Interaction of Murine Dnmt3a with DNA Containing O6-Methylguanine
D. V. Maltseva and E. S. Gromova*
Faculty of Chemistry and Belozersky Institute of Physico-Chemical Biology, Lomonosov Moscow State University, 119991 Moscow, Russia; fax: (495) 939-3181; E-mail: gromova@genebee.msu.ru* To whom correspondence should be addressed.
Received July 13, 2009; Revision received September 16, 2009
O6-Methylguanine (O6meG) is one of the most toxic, mutagenic, and carcinogenic lesions caused by the interaction of DNA with several catabolism products as well as with environmental methylating agents. Carcinogenic impact of O6meG can be conditioned not only by its mutagenic properties but also by alteration in enzymatic methylation of the C5 carbon atom of cytosine residue in CpG sequences. In this study, the effect of O6meG on DNA methylation by the catalytic domain of murine DNA methyltransferase (MTase) Dnmt3a (Dnmt3a-CD) is assessed. Damaged DNA duplexes cooperatively bind with Dnmt3a-CD, and O6meG changes the stability of enzyme–substrate complexes. Kinetic analysis of the methylation reaction revealed that O6meG varies the ratio of productive and nonproductive enzyme–substrate complexes and, depending on localization in substrate, causes decrease or increase in DNA methylation. Dnmt3a-CD is less sensitive to the presence of O6meG in DNA substrate than procaryotic MTase SssI recognizing CpG.
KEY WORDS: cooperative binding, murine DNA methyltransferase Dnmt3a, O6-methylguanine, nonproductive complexDOI: 10.1134/S0006297910020070
Abbreviations: AdoHcy, S-adenosyl-L-homocysteine; AdoMet, S-adenosyl-L-methionine; Dnmt3a-CD, catalytic domain of DNA methyltransferase Dnmt3a; FAM, 6(5)-carboxyfluorescein; MTase, DNA methyltransferase; O6meG, O6-methylguanine; 8-oxoG, 7,8-dihydro-8-oxoguanine.
Living organisms are always under the influence of carcinogenic and
mutagenic substances able to damage DNA and other cell components [1, 2]. Harmful impact can be
caused by environmental pollutants (present in food, tobacco smoke,
etc.) or by metabolic intermediates as well as by a result of various
pathologies including inflammatory processes [1-3]. One such process is DNA alkylation in eukaryotic
cells, which takes place both during exogenous and endogenous impact of
active alkylating particles and is mostly directed to the N7 nitrogen
atom of guanine [4-6]. In
vivo, slight alkylation of the O6 oxygen atom of guanine residue is
observed. However, among products of DNA damage with methylating
agents, O6-methylguanine (O6meG) is one of the
most toxic, mutagenic, and carcinogenic [4, 7-9]. The cytotoxicity of
O6meG is not yet sufficiently studied. However, the presence
of O6meG in DNA had been shown to inhibit the activity of
NF-κB transcription factor, significantly decrease efficiency of
transcription performed by human RNA polymerase II, poison human
topoisomerase I, and induce apoptosis [10-13].
Carcinogenic impact of O6meG was previously thought to be connected just with its mutagenic activity [6-8]. During DNA replication, O6meG can pair with thymine, which is structurally similar to the Watson–Crick G·C pair [14], leading to G·C→A·T mutation in human and animal cells [15-17]. In cancer cells, this mutation is often coupled with activation of oncogenes and inactivation of tumor suppressor genes [8, 16]. However, oncogene mutations are not observed in all types of cancer induced by substances methylating guanine residue in DNA [4]. We suppose that O6meG affects other processes in cells, one of which can be enzymatic DNA methylation.
Enzymatic DNA methylation is one of the most important epigenetic processes involved in genome imprinting, inactivation of X chromosome, regulation of gene transcription, maintenance of genome integrity, etc. [18, 19]. In animal and human cells, it is performed by DNA methyltransferases (MTases) Dnmt1, Dnmt3a, and Dnmt3b, which transfer a methyl group from S-adenosyl-L-methionine (AdoMet) cofactor to the C5 carbon atom of cytosine residue in CpG sequences [20]. Dnmt1 mainly maintains methylation, while Dnmt3a and Dnmt3b perform de novo methylation. Numerous investigations show the interaction of all three enzymes in vivo is necessary in all stages of establishment and maintenance of normal DNA methylation [21-23]. The fact that disorders of epigenetic mechanisms is connected with development of numerous chronic diseases including cancer, cardiovascular diseases, diabetes, and obesity has been proved many times recently [24]. It was previously supposed that change in normal pattern of DNA methylation could follow from lesions of heterocyclic bases in DNA [25]. Treatment of human cells with active methylating agent (N-methyl-N-nitrosourea), also leading to O6meG formation, was shown to decrease DNA methylation [26]. It can be supposed that disturbance of the DNA methylation pattern caused by O6meG can play a significant role in carcinogenesis. There is almost no data about the impact of O6meG on DNA methylation by MTases other than Dnmt1. Introduction of O6meG to the enzyme recognition site was shown to cause significant decrease [27] and blocking of DNA methylation [28].
