YB-1 Is Capable of Forming Extended Nanofibrils
O. M. Selivanova, S. G. Guryanov, G. A. Enin, M. A. Skabkin, L. P. Ovchinnikov, and I. N. Serdyuk*
Institute of Protein Research, Russian Academy of Sciences, 142290 Pushchino, Moscow Region, Russia; E-mail: serdyuk@vega.protres.ru* To whom correspondence should be addressed.
Received December 4, 2009
Here we are the first to report that multifunctional Y-box binding protein 1 (YB-1) forms extended fibrils with a diameter of 15-20 nm. The YB-1 fibrils were visualized by atomic force and electron microscopy after 1-h incubation in solution with 2 M LiCl. Their length grew with incubation time and could exceed 10 µm; their shape is helical or zigzag-like. They possess polarity and tend to associate with one another to give structures of a higher order, like ribbons or bundles. The YB-1 fibrillar architecture has a distinct periodicity with a repeat unit of about 52 nm.
KEY WORDS: YB-1, protein fibrils, atomic force microscopy, electron microscopy, high ionic strengthDOI: 10.1134/S0006297910010153
Abbreviations: AFM, atomic force microscopy; CSD, cold shock domain; EM, electron microscopy; HOPG, highly ordered pyrolytic graphite; YB-1, Y-box binding protein 1.
Y-box binding protein 1 (YB-1) is a mammalian multifunctional DNA- and
RNA-binding protein localized both in the nucleus and the cytoplasm and
involved in many cellular events such as storing, reproduction, and
expression of genetic information [1-3]. In the cell, YB-1 can interact with actin [4] and tubulin cytoskeletons [5].
Besides, it can be actively secreted from cells through a non-classical
pathway, bind to Notch-3 receptors, modulate receptor activation, and
act as an extracellular mitogen [6, 7].
Human YB-1 with a molecular mass of 36 kDa (324 residues) consists of three domains: a small Ala-, Gly-, and Pro-rich N-terminal domain (N-domain), a central cold shock domain (CSD), and a large C-terminal domain (C-domain) formed by alternating clusters of positively and negatively charged residues. Each cluster of the C-domain comprises about 20-30 amino acid residues; there are four clusters of either charge [8]. CSD is an extremely conserved domain. Bacterial proteins consisting solely of this domain are known as major cold shock proteins because low temperature triggers enhanced synthesis of some of them to provide adaptation and growth of bacteria under these conditions [9]. Bacterial cold shock proteins and YB-1 CSD have identical β-barrel spatial structure [10].
YB-1 is rich in proline, glutamine, arginine, and glycine; the first two of these amino acids are known to contribute much to protein disordering in solution [11]. Structural predictions using various algorithms also showed N- and C-domains of YB-1 as disordered [12, 13]. Thus, YB-1 falls in the category of natively disordered proteins.
Previously, it was shown that purified YB-1 in solution, as well as YB-1 in association with RNA at a high YB-1/mRNA ratio, formed multimers up to 800 kDa [14, 15]. According to electron microscopy (EM) and atomic force microscopy (AFM), these multimers are 9-10 nm high, and their diameter is 35-40 nm [16]. The current study revealed that in buffer with 2 M LiCl, commonly used to remove RNA from the protein [17], YB-1 formed vastly extended fibrils. Below, we describe these fibrils.
MATERIALS AND METHODS
YB-1 purification and formation of fibrils. Recombinant YB-1 was synthesized from the plasmid pET-3-1-YB-1 in Escherichia coli and purified as described previously [18] with minor modifications. In brief, the bacterial cells were disrupted by ultrasonication in 20 mM Tris-HCl, pH 7.6, 2 M NaCl, 0.5 mM phenylmethylsulfonyl fluoride. Cell debris and ribosomes were removed by consecutive centrifugation. The supernatant was diluted with three volumes of 20 mM Tris-HCl, pH 7.6, and loaded onto a Heparin-Sepharose column (GE Healthcare, Sweden). YB-1 was eluted with 20 mM Tris-HCl, pH 7.6, 2 M NaCl, dialyzed against 20 mM Hepes-KOH, pH 7.6, 500 mM KCl, and loaded onto a Mono S 4.6/100 PE column (GE Healthcare). The protein was eluted by a 0.5-2 M KCl gradient and finally purified by gel filtration on a Superose 12 10/300 GL column (GE Healthcare) equilibrated with 20 mM Hepes-KOH, pH 7.6, 1 M KCl. Fractions containing pure YB-1 were concentrated and dialyzed against 20 mM potassium-phosphate buffer, pH 7.4, 150 mM KCl. The OD280/OD260 ratio was 2, thus showing the absence of nucleic acids in the protein sample. Protein concentration was determined from absorption at 280 nm, with the extinction coefficient taken as 1.37 mg·ml–1·cm–1.
