Mitochondria as Source of Reactive Oxygen Species under Oxidative Stress. Study with Novel Mitochondria-Targeted Antioxidants – the “Skulachev-Ion” Derivatives
D. S. Izyumov, L. V. Domnina, O. K. Nepryakhina, A. V. Avetisyan, S. A. Golyshev, O. Y. Ivanova, M. V. Korotetskaya, K. G. Lyamzaev, O. Y. Pletjushkina, E. N. Popova, and B. V. Chernyak*
Belozersky Institute of Physico-Chemical Biology and Mitoengineering Center, Lomonosov Moscow State University, 119991 Moscow, Russia; fax: (495) 939-3181; E-mail: bchernyak@yahoo.com* To whom correspondence should be addressed.
Received November 1, 2009
Production of reactive oxygen species (ROS) in mitochondria was studied using the novel mitochondria-targeted antioxidants (SkQ) in cultures of human cells. It was shown that SkQ rapidly (1-2 h) and selectively accumulated in mitochondria and prevented oxidation of mitochondrial components under oxidative stress induced by hydrogen peroxide. At nanomolar concentrations, SkQ inhibited oxidation of glutathione, fragmentation of mitochondria, and translocation of Bax from cytosol into mitochondria. The last effect could be related to prevention of conformational change in the adenine nucleotide transporter, which depends on oxidation of critical thiols. Mitochondria-targeted antioxidants at nanomolar concentrations prevented accumulation of ROS and cell death under oxidative stress. These effects required 24 h or more (depending on the cell type) preincubation, and this was not related to slow induction of endogenous antioxidant systems. It is suggested that SkQ slowly accumulates in a small subpopulation of mitochondria that have decreased membrane potential and produce the major part of ROS under oxidative stress. This population was visualized in the cells using potential-sensitive dye. The possible role of the small fraction of “bad” mitochondria in cell physiology is discussed.
KEY WORDS: oxidative stress, mitochondria-targeted antioxidants, SkQ, mitochondria, apoptosisDOI: 10.1134/S000629791002001X
Abbreviations: ANT, adenine nucleotide translocator; CM-DCF-DA, 5-(-6)-chloromethyl-2′,7′-dichlorodihydrofluorescein diacetate; C12TPP, dodecyltriphenylphosphonium; FCCP, carbonyl cyanide p-trifluoromethoxyphenylhydrazone; ROS, reactive oxygen species; SkQ, cationic derivative of plastoquinone; SkQ1, 10-(6′-plastoquinonyl) decyltriphenylphosphonium; SkQR1, 10-(6′-plastoquinonyl) decylrhodamine 19; TMRM, tetramethylrhodamine methyl ester.
Respiration is a major process of the reduction of oxygen in animal
cells and a major function of mitochondria. Electron transfer reactions
in the respiratory chain are inevitably accompanied by one-electron
reduction of oxygen with superoxide anion formation followed by other
reactive oxygen species (ROS). Mitochondria potentially could be the
major source of ROS in the cell. The role of mitochondria in every
ROS-dependent process needs careful experimental investigation, which
is limited by the absence of efficient instruments.
A whole spectrum of compounds that are selectively addressed to mitochondria was developed after the discovery by V. P. Skulachev, E. A. Liberman, and colleagues [1] of penetrating ions (later dubbed “Skulachev ions”) [2-5]. Mitochondria are the only compartment charged negatively relatively to cytosol. High membrane potential (about –180 mV) at the inner mitochondrial membrane results in many-fold accumulation of penetrating cations and conjugated active compounds. The most successful application of “Skulachev ions” is related to design of novel mitochondria-targeted antioxidants (cationic derivatives of plastoquinone, SkQ) [5]. These compounds are selectively accumulated by mitochondria and regenerated by the respiratory chain after scavenging of ROS. As a result, they can be used as effective antioxidants at nanomolar concentrations. High efficiency of SkQ was confirmed in experiments with artificial lipid membranes, isolated mitochondria, and cells in culture [6]. It was shown that SkQ increased the lifespan of fungi (Podospora), invertebrates (Ceriodaphnia and Drosophila), and mice [7]. Pronounced therapeutic effects of SkQ were observed in the models of ischemic pathology of heart, kidney, and brain and also models of some eye diseases [8, 9].
In the present study, we have used SkQ to investigate participation of mitochondria in development of oxidative stress induced by hydrogen peroxide. The data confirmed the key role of mitochondria in endogenous ROS generation in this model. It is shown that SkQ protects mitochondria against oxidative damage in parallel with accumulation into the cell. However, accumulation of ROS and following apoptosis are prevented only after prolonged incubation with SkQ. It is suggested that a small fraction of mitochondria with decreased membrane potential that accumulate SkQ very slowly and generate the majority of ROS under oxidative stress.
MATERIALS AND METHODS
Human skin fibroblasts and HeLa cells were grown in Dulbecco’s modified Eagle’s medium (DMEM) supplemented with 10% fetal calf serum, streptomycin (100 U/ml), and penicillin (100 U/ml).