The goal of this study was to examine the impact of O6meG on functional properties of the catalytic domain of Dnmt3a (Dnmt3a-CD). It is shown that the character of the impact of O6meG on the DNA methylation level depends on the lesion localization relative to the Dnmt3a-CD recognition site and target cytosine and seems to be due to variation in ratio of productive and nonproductive enzyme–substrate complexes.
MATERIALS AND METHODS
Reagents. For this study, AdoHcy (S-adenosyl-L-homocysteine) was purchased from Sigma and [CH3-3H]AdoMet (S-adenosyl-L-methionine) (77 Ci/mmol, 13 µM) was obtained from Amersham Biosciences. Oligodeoxyribonucleotides (Table 1) were synthesized by Sintol (Moscow) and purified in polyacrylamide gel. The fluorescein label (6-carboxyfluorescein, FAM) was introduced at the ′-end of the oligodeoxyribonucleotides by means of an aminoalkyl linker containing six methyl groups. Oligodeoxyribonucleotide concentrations were estimated spectrophotometrically [29]. The following buffers were used: buffer A (20 mM Hepes-NaOH, pH 7.5, 100 mM KCl, 1 mM EDTA, 0.2 mM dithiothreitol (DTT), and 10% (v/v) glycerol); buffer B (10 mM Tris-HCl, pH 7.9, 50 mM NaCl, 1 mM DTT, and 0.1 mg/ml BSA).
Table 1. Oligodeoxyribonucleotide
sequences*

* M, 5-methylcytosine; X, O6meG; FAM (f),
6-carboxyfluorescein.
Purification of Dnmt3a-CD and M.SssI. Dnmt3a-CD and SssI MTases as derivatives containing a His6 tag on N-terminal were purified as described in [30]. Purity of enzyme preparations was determined using electrophoresis in 12.5% SDS-polyacrylamide gel and was >90%. Protein concentrations determined by the Bradford method were considered as concentrations of MTases monomer form. Active concentration of M.SssI was determined by the fluorescence polarization method as described in [31].
Dnmt3a-CD binding with DNA. Dnmt3a-CD binding with DNA (10 mM) in the presence of AdoHcy (0.1 mM) was examined by fluorescence polarization by direct titration of FAM-labeled oligodeoxyribonucleotide duplexes by Dnmt3a-CD as described in [30]. The signals were measured using a Cary Eclipse spectrofluorimeter (Varian). Fluorescence polarization value, P, was determined according to the equation:
P = (Iv – GIh)/(Iv + Ih),
where Iv and Ih are vertical and horizontal components of irradiated light, respectively; G is the instrumental correction factor. The experimental data are presented as P as a function of Dnmt3a-CD total concentration. Curves of titration of DNA duplexes by MTase were reproduced at least three times and analyzed according to the following Scheme:
![]()
Dissociation constant values Kd1 and Kd2 were estimated by processing of binding curves using the SCIENTIST software (MacroMath) and system of equations (1)-(5) [30]:

This data processing takes into account the decrease in free DNA and enzyme concentrations during complex formation. Equation (1) assumes that P value of the E2S complex is two times higher than that of ES, and that the volume of E2 is nearly two times larger than the volume of E. Equations (2) and (3) are expressions for corresponding Kd values. Equations (4) and (5) describe mass conservation for Dnmt3a-CD and DNA, respectively.
DNA methylation by MTases Dnmt3a-CD and SssI. DNA methylation by Dnmt3a-CD was monitored by measuring the amount of tritium incorporated during transfer of methyl groups from [CH3-3H]AdoMet to cytosine residues. The reaction was performed at 37°C in buffer A in the case of Dnmt3a-CD and in buffer B for M.SssI. Kinetics of DNA methylation by Dnmt3a-CD were examined under conditions where the DNA concentration was 1.7 times greater than that of the enzyme as well as under conditions where the enzyme concentration was 10 times greater than that of DNA (“single turnover” conditions) as described in [30]. In the first case, the reaction mixture contained 1.5 µM DNA, 0.88 µM Dnmt3a-CD, and 2 µM [CH3-3H]AdoMet. Reaction mixture aliquots were taken from 1 to 15 min starting from the beginning of the reaction and applied to DE81 ion-exchanging filters (Whatman), which were treated as described in [32]. Initial rate values of the methylation reaction (vo) were estimated from the linear region slope of DNA methylation dependence on time.
In the second case, the reaction mixture contained 300 nM DNA, 3 µM Dnmt3a-CD, and 1.2 µM [CH3-3H]AdoMet. Reaction mixture aliquots were withdrawn at times from 4 to 90 min. Values of rate constants of the methylation reaction (kst) were estimated assuming pseudo-first order of the reaction (under conditions of saturation by DNA and AdoMet) according to Eq. (6) [33]:
![]()
Methylation of DNA duplexes by MTase SssI was carried out under steady-state conditions as described in [30]. Reaction mixture aliquots containing 500 nM DNA, 20 nM M.SssI, and 1.2 µM [CH3-3H]AdoMet were withdrawn at times from 1 to 3.5 min, and vo values were estimated similarly to that for Dnmt3a-CD.
