Femtosecond Stage of Electron Transfer in Reaction Centers of the Triple Mutant SL178K/GM203D/LM214H of Rhodobacter sphaeroides
A. G. Yakovlev1*, T. A. Shkuropatova2, V. A. Shkuropatova3, and V. A. Shuvalov1,3
1Department of Photobiophysics, Belozersky Institute of Physico-Chemical Biology, Lomonosov Moscow State University, 119991 Moscow, Russia; fax: (495) 939-3181; E-mail: yakov@genebee.msu.su2Department of Biophysics, Huygens Laboratory, Leiden University, P. O. Box 9504, 2300 RA Leiden, The Netherlands
3Institute of Basic Biological Problems, Russian Academy of Sciences, 142290 Pushchino, Moscow Region, Russia; fax: (496) 773-0532; E-mail: shuvalov@issp.serpukhov.su
* To whom correspondence should be addressed.
Received November 19, 2009
Coherent processes in an initial phase of charge transfer in reaction centers (RCs) of the triple mutant S(L178)K/G(M203)D/L(M214)H of Rhodobacter sphaeroides were investigated by difference (light – dark) absorption spectroscopy with 18 fsec time resolution. Electron transfer in the B cofactor branch is activated in this mutant, while the A-branch electron transfer is slowed in comparison with native RCs of Rba. sphaeroides. A bulk of absorption difference spectra was analyzed in the 940-1060 nm range (stimulated emission of excited bacteriochlorophyll dimer P* and absorption of bacteriochlorophyll anions BA– and β–, where β is a bacteriochlorophyll substituting the native bacteriopheophytin HA) and in the 735-775 nm range (bleaching of the absorption band of the bacteriopheophytin HB in the B-branch) in the –0.1 to 4 psec range of delays with respect to the moment of photoexcitation of P at 870 nm. Spectra were measured at 293 and 90 K. The kinetics of P* stimulated emission at 940 nm shows its decay with a time constant of ~14 psec at 90 K and ~18 psec at 293 K, which is accompanied by oscillations with a frequency of ~150 cm–1. A weak absorption band is found at 1018 nm that is formed ~100 fsec after excitation of P and reflects the electron transfer from P* to β and/or BA with accumulation of the P+β– and/or P+BA– states. The kinetics of ΔA at 1018 nm contains the oscillations at ~150 cm–1 and distinct low-frequency oscillations at 20-100 cm–1; also, the amplitude of the oscillations at 150 cm–1 is much smaller at 293 than at 90 K. The oscillations in the kinetics of the 1018 nm band do not contain a 32 cm–1 mode that is characteristic for native Rba. sphaeroides RCs having water molecule HOH55 in their structure. The ΔA kinetics at 751 nm reflects the electron transfer to HB with formation of the P+HB– state. The oscillatory part of this kinetics has the form of a single peak with a maximum at ~50 fsec completely decaying at ~200 fsec, which might reflect a reversible electron transfer to the B-branch. The results are analyzed in terms of coherent nuclear wave packet motion induced in the P* excited state by femtosecond light pulses, of an influence of the incorporated mutations on the mutual position of the energy levels of charge separated states, and of the role of water HOH55 in the dynamics of the initial electron transfer.
KEY WORDS: photosynthesis, charge separation, reaction center, wave packet, electron transferDOI: 10.1134/S0006297910040036
Abbreviations: ΔA, absorption changes (light minus dark); BChl, bacteriochlorophyll; BA and BB, monomeric BChl in A- and B-branch, respectively; BPheo, bacteriopheophytin; HA and HB, BPheo in A- and B-branch, respectively; P, primary electron donor, dimer BChl; PA and PB, BChl molecules forming P; QA and QB, primary and secondary quinone, respectively; RC, reaction center.
The reaction center (RC) of bacterial photosynthesis is a
pigment–protein complex inserted into the photosynthetic
membrane. In RC, a conversion of light energy to the energy of charge
separated states takes place as a result of a series of fast electron
transfer reactions. The three-dimensional structure of the purple
bacteria Blastochloris (Rhodopseudomonas) viridis
and Rhodobacter sphaeroides RC crystals has been studied by
X-ray structure analysis [1, 2]. The Rba. sphaeroides RC consists of three
protein subunits denoted as H (heavy), L (light), and M (middle),
and several cofactors bound to the L and M subunits. The cofactors
form two symmetrical branches denoted as A and B (Fig. 1a), but only the A-branch is photochemically active.
The primary electron donor, dimer bacteriochlorophyll (BChl) P, is
located near the periplasmic side of the membrane and consists of two
common for both branches BChl molecules, PA and
PB. A molecule of monomeric BChl (BA and
BB), a molecule of bacteriopheophytin (BPheo) (HA
and HB), and a molecule of quinone (QA and
QB) are located inside the membrane at successively
increasing distance from the dimer. The quinones are located in the end
of the branches near the cytoplasmic side of the membrane. An atom of
non-heme iron and a carotenoid molecule are also included into the RC
structure. At room temperature, after excitation of P an electron
transits from the first singlet excited level of P* to BA
within ~3 psec with formation of the
P+BA– state [3]. The electron transits further from
BA– to HA within ~1 psec
with formation of the P+HA–
state. The next step consists of electron transfer from
HA– to QA within ~200 psec with
formation of the P+QA– state. At
cryogenic temperatures, all of these reactions are accelerated
2-3-fold.
