REVIEW: Fragmentomics of Natural Peptide Structures
A. A. Zamyatnin1,2
1Bach Institute of Biochemistry, Russian Academy of Sciences, Leninsky pr. 33, 119071 Moscow, Russia; fax: (495) 954-2732; E-mail: aaz@inbi.ras.ru2Universidad Técnica Federico Santa Maria, Departamento de Informática, El Centro Científico Tecnólogico de Valparaíso, Ave. España 1680, Valparaiso, Chile; fax: (5632) 279-75113; E-mail: alexander.zamyatnin@usm.cl
Received January 26, 2009; Revision received June 9, 2009
Natural fragmentation of peptide and other chemical structures is well known. They are a significant object of biochemical investigations. In this connection, the bases and determination are given for the notion of the “fragmentome” as a set of all fragments of a single substance, as well as for global fragmentome of all chemical components of living organisms. It is described how protein-peptide fragments are formed in nature, what experimental and theoretical methods are used for their investigation, as well as mathematical characteristics of fragmentomes. Individual fragmentomes of all subunits and of complete casein fragmentome are considered in detail. Structural and functional variety of its possible fragments was revealed by computer analysis. Formation in an organism of an exogenous–endogenous pool of oligopeptides and correlation of these data with concepts of structure–functional continuum of regulatory molecules is shown on an example of food protein fragments. Possible practical importance of the use of natural fragments in dietology, therapy, as well as in sanitary hygiene and cosmetics is noted.
KEY WORDS: fragmentomics, oligopeptide, protein, casein, structure, function, EROP-Moscow DatabaseDOI: 10.1134/S0006297909130100
Fragmentary structural organization is characteristic of both the simplest and most complex biomolecules. Low molecular weight fragments of biopolymers can be easily seen on various metabolic maps [1]. There are numerous examples showing that relatively small natural physiologically active substances are fragments of larger ones. An obvious example is β-carotene “composed” of two vitamin A molecules [2].
The concept of fragment is most often used in consideration of such polymeric molecules as proteins, nucleic acids, and polysaccharides. Fragments of natural polypeptides are mentioned most frequently. Thus, at present the protein–peptide database Swiss-Prot/TrEMBL (http://www.expasy.org/sprot/) contains information concerning structure of almost one and a half million fragments, and another database, PubMed (http://www.ncbi.nlm.nih.gov/pubmed/), gives information about over 130,000 publications dealing with various fragments (and less than 60,000 publications about metabolites). However, functions of most of these fragments are not known.
In recent years, a steady increase in the number of publications dealing with protein fragment structure and function has been seen. For some proteins there are already hundreds of fragments that have been studied in detail, and it seems that concepts concerning functional importance of the totality of possible fragments of a single protein will be formed. In this connection, we suggested the term fragmentomics for the science studying structure and functions of the set of molecular fragments, and to call fragmentome the whole set of biomolecule fragments [3]. For peptide structures, fragmentomics can be considered as a notion that combines proteomics and peptidomics.
In this work, main ideas of fragmentomics and principles of their usage in studies of natural peptide structures are formulated.
NATURAL PEPTIDE FRAGMENTS
Fragmentation of peptide structures in living organisms is well known. For example, fragments are molecules formed in an organism from specialized precursors due to splitting off of the signal peptide and pre-peptide(s). This process is specific to large protein structures [4] and oligopeptide regulators containing from 2 to ~50 amino acid residues (a.a.) [5, 6]. A simple example of protein fragmentation is bovine serum albumin consisting of 583 a.a. [7]. Its precursor of 607 residues dissociates to fragments: signal peptides (residues 1-18), pre-peptide (19-24 a.a.), and properly serum albumin (25-607 a.a.) [8]. In the case of regulatory oligopeptides, such as human corticoliberin [9], its precursor (194 a.a.) includes signal peptide (1-24 a.a.), pre-peptide (25-153 a.a.), and hormone (154-194 a.a.) [10].
However, precursors often contain information about more than one functionally important structure. A typical representative of such polypeptides is pro-opiomelanocortin (POMC). As follows from its name, it contains different structures having also different functions of nervous (opioids) and endocrine system regulators. The human POMC precursor (Fig. 1) contains 267 a.a. [11], and the signal peptide (1-26) is split off first. The remaining sequence is POMC, which then dissociates into four parts: aldosterone-stimulating peptide (27-102) [12] including the γ-melanotropin sequence (77-87) [13], a peptide with unknown function (105-135) [14], ACTH (138-176) [15] with α-melanotropin site (138-150) [15], and β-lipotropin (179-267) [16] including γ-lipotropin (179-234) with β-melanotropin sequence (217-234) [17], as well as β-endorphin (237-267) [18] with γ- [19] and α-endorphin [20] sequences. Dipeptide regions 104-105, 136-137, and 177-178 are lysyl-arginyl (RK) pairs recognized by the precursor cleaving proteases and are cleaved away [21]. In the given example the figure marking the fragment beginning and end as in the Swiss-Prot/TrEMBL database is given with accounting for the signal peptide. However, the signal peptide is usually not considered in numbering experimentally obtained protein–peptide fragments.