RESULTS
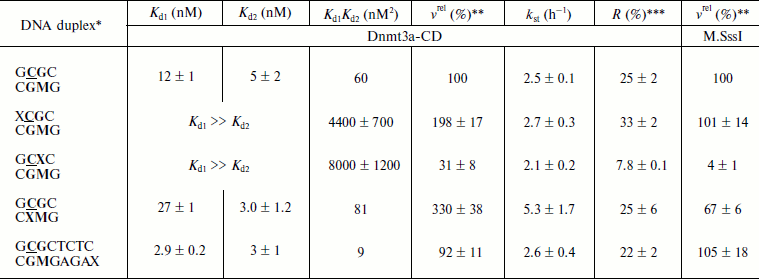
To estimate the impact of O6meG on functional properties of Dnmt3a-CD, binding to 30-mer DNA duplexes containing O6meG and their methylation by Dnmt3a-CD were studied. Dnmt3a-CD can introduce methyl groups into unmethylated DNA as well as into DNA containing a methyl group in one of the strands (hemimethylated DNA). Hemimethylated DNA duplexes were used to estimate the impact of O6meG (X) on methylation of a particular DNA strand. A damaged base was introduced into one of the strands of hemimethylated substrate analogs into the Dnmt3a-CD recognition site (GCXC/CGMG, GCGC/CXMG), at the 5′-side of recognition site (XCGC/CGMG), or four nucleotide residues from it (GCGCTCTC/CGMGAGAX) (Table 2).
Initial rates of methylation of DNA duplexes containing O6meG by Dnmt3a-CD. Initial rates (vo) of methylation of DNA duplexes containing O6meG were estimated using the Dnmt3a catalytic domain under conditions where the DNA concentration was 1.7 times greater than that of the enzyme (Fig. 1), and values of relative initial methylation rates (vrel) were estimated (Table 2). Introduction of O6meG into the recognition site of (GCXC/CGMG) target strand led to 3.2-fold decrease in methylation rate compared with undamaged GCGC/CGMG substrate. At the same time, vo values for duplexes containing O6meG near a CpG site (XCGC/CGMG) and at the recognition site opposite to the target cytosine (GCGC/CXMG) were 2 and 3 times larger, respectively, than vo value of GCGC/CGMG. Replacement of a distant guanine residue with O6meG (GCGCTCTC/CGMGAGAX) had almost no influence on the ability of Dnmt3a-CD to methylate cytosine in the CpG site.
Table 2. Parameters of interaction of Dnmt3a-CD and M.SssI with DNA duplexes containing O6meGFig. 1. Methylation of DNA duplexes by Dnmt3a-CD. CDNA, 1.5 µM; CDnmt3a-CD, 0.9 µM; C[CH3-3H]AdoMet, 1.5 µM. 1) GCGC/CGMG; 2) XCGC/CGMG; 3) GCXC/CGMG; 4) GCGC/CXMG; 5) GCGCTCTC/CGMGAGAX.
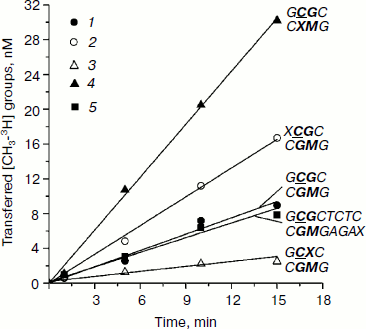
*Designations of oligodeoxyribonucleotides are as in Table 1. Dnmt3a-CD and M.SssI recognition sites are in bold type, the target cytosine is underlined.
**Relative methylation rate. Rate of GCGC/CGMG duplex methylation (0.7 nM/min for Dnmt3a-CD and 11 nM/min for M.SssI) is taken as 100%.
