Chemical and Enzymatic Probing of Spatial Structure of the Omega Leader of Tobacco Mosaic Virus RNA
N. E. Shirokikh, S. Ch. Agalarov, and A. S. Spirin*
Laboratory of Mechanisms of Protein Biosynthesis, Institute of Protein Research, Russian Academy of Sciences, 142290 Pushchino, Moscow Region, Russia; fax: (499) 632-7871; E-mail: spirin@vega.protres.ru* To whom correspondence should be addressed.
Received November 12, 2009; Revision received December 3, 2009
The 5′-untranslated sequence of tobacco mosaic virus RNA – the so-called omega leader – exhibits features of a translational enhancer of homologous and heterologous mRNAs. The absence of guanylic residues, the presence of multiple trinucleotide CAA repeats in its central region, and the low predictable probability of the formation of an extensive secondary structure of the Watson–Crick type were reported as the peculiarities of the primary structure of the omega leader. In this work we performed chemical and enzymatic probing of the secondary structure of the omega leader. The isolated RNA comprising omega leader sequence was subjected to partial modifications with dimethyl sulfate and diethyl pyrocarbonate and partial hydrolyses with RNase A and RNase V1. The sites and the intensities of the modifications or the cleavages were detected and measured by the primer extension inhibition technique. The data obtained have demonstrated that RNase A, which attacks internucleotide bonds at the 3′ side of pyrimidine nucleotides, and diethyl pyrocarbonate, which modifies N7 of adenines not involved in stacking interactions, weakly affected the core region of omega leader sequence enriched with CAA-repeats, this directly indicating the existence of a stable spatial structure. The significant stability of the core region structure to RNase A and diethyl pyrocarbonate was accompanied by its complete resistance against RNase V1, which cleaves a polyribonucleotide chain involved in Watson–Crick double helices and generally all A-form RNA helices, thus being an evidence in favor of a non-Watson–Crick structure. The latter was confirmed by the full susceptibility of all adenines and cytosines of the omega polynucleotide chain to dimethyl sulfate, which exclusively modifies N1 of adenines and N3 of cytosines not involved in Watson–Crick interactions. Thus, our data have confirmed that (1) the regular (CAA)n sequence characteristic of the core region of the omega leader does form stable secondary structure, and (2) the structure formed is not the canonical double helix of the Watson–Crick type.
KEY WORDS: omega leader of TMV RNA, regular (САА)n polyribonucleotide, RNA triple helix, chemical modification of RNA, enzymatic cleavage of RNA, primer extension inhibition, diethyl pyrocarbonate, dimethyl sulfate, RNase A, RNase V1DOI: 10.1134/S0006297910040024
Abbreviations: AMV, avian myeloblastosis virus; cDNA, complementary DNA; CXR, 5(6)-carboxy-X-rhodamine; FAM, 6-carboxy-4′,5′-dichloro-2′,7′-dimethoxyfluorescein; Hepes, 4-(2-hydroxyethyl)-1-piperazineethanesulfonic acid; TMV, tobacco mosaic virus; Tris, tris(hydroxymethyl)aminomethane.
In eukaryotes 5′-untranslated regions of mRNA markedly influence
the translation efficiency of the encoding part of RNA carrying these
regions. The 5′-untranslated region of tobacco mosaic virus (TMV)
genomic RNA contains the so-called omega sequence, a powerful enhancer
of the translation of this RNA (“translational enhancer”)
[1]. In recombinant constructs the leader omega
sequence enhances translation of foreign RNAs both in vivo and
in vitro (in cell-free systems) and provides for efficient
cap-independent translation initiation [2]. Omega
leader can act as a translational enhancer in various cell types, both
plant and animal, as well as in different cell-free translation systems
[3-10].