To obtain YB-1 fibrils, samples containing 0.1 mg/ml YB-1, 20 mM Hepes-KOH, pH 7.6, 2 M LiCl were incubated at 30°C for 15 min and then at 4°C for indicated time periods.
Atomic force microscopy. For AFM studies, 5 µl of YB-1 in 20 mM Hepes-KOH, pH 7.6, 2 M LiCl was applied onto either freshly cleaved mica or highly ordered pyrolytic graphite (HOPG) and left for 15 min (mica) or 5 min (HOPG) in a humid chamber. The substrate-deposited samples were washed twice with bidistilled water and air-dried. The AFM analysis was made using a NTEGRA VITA scanning probe microscope (NT-MDT Company, Russia) with a standard NSG 03 (NT-MDT) silicon cantilever. The resonance frequency of the cantilever was 62-123 kHz, and its length was 100 µm. Tips with an apex curvature radius of 10 nm were used. The samples were probed in semi-contact mode in air.
Electron microscopy. After incubation in 20 mM Hepes-KOH, pH 7.6, 2 M LiCl, YB-1 samples were adsorbed onto copper grids coated with 0.5% formvar in chloroform for 2 min, stained with 1% uranyl acetate (aqueous solution) for 1 min, and examined using a JEM-100C microscope operating at 80 kV.
RESULTS
YB-1 forms fibrils. According to the AFM analysis, YB-1 incubation in 20 mM Hepes-KOH, pH 7.6, 2 M LiCl results in fibril formation. AFM images of these fibrils are presented in Fig. 1. As seen, the length of the fibrils increased from 0.5-1.5 µm (24 h incubation; Figs. 1a and 1b) to 3-6 µm (48 h incubation; Figs. 1c and 1d).
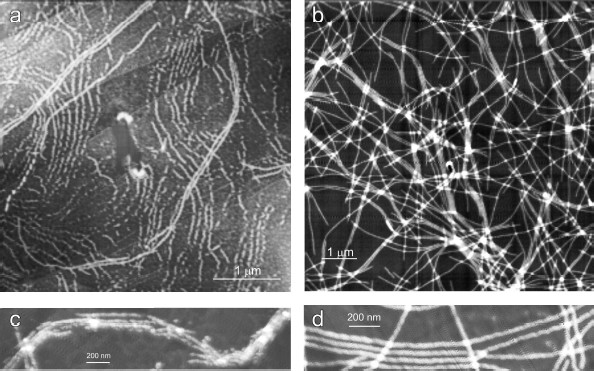
After 30 days of incubation of YB-1 under conditions of high ionic strength, the length of the fibrils exceeded 10 µm (Fig. 2). Magnified images (Figs. 2c and 2d) show that YB-1 fibrils tend to associate with one another to give structures of a higher order, like ribbons or bundles.Fig. 1. AFM images of YB-1 fibrils. Mica-deposited samples after 24-h incubation (a, b) and HOPG-deposited samples after 48-h incubation in 20 mM Hepes-KOH, pH 7.6, 2 M LiCl (c, d).