***Ratio between methylated DNA at the end of the reaction and total DNA in the reaction mixture (Fig. 3).
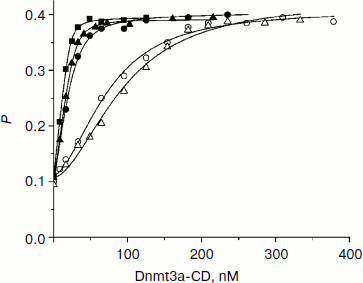
Dnmt3a-CD binding to DNA duplexes containing O6meG. When O6meG is introduced, change in DNA methylation rate might be connected with change in Dnmt3a-CD affinity with damaged DNA. To test this conjecture, binding of FAM-labeled DNA duplexes containing O6meG to Dnmt3a-CD was examined by fluorescence polarization (Fig. 2). An experiment was carried out in the presence of AdoHcy cofactor analog, which promotes formation of a specific complex between DNA and C5-MTases [34] and prevents cytosine deamination [35]. Binding isotherms for fGCGC/CGMG, fGCGC/CXMG, and fGCGCTCTC/CGMGAGAX duplexes were similar to hyperbolic curves commonly observed when proteins bind with DNA in 1 : 1 ratio. However, curves for duplexes XCGC/CGMGf and GCXC/CGMGf had a definite sigmoid shape indicating positive cooperativity. A similar picture, when Dnmt3a-CD bound with DNA containing 7,8-dihydro-8-oxoguanine (8-oxoG), was observed previously [30]. A model of consecutive binding of two Dnmt3a-CD dimers to 30-mer DNA duplex (Scheme) was used for Kd estimation for complexes between Dnmt3a-CD and O6meG-DNA as well as in case of complexes between the enzyme and 8-oxoG-DNA [30]. At that time, a complex between one DNA molecule and two Dnmt3a-CD dimers forms [30]. The assumption that Dnmt3a-CD dimer is a binding protein unit conforms to mutation analysis data showing that Dnmt3a-CD monomer does not possess catalytic activity and is not able to bind DNA [36, 37]. As opposed to the Hill model, in common use for analyzing data on cooperative processes, the model used (see Scheme and Eqs. (1)-(5)) allows taking into consideration a decrease in equilibrium concentrations of free DNA and the enzyme during complex formation. According to this model, Kd1 and Kd2 values were estimated (Table 2). For XCGC/CGMGf and GCXC/CGMGf duplexes, Kd1 was at least 10 times larger than Kd2, which indicates lowering of ES complex to stoichiometrically negligible quantities and prevents estimating definite values of individual constants. In these cases, however, values of the Kd1Kd2 product were calculated with high accuracy (Table 2). From the calculations it follows that Kd1 and Kd2 values depend on each other. This suggests that Dnmt3a-CD binding to XCGC/CGMGf and GCXC/CGMGf has apparent cooperative character. For fGCGC/CGMG substrate and duplexes containing O6meG in a methylated strand (fGCGC/CXMG and fGCGCTCTC/CGMGAGAX), Kd1 was not more than 9 times larger than Kd2. This also indicates that Dnmt3a-CD binding to the given substrate has apparent cooperative character because in the case of independent binding of two enzyme dimers to DNA the macroscopic ratio of constants (Kd2/Kd1) would be equal to 4 [38]. Thus, during formation of enzyme–substrate complex, two Dnmt3a-CD dimers sequentially bind to different sites of 30-mer DNA duplex.
Introduction of O6meG into already methylated DNA strand (GCGC/CXMG and GCGCTCTC/CGMGAGAX) leads to 1.3-fold increase and 6.7-fold decrease in Kd1Kd2 value. In case of duplexes containing damaged guanine in the methylated strand of recognition site (GCXC/CGMGf) or at the 5′-side from it (XCGC/CGMGf), Kd1Kd2 increased 73- and 130-fold. Thus, introduction of O6meG into DNA affects stability of enzyme–substrate complex but it does not explain variations in initial rates of the methylation reaction.Fig. 2. Binding curves of fluorescein-labeled DNA duplexes (10 nM) with Dnmt3a-CD in the presence of AdoHcy (0.1 mM). P, fluorescence polarization. Solid lines represent theoretical curves obtained from experimental data processing by Eqs. (1)-(5). Designations are as in Fig. 1.
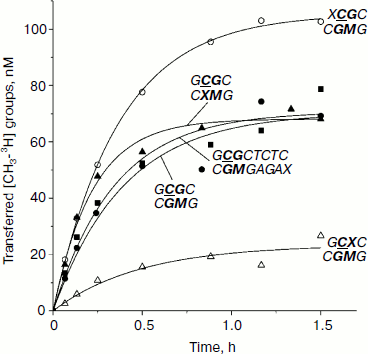
Integral kinetics of methylation of DNA duplexes containing O6meG by Dnmt3a-CD. Further, under conditions of single turnover (under 10-fold excess of the enzyme over the substrate), the kinetics of DNA methylation by Dnmt3a-CD MTase was studied (Fig. 3). The chosen concentration of Dnmt3a-CD (3 µM) satisfied saturation conditions (Fig. 2) and must have promoted formation of a complex between all free DNA and the enzyme. Values of rate constants of the methylation reaction (kst) were estimated using Eq. (6) (see “Materials and Methods”). For all duplexes, kst values were almost the same, except for the GCGC/CXMG duplex for which kst was 2.1 times larger compared with the undamaged substrate GCGC/CGMG (Table 2). However, yield of reaction product [CH3-3H]DNA, R, varied depending the position of the O6meG. At the same time, even in case of undamaged substrate GCGC/CGMG, R value was only 25%. These results relate with data obtained previously when the impact of 8-oxoG on Dnmt3a-CD methylation was studied, where reaction product yield was up to 100% with neither modified substrate due to formation of nonproductive enzyme–substrate complexes [30]. In the studied system, Dnmt3a-CD seems to form stable, slowly dissociating nonproductive complexes with DNA substrates (Fig. 4). In the case of GCXC/CGMG duplex, in which O6meG is located in the recognition site near to the target cytosine, R value decreased 3.2-fold in magnitude that corresponded to vo decrease. And in the case of XCGC/CGMG duplex containing O6meG near the recognition site, increase in vo value with simultaneous 1.4-fold increase of R value was observed.