The nucleotide sequence of the omega leader from RNA of TMV strain U1 is shown in Fig. 1 [11]. This sequence has an unusual primary structure. First, the omega sequence is almost completely deprived of guanylic residues (G): with the exclusion of the first residue, adjacent to the cap-structure at the 5′ end of TMV RNA, there are no other guanylic residues in the almost 70 nucleotide-long leader chain. Second, more than one-third of the omega sequence is comprised by its central part consisting only of adenylic and cytidylic residues. Third, adenylic and cytidylic residues of the central part as well as those of adjacent to 5′ and 3′ proximal parts of the omega sequence are mainly grouped in CAA triplets. Thus, the central part is an almost regular sequence consisting of CAA nucleotide triplets arranged in succession. The CAA-containing part is restricted on both sides by U-rich regions, a short one on the 5′-side and a longer one on the 3′-side. On the whole, the features exhibited by the omega leader nucleotide sequence point to the impossibility of the formation of a stable secondary structure based on canonical Watson–Crick base pairing of G:C and A:U type. This situation was for a long time an argument in favor of the supposition that it is an unstructured nature of the omega leader that provides for its easy accessibility for ribosomes, and as a result, its properties of a translational enhancer [12, 13]. However, it was shown in our laboratory that unstructured sequences, for which the absence of secondary structure in solution was really proved, such as poly(U) [14], do not exhibit properties of translational enhancers, at least in translation systems of higher eukaryotes [15].
On the other hand, we have previously studied physical properties of the RNAs that were synthesized copies of the leader omega sequence of TMV RNA and the regular polyribonucleotide (CAA)19, the latter modeling the nucleotide sequence of the central part of the omega leader [16]. It was shown by UV-spectroscopy and analytical centrifugation that both the complete omega sequence and the homolog of its CAA part have a stable and cooperatively melted structure, and their sedimentation coefficients correspond to those of compactly folded RNAs of the same lengths. At the same time, the polynucleotide of the same composition and of the same length but with random sequence of nucleotides, poly(A2,C), did not exhibit such properties and behaved as a disordered non-compact polymer [16]. These data show that the compact structure of the omega sequence can be formed by the regular nucleotide sequence (CAA)n representing the core part of the omega sequence. Theoretical analysis of possibilities of the regular polyribonucleotide (CAA)n to fold into a compact structure due to formation of hydrogen bond pairs of non-Watson–Crick type resulted in creation of the triple helix model in which bases are bound to each other by hydrogen bond pairs into alternating triads A:C:A, C:A:A, and A:A:C [17].Fig. 1. The 5′-untranslated omega sequence of genomic RNA of TMV strain U1 [11]. The region containing successive (CAA)n repeats is underlined. The initiation AUG codon at the beginning of the open reading frame of the North American firefly luciferase gene is also underlined and shown in bold.
To test the type of possible structural organization of the omega leader RNA, we used standard methods of partial chemical modification of nucleotides and restricted enzymatic degradation, which are widely used in testing RNA spatial structures (see, for example, [18, 19]). The data obtained in this work confirmed experimentally the absence of double helices with Watson–Crick base pairs. At the same time, analysis of the data has shown the presence of an extended region involved in formation of a non-canonical secondary structure in the central part of the omega sequence corresponding to the (CAA)-containing core of the omega leader.
MATERIALS AND METHODS
Purification of plasmid containing omega sequence of TMV RNA. Plasmid construct pTZ10omegaLUC [3] copying omega sequence of TMV RNA (Fig. 2) was amplified in Escherichia coli DH5α cells. Cells were lysed and the lysate was neutralized using appropriate solutions from the PureYield Plasmid Midiprep System kit (Promega, USA). Cell debris, denatured proteins, and genomic DNA were pelleted by centrifugation, and the resulting supernatant was applied on adsorption columns from the same reagent kit. Plasmid DNA was eluted from the columns by deionized water and purified from protein contaminants by phenol extraction. Sodium acetate (pH 5.0) up to 300 mM and 2.5 volumes of 96% ethanol were added to the solution and DNA was collected by centrifugation at 15,000g for 15 min, dried, and dissolved in deionized water. The integrity and purity of the pTZ10omegaLUC plasmid were checked by electrophoresis in 1.5% agarose gel (stained with ethidium bromide).