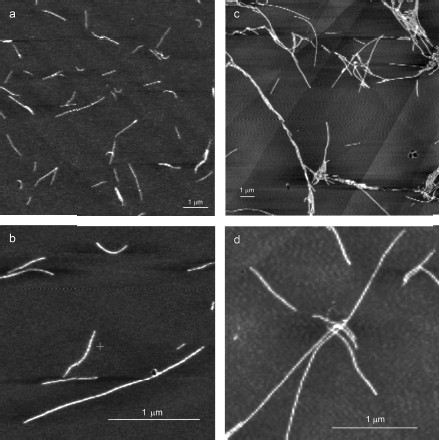
As follows from all these AFM images, there is a distinct periodicity in the YB-1 fibrillar architecture. On both graphite and mica as substrates, the average repeat unit was 50-52 nm, and the fibril diameter was about 20 nm.Fig. 2. AFM images of YB-1 fibrils after 30-day incubation in 20 mM Hepes-KOH, pH 7.6, 2 M LiCl. HOPG- (a, c) and mica-deposited (b, d) samples.
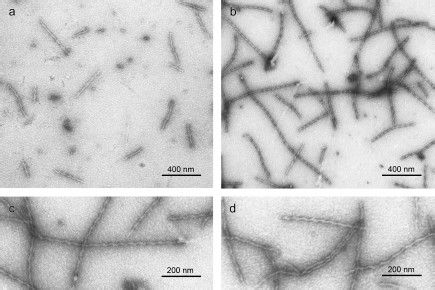
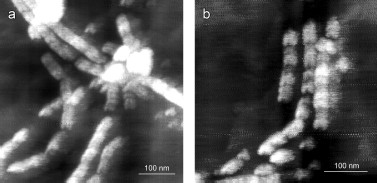
We also applied EM to study YB-1 fibrils. As soon as after 1 h incubation of YB-1 in 20 mM Hepes-KOH, pH 7.6, 2 M LiCl, EM revealed 1 µm long fibrils (Fig. 3a), as well as small aggregates. After 24 h incubation, the fibril length increased to 1.5 µm, with concurrent decrease in the number of small aggregates (Fig. 3b). The fibril diameter was about 15 nm.
Peculiarities of YB-1 fibrillar architecture. The magnified EM images (Figs. 3c and 3d) of YB-1 fibrils after 24 h incubation clearly demonstrate their helical or zigzag-like shape. Like in AFM images, the repeat unit is seen to be about 50-52 nm.Fig. 3. EM images of negatively stained YB-1 after 1-h (a) and 24-h (b) incubation in 20 mM Hepes-KOH, pH 7.6, 2 M LiCl. Magnified images (c, d) show YB-1 after 24-h incubation.
According to EM, the fibrils are polar, because there is a difference between their ends (Figs. 4a and 4b): one end (“closed”) is much better stained with uranyl acetate than the other (“open”).
The magnified AFM images (Fig. 5) show some details of the YB-1 fibrillar architecture. As seen, the filaments comprise separate asymmetric units of 20 × 25 nm each.Fig. 4. EM images of terminal fragments of YB-1 fibrils: “closed (a) and “open” (b) fibril ends.
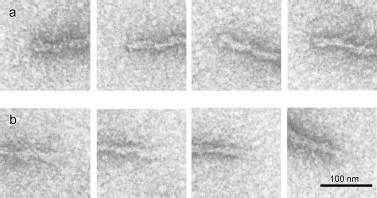
Fig. 5. Magnified AFM images of HOPG-deposited YB-1 fibrils demonstrating their modular structure (a, b).
DISCUSSION
This paper is the first report that in solution YB-1 can form extended fibrils with a length amounting to >10 µm. Almost as soon as the ionic strength is increased to 2 M LiCl, YB-1 polymerization begins to give particles of various size seen on EM images (Fig. 3a). With time, small protofibrils virtually disappear to give place to fibrils reaching 10 µm in length (Fig. 2). Note that the formation of YB-1 multimers is a two-step process: first, they mainly gain length, and then, they get drawn up “in parallel” with one another. This means that their “head-tail” interactions are much more pronounced than “side-by-side” ones.
The helical or zigzag-like shape of the fibrils (with a repeat unit of 50-52 nm), clearly seen on EM images, is shown by AFM as a finer structure composed by separate asymmetric 20 × 25 nm units (Figs. 5a and 5b).
According to EM, the fibrils display distinct polarity, since there is seen a difference between their ends. One of these, presumably growth initiating, shows much better staining with uranyl acetate, while the other, probably length gaining, demonstrates a lower staining ability (Fig. 4).