Accumulation of ROS in fibroblasts was analyzed after staining of the cells with 5 µM CM-DCF-DA (5-(-6)-chloromethyl-2′,7′-dichlorodihydrofluorescein diacetate) (Molecular Probes, USA) for 15 min at 37°C. Fluorescence was analyzed using a Beckman-Coulter FC500 flow cytometer (USA) equipped with a 488-nm argon laser.
Viability of fibroblasts was analyzed using Cell Titer Blue (Promega, USA). Fluorescence (excitation at 560 nm, emission at 590 nm) was measured in Thermoscan plate reader (ThermoLab Systems, Finland).
Mitochondria in cells were stained with 300 nM Mitotracker Green (Molecular Probes) or 200 nM TMRM (tetramethylrhodamine methyl ester) (Molecular Probes) for 15 min at 37°C. For immunostaining, cells grown on glass cover slips were fixed with 3.7% formaldehyde in phosphate-buffered saline solution (PBS) for 15 min at room temperature and stained with monoclonal antibodies against cytochrome c (6H2.B4; BD Pharmingen, USA) or with anti-BAX polyclonal antibodies (13666E; BD Pharmingen). The secondary antibodies conjugated with Oregon Green or Texas Red-X (BD Pharmingen) were used. Cells on cover slips were embedded into Vectashield medium (Vector Labs, USA). Images were analyzed with an Axiovert microscope (Carl Zeiss, Germany) and with an LSM 510 confocal microscope (Carl Zeiss). In all experiments on fragmentation, more than 100 cells were counted in each sample.
For analysis of SkQR1 distribution in fibroblasts, cells were co-stained with Mitotracker Green, and confocal images were analyzed using ImageJ software (http://rsb.info.nih.gov/ij/). The ratio of Mitotracker Green (300 nM) and SkQR1 (10-(6′-plastoquinonyl) decylrhodamine 19) (50 nM) fluorescence was analyzed in the area stained with Mitotracker. When quenching of Mitotracker Green (200 nM, λex 490 nm, λem 516 nm) fluorescence with TMRM (400 nM, λex 549 nm, λem 573 nm) was studied, the areas stained with any of the dyes were analyzed. The distribution of the ratio of green fluorescence to red fluorescence is presented in a histogram. The area of high fluorescence of Mitotracker Green indicated in a histogram corresponded to the mitochondria with decreased membrane potential.
RESULTS
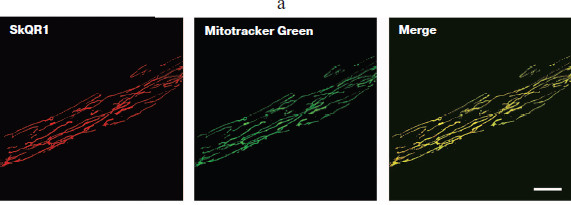
Accumulation of SkQ in cells and protective action. Accumulation of SkQ in the cells was studied using fluorescent analog SkQR1, which contained the cation of rhodamine 19 conjugated with plastoquinone. SkQR1 was selectively accumulated in mitochondria of human fibroblasts (Fig. 1a; see color insert). The analysis of the images did not reveal any detectable accumulation of SkQR1 in other cellular compartments. Dissipation of the membrane potential by protonophoric uncoupler prevented accumulation of SkQR1 in the cells. The small residual accumulation of SkQR1 in the presence of the uncoupler was probably related to a small membrane potential persisting in mitochondria. Distribution of SkQR1 in the mitochondrial population of the cell will be discussed in details below.
Kinetics of SkQR1 accumulation in human fibroblasts is shown in Fig. 1b. The slow rate of accumulation could be related to high lipophilicity of this compound and slow redistribution between different cellular membranes. The level of SkQ1 in the cells reached a plateau during 1.5-2 h and did not rise for several more hours. Moreover, prolonged (20-24 h) incubation resulted in some decrease of SkQR1 fluorescent in the cells, which could be related to decomposition of the compound in the aqueous phase. A similar effect was even more pronounced in HeLa cells, but it was partially due to proliferation of the cells during the experiment. These data did not exclude possible slow accumulation of additional SkQR1, but this effect could be only very small in comparison with accumulation during the first 2 h. After washing only a part of the SkQR1 was released from the cell (Fig. 1b).
Fig. 1. Interaction of SkQ with fibroblasts in culture. a) SkQR1 selectively accumulates in mitochondria of the cells. Fibroblasts were incubated with 50 nM SkQR1 and 300 nM Mitotracker Green for 15 min at 37°C. Confocal images are shown. Bar, 15 µm.