Transcription of RNA using T7 RNA polymerase and its purification. Purified plasmid pTZ10omegaLUC was cleaved by restriction endonuclease XbaI in the region following the luciferase gene of the American firefly Photinus pyralis (Fig. 2). A sample of 200 µg of plasmid pTZ10omegaLUC was incubated with 400 units of endonuclease XbaI (Fermentas, Lithuania) at 37°C for 4 h in Tango 1X buffer (Fermentas), after which the reaction mixture was twice extracted with phenol. After phenol extraction, DNA was precipitated from the reaction mixture by addition of sodium acetate (pH 5.0) to 300 mM, and 2.5 volumes of 96% ethanol and the precipitate was centrifuged at 15,000g for 15 min, after which the pellet was dried and dissolved in deionized water. Completeness of the cleavage reaction was estimated by electrophoresis in 2% agarose gel (ethidium bromide staining).Fig. 2. Scheme of the use of pTZ10omegaLUC construct for the synthesis of RNA containing omega sequence of RNA from TMV strain U1 (shown in bold). The relative position of the restriction site of endonuclease XbaI used for linearization of the plasmid is indicated. Nucleotide sequence of the 5′-untranslated RNA region (total RNA length is 136 nucleotides) containing the omega leader RNA is shown at the bottom; the omega sequence within the 5′-untranslated RNA region is shown in bold, and the initiation codon of the luciferase gene is shown in bold and underlined.
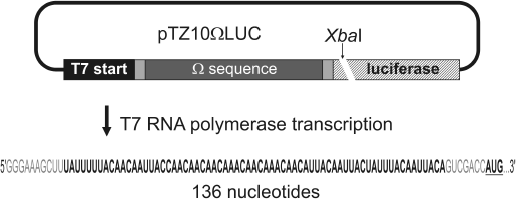
RNA of total length of 136 nt containing the omega sequence was obtained by transcription of the above-mentioned template DNA by T7 RNA polymerase (Fig. 2) under the conditions recommended in the literature [20, 21] with slight modifications. The mixture for the transcription reaction contained 80 mM Tris-acetate, pH 7.5, 10 mM KCl, 22.2 mM Mg(CH3COO)2, 20 mM dithiothreitol, 20 mM β-mercaptoethanol, 2 mM spermidine, 0.01% (v/v) Triton X-100, 0.2 mM EDTA, 4 mM each nucleoside triphosphate (ATP, GTP, CTP, UTP) (Fermentas), 1 unit/µl RNase inhibitor RiboLock (Fermentas), 0.1 µg/µl template DNA, and 12 units/µl T7 RNA polymerase (Fermentas) in 500 µl total volume. The reaction mixture was incubated for 3 h at 37°C, then extracted twice by acidic (pH 5.5) phenol, and nucleic acids were precipitated by addition of ammonium acetate to 2 M and 2.5 volumes of 96% ethanol. The precipitated nucleic acids were collected by centrifugation at 15,000g for 15 min in the cold, and the pellets were dried under vacuum and dissolved in 250 µl of deionized water. To purify the preparation from the initial template DNA, the solution was brought to 10 mM Tris-acetate, pH 7.5, 2.5 mM Mg(CH3COO)2, 0.1 mM CaCl2, 2 units/µl RNase inhibitor RiboLock (Fermentas), and 1.5 units/µl DNase I (Boehringer Mannheim, Germany). The reaction mixture was incubated for 1 h at 37°C, then it was extracted twice with acidic phenol (pH 5.5), and nucleic acids were precipitated by addition of 300 mM sodium acetate (pH 5.0) and 2.5 volumes of 96% ethanol. Precipitated nucleic acids were collected by centrifugation at 15,000g for 15 min in the cold, and pellets were dissolved in 200 µl of deionized water. The solution was supplemented with 68 µl of saturated LiCl solution, the mixture was incubated for 1 h at 0°C, and the precipitate was collected by centrifugation at 15,000g for 15 min in the cold. The pellet was dissolved in 500 µl of deionized water, then the solution was brought to 2 M ammonium acetate and 2.5 volumes of 96% ethanol were added. The resulting mixture was centrifuged at 15,000g for 15 min in the cold, the pellet after centrifugation was dissolved in 500 µl of deionized water, and the resulting solution was brought to 300 mM sodium acetate (pH 5.0) and 2.5 volumes of 96% ethanol were added. The resulting mixture was centrifuged at 15,000g for 15 min in the cold, the pellet was washed with 80% ethanol and dissolved in 150 µl of deionized water, and the solution was frozen. The purity and homogeneity of the RNA preparation was estimated by electrophoresis in 6% denaturing polyacrylamide gel (staining by toluidine blue).