It should be noted that the first 5 a.a. of β-endorphin are the met-enkephalin sequence [22], but this oligopeptide is not cleaved off the POMC structure and is formed from its own specialized precursor [23]. This precursor is organized according to the same above-described principles and contains sequences of one leu- and six met-enkephalins. The presence of more than one copy of the same oligopeptide is not rare and the number of them can be quite high. Thus, the precursor of the mollusk Aplysia californica neuropeptides contains 28 structures of FMRF tetrapeptide and additionally one FLRF structure [24]. Most of these oligopeptides are separated (flanked) by lysyl-arginyl pairs with different combinations of such amino acid residues.Fig. 1. Complete amino acid sequence of human pro-opiomelanocortin precursor (underlined) and primary structures of its natural fragments. The POMC structure is shown in bold, while signal peptide and cleaved off lysyl-arginyl pairs (KR) are shown in usual type. The standard single-letter code is used to designate amino acid residues. Other details are in the text.

After cleaving off from precursors, in many cases the process of reduced structure formation is not complete. For example, co-existence in oligopeptides of multiple forms resulting from enzymic reactions leading to formation of shorter fragments was noted long ago. Thus, deciphering amino acid sequence of one of the first known oligopeptides, angiotensin (formerly called hypertensin), immediately revealed two structures of ten (1-10) and eight (1-8) amino acid residues which were named angiotensins I and II, respectively [25]. However, it was shown later that in addition there are at least four shorter natural forms of these oligopeptides, angiotensins III (2-8), IV (2-10), V (3-8), and VI (4-8) [26]. Detailed investigations revealed enzymes involved in formation of the latter, and it was shown that such structures could exist simultaneously. It became clear later that oligopeptide polymorphism is a common event, it has been found in somatostatin [27-29], atrial natriuretic peptide [30, 31], and many other oligopeptide regulators. Therefore, among thousands of already discovered oligopeptides there may be many characterized by polymorphism, but so far their fragments are not identified.
Natural fragments of larger peptide molecules like endorphins and enkephalins can retain their functions. However, they can be devoid of the initial molecule function, exhibiting different functional properties. For example, the N-terminal fragment (13 a.a.) of the above-mentioned bovine adrenocorticotropic hormone consisting of 39 residues has no cortisol-releasing function of ACTH, but it exhibits activity of melanocyte-stimulating hormone (α-melanotropin) [32, 33].
Besides, evidence is accumulating showing that in addition, in different organs and tissues of living organisms there are peptide structures that are not formed from specialized precursors, but are natural fragments of well-known proteins. Numerous fragments of α- and β-hemoglobin chains have been found in bovine brain [34-37] and epiphysis [38], in casein of cow milk [39, 40], and in other sources.
Thus, already many natural peptide fragments formed during biogenesis are known, and their number continues to grow.
METHODS OF FRAGMENT AND FRAGMENTOME INVESTIGATION
There are several approaches to obtaining fragments of peptide structures. The main experimental approaches are analytical extraction, purification, and sequencing. A powerful tool for protein and oligopeptide isolation is two-dimensional electrophoresis [41, 42], while for primary structure determination it is mass-spectrometry [43] when initial peptide structure is cleaved to fragments. Use of these methods together has resulted in explosive development of proteomics and peptidomics [44].
The method of artificial fragmentation is also widespread; it is based on degradation of natural peptide structures (such as Edman chemical degradation [45, 46]). The use of such approach revealed structures of translated peptide structures oxytocin [47], bovine [Arg-8]vasopressin and porcine [Lys-8]vasopressin [48], as well as of bovine insulin [49]. However, in such methods the resulting fragments are used only for further detection of primary structure of the studied substances, while their functional properties are usually not studied.
Functional properties of fragments are often studied for detection of a minimal site retaining physiological activity specific of the initial structure. For this purpose, chemical synthesis of different fragments of the same peptide structure is used. For example, many fragments of α-melanotropin [50] and bradykinin [51] have been synthesized. Moreover, in the case of bradykinin actually the whole fragmentome was synthesized (Fig. 2). After synthesis, all of its fragments were studied in a standard test for ability to contract a strip of guinea pig ileum smooth muscle. It was shown that just a single fragment (5-9) exhibited any noticeable activity compared to the complete bradykinin molecule.
It should be noted that chemical synthesis is widely used for creation of numerous chemical analogs of natural oligopeptides to search for structures more active than the natural ones. For example, almost 200 bradykinin analogs have synthesized [52].Fig. 2. Results of bradykinin (BK) fragment syntheses and study of their effects on contraction of guinea pig ileum smooth muscle strips (GPI-test) [51] (n is the number of amino acid residues).
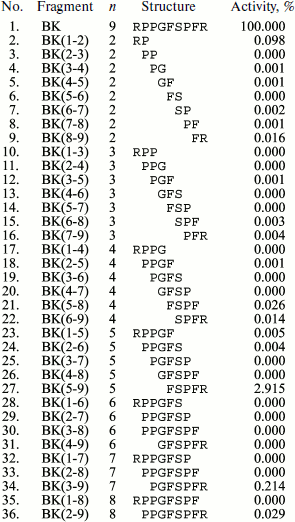
Fragments are also used in different theoretical analyses, in particular, in detection of protein homologs [53-55]. For this purpose computer programs are created which can generate fragments of one compared structure and to reveal homology by their scanning along the amino acid sequence of another. The use of specially developed computer programs allows comparison of peptide fragments of a certain origin, such as bovine hemoglobin, with structures of known oligopeptides obtained from different sources [3, 56]. This method is used to reveal protein regions identical or homologous to a regulatory oligopeptide with known functions. Fragments are generated using these programs in the succession shown in Fig. 2, from dipeptide to the largest peptides. Theoretical analysis of experimental data can reveal new information about structure–functional properties of still unstudied fragments of natural peptide structures.