When the cells were treated with hydrogen peroxide, accumulation of ROS took 1-2 h after addition of the peroxide. It was shown earlier that hydrogen peroxide is decomposed in the cell culture during 30 min after addition due to interaction with the components of the medium (metal ions) and the cells (catalase) [10]. Thus, accumulation of ROS was a result of endogenous processes induced by exogenous peroxide. The role of mitochondria in these processes was confirmed in experiments where inhibitors of respiration stimulated accumulation of ROS in the same model [11]. Preincubation of the fibroblasts with nanomolar concentrations of SkQ prevented accumulation of ROS induced by hydrogen peroxide (Fig. 1c). This effect was not observed in the presence of the uncoupler or when the analogous cations without antioxidant (plastoquinone) residue were applied. The traditional antioxidants N-acetylcysteine (NAC) and Trolox (water-soluble analog of vitamin E) also prevented accumulation of ROS but at concentrations that were 2,500,000- and 100,000-fold higher then for SkQ, respectively. It was concluded that mitochondria were the major source of ROS in the cells where oxidative stress was induced by hydrogen peroxide.Fig. 1. Interaction of SkQ with fibroblasts in culture. b) Kinetics of SkQR1 accumulation in fibroblasts. Cells were incubated with 50 nM SkQR1 and analyzed using fluorescent flow cytometry. SkQR1 was washed out after 3 h (dotted line) and measurements were continued. Carbonyl cyanide p-trifluoromethoxyphenylhydrazone (FCCP) (10 µM) was added 30 min before SkQR1 where indicated. c) Time course of development of SkQ antioxidant action. Fibroblasts were incubated with 2 nM SkQ1 (10-(6′-plastoquinonyl) decyltriphenylphosphonium) (light columns) or 2 nM SkQR1 (dark columns), then 0.6 mM H2O2 was added and ROS level was measured after 2 h. The mean fluorescence of CM-DCF-DA in the cell population is shown (the results of a typical experiment). d) Concentration dependence of the protective effect of SkQ. Fibroblasts were incubated with SkQ1 (diamonds) or SkQR1 (squares) or C12TPP (dodecyltriphenylphosphonium) (triangles) for 24 h, then 0.5 mM H2O2 was added and viability was analyzed after 18 h. e) Trolox does not interfere with the protective effect of SkQ1. Fibroblasts were incubated for 1 h with 0.2 mM Trolox, then 2 nM SkQ1 was added and after 24 h both antioxidants were washed out. Fibroblasts were treated with 0.5 mM H2O2 and viability was analyzed after 18 h. Dashed line designates experiment where no antioxidants were added.
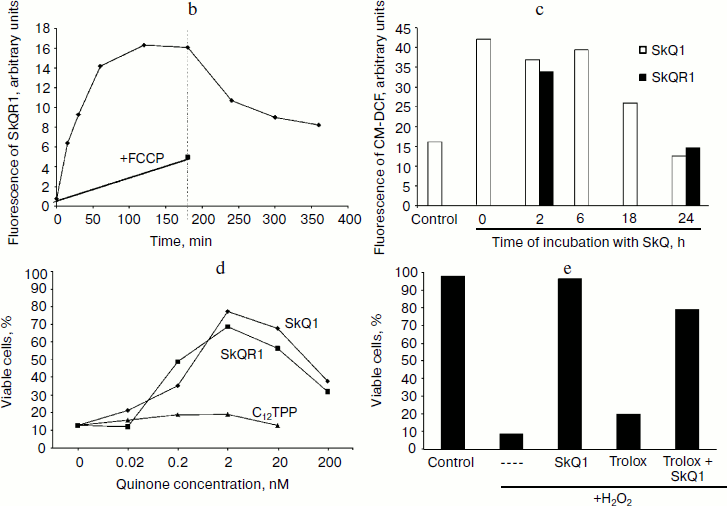
Antioxidant effect of SkQ was developed slowly (during 24 h) in comparison with their accumulation in the cells (compare Figs. 1b and 1c). The complete protection against cell death induced by hydrogen peroxide also was reached only after 24 h of incubation with SkQ (Fig. 1d). Increasing SkQ concentration 10-fold did not shorten the lag period, and further increase in SkQ concentration resulted in a toxic effect related to the prooxidant action of these compounds [6]. Multiple additions of low doses of SkQ also did not accelerate development of the protective effect (not shown).
One could suggest that delayed effects of SkQ were related to induction of the endogenous antioxidant systems in the cell. This effect is well known in the case of weak prooxidant stimuli, such as ischemic “preconditioning”, which induced strong protection against ischemia/reperfusion and other oxidative insults [12]. To test this possibility we analyzed the effect of SkQ1 on the basal level of ROS in fibroblasts. It was shown that SkQ1 at 2 nM (which was optimal for the antioxidant and protective effects) did not affect the level of ROS during 24 h after addition. However, at 20 nM SkQ1 a small (15-25%) statistically significant increase in ROS was observed after 1 h. This effect was prevented by Trolox (not shown). We took advantage of rapid reversibility of the antioxidant effect of Trolox and long (1-2 days) persistence of the effect of SkQ. To verify the hypothesis of induction, we added Trolox before SkQ1 and then both agents were removed after 20 h. The effect of Trolox was not observed, while the protective action of SkQ1 remained unaffected (Fig. 1e). These data indicated that the initial increase of ROS after addition of SkQ1 could not be a stimulus for induction of antioxidant systems.
