32, 625-628.
315.Shikata, Y., Watanabe, T., Teramoto, T., Inoue,
A., Kawakami, Y., Nishizawa, Y., Katayama, K., and Kuwada, M.
(1995) J. Biol. Chem., 270, 16719-16723.
316.Jilek, A., Mollay, C., Tippelt, C., Grassi, J.,
Mignogna, G., Mullegger, J., Sander, V., Fehrer, C., Barra, D., and
Kreil, G. (2005) Proc. Natl. Acad. Sci. USA, 102,
4235-4239.
317.Heck, S. D., Faraci, W. S., Kelbaugh, P. R.,
Saccomano, N. A., Thadeio, P. F., and Volkmann, R. A. (1996)
Proc. Natl. Acad. Sci. USA, 93, 4036-4039.
318.Bansal, P. S., Torres, A. M., Crossett, B.,
Wong, K. K., Koh, J. M., Geraghty, D. P., Vandenberg, J. I.,
and Kuchel, P. W. (2008) J. Biol. Chem., 283,
8969-8975.
319.Ramos-Cerrillo, B., Olvera, A., Odell, G. V.,
Zamudio, F., Paniagua-Solis, J., Alagon, A., and Stock, R. P. (2004)
Toxicon, 44, 507-514.
320.Futrell, J. M. (1992) Am. J. Med. Sci.,
304, 261-267.
321.Mota, I., and Barbaro, K. C. (1995) J.
Toxicol. Tox. Rev., 14, 401-421.
322.Babcock, J. L., Civello, D. J., and Geren, C.
R. (1981) Toxicon, 19, 677-689.
323.Kurpiewski, G., Forrester, L. J., Barrett,
J. T., and Campbell, B. J. (1981) Biochim. Biophys. Acta,
678, 467-476.
324.Lee, S., and Lynch, K. R. (2005) Biochem.
J., 391, 317-323.
325.Murakami, M. T., Fernandes Pedrosa, M. F.,
Tambourgi, D. V., and Arni, R. K. (2005) J. Biol. Chem.,
280, 13658-13664.
326.Machado, L. F., Laugesen, S., Botelho, E.
D., Ricart, C. A., Fontes, W., Barbaro, K. C., Roepstorff, P., and
Sousa, M. V. (2005) Proteomics, 5, 2167-2176.
327.Braz, A., Minozzo, J., Abreu, J. C., Gubert, I.
C., and Chavez-Olortegui, C. (1999) Toxicon, 37,
1323-1328.
328.Fernandes Pedrosa, M. F., Junqueira de
Azevedo, I. L., Goncalves-de-Andrade, R. M., van den Berg, C. W.,
Ramos, C. R., Ho, P. L., and Tambourgi, D. V. (2002) Biochem.
Biophys. Res. Commun., 298, 638-645.
329.Tambourgi, D. V., Fernandes Pedrosa, M. F., van
den Berg, C. W., Goncalves-de-Andrade, R. M., Ferracini, M.,
Paixao-Cavalcante, D., Morgan, B. P., and Rushmere, N. K. (2004)
Mol. Immunol., 41, 831-840.
330.Binford, G. J., Cordes, M. H., and Wells, M. A.
(2005) Toxicon, 45, 547-560.
331.Pauli, I., Puka, J., Gubert, I. C., and
Minozzo, J. C. (2006) Toxicon, 48, 123-137.
332.Ribeiro, R. O., Chaim, O. M., da Silveira, R.
B., Gremski, L. H., Sade, Y. B., Paludo, K. S., Senff-Ribeiro, A., de
Moura, J., Chavez-Olortegui, C., Gremski, W., Nader, H. B., and Veiga,
S. S. (2007) Toxicon, 50, 1162-1174.
333.Appel, M. H., da Silveira, R. B., Chaim, O. M.,
Paludo, K. S., Silva, D. T., Chaves, D. M., da Silva, P. H., Mangili,
O. C., Senff-Ribeiro, A., Gremski, W., Nader, H. B., and Veiga, S. S.
(2008) Biochim. Biophys. Acta, 1780, 167-178.
334.Senff-Ribeiro, A., Henrique da Silva, P.,
Chaim, O. M., Gremski, L. H., Paludo, K. S., Bertoni da Silveira, R.,
Gremski, W., Mangili, O. C., and Veiga, S. S. (2008) Biotechnol.
Adv., 26, 210-218.
335.Binford, G. J., Bodner, M. R., Cordes, M. H.,
Baldwin, K. L., Rynerson, M. R., Burns, S. N., and Zobel-Thropp, P. A.