RNA modification by dimethyl sulfate. In the case of modification by dimethyl sulfate, 0.5 µl dimethyl sulfate was added to 50 µl solution containing buffer A (40 mM Hepes-KOH, pH 7.5, 50 mM KCl, 0.5 mM EDTA) and 2 µg RNA. The reaction was carried out for 30 sec at 25°C, then stopped by addition of 200 mM Tris-HCl, 10 µg total tRNA from Escherichia coli, and 2.5 volumes of 96% ethanol, and the mixture was cooled in liquid nitrogen.
RNA modification by diethyl pyrocarbonate. For modification by diethyl pyrocarbonate, 4 µl diethyl pyrocarbonate was added to 20 µl solution containing buffer A and 2 µg RNA. The reaction was run for 5 min at 25°C with continuous mixing, after which it was stopped by addition of 10 µg total tRNA of Escherichia coli and 2.5 volumes of 96% ethanol, and then cooled in liquid nitrogen.
RNA cleavage by RNase V1. For partial cleavage by RNase V1, reaction mixture (50 µl) containing buffer A, 10 mM Mg(OAc)2, 4 µg RNA, and 0.075 ng/µl RNase was kept for 20 min at 25°C, then the solution was twice extracted with phenol. Acidic (pH 5.0) 300 mM sodium acetate and 2.5 volumes of 96% ethanol were added to the aqueous phase, and the mixture was cooled to –20°C.
RNA cleavage by RNase A. In the case of partial cleavage by RNase A, reaction mixture (50 µl) containing buffer A, 4 µg RNA, and 10–6 unit/µl RNase was incubated for 5 min at 25°C, and then the solution was twice extracted by phenol. Acidic sodium acetate (pH 5.0) (final concentration 300 mM) and 2.5 volumes of 96% ethanol were added to the aqueous phase, and then the mixture was cooled to –20°C.
RNA purification after modification or cleavage. After precipitation with ethanol, RNA in all cases was collected by centrifugation at 14,000g in the cold for 15 min, and the pellet was washed with 80% ethanol, dried in vacuum, and dissolved in 10 µl deionized water.
Reverse transcription. The reaction of reverse transcription was carried out mainly as described previously [22, 23]. Reaction mixture for reverse transcription of the total volume 20 µl contained 1 µg of untreated RNA (control) or RNA after treatment (as described previously), 100 pmol of fluorescence-labeled DNA primer (5′-FAM-GGGCCTTTCTTTATG), 1× buffer for reverse transcriptase of avian myeloblastosis virus (AMV) recommended by Promega (USA) (50 mM Tris-HCl, pH 8.3 at 25°C, 50 mM KCl, 10 mM MgCl2, 0.5 mM spermidine, 10 mM dithiothreitol), 0.5 mM of each deoxynucleotide triphosphate (dATP, dGTP, dTTP, dCTP), 1 unit/µl RNase inhibitor RiboLock (Fermentas), and 0.33 unit/µl AMV reverse transcriptase from Promega. The mixture was incubated for 20 min at 37°C, then 130 µl solution for reaction stopping (1% SDS, 10 mM EDTA) was added, and the mixture was extracted by an equal volume of phenol. The nucleic acid fraction was precipitated from the aqueous phase by 300 mM acidic (pH 5.0) sodium acetate and 2.5 volumes of 96% ethanol and collected by centrifugation at 14,000g for 15 min in the cold; 20-30 µg of total purified tRNA of Escherichia coli from Serva (USA) was used as co-precipitator. The nucleic acid precipitates were dried at 45°C for 15 min and dissolved in 50 µl 90% formamide with 1× TBE buffer (89 mM Tris-base, 89 mM boric acid, and 2 mM EDTA).