An alternative explanation of delayed antioxidant and protective effects of SkQ suggested slow redistribution of this compound in the population of mitochondria in the cell. It seems possible that there is a small fraction of mitochondria that accumulates SkQ slowly (due to low membrane potential, for example) and produces the major part of the ROS under oxidative stress. To test this hypothesis, we analyzed the effects of SkQ on the mitochondrial population in the cells.
Protection of mitochondria from fragmentation and oxidation by SkQ. Mitochondria in the cell form a dynamic structure very sensitive to external stimuli. Equilibration between fusion and fission in the majority of cells (including fibroblasts and HeLa cells) results in the formation of reticular mitochondrial networks [13]. Various stresses (including oxidative stress induced by hydrogen peroxide) induce fragmentation of mitochondria. Short-term (2 h) incubation of fibroblasts with SkQ prevented mitochondrial fragmentation (Fig. 2a; see color insert). Half-maximal protective effect of SkQ1 was observed at 2 nM, while the efficiency of SkQR1 was even higher (C1/2 = 0.05 nM). SkQ were ineffective if fragmentation of mitochondria was induced by non-oxidative stimulus (an inhibitor of protein kinases, staurosporine, for example). The traditional antioxidants NAC and Trolox also prevented fragmentation of mitochondria induced by hydrogen peroxide but only at 5 and 0.1 mM, respectively. It is important that 2 h of preincubation with SkQ did not decrease the level of ROS in the cell (Fig. 1c). It was concluded that ROS localized in mitochondria were the major stimuli for their fragmentation in our model of oxidative stress.
Unfortunately, methods for measurement of ROS in mitochondria are not yet developed. One of the most popular dyes, MitoSOX (Invitrogen, USA), is a conjugate of dihydroethidine (ROS-sensitive fluorophore) with triphenylphosphonium (“Skulachev-cation”). In our experiments, MitoSOX was accumulated in mitochondria but responded only to high doses of added H2O2. This effect was related to depolarization of mitochondria, release of MitoSOX, and following accumulation of MitoSOX in the nucleus.Fig. 2. Protection of mitochondrial population in cell by SkQ. a) SkQ1 prevents fragmentation of mitochondria in fibroblasts induced by hydrogen peroxide. Cells were incubated with 2 nM SkQ1 for 2 h, then 0.4 mM H2O2 was added and mitochondria stained with Mitotracker Green were analyzed after 3 h using confocal microscopy. Bar, 15 µm. b) SkQ1 and bongkrekate prevented translocation of Bax from cytosol into mitochondria induced by hydrogen peroxide. HeLa cells were incubated for 2 h with 2 nM SkQ1 or 10 µM bongkrekate (BK) and then treated with 0.25 mM H2O2. Distribution of Bax (red) and cytochrome c (green, mitochondrial marker) was analyzed by immunofluorescence microscopy after 18 h. Yellow staining of mitochondria indicated co-localization of Bax and cytochrome c. Red staining of the nucleus was an artifact of immunostaining. Bar, 10 µm.
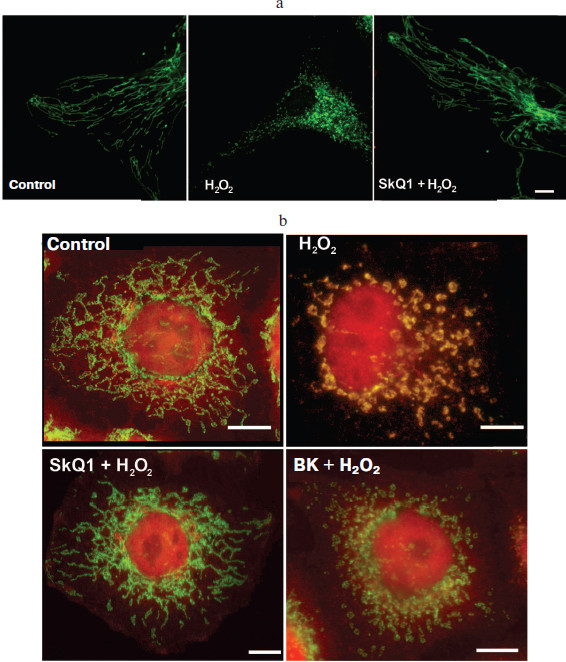
Fragmentation of mitochondria during apoptosis or mitosis depends on translocation of Drp1 from cytosol to the outer mitochondrial membrane [14]. We did not detect any significant translocation of Drp1 under oxidative stress induced by sublethal doses of H2O2 in HeLa cells (not shown). However, in this model massive translocation of Bax was detected (Fig. 2b; see color insert). This protein from the Bcl-2 family plays an important role in apoptosis, stimulating the release of cytochrome c from mitochondria into cytosol. During apoptosis Bax changes conformation, translocates to the outer mitochondrial membrane, and forms multimeric complexes and large pores that allow the release of proteins from intermembrane space into cytosol [15]. In our model of oxidative stress, translocation of Bax was not accompanied by release of cytochrome c and following cell death. Short term (2 h) preincubation with SkQ prevented translocation of Bax (Fig. 2b). Since SkQ under these conditions did not decrease the cellular ROS, we concluded that translocation of Bax was induced by oxidative events inside mitochondria. Taking into account localization SkQ in the inner mitochondrial membrane, one can suggest that oxidation of components of this membrane was critical for Bax translocation. It was shown earlier that Bax could interact with mitochondria at so-called “contact sites” [16, 17]. These supercomplexes of variable composition contain the proteins of the outer membrane (VDAC and peripheral benzodiazepine receptor) as well as the inner membrane proteins (adenine nucleotide translocator, ANT). It was shown that the structure of “contact sites” depended on conformation of ANT, which in turn could be changed by oxidation of reactive thiols [18].