(2009) Mol. Biol. Evol., 26, 547-566.
336.Cordes, M. H., and Binford, G. J. (2006)
Bioinformatics, 22, 264-268.
337.Morgan, P. N. (1969) Toxicon, 6,
161-165.
338.Rees, R. S., Gates, C., Timmons, S., des Prez,
R. M., and King, L. E., Jr. (1988) Toxicon, 26,
1035-1045.
339.Tambourgi, D. V., Pedrosa, M. F., de Andrade,
R. M., Billington, S. J., Griffiths, M., and van den Berg, C. W. (2007)
Mol. Immunol., 44, 576-582.
340.Tambourgi, D. V., Paixao-Cavalcante, D.,
Goncalves de Andrade, R. M., Fernandes-Pedrosa, M. F., Magnoli, F. C.,
Paul Morgan, B., and van den Berg, C. W. (2005) J. Invest.
Dermatol., 124, 725-731.
341.Tambourgi, D. V., de Sousa da Silva, M.,
Billington, S. J., Goncalves de Andrade, R. M., Magnoli, F. C.,
Songer, J. G., and van den Berg, C. W. (2002) Immunology,
107, 93-101.
342.Ramu, Y., Xu, Y., and Lu, Z. (2006)
Nature, 442, 696-699.
343.Longenecker, H. E., Jr., Hurlbut, W. P., Mauro,
A., and Clark, A. W. (1970) Nature, 225, 701-703.
344.Frontali, N., Ceccarelli, B., Gorio, A., Mauro,
A., Siekevitz, P., Tzeng, M. C., and Hurlbut, W. P. (1976) J. Cell.
Biol., 68, 462-479.
345.Graudins, A., Gunja, N., Broady, K. W., and
Nicholson, G. M. (2002) Toxicon, 40, 767-775.
346.Rosenthal, L., and Meldolesi, J. (1989)
Pharmacol. Ther., 42, 115-134.
347.Misler, S., and Hurlbut, W. P. (1979) Proc.
Natl. Acad. Sci. USA, 76, 991-995.
348.Storchak, L. G., Pashkov, V. N.,
Pozdnyakova, N. G., Himmelreich, N. H., and Grishin, E. V. (1994)
FEBS Let., 351, 267-270.
349.Fritz, L. C., Tzen, M. C., and Mauro, A. (1980)
Nature, 283, 486-487.
350.Grishin, E. (1994) Pure Appl. Chem.,
66, 783-790.
351.Kovalevskaya, G. I., Pashkov, V. N., Bulgakov,
O. V., Fedorova, I. M., Magazanik, L. G., and Grishin, E. V. (1990)
Bioorg. Khim., 16, 1013-1018.
352.Krasnoperov, V. G., Shamotienko, O. G., and
Grishin, E. V. (1990) Bioorg. Khim., 16, 1138-1140.
353.Krasnoperov, V. G., Shamotienko, O. G., and
Grishin, E. V. (1990) Bioorg. Khim., 16, 1567-1569.
354.Magazanik, L. G., Fedorova, I. M.,
Kovalevskaya, G. I., Pashkov, V. N., Bulgakov, O. V., and Grishin, E.
V. (1992) Neuroscience, 46, 181-188.
355.Finkelstein, A., Rubin, L. L., and Tzeng, M. C.
(1976) Science, 193, 1009-1011.
356.Shatursky, O., Pashkov, V. N.,
Bulgacov, O. V., and Grishin, E. V. (1995) Biochim.
Biophys. Acta, 1233, 14-20.
357.Kiyatkin, N. I., Dulubova, I. E.,
Chekhovskaya, I. A., and Grishin, E. V. (1990) FEBS
Lett., 270, 127-131.
358.Kiyatkin, N., Dulubova, I., and Grishin, E.
(1993) Eur. J. Biochem., 213, 121-127.
359.Dulubova, I. E., Krasnoperov, V. G., Khvotchev,
M. V., Pluzhnikov, K. A., Volkova, T. M., Grishin, E. V., Vais, H.,
Bell, D. R., and Usherwood, P. N. (1996) J. Biol. Chem.,
271, 7535-7543.
360.Danilevich, V. N., Luk’yanov, S. A., and
Grishin, E. V. (1999) Bioorg. Khim., 25, 537-547.
361.Voronin, D. A., and Kiseleva, E. V. (2007)
Tsitologiya, 49, 989-999.