Fig. 3. Methylation of DNA duplexes by Dnmt3a-CD under “single turnover” conditions. CDNA, 0.3 µM; CDnmt3a-CD, 3 µM; C[CH3-3H]AdoMet,1.2 µM. Solid lines represent theoretical curves obtained from experiment data processing by Eq. (6). Designations are as in Fig. 1.
Methylation of DNA duplexes containing O6meG by M.SssI. Procaryotic MTase SssI and Dnmt3a recognize the same DNA sequence [39]. To examine the impact of O6meG on the DNA methylation by M.SssI, vo values of methylation of DNA duplexes containing O6meG were measured using the given enzyme under steady-state kinetic conditions (DNA concentration was 25 times higher than that of M.SssI) (Fig. 5 and Table 2). As it is clear from the figure, M.SssI is more sensitive to the presence of O6meG in the recognition site of the enzyme. Thus, in case of GCXC/CGMG duplex containing damaged guanine residue in the recognition site at the 3′-side from the target cytosine, vo value was 25 times smaller than that for GCGC/CGMG. Replacement of guanine residue in the recognition site opposite to the methylated cytosine (GCGC/CXMG) led to 1.5-fold decrease in vo value. Introduction of O6meG into the outer region of the enzyme recognition site had practically no influence on vo values of corresponding DNA duplexes (XCGC/CGMG and GCGCTC/CGMGAGAX).Fig. 4. Binding of Dnmt3a-CD to DNA in productive (a) and nonproductive (b and c) orientations.
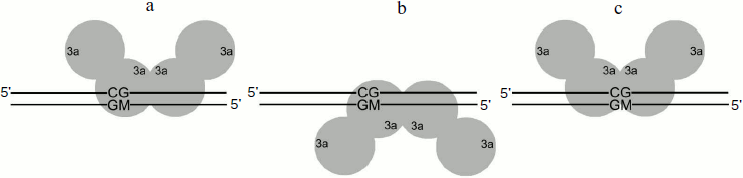
Fig. 5. Methylation of DNA duplexes by M.SssI. CDNA, 0.5 µM; CM.SssI, 20 nM; C[CH3-3H]AdoMet,1.2 µM. Designations are as in Fig. 1.

DISCUSSION
In the present work, it is shown that the appearance of very cytotoxic O6meG in DNA influences the functioning of murine Dnmt3a-CD. The effect of O6meG on the initial rate of methylation of 30-mer DNA duplexes catalyzed by murine Dnmt3a-CD MTase depends on the position of the damaged guanine residue relative to the CpG sequence and methylated cytosine. Introduction of O6meG into the CpG recognition site lowers methylation rate of the target cytosine located near the O6meG and raises methylation rate of the target cytosine opposed to the lesion. The presence of O6meG near the recognition site in the methylated strand increases the DNA methylation level. The ability of O6meG to stimulate DNA methylation opposite to the target cytosine correlates with data obtained for human Dnmt1 [27]. However, in contrast to Dnmt3a-CD, Dnmt1 loses its DNA methylation ability when O6meG is in a CpG region near the methylated cytosine [27]. In the case of M.SssI, a procaryotic C5 MTase recognizing CpG, this position also appears to be the most critical (Table 2). The presented data indicate the importance of the guanine residue adjoined with the methylated cytosine for recognition of the CpG sequence by C5-MTases.
Studying a stage of Dnmt3-CD binding to 30-mer DNA duplexes containing and not containing O6meG indicate cooperative character of the process. This observation conforms to recently obtained data about cooperative character of interaction between 146-mer DNA and Dnmt3a-CD/Dnmt3L (Dnmt3L is a regulatory factor) complex [37] and interaction between 208-mer DNA and full-size Dnmt3a [40]. The data (Fig. 2) were well described by a previously proposed model of sequential binding of two Dnmt3a-CD dimers with two different DNA sites (Scheme) [30]. The model was developed based on X-ray structure analysis data about homoassociation of Dnmt3a-CD or Dnmt3a-CD/Dnmt3L complex [36]. As on the model, Dnmt3a-CD possesses practically identical affinity with both binding sites in the case of undamaged substrate (GCGC/CGMG) and DNA duplex containing a damaged guanine residue at the distance of four nucleotide residues from the recognition site (GCGCTC/CGMGAGAX) (Table 2). Introduction of O6meG into the CpG site (GCXC/CGMG and GCGC/CXMG) or at the 5′-side from it (XCGC/CGMG) leads to reduction in Dnmt3a-CD affinity with the first binding site relative to the second (Kd1 > Kd2) (Table 2). At the same time, both deceleration (GCXC/CGMG) and acceleration (XCGC/CGMG and GCGC/CXMG) of methylation was observed for the given substrates. It should be noted that the substrate concentration (1.5 µM) used for vo value estimations (Fig. 1) was saturating in all cases. It follows that vo variations caused by replacement of guanine residues with O6meG in DNA are not due to alteration of Dnmt3a-CD affinity with damaged DNA.