Biological reasons for and consequences of the fibril-forming capability of YB-1 are at the interface between two sets of available data. One of these is connected with multifunctionality of the protein, i.e. its ability to interact with many cellular partners, which has been widely reported in the literature [1, 19]. The other deals with protein fibrils and their role in the cell’s life, which has never been discussed as applied to YB-1.
Subcellular fibrils are known to be vital for the cell’s existence. For example, elements of the cytoskeleton, such as microfilaments (made of actin), microtubules (made of tubulins), and intermediate filaments (made of proteins that differ from tissue to tissue), are responsible for a number of basic cell functions, namely, the cell shape and movement, as well as subcellular compartmentalization and transport of large cell particles and organelles. Apart from these, in the cell there can appear the so-called amyloid fibrils that embarrass the cell’s life and provoke a number of diseases [20]. Amyloid fibrils are insoluble aggregates that originate from some soluble proteins. Besides, quite a number of structurally different proteins can in vitro undergo polymerization to give the so-called amyloid-like insoluble fibrils. Although such polymerization usually requires conditions other than physiological ones, two mechanisms sufficiently well describing the process of appearing of amyloid-like fibrils (refolding and gain-of-interactions) have been proposed [21].
According to the latest reports, there are amyloids unrelated to diseases. These are called functional amyloids [20]. A well-studied example of a functional amyloid is the protein curlin from E. coli used by it to colonize inert surfaces and mediate binding to host proteins [22]. A second example involves amyloid fibrils formed in aerial hyphae of the filamentous bacterium Streptomyces coelicolor from proteins secreted by this bacterium and called chaplins [23]. As well as in bacteria, the formation of similar fibrils was observed in animal cells. Specifically, the protein Pmel17 responsible for the epidermal production of the melanin pigment has been reported to form a functional amyloid [24]. In secretory granules of the endocrine system, a number of peptide and protein hormones are kept for a long time in the form of amyloid-like structures without being toxic for the organism [25]. Consequently, living systems can use the amyloid structure as a functional state of some proteins. In its turn, this means that within a functionally normal system proteins can exist in either soluble or aggregated amyloid-like form.
As far as we know, YB-1 can hardly be assigned to either of the above groups. It belongs to natively disordered proteins that are either fully disordered or contain large disordered sequences. This type is represented by prion proteins Ure2p [26], Sup35p [27], and Rnq1p [28] from Saccharomyces cerevisiae, as well as HET-s from Podospora anserine [29], an unfolded β-amyloid polypeptide (42 amino acid residues) responsible for Alzheimer’s disease, and α-synuclein (140 residues) responsible for Parkinson’s disease and other forms of senile dementia [20]. These proteins and YB-1 have the following common features, although their functions are completely different.
First, the domain composition: in prions, there are “prion” and “functional” domains [30]; in YB-1, there are the N-domain, CSD, and the C-domain. It is reasonable to suggest that one of the YB-1 domains can be a “prion” (“functional”) one, although which of them can be believed to act as such is still an open question.
Second, polarity: prion filaments are always polar, and YB-1 fibrils possess polarity as well.
Third, the amino acid composition: the prion domains Ure2p, Sup35p, and Rnq1p are extremely rich in asparagine and glutamine and have a relatively low level of charged and hydrophobic residues, and YB-1 is very rich in glutamine too.
Fourth, the fibril shape: as seen in microphotos, prion protofibrils are surrounded by globular domains. These are asymmetric relative to the fibril axis and form precessing projections that make the filament zigzag-like in shape [30]. Here we report that YB-1 fibrils are helical or zigzag-like in shape with a repeat unit of 50-52 nm, and have a finer architecture shown by AFM and EM microscopy.
Lastly, the key factor of prion aggregation is their charge. A decreased charge increases the rate of fibril formation [31]. Sometimes, this results from interaction with molecules possessing a compensating charge [32, 33]. Although a partial neutralization of the negative charge can stimulate aggregating at the purely electrostatic level, α-synuclein containing paired clusters of the opposite charges undergoes aggregation much faster, provided its NAC-sequence (functional sequence) is unprotected [34]. Similarly, the C-domain of YB-1 comprises alternating clusters of positively and negatively charged amino acids. Supposedly, the role of 2 M LiCl buffer consists in shielding these clusters.