MATHEMATICAL PRINCIPLES OF FRAGMENTOMICS
Only linear peptide structures are always formed first during translation. Later they can establish intramolecular S–S bonds, transforming them into nonlinear molecules. However, disulfide bonds are absent from practically all experimentally detected fragments of larger structures; therefore, at this stage it is possible consider only the simplest mathematical analysis of linear fragments.
It is apparent that the number of possible natural peptide structures P composed of different amino acid residues and including amino acid repeats is described by formula:
where A is the number of different amino acid residues, and n is the number of residues in the structure. Since the number of canonical translated residues is 20, then according to formula (1) the maximal number of different dipeptides is 400, tripeptides 8000, tetrapeptides 160,000, and so on. Thus, as the length of peptide structure increases, the number of possible combinations between residues quickly grows. However, it follows from the same formula that in the case of usage by nature of all possible combinations, in the whole human genome (~3·109 bases [57]) all different structures containing no more than eight residues could be described because 208 = ~3·1010. However, since the translated part makes up only several percent of the whole genome, different amino acid combinations should be even shorter. Analysis of all known natural peptide structures has shown that as their length increases, the fraction of existing combinations of all possible quickly decreases, and for octapeptides it makes up only 0.0016% [58]. Obviously, in structures translated from a single genome the number of such combinations will be even lower.
The theoretically possible maximal number of all (including identical) overlapped fragments of peptide structure Nktheor with assigned number of amino acid residues k is described by the expression [3]:
and maximal total number of all possible overlapping peptide fragments of the protein (also including identical) for all k, i.e. from k = 2 (dipeptides) to k = n – 1, can be obtained using the following expression:
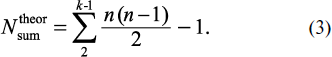
These formulas characterize the upper level of calculated values. In particular, as follows from formula (2), a hypothetical sequence containing one of each dipeptide fragment combinations should consist of 401 a.a., while according to formula (3) the total number of all possible overlapped fragments in this sequence will be 80,199 items.
We have used formula (3) to calculate values of theoretically possible fragments Nsumtheor for a number of different natural oligopeptides and proteins (Table 1). However, results of calculations using these formulas are valid only in the case of lack of repetition of the primary structure of the fragment in the initial molecule. Primary structures of pentapeptide met-enkephalin (YGGFM) [22] and nonapeptide bradykinin (RPPGFSPFR) [59] meet these requirements. However, for example, in ACTH structure [15] there are two copies of dipeptide fragment GK, while in α- and β-hemoglobins [60] there are multiple fragment repeats. The existence of repetitive fragments is possible even in natural pentapeptides as is seen in primary structure of one of the cricket hormones (AAAPF) [61] in which the dipeptide fragment AA is repeated twice. As the number of amino acid residues in the initial structure of larger molecules increases, repetitive amino acid sequences become more and more frequent, and this results in lowering of the real number of different fragments below the Nktheor and Nsumtheor values. Therefore formulas (2) and (3) are not applicable to calculation of the real characteristics of the protein fragmentome, which can be designated as Nkexp and Nsumexp.
Table 1. Maximal possible number of
Nsumtheor fragments in a number of natural
oligopeptides and proteins without considering repeats

*n is the number of amino acid residues in complete
structure.
Thus, the number of different fragments Nkexp for each k should be lower than Nktheor for some value Rk, i.e.
In this case, even the equal length fragments (i.e. at the same k value) can have different amino acid sequence. Owing to this, it is necessary to introduce additional values i (the number of identical structures at one k) and m (the number of different structures also at one k) in order to calculate Rk. Then summing up by m will give:
i.e. the number of different equal-length fragments of all k values is:
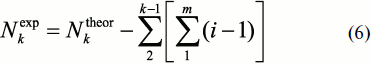
or in complete form:
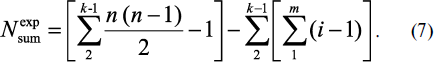
In extended peptide structures, the variety of amino acid sequences in repetitive overlapping fragments with the same k value is practically always observed. Therefore, formulas (6) and (7) should be used to calculate real content of different fragments. In these expressions first summing the repetitive fragments with equal number and identical sequences of amino acid residues takes place (summing up by m at the same k), and then summing is carried out for all k, i.e. for any length fragments having identical and different amino acid sequences. The final result, i.e. determination of the value of Nsumexp, is obtained after subtraction of the double summing result from Nsumtheor value produced by formula (3). It is reasonable to carry out these cumbersome calculations using a special computer structural and functional analysis of natural peptide structure, examples of which are given below.
As follows from data of the oligopeptide database EROP-Moscow (http://erop.inbi.ras.ru/) [62], at present the highest number of fragments was experimentally revealed and functionally characterized in bovine hemoglobin (62 fragments) and casein (72 fragments). We described previously structure–functional analysis of hemoglobin fragments [3, 63]; therefore, in this work other peptide structures are chosen for description of fragmentome properties.
STRUCTURAL CHARACTERISTICS OF A FRAGMENTOME
We shall consider the bovine casein fragmentome as an example. This protein (Fig. 3) consists of four subunits: α-s1 [64], α-s2 [65], β [65], and κ [64], containing from 169 to 209 a.a. (without signal peptides). The structure of each subunit precursor includes signal peptide and the protein amino acid sequence proper. Amino acid composition of α-2s and κ subunits includes all 20 standard amino acid residues, while no cysteine residue is present in subunits α-s1 and β.