Capillary electrophoresis, fluorescence detection, and analysis of signal from separated cDNA transcripts. Immediately before separation the samples (see above “Reverse transcription”) were additionally diluted 2 to 10 times (depending on the amount of cDNA corresponding to the maximal fluorescence peak) by 90% formamide containing 1× TBE buffer and supplemented with Fluorescent Ladder 60-400 bases (CXR) fluorescent DNA length markers (Promega) (0.3-0.6 µl marker per 15 µl sample). Then samples were heated at 99°C for 2 min. The total volume of each sample introduced in one well of a standard 96-well plate was 15 µl. Fluorescence-labeled cDNA in samples was separated and analyzed on ABI PRISM 3100-Avant Genetic Analyzer apparatus for capillary electrophoresis from Applied Biosystems (USA), mainly according to recommendations of the manufacturer, as described in [22, 23]. The data were processed using GeneMarker 1.5 software from SoftGenetics (USA). The fluorescence distribution profiles of separated cDNA were aligned with initial RNA sequence using calibration of separation by the lengths of marker DNA.
RESULTS AND DISCUSSION
Experiments were performed according to the following scheme. The RNA containing nucleotide sequence of the omega leader (omega RNA) was treated with modifying or cleaving agents at room temperature (25°C) in buffer conditions identical to those described in the literature [16] (40 mM Hepes-KOH, pH 7.5, 50 mM KCl, 0.5 mM EDTA) except for reactions with RNase V1. Since RNase V1 is active only in the presence of magnesium ions, 10 mM magnesium acetate was additionally introduced into the corresponding reaction buffer. Conditions for all reactions were adjusted to provide for modification or hydrolysis of less than half of the RNA molecules in each reaction mixture. Positions of modification or cleavage sites were determined using analysis of lengths of the cDNA-products of reverse transcription (method of inhibition of primer extension) synthesized on the modified or partially cleaved omega RNA template. To achieve this, the DNA primer carrying a fluorescent group at the 5′ terminus was added to the reaction mixture containing all four deoxyribonucleotide triphosphates and AMV reverse transcriptase. The primer annealing site was located within the coding region of the luciferase gene at the 3′ end of the RNA molecule and was designed to provide efficient resolution of reverse transcription products. Extension of the DNA primer on the control RNA chain led to reading out of the whole remaining RNA sequence up to its 5′ end. In the case of partially cleaved or modified omega RNA, additional stops of reverse transcriptase occurred at the sites of chain breaks or near the sites of nucleotide modifications. In such cases shortened DNA copies of omega RNA were formed. Lengths of synthesized shortened cDNAs directly indicated sites of cleavage or modification in the initial RNA, while the amount of shortened cDNA molecules showed intensity of the cleavage or modification at the corresponding sites. Capillary electrophoresis with detection of fluorescent signal of reverse transcription cDNA products in gel was used for analysis of lengths and amounts of cDNA generated during reverse transcription [22-26]. The distribution profiles of fluorescence of separated cDNAs were aligned with initial RNA sequence using calibration of separation by the DNA markers of different lengths. Figure 3 (a-e) shows the dependence of the fluorescence intensities of the cDNAs of particular lengths on the initial RNA nucleotide position after the data processing.
Figure 3a shows the dependence of cDNA fluorescence of the initial RNA sequence after primer elongation by reverse transcriptase on untreated (control) RNA. In this experiment a group of peaks corresponding to formation of the full-size cDNA transcript is prevalent. There are no additional sites of polymerase stops along the RNA chain.Fig. 3. Results of chemical modifications and enzymatic cleavages of omega RNA. Control unmodified omega RNA (a) or omega RNA after modification (b-e; see text) were used for reverse transcription, where fluorescence-labeled DNA primer (FAM) was used. The fluorescence-labeled cDNA was separated by gel electrophoresis in capillary tubes. Fluorescence profiles of separated cDNAs were read from the gel and compared with the initial RNA sequence using fluorescent length markers (CXR) added to the samples. The figure shows the cDNA fluorescence plotted against the corresponding nucleotide positions of the initial omega RNA. All plots are normalized to total FAM fluorescence in the gel. The scale on ordinate axis is chosen so that the shortened cDNA formed as a result of reverse transcriptase stopping at the modification or cleavage sites were well visualized. The fluorescence peak corresponding to the full-size cDNA (at 5′-end of omega RNA sequence) is shown incompletely to meet scaling requirements. Numbers near the peak of the full-size cDNA show the amount of full-size transcripts of omega RNA (free of hydrolysis or modifications) as % of total amount of transcripts from this RNA. The gray area shows the region of omega RNA containing successive (CAA)n repeats. a) Unmodified (control) omega RNA; b) omega RNA modified by dimethyl sulfate; c) omega RNA modified by diethyl pyrocarbonate; d) partial cleavage of omega RNA by RNase V1; e) partial cleavage of omega RNA by RNase A.