As it follows from the above, there are some common features in the mechanisms of fibril formation by all natively disordered proteins [35], and hence, the available information on this group of proteins can be most helpful in studying YB-1. Presently, it is necessary to understand (i) the mechanism of YB-1 fibril formation and (ii) the relationship between the reported here ability of YB-1 to form extended fibrils and its canonic function of interaction with many cellular partners. Experimental studies in these directions can serve as a good basis for revision of the present concept of relationship between protein aggregation and protein functions.
In spite of numerous differences in structural details of amyloid and amyloid-like fibrils reported in the literature, they share a number of common well-established features, among which the most distinguished are growth in one direction, an unbranched shape, peculiar staining with Congo red and thioflavin T, and availability of the cross-β-structure that manifests itself through specific fibril-typical diffraction [21]. So far, we have provided evidence that two of these, the growth in one direction and the unbranched shape, are true for YB-1.
The available data on YB-1 fibril formation hardly allow proposing a mechanism of this process, since neither the role of the disordered N- and C-ends of YB-1 (3/4 of the molecule length), nor that of its cold chock domain is known. A certain contribution can be made by studies of mutants containing various YB-1 fragments and temperature dependence of the fibril formation process, as well as polymerization-induced changes in the YB-1 secondary structure. It is these problems that are currently in the focus of our attention.
The authors thank A. Machulin for technical assistance in EM and AFM measurements.
This work was supported in part by grants of the program on “Molecular and Cellular Biology” and the program on “Basic Sciences in Medicine” from the Russian Academy of Sciences (SGG and LPO).
REFERENCES
1.Kohno, K., Izumi, H., Uchiumi, T., Ashizuka, M.,
and Kuwano, M. (2003) Bioessays, 25, 691-698.
2.Skabkin, M. A., Lyabin, D. N., and Ovchinnikov, L.
P. (2006) Mol. Biol. (Moscow), 40, 551-563.
3.Evdokimova, V., Ovchinnikov, L. P., and Sorensen,
P. H. (2006) Cell Cycle, 5, 1143-1147.
4.Ruzanov, P. V., Evdokimova, V. M., Korneeva, N. L.,
Hershey, J. W., and Ovchinnikov, L. P. (1999) J. Cell Sci.,
112 (Pt. 20), 3487-3496.
5.Chernov, K. G., Mechulam, A., Popova, N. V.,
Pastre, D., Nadezhdina, E. S., Skabkina, O. V., Shanina, N. A.,
Vasiliev, V. D., Tarrade, A., Melki, J., Joshi, V., Baconnais, S.,
Toma, F., Ovchinnikov, L. P., and Curmi, P. A. (2008) BMC
Biochem., 9, 23.
6.Frye, B. C., Halfter, S., Djudjaj, S., Muehlenberg,
P., Weber, S., Raffetseder, U., En-Nia, A., Knott, H., Baron, J. M.,
Dooley, S., Bernhagen, J., and Mertens, P. R. (2009) EMBO Rep.,
10, 783-789.
7.Rauen, T., Raffetseder, U., Frye, B. C., Djudjaj,
S., Muehlenberg, P. J., Eitner, F., Lendahl, U., Bernhagen, J., Dooley,
S., and Mertens, P. R. (2009) J. Biol. Chem., 284,
26928-26940.
8.Skabkin, M. A., Skabkina, O. V., and Ovchinnikov,
L. P. (2004) Usp. Biol. Khim., 44, 3-52.
9.Yamanaka, K., Fang, L., and Inouye, M. (1998)
Mol. Microbiol., 27, 247-255.
10.Kloks, C. P., Spronk, C. A., Lasonder, E.,
Hoffmann, A., Vuister, G. W., Grzesiek, S., and Hilbers, C. W. (2002)
J. Mol. Biol., 316, 317-326.
11.Serdyuk, I. N. (2007) Mol. Biol. (Moscow),
41, 262-277.
12.Tompa, P., and Csermely, P. (2004) FASEB
J., 18, 1169-1175.
13.Obradovic, Z., Peng, K., Vucetic, S., Radivojac,
P., and Dunker, A. K. (2005) Proteins, 61 (Suppl. 7),
176-182.