Computer fragmentation of all subunits and analysis of so obtained fragments have shown that many small fragments are repeated both in a separate and in different casein subunits. Tens of different fragments were repeated. Most frequent (i = 13) were the shortest (dipeptide) EE fragments in three subunits α-s1, α-s2, and β. Next by frequency were fragments PF (one in α-s1 subunit and six in β subunit), QS (six in subunit β) as well as PT (one in α-s2, one in β, and four in κ-subunits). The largest repetitive structures were heptapeptide fragments SSSEESI that are present twice in β subunit.Fig. 3. Amino acid sequences of precursors of bovine casein subunits. Total number of amino acid residues is given after the subunit name. Signal peptides are shown in italics and are underlined. Some repeated structures are shown in bold (explanations are in the text).

Table 2 shows the results of computer analysis of real content of dipeptide and all fragments compared to corresponding values obtained using formulas (2) and (3) for fragmentomes of separate subunits and for the complete casein fragmentome. In both cases, real values were below the calculated ones due to the presence of repeated structure. Thus, in the α-s1 subunit the number of different repetitive dipeptide fragments (with the number of repeats from 1 to 4) was equal to 64 just due to which N2theor and N2exp are equal to 198 and 134, respectively. It should also be noted that values characterizing the complete casein fragmentome are not the sum of real values for subunits, because structure recurrence appears not only within subunits, but in different subunits as well. Taking this into consideration in the case of different dipeptide fragments of the complete casein fragmentome, N2exp was almost two times lower than the sum of N2exp obtained for all subunits (266 instead of 507).
Table 2. Comparison of real number of
different dipeptide (k = 2) fragments
N2exp and all (at all k)
Nsumexp fragments with
N2theor and
Nsumtheor, respectively, in bovine
casein
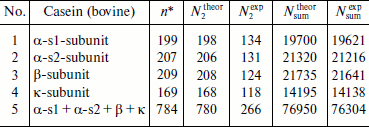
*Number of amino acid residues in complete structure.
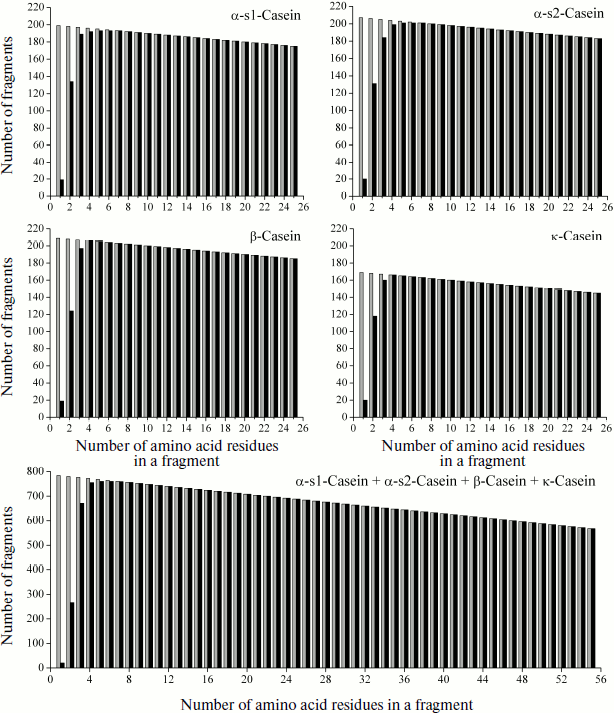
Length distribution of casein fragments is shown in Fig. 4 as a graph of all initial regions of the fragmentome of four subunits (in equal scale) and of the complete casein fragmentome. It is seen how real and theoretical values of the fragment number correlate with each other and at which fragment length (k) the Nktheor and Nkexp values become identical. It is also seen that dimensions of repetitive fragments of the considered protein are relatively low.
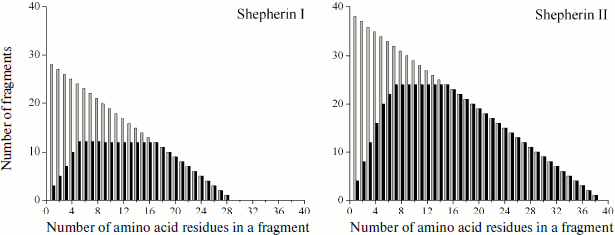
However, there are natural peptide structures composed of a few different amino acid residues, due to which their repeating fragments can be significantly longer. An example of this are highly homologous antimicrobial oligopeptides Shepherins from roots of officinal plant Shepherd’s purse Capsella bursa-pastoris [66], formed by only three and four different amino acid residues and only of 28 and 38 residues in length:Fig. 4. Graphic representation of fragmentomes of different bovine casein subunits and its complete protein molecule. Theoretical values of Nktheor are shown in gray, experimental Nkexp are in black. In addition to peptide structures, data on “monopeptides” are also shown (k = 1).
![]()
Fig. 5. Graphic representation of fragmentomes of natural antimicrobial oligopeptides Shepherins I and II.