We have suggested that oxidation of thiols in ANT was critical for translocation of Bax in our model. To test this hypothesis, we applied the specific inhibitors of ANT atractyloside and bongkrekate that fixed ANT in the different conformations [19]. Bongkrekate completely abolished translocation of Bax induced by H2O2 (Fig. 2b), while atractyloside stimulated translocation of Bax even in the absence of hydrogen peroxide. In the presence of atractyloside, SkQ failed to prevent translocation of Bax (not shown). These data indicated that under oxidative stress SkQ prevented oxidation and conformational change of ANT, which are necessary for translocation of Bax. It is interesting that fragmentation of mitochondria induced by H2O2 was not prevented by bongkrekate (not shown). It seems possible that fragmentation of mitochondria under oxidative stress did not depend on oxidation of ANT and translocation of Bax but rather was related to other unknown oxidative events. Consistent with this conclusion, it was found recently that translocation of Bax was not necessary for fragmentation of mitochondria during apoptosis [20].
The data described above indicated that SkQ accumulated in mitochondria effectively protected their components against oxidation, and this effect did not need a long induction period. The following studies were directed to verification of the hypothesis of heterogeneous distribution of SkQ in the mitochondrial population.
Analysis of heterogeneity of mitochondrial population in the cell. Heterogeneity of mitochondrial population is well recognized in specialized cells (myocytes, neurons) with complex internal spatial organization [21]. Appearance of mitochondria with decreased membrane potential has also been described in hepatocytes [22], β-cells [23], and HeLa cells [24]. To analyze the distribution of SkQR1 in mitochondria of fibroblasts, we measured the ratio of SkQR1 fluorescence and fluorescence of Mitotracker Green (Molecular Probes). This potential-insensitive mitochondrial dye was presumably distributed homogenously in the mitochondrial population. This approach allowed to compensate possible artifacts related to location of mitochondria in confocal optical slices. Analysis of the images using ImageJ software resulted in a histogram of SkQR1 distribution in the population of mitochondria (Fig. 3a). These data showed that practically all the mitochondria accumulated SkQR1. Deviation of the distribution from Gaussian indicated that some fraction of mitochondria could have decreased content of SkQR1, but the further analysis did not allow us to identify these mitochondria in individual cells.
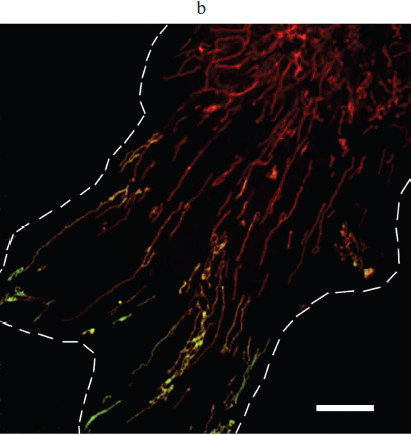
For more precise analysis, we took an advantage of quenching of Mitotracker Green fluorescence with rhodamine. This approach was applied earlier for identification of depolarized mitochondria in hepatocytes [22]. Unfortunately, it was not possible to analyze quenching of Mitotracker Green with SkQR1 due to its high green fluorescence. As a potential-sensitive dye, we used TMRM that effectively quenched Mitotracker Green fluorescence in the major part of mitochondria. However, in a small fraction of mitochondria the effect of quenching was not pronounced. This fraction was clearly visible in confocal images of individual fibroblasts and was quantified by analysis of these images (Fig. 3b (color insert) and Fig. 3, c and d). Interestingly these mitochondria were localized in the lamellar part of the cell in the vicinity of the active edge but not in the perinuclear area.Fig. 3. Study of heterogeneity of mitochondrial population in the cell. a) Distribution of SkQR1 in mitochondrial population of fibroblast. Cells were incubated with 50 nM SkQR1 for 2 h, then 300 nM Mitotracker Green was added for 15 min and fluorescence was analyzed using confocal microscopy. The ratio of green (Mitotracker) to red (SkQR1) fluorescence was measured in the area stained with Mitotracker and distribution of this parameter is presented. The Gaussian distribution is shown with the dashed line. c) Analysis of mitochondria with decreased membrane potential in fibroblasts. Cells were incubated with 200 nM Mitotracker Green and 400 nM TMRM (conditions of quenching of Mitotracker fluorescence) for 15 min and analyzed using confocal microscopy. The ratio of green (Mitotracker) to red (TMRM) fluorescence was measured in the area stained with both dyes. The dashed line indicates the values of this parameter for pixels analyzed below. The same cell as in Fig. 3b (color insert) was analyzed. d) Localization of the mitochondria with decreased membrane potential in the cell. The area, which was analyzed as described in Fig. 3b (color insert), is indicated with solid contour. The darkened area corresponds to the pixels that lie to the right from the dashed line on the distribution in panel (c). The cell borders are marked with a dashed line. The mitochondria with decreased membrane potential are localized in the lamellar area close to the active edge. Bar, 15 µm.