14.Evdokimova, V. M., Wei, C. L., Sitikov, A. S.,
Simonenko, P. N., Lazarev, O. A., Vasilenko, K. S., Ustinov, V. A.,
Hershey, J. W., and Ovchinnikov, L. P. (1995) J. Biol. Chem.,
270, 3186-3192.
15.Tafuri, S. R., and Wolffe, A. P. (1992) New
Biol., 4, 349-359.
16.Skabkin, M. A., Kiselyova, O. I., Chernov, K. G.,
Sorokin, A. V., Dubrovin, E. V., Yaminsky, I. V., Vasiliev, V. D., and
Ovchinnikov, L. P. (2004) Nucleic Acids Res., 32,
5621-5635.
17.Littlechild, J. A., and Malcolm, A. L. (1978)
Biochemistry, 17, 3363-3369.
18.Evdokimova, V. M., Kovrigina, E. A., Nashchekin,
D. V., Davydova, E. K., Hershey, J. W., and Ovchinnikov, L. P. (1998)
J. Biol. Chem., 273, 3574-3581.
19.Sorokin, A. V., Selyutina, A. A., Skabkin, M. A.,
Guryanov, S. G., Nazimov, I. V., Richard, C., Th’ng, J., Yau, J.,
Sorensen, P. H., Ovchinnikov, L. P., and Evdokimova, V. (2005) EMBO
J., 24, 3602-3612.
20.Chiti, F., and Dobson, C. M. (2006) Annu. Rev.
Biochem., 75, 333-366.
21.Nelson, R., and Eisenberg, D. (2006) Adv.
Protein Chem., 73, 235-282.
22.Chapman, M. R., Robinson, L. S., Pinkner, J. S.,
Roth, R., Heuser, J., Hammar, M., Normark, S., and Hultgren, S. J.
(2002) Science, 295, 851-855.
23.Claessen, D., Rink, R., de Jong, W., Siebring,
J., de Vreugd, P., Boersma, F. G., Dijkhuizen, L., and Wosten, H. A.
(2003) Genes Dev., 17, 1714-1726.
24.Berson, J. F., Theos, A. C., Harper, D. C.,
Tenza, D., Raposo, G., and Marks, M. S. (2003) J. Cell Biol.,
161, 521-533.
25.Maji, S. K., Perrin, M. H., Sawaya, M. R.,
Jessberger, S., Vadodaria, K., Rissman, R. A., Singru, P. S., Nilsson,
K. P., Simon, R., Schubert, D., Eisenberg, D., Rivier, J., Sawchenko,
P., Vale, W., and Riek, R. (2009) Science, 325,
328-332.
26.Chien, P., Weissman, J. S., and DePace, A. H.
(2004) Annu. Rev. Biochem., 73, 617-656.
27.Eaglestone, S. S., Cox, B. S., and Tuite, M. F.
(1999) EMBO J., 18, 1974-1981.
28.Giaever, G., Chu, A. M., Ni, L., Connelly, C.,
Riles, L., Veronneau, S., Dow, S., Lucau-Danila, A., Anderson, K.,
Andre, B., Arkin, A. P., Astromoff, A., El-Bakkoury, M., Bangham, R.,
Benito, R., et al. (2002) Nature, 418, 387-391.
29.Coustou, V., Deleu, C., Saupe, S., and Begueret,
J. (1997) Proc. Natl. Acad. Sci. USA, 94, 9773-9778.
30.Baxa, U., Taylor, K. L., Wall, J. S., Simon, M.
N., Cheng, N., Wickner, R. B., and Steven, A. C. (2003) J. Biol.
Chem., 278, 43717-43727.
31.Fandrich, M., and Dobson, C. M. (2002) EMBO
J., 21, 5682-5690.
32.Giasson, B. I., Lee, V. M., and Trojanowski, J.
Q. (2003) Neuromol. Med., 4, 49-58.
33.Konno, T. (2001) Biochemistry, 40,
2148-2154.
34.Hoyer, W., Cherny, D., Subramaniam, V., and
Jovin, T. M. (2004) Biochemistry, 43, 16233-16242.
35.Uversky, V. N. (2008) Curr. Alzheimer
Res., 5, 260-287.