FUNCTIONAL CHARACTERISTICS OF FRAGMENTOME
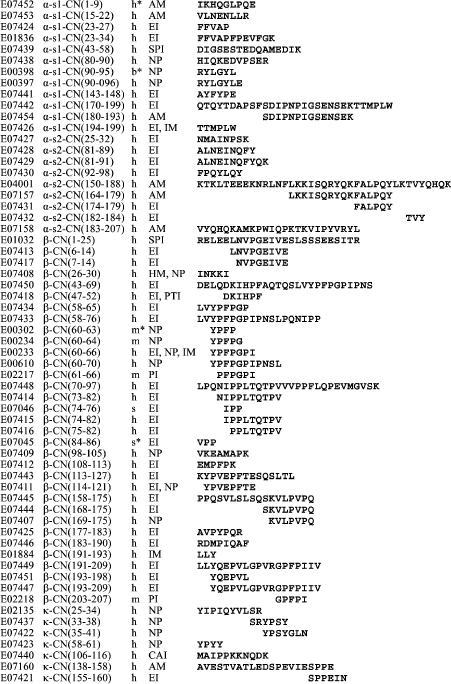
It can be supposed that the variety of fragmentome structures is the basis for variety of the fragment functions. In the case of casein, this can be seen in Fig. 6 showing structures of its 60 fragments obtained experimentally by different researchers and included in the EROP-Moscow database together with functional characteristics. Some of these fragments can be considered as natural because they were isolated either from a cow’s body or from her milk. However, in most cases fragments were obtained by artificial proteolysis. Certainly, these fragments comprise just a small part of the casein fragmentome (~0.1%) but a high variety is also observed in their functions. Most of these fragments are enzyme inhibitors (angiotensin-converting enzyme inhibitor and cathepsin). The most representative are fragments of β-subunit. A special group of four fragments formed of this subunit region (60-70) was named casomorphins according to their opioid activity [67-69]. In the same subunit, the function of enzyme inhibitors was detected in four fragments of the region 43-66 (see database EROP-Moscow under identification numbers shown in the figure).
These data can be supplemented with results of investigations of a number of synthetic casein fragments. As shown in Fig. 7, all 12 synthesized fragments representing the 48-61 region of β-subunit have enzyme inhibitor function (Fig. 7) [70], like the experimentally obtained fragments 47-52 and 58-65 belonging to the same region of the same subunit (Fig. 6). In addition to the above-mentioned functions, fragments of different casein subunits exhibit antimicrobial activity, properties of hormones and other important peptide regulators, and more than one type of activity were registered for some.Fig. 6. Experimentally obtained casein (CN) fragments. Identification numbers from EROP-Moscow database are given consecutively, including numbers of the first and last residue in the subunit (without regard for the signal peptide), sources, functional properties, and primary structures. The following abbreviations are used for functional properties: AM, antimicrobial; CAI, cell aggregation inhibitor; EI, enzyme inhibitor; HM, hormone; IM, immunomodulator; NP, neuropeptide; PI, protein inhibitor; PTI, protein transport inhibitor; SPI, salt precipitation inhibitor.
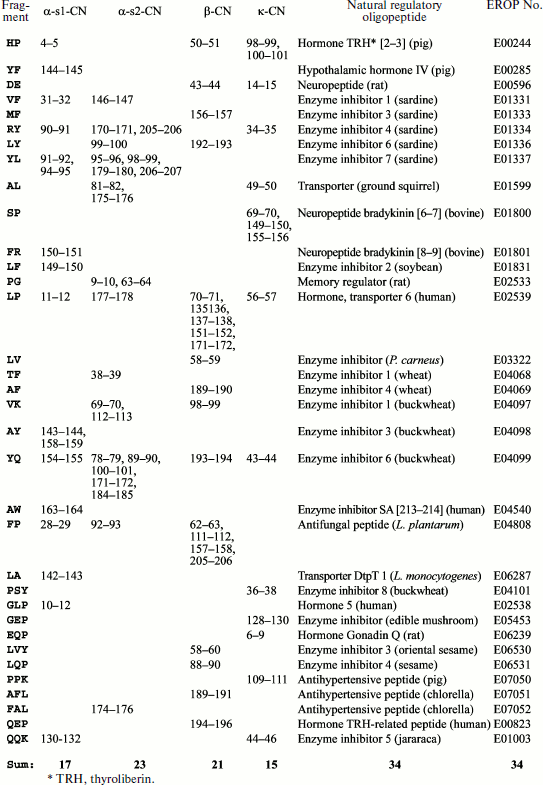
Computer analysis allows detection of functional properties in different fragments. We have performed computer comparison of all casein fragments with all known functionally characterized oligopeptides included in EROP-Moscow database. It appeared that (Fig. 8) 22 different dipeptide and 12 tripeptide casein fragments were fully identical to natural non-casein oligopeptides obtained from different kingdoms of living organisms (animals, plants, bacteria, and fungi). In total, they present 77 regions in all casein subunits and many of them are beyond the limits of amino acid sequences of experimentally obtained structures (Fig. 6). Spectrum of their functions is also diverse. Comparison of data shown in Figs. 6 and 8 in some cases confirms retention of functional properties after fragment shortening. Thus, fragment 31-32 (VF) of α-s1 subunit (Fig. 8) obtained from muscle of Sardinops melanostictus [71], is a part of fragment 23-34 of the same casein subunit (Fig. 6) and both exhibit function of enzyme inhibitor. However, fragment subdivision is accompanied by alteration of functional properties both in the case of fragment 34-35 (RY) of κ-subunit (Fig. 8) obtained from the same sardines [72] and of fragment 33-38 (Fig. 6) of the same casein subunit, for which functions of enzyme inhibitor and neuropeptide were found, respectively.Fig. 7. Angiotensin-converting enzyme inhibition by artificially synthesized bovine β-casein (β-CN) fragments. nd*, fragment 4 is insoluble at concentrations above 1 µM [70].