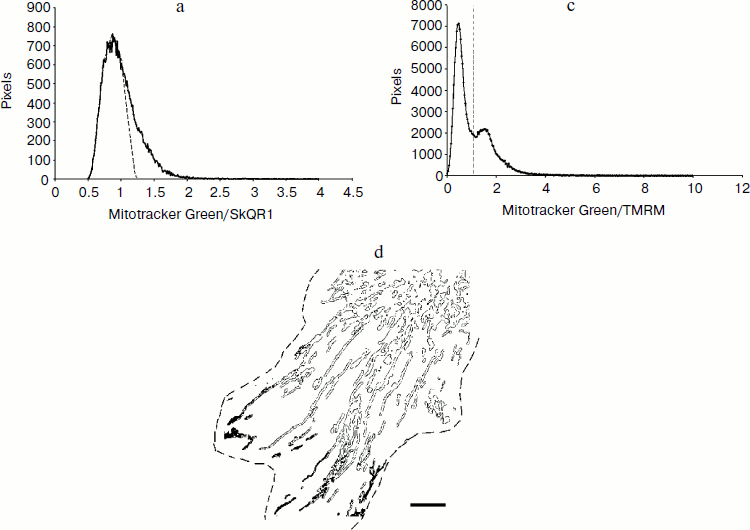
The nature of this special distribution remained enigmatic as well as the mechanism(s) of depolarization of these mitochondria. One could suggest that ΔpH was increased in these mitochondria and membrane potential was decreased as far as electrochemical potential (ΔµH+) remained constant. However, addition of K+/H+ exchanger nigericin that dissipated ΔpH neither caused homogenization of the mitochondrial population nor increased accumulation of SkQR1 in the cell. Inhibition of ATP synthase in mitochondria with oligomycin also did not affect heterogeneity of mitochondria. We suggested that the heterogeneity was related to excessive ROS production in some mitochondria. If so, prolonged incubation with antioxidants could restore the membrane potential and cause additional accumulation of SkQR1. However, incubation of fibroblasts with Trolox (0.2 mM, 24 h) neither increased following accumulation of SkQR1 nor decreased the content of depolarized mitochondria (not shown).Fig. 3. Study of heterogeneity of mitochondrial population in the cell. b) Identification of mitochondria with decreased membrane potential in the cell. Fibroblasts were incubated with 200 nM Mitotracker Green and 400 nM TMRM for 15 min and analyzed using confocal microscopy. TMRM caused quenching of Mitotracker Green fluorescence only in mitochondria with high membrane potential where it was accumulated to high extent. Bright green fluorescence indicates mitochondria with decreased membrane potential. The cell borders are marked with a dashed line. Bar, 15 µm.
Our data clearly demonstrate the existence of a fraction of mitochondria with decreased membrane potential in the cell, but their origin remains obscure. It was shown earlier in different models that low membrane potential correlated with excessive ROS production in individual mitochondria [24, 25]. These data allow us suggest that the fraction of depolarized mitochondria is a major source of endogenous ROS produced under oxidative stress induced by hydrogen peroxide. This hypothesis explained slow development of antioxidant and protective effects of SkQ, which accumulated in mitochondria due to membrane potential. Antioxidant effect of SkQ in individual mitochondria with high membrane potential (the major mitochondrial population) developed much faster, in agreement with the rate of accumulation of SkQ in the cell.
DISCUSSION
The studies on interaction of the novel mitochondria-targeted antioxidants (SkQ) with cells in culture confirmed their high efficiency. It was shown that SkQ accumulated selectively in mitochondria of different cells and the process was completed in 1-2 h. These antioxidants at nanomolar concentrations inhibited the processes induced by ROS in mitochondria and prevented their fragmentation under oxidative stress. Oxidation of mitochondrial components (presumably of adenine nucleotide antiporter in the inner membrane) was necessary for translocation of proapoptotic protein Bax from cytosol into mitochondria under oxidative stress. The mitochondria-targeted antioxidants inhibited this process, and this probably was important for prevention of apoptosis induced by prooxidants.