Analysis of dependences of methylation level of DNA duplexes on time under single turnover conditions (Fig. 3) and R values (Table 2) allowed making suggestions about reasons for changes in vo values. Dnmt3a-CD was shown to form stable nonproductive complexes with DNA duplexes containing O6meG as well as with previously studied DNA duplexes containing another type of damaged guanine, 7,8-dihydro-8-oxoguanine [30]. In the case of hemimethylated substrates, formation of nonproductive complexes, in contrast to that of productive ones (Fig. 4a), seems to be due to Dnmt3a-CD binding to DNA at the methylated strand side (Fig. 4b). Dnmt3a-CD is able to bind with 30-mer substrate containing fully methylated CpG region [30]. Another kind of nonproductive Dnmt3a-CD binding to DNA is also possible when both of the enzyme dimers bind nonspecifically, i.e. none of the dimers interact with a CpG region (Fig. 4c). This supposition is confirmed by Dnmt3a-CD complex formation with 30-mer DNA duplex not containing recognition site (data not provided). The ratio of possible complex types depends on affinity between the enzyme and DNA site binding the first and, too, is likely to depend on binding rate, as formed complex between the enzyme and DNA dissociate very slowly [41] and Dnmt3a-CD does not seem to redistribute during the methylation reaction.
For GCGC/CXMG duplex, when a damaged guanine residue is located in already methylated strand opposite to the target cytosine, R value is similar to that of GCGC/CGMG, while vo value rises (Table 2). For GCXC/CGMG duplex, whose affinity to the enzyme and vo value were observed to decrease, R value was also observed to decrease. For (XCGC/CGMG) duplex R value increased, which corresponded to vo value increase. Except for GCGC/CXMG duplex, whose kst value increased, kst values of all other duplexes did not change. Variations in vo values observed when O6meG was in the methylated strand (GCXC/CGMG and XCGC/CGMG) seemed to be due to variations in the ratio of productive and nonproductive enzyme–substrate complexes, while when O6meG was in the strand opposite to the target cytosine kst increased (acceleration of the methylation reaction itself).
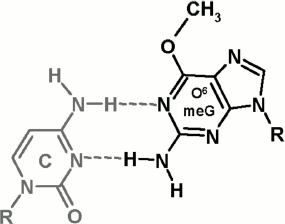
It is important to investigate molecular bases for the impact of O6meG on functioning of Dnmt3a-CD and M.SssI. The presence of O6meG opposite to cytosine in DNA double helix breaks its structure quite strongly: between the bases in C·O6meG pair, only two hydrogen bonds are formed (Fig. 6), cytosine and O6meG residues drift into the minor and major groove, respectively, and the structure of the sugar-phosphate backbone changes [42, 43]. The methyl group in O6 position is exhibited to the major groove. When bound with DNA, M.SssI makes contact with the N7 nitrogen atom of the guanine residue adjoined to the target cytosine and with internucleotide phosphates in the recognition site and near to it [44, 45]. From data of computer modeling of the complex between M.SssI, DNA, and AdoHcy, it follows that M.SssI contacts with the O6 oxygen atom of the guanine residue located opposite to the target cytosine [46]. Thus, the impact of O6meG on DNA methylation by MTase can be related with breakdown of M.SssI contacts with damaged DNA caused by local deformations of double helix structure as well as by steric barriers caused by the methyl group at O6 position. Relying on the fact that M.SssI and Dnmt3a-CD possess identical sequence specificity as well as on high homology of primary structure of C5 MTases and Dnmt3a-CD [20, 39], it can be supposed that deterioration of GCXC/CGMG methylation by Dnmt3a-CD is due to breakdown of the enzyme contact with the N7 nitrogen atom of the guanine residue located near the target cytosine.