In addition to identical structures, numerous natural oligopeptides were also distinguished in the oligopeptide database whose structures were highly homologous to natural casein fragments and functions were identical. However, similar structures could have different functions.Fig. 8. Bovine casein (CN) fragments, structurally identical to functionally characterized natural oligopeptides obtained from different living organisms.
Due to the presence of proteolytic enzymes within cells and in extracellular medium of a living organism, continuous degradation of peptide structures takes place. Four hundred different types of peptide bonds are cleaved with different probability, and this can result in a continuously changing mosaic of numerous fragments of endogenous proteins. In a limiting case, formation of a complete fragmentome of each of them is possible. Formation of a complete fragmentome is quite probable during digestion, when exogenous proteins supplied with food, including those exhibiting enzymic activity, are cleaved by a large totality of enzymes [56]. Thus, exogenous fragments, among which regulatory oligopeptides can be present, are added to the pool of endogenous fragments. Due to partial repetition of amino acid sequences inside and outside protein subunit fragments as well as in different proteins, these regulators can be formed in significant amounts and noticeably influence different processes of metabolism. In particular, detection among them of enzyme inhibitors shows that the process of food protein cleavage can be inhibited by proteolysis products. Besides, fragments only just formed in the gastrointestinal tract and exhibiting antimicrobial properties are able to take part in regulation of the microflora balance and so to be a component of immune regulation [73]. Thus, fragmentation within an organism can result in generation of a dynamically developing pool of exogenous regulatory oligopeptides, functions of which can change during formation of smaller and smaller fragments. Probably the existence of the endogenous–exogenous pool of regulatory molecules makes wider the sense and content of the hypothesis concerning a functionally continuous totality (continuum) of natural oligopeptides [74].
CONCLUSION
A single protein fragmentome is only a part of the totality of fragmentomes of all cellular proteins. All are components of a global fragmentome formed by efforts of numerous researchers in the protein–peptide databases. This protein–peptide fragmentome, in turn, is presented as a component of the global fragmentome of all chemical substances of all living organisms, just which is an object of biochemical investigations.
By the present time only a small part of the possible natural protein fragments are identified experimentally and even less is known about their functional properties. In particular, this concerns generation and functions of fragments formed in the gastrointestinal tract upon nutritive product cleavage by enzymes (there are not many examples similar to investigation of the sardine muscle fragments [71], shown in Fig. 8). Detection of natural oligopeptides formed of specialized precursors is also far from completion. Moreover, only a single type function was studied in most of them, whereas they may be multifunctional.
Accumulation of data on structure and functions will make it possible to characterize more completely the functional abilities of numerous, still unstudied protein fragments, to approach understanding their role in evolution and to use all these data in practice. Thus, recording functional properties of nutritive protein fragments can suggest to dieticians what food is preferable for patients, and to pharmacologists what peptide fragments are reasonable to use as drugs and food additives. Some amino acids like glycine, exhibiting sedative effect [75-81], glutamate, widely used in food industry, etc. are already used as remedies.
Due to improvement of research methods, including computer analysis, our knowledge of structural and functional properties of the global fragmentome is intensively growing. This knowledge is still not enough for complete understanding of the regulatory role of fragments in living organisms. Nevertheless, already now it is possible to formulate ideas concerning structure–functional fragmentomics of natural peptides and other substances.
The author is indebted to B. I. Kurganov and A. G. Malygin for useful discussion of this work and to A. S. Borchikov for modifications of computer programs.
This work was supported by the Russian Academy of Sciences Presidium program “Molecular and Cell Biology” and by the Chilean National Research Foundation FONDECYT (grant No. 1080504).
REFERENCES
1.Malygin, A. G. (1999) Metabolism of Carboxylic
Acids (a periodical scheme), International Education Program,
Moscow.
2.Capper, N. S. (1930) Biochem. J., 24,
980-982.
3.Zamyatnin, A. A. (2008) Biophysics,
53, 329-355.
4.Zamyatnin, A. A. (2004) Biochemistry
(Moscow), 59, 1276-1282.
5Zamyatnin, A. A. (1984) Ann. Rev. Biophys. Bioeng., 13,
145-165.
6.Zamyatnin, A. A. (1991) Prot. Seq. Data
Anal., 4, 49-52.
7.Holowachuk, E. W., Stoltenborg, J. K., Reed, R. G.,
and Peters, T., Jr. (1991) Submitted (AUG-1991) to the
EMBL/GenBank/DDBJ databases.
8.Brown, J. R. (1974) Fed. Proc., 33,
1389-1389.
9.Shibahara, S., Morimoto, Y., Furutani, Y.,
Notake, M., Takahashi, H., Shimizu, S., Horikawa, S., and Numa, S.
(1983) EMBO J., 2, 775-779.
10.Romier, C., Bernassau, J.-M., Cambillau, C.,
and Darbon, H. (1993) Protein Eng., 6, 149-156.
11.Takahashi, H., Teranishi, Y., Nakanishi, S.,
and Numa, S. (1981) FEBS Lett., 135, 97-102.
12.Seidah, N. G., Rochemont, J., Hamelin, J.,
Lis, M., and Chretien, M. (1981) J. Biol. Chem., 256,
7977-7984.