The novel mitochondria-targeted antioxidants prevented excessive ROS production and cell death induced by hydrogen peroxide. These effects were observed at the same low (nanomolar) concentrations but were developed with significant delay (~20 h) in the case of fibroblasts. A hypothesis on induction of endogenous antioxidant systems by SkQ was not supported by our experiments. We suggested that a small fraction of depolarized mitochondria could accumulate SkQ very slowly in parallel with restoration of the membrane potential. This subpopulation of mitochondria was identified in fibroblasts for the first time in this work. By analogy with the other cellular models [24, 25], we suggested that this small fraction of “bad” mitochondria intensively generated ROS. Probably these “bad” mitochondria generated the major part of ROS determining the fate of the cells under oxidative stress. This conclusion clarified the postulate on mitochondria as a major source of ROS and, according to V. P. Skulachev, “the dirtiest place in the cell” [26].
The small fraction of mitochondria that generate a large amount of ROS could play an important role in cell physiology. These mitochondria could serve as sensitive local sensors that respond with ROS release to changes in their microenvironment in different parts of the cell. These signals could be locally registered by signaling molecules (protein kinases, phosphatases, etc.), which (as it was found recently) are specifically associated with mitochondria (see [27] for review). A very stimulating concept of programmed aging mediated by mitochondrial ROS was recently introduced by V. P. Skulachev [5, 28]. It could be suggested that the signals from a small fraction of “sensory” mitochondria are critical for execution of the program of aging. This hypothesis could help to find agreement between the “mitochondrial theory of aging” [29] and the data on small amount of mutations accumulated in mitochondrial DNA with age [30].
The authors express their heartfelt gratitude to V. P. Skulachev for constant interest and support. We wish Vladimir Petrovich Happy Birthday and many happy years of work for the benefit of science.
This work was supported by the Russian Foundation for Basic Research (grant Nos. 07-04-00335, 09-04-00667, 09-04-01454).
REFERENCES
1.Liberman, E. A., Topali, V. P., Tsofina, L. M.,
Jasaitis, A. A., and Skulachev, V. P. (1969) Nature, 222,
1076-1078.
2.Cocheme, H. M., Kelso, G. F., James, A. M., Ross,
M. F., Trnka, J., Mahendiran, T., Asin-Cayuela, J., Blaikie, F. H.,
Manas, A. R., Porteous, C. M., Adlam, V. J., Smith, R. A., and Murphy,
M. P. (2007) Mitochondrion, 7, 94-102.
3.Armstrong, J. S. (2008) Antioxid. Redox.
Signal., 10, 575-578.
4.Antonenko, Y. N., Roginsky, V. A., Pashkovskaya, A.
A., Rokitskaya, T. I., Kotova, E. A., Zaspa, A. A., Chernyak, B. V.,
and Skulachev, V. P. (2008) J. Membr. Biol., 222,
141-149.
5.Skulachev, V. P., Anisimov, V. N., Antonenko, Y.
N., Bakeeva, L. E., Chernyak, B. V., Erichev, V. P., Filenko, O. F.,
Kalinina, N. I., Kapelko, V. I., Kolosova, N. G., Kopnin, B. P.,
Korshunova, G. A., Lichinitser, M. R., Obukhova, L. A., Pasyukova, E.
G., Pisarenko, O. I., Roginsky, V. A., Ruuge, E. K., Senin, I. I.,
Severina, I. I., Skulachev, M. V., Spivak, I. M., Tashlitsky, V. N.,
Tkachuk, V. A., Vyssokikh, M. Y., Yaguzhinsky, L. S., and Zorov, D. B.
(2009) Biochim. Biophys. Acta, 1787, 437-461.
6.Antonenko, Y. N., Avetisyan, A. V., Bakeeva, L. E.,
Chernyak, B. V., Chertkov, V. A., Domnina, L. V., Ivanova, O. Y.,
Izyumov, D. S., Khailova, L. S., Klishin, S. S., Korshunova, G. A.,
Lyamzaev, K. G., Muntyan, M. S., Nepryakhina, O. K., Pashkovskaya, A.
A., Pletjushkina, O. Y., Pustovidko, A. V., Roginsky, V. A.,
Rokitskaya, T. I., Ruuge, E. K., Saprunova, V. B., Severina, I. I.,
Simonyan, R. A., Skulachev, I. V., Skulachev, M. V., Sumbatyan, N. V.,
Sviryaeva, I. V., Tashlitsky, V. N., Vassiliev, J. M., Vyssokikh, M.
Y., Yaguzhinsky, L. S., Zamyatnin, A. A., Jr., and Skulachev, V. P.
(2008) Biochemistry (Moscow), 73, 1273-1287.
7.Anisimov, V. N., Bakeeva, L. E., Egormin, P. A.,
Filenko, O. F., Isakova, E. F., Manskikh, V. N., Mikhelson, V. M.,
Panteleeva, A. A., Pasyukova, E. G., Pilipenko, D. I., Piskunova, T.
S., Popovich, I. G., Roshchina, N. V., Rybina, O. Y., Saprunova, V. B.,
Samoylova, T. A., Semenchenko, A. V., Skulachev, M. V., Spivak, I. M.,
Tsybul’ko, E. A., Tyndyk, M. L., Vyssokikh, M. Y., Yurova, M. N.,
Zabezhinsky, M. A., and Skulachev, V. P. (2008) Biochemistry
(Moscow), 73, 1329-1342.