Comparing damaging impact on Dnmt3a-CD and M.SssI functioning, M.SssI can be said to be more sensitive to the presence of O6meG in DNA as well as to its location relative to the recognition site and methylated cytosine. This fact suggests that the Dnmt3a-CD active center possesses better ability to adjust to damaged DNA than the procaryotic M.SssI. In the case of O6meG, such adjustment can be realized by steric tension reduction in the Dnmt3a-CD active center as a consequence of methyl group displacement relative to the N1–C6 bond of the damaged guanine residue. The possibility of such alterations in orientation of the O6meG methyl group was shown for human RNA polymerase II [13]. Thus, introduction of an O6meG residue, depending on its location, into DNA leads to small decrease or increase in Dnmt3a-CD DNA methylation. We cannot accept as fact that in vivo in the presence of Dnmt3L regulatory factor, which stabilizes Dnmt3a-CD catalytic loop [36, 42], the impact of O6meG on functioning of full-size Dnmt3a can be greater. For instance, despite the fact that in several cases in vitro, O6meG stimulates DNA methylation by Dnmt1 MTase [27], DNA hypomethylation was observed when human lymphoblasts were treated with methylating agent [26]. This can be connected with the presence of a regulatory mechanism of DNA methylation in cells, in which other DNA binding proteins are involved. Recently, it was established that, in cells, hemimethylated CpG sites are recognized by UHRF1 protein, which then attracts Dnmt1 to these sites [47]. Interacting with DNA, UHRF1 was shown to form numerous contacts with internucleotide phosphates both in the recognition site and near to it (from 5′- and 3′-side) and to the bases (also to O6 and N7 atoms of guanine opposite to the target cytosine) [48, 49]. Obviously, replacement of guanine in the CpG site or near to it with O6meG can break down the interaction of UHRF1 with DNA and, thereby, prevent the interaction of Dnmt1 with the given site. DNA hypomethylation observed in work [26] seems to be also caused by this mechanism. The mechanism of attraction of Dnmt3a to particular DNA sites in a cell is not studied yet; therefore, in the case of Dnmt3a, disturbance of DNA methylation through interaction with mediator proteins is also possible and requires further investigations.Fig. 6. Structure of C·O6meG nucleotide pair (from [42, 43]).
We thank Prof. A. Jeltsch for the plasmid containing the Dnmt3a-CD gene, Dr. B. Jack from New England BioLabs for the plasmid containing the M.SssI gene, as well as Prof. A. A. Baykov for valuable discussion.
This work was supported by the Russian Foundation for Basic Research (07-04-00583 and 08-04-01096) and joint program of US Civilian Research & Development Foundation and the Russian Foundation for Basic Research (RUB1-2919-MO-07/08-04-91109).
REFERENCES
1.Wogan, G. N., Hecht, S. S., Felton, J. S., Conney,
A. H., and Lawrence, A. L. (2004) Cancer Biol., 14,
473-486.
2.Martinez, G. R., Loureiro, A. P. M., Marques, S.
A., Miyamoto, S., Yamaguchi, L. F., Onuki, J., Almeida, E. A., Garcia,
C. C. M., Barbosa, L. F., Medeiros, M. H. G., and Mascio, P. D. (2003)
Mutat. Res., 544, 115-127.
3.Marnett, L. J., Riggins, J. N., and West, J. D.
(2003) J. Clin. Invest., 111, 583-593.
4.Povey, A. C., Badawi, A. F., Cooper, D. P., Hall,
C. N., Harrison, K. L., Jackson, P. E., Lees, N. P., O’Connor, P.
J., and Margison, G. P. (2002) J. Nutr., 132 (11
Suppl.), 3518S-3521S.
5.Xiao, W., and Samson, L. (1993) Proc. Natl.
Acad. Sci. USA, 90, 2117-2121.
6.Beranek, D. T. (1990) Mutat. Res.,
231, 11-30.
7.Abdulnur, S. F., and Flurry, R. L. (1976)
Nature, 264, 369-370.
8.Saffhill, R., Margison, G. P., and O’Connor,
P. J. (1985) Biochim. Biophys. Acta, 823, 111-145.
9.Margison, G. P., and Santibanez-Koref, M. F. (2002)
Bioessays, 24, 255-266.
10.Kaina, B., Ziouta, A., Ochs, K., and Coquerelle,
T. (1997) Mutat. Res., 381, 227-241.
11.Pourquier, P., Waltman, J. L., Urasaki, Y.,
Loktionova, N. A., Pegg, A. E., Nitiss, J. L., and Pommier, Y. (2001)
Cancer Res., 61, 53-58.
12.Yamini, B., Yu, X., Dolan, M. E., Wu, M. H.,
Kufe, D. W., and Weichselbaum, R. R. (2007) Cancer Res.,
67, 6889-6898.
13.Dimitri, A., Burns, J. A., Broyde, S., and
Scicchitano, D. A. (2008) Nucleic Acids Res., 36,
6459-6471.
14.Leonard, G. A., Thomson, J., Watson, W. P., and
Brown, T. (1990) Proc. Natl. Acad. Sci. USA, 87,
9573-9576.
15.Pauly, G. T., and Moschel, R. C. (2001) Chem.
Res. Toxicol., 14, 894-900.
16.Topal, M. D. (1988) Carcinogenesis,
9, 691-696.
17.Mitra, G., Pauly, G. T., Kumar, R., Pei, G. K.,
and Hughes, S. H. (1989) Proc. Natl. Acad. Sci. USA, 86,
8650-8654.
18.Bird, A. (2002) Genes Dev., 16,
6-21.
19.Lichtenstein, A. V., and Kisseljova, N. P. (2001)
Biochemistry (Moscow), 66, 235-255.
20.Goll, M. G., and Bestor, T. H. (2005) Annu.
Rev. Biochem., 74, 481-514.
21.Kim, G. D., Ni, J., Kelesoglu, N., Roberts, R.