362.Li, J., Mahajan, A., and Tsai, M. D. (2006)
Biochemistry, 45, 15168-15178.
363.Kiyatkin, N., Dulubova, I. E., Volkova, T. M.,
Chekhovskaya, I. A., Lipkin, A. V., and Grishin, E. V. (1992)
Dokl. Akad. Nauk SSSR, 323, 178-180.
364.Kiyatkin, N., Dulubova, I., Chekhovskaya,
I., Lipkin, A., and Grishin, E. (1992) Toxicon, 30,
771-774.
365.Volkova, T. M., Pluzhnikov, K. A., Woll, P. G.,
and Grishin, E. V. (1995) Toxicon, 33, 483-489.
366.Gasparini, S., Kiyatkin, N., Drevet, P.,
Boulain, J. C., Tacnet, F., Ripoche, P., Forest, E., Grishin, E.,
and Menez, A. (1994) J. Biol. Chem., 269,
19803-19809.
367.Kiyatkin, N. I., Kulikovskaya, I. M., Grishin,
E. V., Beadle, D. J., and King, L. A. (1995) Eur. J. Biochem.,
230, 854-859.
368.Lunev, A. V., Demin, V. V., Zaitsev, O. I.,
Spadar, S. I., and Grishin, E. V. (1991) Bioorg. Khim.,
17, 1021-1026.
369.Orlova, E. V., Rahman, M. A., Gowen, B.,
Volynski, K. E., Ashton, A. C., Manser, C., van Heel, M., and
Ushkaryov, Y. A. (2000) Nat. Struct. Biol., 7,
48-53.
370.Ashton, A. C., Rahman, M. A., Volynski, K.
E., Manser, C., Orlova, E. V., Matsushita, H., Davletov, B. A.,
van Heel, M., Grishin, E. V., and Ushkaryov, Y. A. (2000)
Biochimie, 82, 453-468.
371.Petrenko, A. G., Kovalenko, V. A., Shamotienko,
O. G., Surkova, I. N., Tarasyuk, T. A., Ushkaryov, Yu. A., and Grishin,
E. V. (1990) EMBO J., 9, 2023-2027.
372.Surkova, I. N., and Grishin, E. V. (1991)
Biomed. Sci., 2, 417-420.
373.O’Connor, V. M., Shamotienko, O.,
Grishin, E., and Betz, H. (1993) FEBS Lett., 326,
255-260.
374.Ushkaryov, Y. A., Petrenko, A. G., Geppert, M.,
and Sudhof, T. C. (1992) Science, 257, 50-56.
375.Davletov, B. A., Shamotienko, O. G., Lelianova,
V. G., Grishin, E. V., and Ushkaryov, Y. A. (1996) J. Biol.
Chem., 271, 23239-23245.
376.Lelianova, V. G., Davletov, B. A.,
Sterling, A., Rahman, M. A., Grishin, E. V., Totty, N. F.,
and Ushkaryov, Y. A. (1997) J. Biol. Chem., 272,
21504-21508.
377.Krasnoperov, V. G., Beavis, R., Chepurny,
O. G., Little, A. R., Plotnikov, A. N., and Petrenko, A. G. (1996)
Biochem. Biophys. Res. Commun., 227, 868-875.
378.Krasnoperov, V., Bittner, M. A., Mo, W.,
Buryanovsky, L., Neubert, T. A., Holz, R. W., Ichtchenko, K., and
Petrenko, A. G. (2002) J. Biol. Chem., 277,
35887-35895.
379.Liu, J., Wan, Q., Lin, X., Zhu, H., Volynski,
K., Ushkaryov, Y., and Xu, T. (2005) Traffic, 6,
756-765.
380.Ushkaryov, Y. A., Rohou, A., and Sugita, S.
(2008) Handb. Exp. Pharmacol., 184, 171-206.
381.Andreev, Ya. A., Danilevich, V. N., and
Grishin, E. V. (2005) Bioorg. Khim., 31, 175-185.
382.Mee, C. J., Tomlinson, S. R., Perestenko,
P. V., de Pomerai, D., Duce, I. R., Usherwood, P. N., and Bell, D. R.
(2004) Biochem. J., 378, 185-191.
383.Ushkaryov, Y. A., Volynski, K. E., and Ashton,
A. C. (2004) Toxicon, 43, 527-542.
384.Krapcho, K. J., Kral, R. M., Jr.,
Vanwagenen, B. C., Eppler, K. G., and Morgan, T. K. (1995)
Insect Biochem. Mol. Biol., 25, 991-1000.