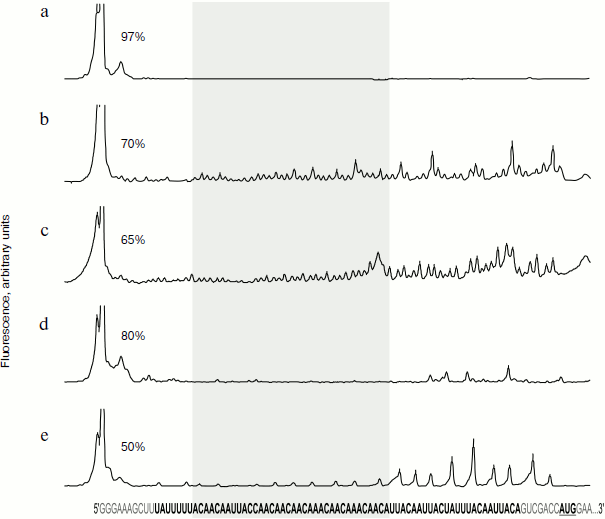
Dimethyl sulfate can attack the nitrogen atom in the first position of adenine or third position of cytosine. These atoms are necessary for hydrogen bond formation upon canonical (Watson–Crick) base pairing. Thus, dimethyl sulfate has to modify RNA only in sites free of canonical base pairing [27-29]. Numerous additional sites of reverse transcriptase stops are generated on RNA in the case of omega RNA modification by dimethyl sulfate (Fig. 3b). Modifications are distributed here along the whole RNA strand and practically all adenines and cytosines are modified. Thus, there are no extended regions with Watson–Crick base interactions in the omega RNA structure.
Diethyl pyrocarbonate can modify the adenine nitrogen atom in the seventh position if the base is not involved in stacking interactions, for example, in the case of the absence of A-form helical structure of the RNA [27-32]. The distribution of modification sites along the RNA after its treatment by diethyl pyrocarbonate is shown in Fig. 3c. In this case mainly the 3′-proximal part of the omega RNA is modified. The 5′ region of omega RNA is relatively weakly modified. This implies that in the 5′ region the stacking of bases is possible on an extended part. On the other hand, the region of relatively low modification includes almost the entire regular omega RNA part with CAA repeats.
RNase V1 is able to catalyze cleavage of the phosphoester bond of any nucleotide residues incorporated in A-form helices [28, 29, 33]. One can see that upon RNase V1 cleavage of omega RNA (Fig. 3d), hydrolysis of the sugar-phosphate chain near the 5′-terminal end of the omega sequence occurs, and a group of isolated peaks is formed, which are indicative of chain cleavage in the 3′ part of the molecule. The complete absence of cleavage sites along the whole length of the central part, containing regular CAA repeats also confirms the absence of an A-form RNA helix in this region. At the same time, formation of some short A-form helices, separated by non-helical RNA regions, is probable in the 3′-proximal part of the omega sequence.