13.Shibasaki, T., Ling, N., and Guillemin, R. (1980)
Biochem. Biophys. Res. Commun., 96, 1393-1399.
14.Seidah, N. G., Rochemont, J., Hamelin, J.,
Benjannet, S., and Chretien, M. (1981) Biochem. Biophys. Res.
Commun., 102, 710-716.
15.Harris, J. I., and Lerner, A. B. (1957)
Nature, 179, 1346-1347.
16.Li, C. H., and Chung, D. (1976) Nature,
260, 622-624.
17.Harris, J. I. (1959) Nature, 184,
167-169.
18.Dragon, N., Seidah, N. G., Lis, M., Routhie, R.,
and Chretien, M. (1977) Can. J. Biochem., 55,
666-670.
19.Ling, N., Burgus, R., and Guillemin, R. (1976)
Proc. Natl. Acad. Sci. USA, 73, 3942-3946.
20.Guillemin, R., Ling, N., and Burgus, R. (1976)
Compt. Rend. Acad. Sci., 282D, 783-785.
21.Loh, Y. P., Parish, D. C., and Tuteja, R. (1985)
J. Biol. Chem., 260, 7194-7205.
22.Hughes, J., Smyth, T. W., Kosterlitz, H. W.,
Fothergill, L. A., Morgan, B. A., and Morris, H. R. (1975)
Nature, 258, 577-579.
23.Noda, M., Teranishi, Y., Takahashi, H., Toyosato,
M., Notake, M., Nakanishi, S., and Numa, S. (1982) Nature,
297, 431-434.
24.Sheller, R. H., and Kirk, M. D. (1987) Trends
Neurosci., No. 10, 46-52.
25.Skeggs, L. T., Lentz, K. L., Kahn, J. R.,
Shumway, N. P., and Woods, K. R. (1956) J. Exp. Med.,
104, 193-197.
26.Tsai, B. S., Peach, M. J., Khoshla, M. C., and
Bumpus, F. M. (1975) J. Med. Chem., 18, 1180-1183.
27.Brazeau, P., Vale, W., Burgus, R., Ling, N.,
Butcher, M., Rivier, J., and Guillemin, R. (1973) Science,
179, 77-79.
28.Arakawa, Y., and Tachibana, S. (1984) Life
Sci., 35, 2529-2536.
29.Pradayrol, L., Jornvall, H., Mutt, V., and Ribet,
A. (1980) FEBS Lett., 109, 55-58.
30.Geller, D. M., Currie, M. G., Wakitani, K., Cole,
B. R., Adams, S. P., Fok, F. K., Siegel, N. R., Eubanks, S. R.,
Galluppi, G. R., and Needleman, P. (1984) Biochem. Biophys. Res.
Commun., 120, 333-338.
31.Iimura, O., Shimamoto, K., Ando, T., Ura, N.,
Ishida, H., Nakagawa, M., Yokoyama, T., Fukuyama, S., Yamaguchi, Y.,
and Yamaji, I. (1987) Can. J. Physiol. Pharm., 65,
1701-1705.
32.Lee, T. H., Lerner, A. B., and Buettner-Janusch,
V. (1961) J. Biol. Chem., 236, 2970-2974.
33.Graf, L., Bajusz, S., Patty, A., Barat, E., and
Cseh, G. (1971) Acta Biochim. Biophys. Acad. Sci. Hung.,
6, 415-418.
34.Takagi, H., Shiomi, H., Ueda, H., and Amano, H.
(1979) Nature, 282, 410-412.
35.Takagi, H., Shiomi, H., Fukui, K., Hayashi, K.,
Kiso, Y., and Kitagawa, K. (1982) Life Sci., 31,
1733-1736.
36.Barkhudaryan, N., Kellerman, I., Lottspeich, F.,
and Galoyan, A. A. (1991) Neirokhimiya, 10, 146-154.
37.Ivanov, V. T., Karelin, A. A., Mikhaleva, I. I.,
Vas’kovskii, B. V., Sviryaev, V. I., and Nazimov, I. V. (1992)
Bioorg. Khim., 18, 1271-1311.
38.Benson, B., and Ebels, L. (1993) Life
Sci., 54, PL437-PL443.
39.Ekeke, N. U., Shaw, C., Johnston, C. F., and
Thim, L. (1992) Regul. Peptides, 40, 140-140.
40.Nakamura, Y., Yamamoto, N., Sakai, K., and
Takano, T. (1995) J. Dairy Sci., 78, 1253-1257.
41.Kenrick, K. G., and Margolis, J. (1970) Anal.
Biochem., 33, 204-207.
42.Malygin, A. G. (1993) Uspekhi Biol. Khim.,
33, 173-213.
43.Chapman, J. R. (ed.) (1996) Protein and
Peptide Analysis by Mass-Spectrometry, Humana Press.
44.Wilkins, M. R., Williams, K. L., Appel, R. D.,
and Hochstrasser, D. F. (eds.) (1997) Proteome Research: New
Frontiers in Functional Genome, Springer, Berlin.
45.Edman, P. (1949) Arch. Biochem. Biophys.,
22, 475-476.
46.Edman, P. (1970) in Protein Sequence
Determination (Needleman, S. B., ed.) Springer, Berlin, pp.
211-215.
47.Tuppy, H. (1953) Biochim. Biophys. Acta,
11, 449-450.
48.Du Vigneaud, V., Lowler, H. C., and
Popenoe, E. A. (1953) J. Am. Chem. Soc., 75,
4880-4881.