385.Qiao, P., Zuo, X. P., Chai, Z. F., and Ji, Y.
H. (2004) Acta Biochim. Biophys. Sin. (Shanghai), 36,
656-660.
386.Jiang, L., Chen, J., Peng, L., Zhang, Y.,
Xiong, X., and Liang, S. (2008) Peptides, 29,
1679-1684.
387.Danilevich, V. N., and Grishin, E. V. (2000)
Bioorg. Khim., 26, 933-939.
388.Shlyapnikov, Y. M., Andreev, Y. A.,
Kozlov, S. A., Vassilevski, A. A., and Grishin, E. V. (2008)
Protein Expr. Purif., 60, 89-95.
389.Vassilevski, A. A., Kozlov, S. A., and
Grishin, E. V. (2008) Recent Pat. Inflamm. Allergy Drug
Discov., 2, 58-63.
390.Paetzel, M., Karla, A., Strynadka, N. C., and
Dalbey, R. E. (2002) Chem. Rev., 102, 4549-4580.
391.Schechter, I., and Berger, A. (1967)
Biochem. Biophys. Res. Commun., 27, 157-162.
392.Seidah, N. G., and Chretien, M. (1999) Brain
Res., 848, 45-62.
393.Collins, S. P., Comis, A., Tyler, M. I.,
Marshall, M., and Howden, M. E. (1995) Comp. Biochem. Physiol. C:
Pharmacol. Toxicol. Endocrinol., 110, 89-93.
394.Liang, S., Li, X., Cao, M., Xie, J., Chen, P.,
and Huang, R. (2000) J. Protein Chem., 19,
225-229.
395.Corzo, G., Gilles, N., Satake, H., Villegas,
E., Dai, L., Nakajima, T., and Haupt, J. (2003) FEBS Lett.,
547, 43-50.
396.Milne, T. J., Abbenante, G., Tyndall, J. D.,
Halliday, J., and Lewis, R. J. (2003) J. Biol. Chem.,
278, 31105-31110.
397.Eipper, B. A., Stoffers, D. A., and Mains, R.
E. (1992) Annu. Rev. Neurosci., 15, 57-85.
398.Fricker, L. D. (1988) Annu. Rev.
Physiol., 50, 309-321.
399.Xin, X., Varlamov, O., Day, R., Dong, W.,
Bridgett, M. M., Leiter, E. H., and Fricker, L. D. (1997) DNA Cell
Biol., 16, 897-909.
400.Salamoni, S. D., Costa da Costa, J., Palma, M.
S., Konno, K., Nihei, K., Tavares, A. A., de Abreu, D. S., Venturin, G.
T., de Borba Cunha, F., de Oliveira, R. M., and Breda, R. V. (2005)
Brain Res., 1048, 170-176.
401.Mortari, M. R., Cunha, A. O., Ferreira, L.
B., and dos Santos, W. F. (2007) Pharmacol. Ther., 114,
171-183.
402.Hancock, R. E., and Sahl, H. G. (2006) Nat.
Biotechnol., 24, 1551-1557.
403.Piers, K. L., Brown, M. H., and Hancock, R. E.
(1993) Gene, 134, 7-13.
404.Zhang, L., Falla, T., Wu, M., Fidai, S.,
Burian, J., Kay, W., and Hancock, R. E. (1998) Biochem. Biophys.
Res. Commun., 247, 674-680.
405.Kass, R. S. (2005) J. Clin. Invest.,
115, 1986-1989.
406.Cannon, S. C. (2006) Annu. Rev.
Neurosci., 29, 387-415.
407.Catterall, W. A., Dib-Hajj, S., Meisler,
M. H., and Pietrobon, D. (2008) J. Neurosci., 28,
11768-11777.
408.Pluzhnikov, K., Vassilevski, A.,
Korolkova, Y., Fisyunov, A., Iegorova, O., Krishtal, O., and
Grishin, E. (2007) Toxicon, 50, 993-1004.
409.Khan, S. A., Zafar, Y., Briddon, R. W., Malik,
K. A., and Mukhtar, Z. (2006) Transgenic Res., 15,
349-357.
410.Hernandez-Campuzano, B., Suarez, R., Lina, L.,
Hernandez, V., Villegas, E., Corzo, G., and Iturriaga, G. (2009)
Toxicon, 53, 122-128.
411.Fitches, E., Edwards, M. G., Mee, C., Grishin,
E., Gatehouse, A. M., Edwards, J. P., and Gatehouse, J. A. (2004)
J. Insect Physiol., 50, 61-71.
412.Osusky, M., Zhou, G., Osuska, L., Hancock, R.