J., and Pradhan, S. (2002) EMBO J., 21, 4183-4195.
22.Fatemi, M., Hermann, A., Gowher, H., and Jeltsch,
A. (2002) Eur. J. Biochem., 269, 4981-4984.
23.Metivier, R., Gallais, R., Tiffoche, C., le
Peron, C., Jurkowska, R. Z., Carmouche, R. P., Ibberson, D., Barath,
P., Demay, F., Reid, G., Benes, V., Jeltsch, A., Gannon, F., and
Salbert, G. (2008) Nature, 452, 45-50.
24.Jirtle, R. L., and Skinner, M. K. (2007) Nat.
Rev. Genet., 8, 253-262.
25.Holliday, R. (1987) Mutat. Res.,
181, 215-217.
26.Boehm, T. L., and Drahovsky, D. (1981)
Carcinogenesis, 2, 39-42.
27.Tan, N. W., and Li, B. F. (1990)
Biochemistry, 29, 9234-9240.
28.Hepburn, P. A., Margison, G. P., and Tisdale, M.
J. (1991) J. Biol. Chem., 266, 7985-7987.
29.Baskunov, V. B., Subach, F. V., Kolbanovskiy, A.,
Kolbanovskiy, M., Eremin, S. A., Johnson, F., Bonala, R., Geacintov, N.
E., and Gromova, E. S. (2005) Biochemistry, 44,
1054-1066.
30.Maltseva, D. V., Baykov, A. A., Jeltsch, A., and
Gromova, E. S. (2009) Biochemistry, 48, 1361-1368.
31.Darii, M. V., Kirsanova, O. V., Drutsa, V. L.,
Kochetkov, S. N., and Gromova, E. S. (2007) Mol. Biol. (Moscow),
41, 110-117.
32.Subach, O. M., Khoroshaev, A. V., Gerasimov, D.
N., Baskunov, V. B., Shchyolkina, A. K., and Gromova, E. S. (2004)
Eur. J. Biochem., 271, 2391-2399.
33.Fersht, A. (1999) Structure and Mechanism in
Protein Science: a Guide to Enzyme Catalysis and Protein Folding,
2nd Edn., W. H. Freeman and Company, USA, pp. 199-201.
34.Wyszynski, M. W., Gabbara, S., Kubareva, E. A.,
Romanova, E. A., Oretskaya, T. S., Gromova, E. S., Shabarova, Z. A.,
and Bhagwat, A. S. (1993) Nucleic Acids Res., 21,
295-301.
35.Wyszynski, M., Gabbara, S., and Bhagwat, A. S.
(1994) Proc. Natl. Acad. Sci. USA, 91, 1574-1578.
36.Jia, D., Jurkowska, R. Z., Zhang, X., Jeltsch,
A., and Cheng, X. (2007) Nature, 449, 248-251.
37.Jurkowska, R. Z., Anspach, N., Urbanke, C., Jia,
D., Reinhardt, R., Nellen, W., Cheng, X., and Jeltsch, A. (2008)
Nucleic Acids Res., 36, 6656-6663.
38.Dixon, M., Webb, E. C., Thorne, C. J. R., and
Tipton, K. F. (1979) Enzymes, 3rd Edn., Longman Group Ltd.,
London, pp. 140-148.
39.Nur, I., Szyf, M., Razin, A., Glaser, G., Rottem,
S., and Razin, S. (1985) J. Bacteriol., 164, 19-24.
40.Robertson, A. K., Geiman, T. M., Sankpal, U. T.,
Hager, G. L., and Robertson, K. D. (2004) Biochem. Biophys. Res.
Commun., 322, 110-118.
41.Gowher, H., Liebert, K., Hermann, A., Xu, G., and
Jeltsch, A. (2005) J. Biol. Chem., 280,
13341-13348.
42.Swann, P. F. (1990) Mutat. Res.,
233, 81-94.
43.Spratt, T. E., and Levy, D. E. (1997) Nucleic
Acids Res., 25, 3354-3361.
44.Renbaum, P., and Razin, A. (1995) J. Mol.
Biol., 248, 19-26.
45.Darii, M. V., Cherepanova, N. A., Subach, O. M.,
Kirsanova, O. V., Rasko, T., Slaska-Kiss, K., Kiss, A., Deville-Bonne,
D., Reboud-Ravaux, M., and Gromova, E. S. (2009) Biochim. Biophys.
Acta, 1794, 1654-1662.
46.Koudan, E. V., Bujnicki, J. M., and Gromova, E.
S. (2004) J. Biomol. Struct. Dyn., 22,
339-345.
47.Jeltsch, A. (2008) Nat. Struct. Mol.
Biol., 15, 1003-1004.
48.Hashimoto, H., Horton, J. R., Zhang, X., Bostick,
M., Jacobsen, S. E., and Cheng, X. (2008) Nature, 455,
826-829.
49.Arita, K., Ariyoshi, M., Tochio, H., Nakamura,
Y., and Shirakawa, M. (2008) Nature, 455, 818-821.