49.Ryle, A. P., Sanger, F., Smith, L. F., and Kitai,
R. (1955) Biochem. J., 60, 541-556.
50.Hruby, V. J., Wilkes, B. C., Hadley, M. E., Al-Obeidi, F.,
Sawyer, T. K., Staples, D. J., de Vaux, A. E., Dym, O., Castrucci, A.
M., Hintz, M. F., Riehm, J. P., and Rao, K. R. (1987) J. Med.
Chem., 30, 2126-2130.
51.Suzuki, K., Abiko, T., and Endo, N. (1969)
Chem. Pharm. Bull., 17, 1671-1678.
52.Erdos, E. G. (ed.) (1970) Bradykinin,
Kallidin, Kallikrein, Springer-Verlag, Berlin-Heidelberg-New
York.
53.Nazipova, N. N., Shabalina, S. A., Ogurtsov, A.
Yu., Kondrashov, A. S., Roytberg, M. A., Buryakov, G. V., and
Vernoslov, S. E. (1995) Comput. Appl. Biosci.,
11, 423-426.
54.Panchenko, A. R., and Bryant, S. H. (2002)
Protein Sci., 11, 361-370.
55.Huska, M. R., Buschmann, H., and
Andrade-Navarro, M. A. (2007) Bioinformatics,
23, 3093-3094.
56.Zamyatnin, A. A. (2007) Neurochem. J.,
1, 188-195.
57.International Human Genome Sequencing
Consortium (2004) Nature, 431, 931-945.
58.Orcutt, B. C., and Barker, W. C. (1984)
Bull. Math. Biol., 46, 545-552.
59.Elliott, D. F., Lewis G. P., and Horton, E. W.
(1960) Biochem. Biophys. Res. Commun., 3, 87-91.
60.Braunitzer, G., Gehring-Muller, R., Hilschmann,
N., Hilse, K., Hobom, G., Rudloff, V., and Wittmann-Liebold, B. (1961)
Hoppe-Seyler’s Z. Physiol. Chem., 325, 283-286.
61.Wicker, C. L., and Wicker, C. A. (1987) Comp.
Biochem. Physiol., 88C, 185-187.
62.Zamyatnin, A. A., Borchikov, A. S., Vladimirov,
M. G., and Voronina, O. L. (2006) Nucleic Acids Res., 34,
D261-D266.
63.Zamyatnin, A. A. (2009) Biochemistry
(Moscow), 74, 201-208.
64.Nagao, M., Maki, M., Sasaki, R., and Chiba, R.
(1984) Agric. Biol. Chem., 48, 1663-1667.
65.Stewart, A. F., Bonsing, J., Beattie, C. W.,
Shah, F., Willis, I. M., and Mackinlay, A. G. (1987) Mol. Biol.
Evol., 4, 231-241.
66.Park, C. J., Park, C. B., Hong, S.-S., Lee,
H.-S., Lee, S. Y., and Kim, S. C. (2000) Plant Mol. Biol.,
44, 187-197.
67.Henschen, A., Lottspeich, F., Brantl, V., and
Teschemacher, H. (1979) Hoppe-Seyler’s Z. Physiol. Chem.,
360, 1217-1224.
68.Meisel, H. (1986) FEBS Lett., 196,
223-227.
69.Brantl, V., Teschemacher, H., Blasig, J.,
Henschen, A., and Lottspeich, F. (1981) Life Sci., 28,
1903-1909.
70.Kohmura, M., Nio, N., and Ariyoshi, Y. (1990)
Agric. Biol. Chem., 54, 1101-1102.
71.Matsuda, H., Ishizaki, T., Morita, H., Nagaoka,
T., Osajima, K., and Osajima, Y. (1992) J. Jap. Soc. Biosci.
Biotechnol. Agrochem., 66, 1645-1647.
72.Matsufuji, H., Matsui, T., Seki, E., Osajima, K.,
Nakashima, M., and Osajima, Y. (1994) Biosci. Biotechnol.
Biochem., 58, 2244-2245.
73.Zamyatnin, A. A., and Voronina, O. L. (1998)
Uspekhi Biol. Khim., 38, 165-197.
74.Ashmarin, I. P., and Obukhova, M. F. (1986)
Biokhimiya, 51, 531-544.
75.Asechi, M., Kurauchi, I., Tomonaga, S., Yamane,
H., Suenaga, R., Tsuneyoshi, Y., Denbow, D. M., and Furuse, M.
(2008) Amino Acids, 34, 55-60.
76.Dolu, N. (2007) J. Basic Clin. Physiol.
Pharmacol., 18, 141-147.
77.Chojnacka-Wojcik, E., Tatarczynska, E., and
Deren-Wesołek, A. (1996) Pol. J. Pharmacol., 48,
627-629.
78.Javitt, D. C., Silipo, G., Cienfuegos, A.,
Shelley, A. M., Bark, N., Park, M., Lindenmayer, J. P., Suckow, R., and
Zukin, S. R. (2001) Int. J. Neuropsychopharmacol.,
4, 385-391.
79.File, S. E., Fluck, E., and Fernandes, C. (1999)
J. Clin. Psychopharmacol., 19, 506-512.
80.Ohno, M., Yamamoto, T., and Watanabe, S. (1994)
Eur. J. Pharmacol., 253, 183-187.
81.Schwartz, B. L., Hashtroudi, S., Herting, R. L.,
Handerson, H., and Deutsch, S. I. (1991) Neurology, 41,
1341-1343.