RNase A mainly cleaves single-stranded RNA in the sites following pyrimidine nucleotide residues, which are not incorporated into helices and form no tertiary interactions [34, 35]. The result of omega RNA cleavage by RNase A is shown in Fig. 3e. The most intensive omega RNA cleavage occurs in the 3′-proximal third of the chain rich in uridylic residues. In the 5′-proximal and central parts of the molecule the amount of RNase A cleavage sites is relatively low. This part of omega RNA includes the regular region of the molecule with CAA repeats. The resistance of 5′-proximal and central parts of omega RNA to RNase A directly points to existence of some stable secondary structure in this region, whereas the 3′-proximal part is evidently single-stranded and available for the cleavage. It should be noted that diethyl pyrocarbonate and RNase A affect approximately the same, 3′-proximal, part of omega RNA polyribonucleotide chain. The 5′-proximal half and central part of the omega sequence, including the ordered regular region with CAA repeats, are weakly susceptible to their attack. Relative structural stability of the central part against these agents is also accompanied by its complete resistance to RNase V1, cleaving polynucleotide strands within Watson–Crick double helices.
Hence, the experimental data shown in this paper well agree with the recently proposed model of structural organization of polyribonucleotides containing consecutive CAA repeats [17]. They confirm that, first, regular sequence (CAA)n characteristic of the omega leader central part is able to generate a stable secondary structure, and second, the structure formed is not a canonical double helix of a Watson–Crick type or anything similar in the character of base interactions. The model suggests the folding of RNA regions containing successive (CAA)n repeats into a relatively stable and compact triple helix. Data of this work do not contradict the proposed model. The high sedimentation coefficient of the omega leader and a higher thermodynamic stability of this RNA, as compared to the polyribonucleotide consisting of only regular (CAA)n repeats [16], agree with the previously published data on enzymatic testing showing a higher, compared to polyribonucleotide (CAA)n, resistance of omega RNA to nucleases cleaving single-stranded RNA [9]. Additional structural stabilization of the omega RNA region with CAA repeats can be attained by the interactions with 3′-terminal U-rich region of omega RNA. On the other hand, the described type of triple helices have a relatively short length [17], which should result in inevitable structural fluctuations making nitrogen atoms (N1 of adenine and N3 of cytosine) in the omega RNA accessible for dimethyl sulfate attack.
The authors are grateful to A. B. Poltaraus for methodical help, to M. V. Kryuchkov for assistance in some experiments, and to A. V. Efimov for discussion and advice during experiments and manuscript preparation.
This work was supported by the Russian Foundation for Basic Research (grants 06-04-48964-a, 09-04-01729-a, 09-04-00537-a, and 09-04-01726-a), by the Russian Federation State Program for Support of Leading Research Schools NSh-4610.2008.4, and by a grant from the Cell and Molecular Biology Program of the Presidium of the Russian Academy of Sciences.
REFERENCES
1.Sleat, D. E., Gallie, D. R., Jefferson, R. A.,
Bevan, M. W., Turner, P. C., and Wilson, T. M. (1987) Gene,
60, 217-225.
2.Gallie, D. R. (2002) Nucleic Acids Res.,
30, 3401-3411.
3.Alekhina, O. M., Vassilenko, K. S., and Spirin, A.
S. (2007) Nucleic Acids Res., 35, 6547-6559.
4.Zeyenko, V. V., Ryabova, L. A., Gallie, D. R., and
Spirin, A. S. (1994) FEBS Lett., 354, 271-273.
5.Saejung, W., Fujiyama, K., Takasaki, T., Ito, M.,
Hori, K., Malasit, P., Watanabe, Y., Kurane, I., and Seki, T. (2007)
Vaccine, 25, 6646-6654.
6.Schmitz, J., Prufer, D., Rohde, W., and Tacke, E.
(1996) Nucleic Acids Res., 24, 257-263.
7.Gallie, D. R., Walbot, V., and Hershey, J. W.
(1988) Nucleic Acids Res., 16, 8675-8694.
8.Gallie, D. R., Sleat, D. E., Watts, J. W., Turner,
P. C., and Wilson, T. M. (1988) Nucleic Acids Res., 16,
883-893.
9.Tzareva, N. V., Makhno, V. I., and Boni, I. V.
(1994) FEBS Lett., 337, 189-194.
10.Kopeina, G. S., Afonina, Z. A., Gromova, K. V.,
Shirokov, V. A., Vasiliev, V. D., and Spirin, A. S. (2008) Nucleic
Acids Res., 36, 2476-2488.