8.Bakeeva, L. E., Barskov, I. V., Egorov, M. V.,
Isaev, N. K., Kapelko, V. I., Kazachenko, A. V., Kirpatovsky, V. I.,
Kozlovsky, S. V., Lakomkin, V. L., Levina, S. B., Pisarenko, O. I.,
Plotnikov, E. Y., Saprunova, V. B., Serebryakova, L. I., Skulachev, M.
V., Stelmashook, E. V., Studneva, I. M., Tskitishvili, O. V.,
Vasilyeva, A. K., Victorov, I. V., Zorov, D. B., and Skulachev, V. P.
(2008) Biochemistry (Moscow), 73, 1288-1299.
9.Neroev, V. V., Archipova, M. M., Bakeeva, L. E.,
Fursova, A. Z., Grigorian, E. N., Grishanova, A. Y., Iomdina, E. N.,
Ivashchenko, Z., Katargina, L. A., Khoroshilova-Maslova, I. P., Kilina,
O. V., Kolosova, N. G., Kopenkin, E. P., Korshunov, S. S., Kovaleva, N.
A., Novikova, Y. P., Philippov, P. P., Pilipenko, D. I., Robustova, O.
V., Saprunova, V. B., Senin, I. I., Skulachev, M. V., Sotnikova, L. F.,
Stefanova, N. A., Tikhomirova, N. K., Tsapenko, I. V., Shchipanova, A.
I., Zinovkin, R. A., and Skulachev, V. P. (2008) Biochemistry
(Moscow), 73, 1317-1328.
10.Pletjushkina, O. Y., Fetisova, E. K., Lyamzaev,
K. G., Ivanova, O. Y., Domnina, L. V., Vyssokikh, M. Y., Pustovidko, A.
V., Vasiliev, J. M., Murphy, M. P., Chernyak, B. V., and Skulachev, V.
P. (2005) Cell Death Differ., 12, 1442-1444.
11.Chernyak, B. V., Izyumov, D. S., Lyamzaev, K. G.,
Pashkovskaya, A. A., Pletjushkina, O. Y., Antonenko, Y. N., Sakharov,
D. V., Wirtz, K. W., and Skulachev, V. P. (2006) Biochim. Biophys.
Acta, 1757, 525-534.
12.Saini, H. K., Machackova, J., and Dhalla, N. S.
(2004) Antioxid. Redox. Signal., 6, 393-404.
13.Skulachev, V. P. (2001) Trends Biochem.
Sci., 26, 23-29.
14.Benard, G., and Karbowski, M. (2009) Semin.
Cell Dev. Biol., 20, 365-374.
15.Adams, J. M., and Cory, S. (2007) Curr. Opin.
Immunol., 19, 488-496.
16.Crompton, M. (2000) J. Physiol.,
15, 11-21.
17.Vyssokikh, M. Y., and Brdiczka, D. (2003) Acta
Biochim. Pol., 50, 389-404.
18.Halestrap, A. P., and Brennerb, C. (2003)
Curr. Med. Chem., 10, 1507-1525.
19.Klingenberg, M. (2008) Biochim. Biophys.
Acta, 1778, 1978-2021.
20.Sheridan, C., Delivani, P., Cullen, S. P., and
Martin, S. J. (2008) Mol. Cell, 22, 570-585.
21.Kuznetsov, A. V., and Margreiter, R. (2009)
Int. J. Mol. Sci., 10, 1911-1929.
22.Lemasters, J. J., and Ramshesh, V. K. (2007)
Methods Cell Biol., 80, 283-295.
23.Twig, G., Elorza, A., Molina, A. J., Mohamed, H.,
Wikstrom, J. D., Walzer, G., Stiles, L., Haigh, S. E., Katz, S., Las,
G., Alroy, J., Wu, M., Py, B. F., Yuan, J., Deeney, J. T., Corkey, B.
E., and Shirihai, O. S. (2008) EMBO J., 27,
433-446.
24.Belousov, V. V., Fradkov, A. F., Lukyanov, K. A.,
Staroverov, D. B., Shakhbazov, K. S., Terskikh, A. V., and Lukyanov, S.
(2006) Nat. Methods, 3, 281-286.
25.Zorov, D. B., Filburn, C. R., Klotz, L. O.,
Zweier, J. L., and Sollott, S. J. (2000) J. Exp. Med.,
192, 1001-1014.
26.Skulachev, V. P. (2005) IUBMB Life,
57, 305-310.
27.Storz, P. (2007) Trends Cell Biol.,
17, 13-18.
28.Skulachev, V. P., and Longo, V. D. (2005) Ann.
N. Y. Acad. Sci., 1057, 145-164.
29.Harman, D. (1956) J. Gerontol., 11,
298-300.
30.Greaves, L. C., Beadle, N. E., Taylor, G. A.,
Commane, D., Mathers, J. C., Khrapko, K., and Turnbull, D. M. (2009)
Aging Cell, 8, 566-572.