E., Kay, W. W., and Misra, S. (2000) Nat. Biotechnol.,
18, 1162-1166.
413.Montesinos, E. (2007) FEMS Microbiol.
Lett., 270, 1-11.
414.Hughes, S. R., Dowd, P. F., Hector, R. E.,
Panavas, T., Sterner, D. E., Qureshi, N., Bischoff, K. M., Bang, S. S.,
Mertens, J. A., Johnson, E. T., Li, X. L., Jackson, J. S., Caughey, R.
J., Riedmuller, S. B., Bartolett, S., Liu, S., Rich, J. O., Farrelly,
P. J., Butt, T. R., Labaer, J., and Cotta, M. A. (2008) J. Pept.
Sci., 14, 1039-1050.
415.Hughes, S. R., Sterner, D. E., Bischoff, K. M.,
Hector, R. E., Dowd, P. F., Qureshi, N., Bang, S. S., Grynaviski, N.,
Chakrabarty, T., Johnson, E. T., Dien, B. S., Mertens, J. A., Caughey,
R. J., Liu, S., Butt, T. R., Labaer, J., Cotta, M. A., and Rich, J. O.
(2008) Plasmid, 61, 22-38.
416.Billen, B., Vassilevski, A., Nikolsky, A.,
Tytgat, J., and Grishin, E. (2008) Toxicon, 52,
309-317.
417.Foelix, R. F. (1996) Biology of
Spiders, Oxford University Press, New York-Oxford, 330 p.
418.Ruppert, E. E., Fox, R. S., and Barnes, R. D.
(2003) Invertebrate Zoology: a Functional Evolutionary
Approach, Brooks/Cole, Pacific Grove, London, 963 p.
419.Nentwig, W., and Aitchison-Benell, C. W. (1987)
Ecophysiology of Spiders, Springer-Verlag, Berlin-New York,
448 p.
420.Oxford, G. S., and Gillespie, R. G. (1998)
Annu. Rev. Entomol., 43, 619-643.
421.Whitehousel, M. E., and Lubin, Y. (2005)
Biol. Rev. Camb. Philos. Soc., 80, 347-361.
422.Cushing, P. E. (1997) Florida
Entomologist, 80, 165-193.
423.Jackson, R. R., and Pollard, S. D. (1996)
Annu. Rev. Entomol., 41, 287-308.
424.Allan, R. A., Capon, R. J., Brown, W. V., and
Elgar, M. A. (2002) J. Chem. Ecol., 28, 835-848.
425.Elgar, M. A., and Allan, R. A. (2004)
Naturwissenschaften, 91, 143-147.
426.Eberhard, W. G. (1977) Science,
198, 1173-1175.
427.Stowe, M. K., Tumlinson, J. H., and Heath, R.
R. (1987) Science, 236, 964-967.
428.Gemeno, C., Yeargan, K. V., and Haynes, K. F.
(2000) J. Chem. Ecol., 26, 1235-1243.
429.Harland, D. P., and Jackson, R. R. (2000)
Cimbebasia, 16, 231-240.
430.Eisner, T., and Camazine, S. (1983) Proc.
Natl. Acad. Sci. USA, 80, 3382-3385.
431.Arzt, E., Gorb, S., and Spolenak, R. (2003)
Proc. Natl. Acad. Sci. USA, 100, 10603-10606.
432.Kesel, A. B., Martin, A., and Seidl, T. (2003)
J. Exp. Biol., 206, 2733-2738.
433.Gorb, S. N., Niederegger, S., Hayashi, C. Y.,
Summers, A. P., Votsch, W., and Walther, P. (2006) Nature,
443, 407.
434.Craig, C. L. (2003) Spiderwebs and Silk:
Tracing Evolution from Molecules to Genes to Phenotypes, Oxford
University Press, New York, 230 p.
435.Vollrath, F. (2000) J. Biotechnol.,
74, 67-83.
436.Rising, A., Nimmervoll, H., Grip, S.,
Fernandez-Arias, A., Storckenfeldt, E., Knight, D. P., Vollrath, F.,
and Engstrom, W. (2005) Zoolog. Sci., 22,
273-281.
437.Vollrath, F. (2005) Curr. Biol.,
15, R364-R365.
438.Kluge, J. A., Rabotyagova, O., Leisk, G. G.,
and Kaplan, D. L. (2008) Trends Biotechnol., 26,
244-251.