11.Kukla, B. A., Guilley, H. A., Jonard, G. X.,
Richards, K. E., and Mundry, K. W. (1979) Eur. J. Biochem.,
98, 61-66.
12.Gallie, D. R., and Walbot, V. (1992) Nucleic
Acids Res., 20, 4631-4638.
13.Mundry, K. W., Watkins, P. A., Ashfield, T.,
Plaskitt, K. A., Eisele-Walter, S., and Wilson, T. M. (1991) J. Gen.
Virol., 72 (Pt. 4), 769-777.
14.Saenger, W. (1984) in Principles of Nucleic
Acid Structure, Springer, New York.
15.Gudkov, A. T., Ozerova, M. V., Shiryaev, V. M.,
and Spirin, A. S. (2005) Biotechnol. Bioeng., 91,
468-473.
16.Kovtun, A. A., Shirokikh, N. E., Gudkov, A. T.,
and Spirin, A. S. (2007) Biochem. Biophys. Res. Commun.,
358, 368-372.
17.Efimov, A. V., and Spirin, A. S. (2009)
Biochem. Biophys. Res. Commun., 388, 127-130.
18.Huntzinger, E., Possedko, M., Winter, F., Moine,
H., Ehresmann, C., Romby, P., (2008) in Handbook of RNA
Biochemistry (Hartmann, R. K., Bindereif, A., Schon, A., and
Westhof, E., eds.) Wiley-VCH Verlag, Weinheim, pp. 151-171.
19.Merryman, C., and Noller, H. F. (1998) in
RNA:Protein Interactions, A Practical Approach (Smith, C. W. J.,
ed.) Oxford University Press, New York, pp. 237-253.
20.Gurevich, V. V. (1996) Meth. Enzymol.,
275, 382-397.
21.Gurevich, V. V., Pokrovskaya, I. D., Obukhova, T.
A., and Zozulya, S. A. (1991) Anal. Biochem., 195,
207-213.
22.Shirokikh, N. E., and Spirin, A. S. (2008)
Proc. Natl. Acad. Sci. USA, 105, 10738-10743.
23.Shirokikh, N. E., Alkalaeva, E. Z., Vassilenko,
K. S., Afonina, Z. A., Alekhina, O. M., Kisselev, L. L., and Spirin, A.
S. (2009) Nucleic Acids Res., doi: 10.1093/nar/gkp1025.
24.Fekete, R. A., Miller, M. J., and Chattoraj, D.
K. (2003) Biotechniques, 35, 90-98.
25.Yindeeyoungyeon, W., and Schell, M. A. (2000)
Biotechniques, 29, 1034-1041.
26.Gould, P. S., Bird, H., and Easton, A. J. (2005)
Biotechniques, 38, 397-400.
27.Peattie, D. A., and Gilbert, W. (1980) Proc.
Natl. Acad. Sci. USA, 77, 4679-4682.
28.Mandiyan, V., and Boublik, M. (1990) Nucleic
Acids Res., 18, 7055-7062.
29.Mougel, M., Eyermann, F., Westhof, E., Romby, P.,
Expert-Bezancon, A., Ebel, J. P., Ehresmann, B., and Ehresmann, C.
(1987) J. Mol. Biol., 198, 91-107.
30.Maxam, A. M., and Gilbert, W. (1977) Proc.
Natl. Acad. Sci. USA, 74, 560-564.
31.Leonard, N. J., McDonald, J. J., Henderson, R.
E., and Reichmann, M. E. (1971) Biochemistry, 10,
3335-3342.
32.Weeks, K. M., and Crothers, D. M. (1993)
Science, 261, 1574-1577.
33.Lockard, R. E., and Kumar, A. (1981) Nucleic
Acids Res., 9, 5125-5140.
34.Kop, J., Kopylov, A. M., Magrum, L., Siegel, R.,
Gupta, R., Woese, C. R., and Noller, H. F. (1984) J. Biol.
Chem., 259, 15287-15293.
35.Hartshorne, T., and Agabian, N. (1994) Nucleic
Acids Res., 22, 3354-3364.