In native RC, the P+BA– state is short lived (a few picoseconds) because it is depleted several times faster than it is populated. This circumstance makes registration of this state much more difficult. Nevertheless, the data obtained during the registration of the radical-anion BA– absorption band dynamics at 1020 nm characterized the BA molecule as the primary electron acceptor [3, 4]. In pheophytin-modified RC of Rba. sphaeroides R-26, the long lived (~1 nsec) P+BA– state is registered at 5 K with complete transformation of the P* state to the P+BA– state, which shows the main direction of the electron transfer reaction [4]. Recently, the long-lived (hundreds of picoseconds) P+BA– state was registered at 295 K in RC of DLL-mutant of Rba. capsulatus, in the structure of which the bacteriopheophytin acceptor HA is absent and tyrosine M208 is introduced in addition, which stabilizes the P+BA– state and creates the possibility of thermodynamic population of this state from the P* state [5]. According to various estimates, the free energy level of the P+BA– state is 40-70 meV lower than those of the P* state [3, 6]. These results do not exclude the possibility of direct electron transfer from P* to HA with a virtual participation of the BA molecule by a super exchange mechanism, or simultaneous action of both mechanisms [7, 8]. The quantum yield of the primary process of charge separation is close to 1 at both room and low temperatures [3]. Each further stage of electron transfer is accompanied by free energy loss compensated by increasing dissipation time of accumulated energy from ~300 psec for the P* state to ~0.1 sec for the P+QA– state.Fig. 1. Space structure of Rba. sphaeroides RCs (a) and a fragment of this structure (b) according to the Protein Data Bank (PDB, file 1AIJ). BChl molecules PA and PB forming the dimer P, molecules of monomer BChl BA and BB, BPheo molecules HA and HB, molecules of quinone QA and QB, the iron atom, the molecule of crystallographic water HOH55, and histidine M202 are shown. BChl phytol side chains are not shown for simplicity of the picture. The chain of atoms N-Mg(PB)-N-C-N(HisM202)-HOH55-O=(BA) forms a direct space bonding between P and BA.
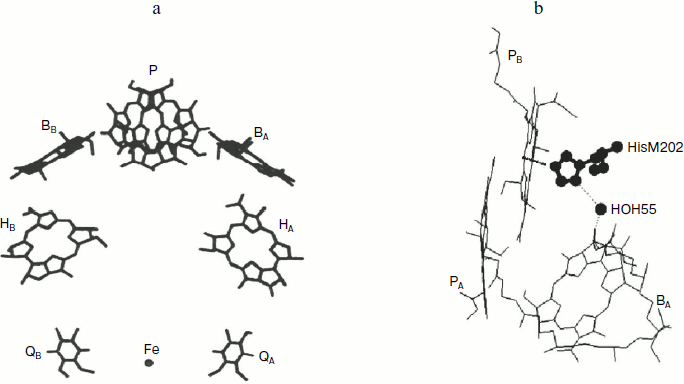
A molecule of crystallographically defined water HOH55, located between PB and BA, is also included in the RC structure (file 1AIJ of Protein Data Bank and Fig. 1b). A symmetrically located water molecule HOH30 is in the B-branch of the RC. The water HOH55 is located within the distance of hydrogen bond formation both from the oxygen of the 131-ketocarbonyl group of BA and from the nitrogen atom of the His M202 residue, which provides an axial liganding of the magnesium central atom in BChl PB [9]. Thus, a direct connection through space between PB and BA via HOH55 and His M202 is seen.
Coherent optical spectroscopy with femtosecond temporal resolution reveals nuclear motions of cofactors and protein that accompany electron transfer reactions. It was found that excitation of P from purple bacterial RCs by laser pulses of less than 30 fsec pulse-width leads to oscillations in the decay kinetics of P* stimulated emission with frequencies within the range of 10-400 cm–1 [10]. Oscillations with the same frequencies are observed in spontaneous emission of RCs [11]. The appearance of the oscillations can be explained by coherent nuclear motion along the potential energy surface of the P* excited state [10]. These oscillations are maximal at ~900 and 930-940 nm, which is explained by relative shift of the P*/P potential surfaces and by temporal dependence of nuclear wave packet position on the P* potential surface. The oscillations at the two maxima have opposite phases but have similar frequency spectra. Coherent oscillations have been found in RCs of Rba. sphaeroides R-26 in the kinetics of the absorption band at 1020 nm characteristic for radical-anion BA– [12-14], and in the kinetics of BA absorption band bleaching at 800 nm [15]. Coherent effects in Rba. sphaeroides RCs are revealed also in the kinetics of the Qy absorption band bleaching of HA at 760 nm [13, 14] and in the kinetics of the electrochromic shift of monomer BChl [16]. Femtosecond oscillations in the kinetics of P* stimulated emission at 945 nm and of BA– absorption at 1028 nm have been found in RCs of Chloroflexus aurantiacus at 90 K [17].
The Fourier spectrum of the oscillations in the kinetics of charge separated states P+BA– and P+HA– in Rba. sphaeroides R-26 RCs contains a dominated mode at 32 cm–1 and a progression of several bands with frequencies which are approximately multiples of 32 cm–1 [14]. Exclusion of water HOH55 from the RC structure in the GM203L Rba. sphaeroides mutant [18] is accompanied by disappearance of the oscillation band at 32 cm–1 in the kinetics at 1020 nm (BA– absorption band) and at 760 nm (HA band bleaching) and leads to the slowdown of the P* stimulated emission decay by ~4-fold [19]. Probably, with the exclusion of the HOH55 water from the mutant GM203L RC the electron transfer from P* to BA is stopped along a most effective path formed by a chain of polar atoms N-Mg(PB)-N-C-N(HisM202)-HOH55-O=(BA) (Fig. 1b). Steric exclusion of water HOH55 from the RC structure was shown also in the double mutant FM197R/GM203D Rba. sphaeroides [20]. The absence of HOH55 is supposed also in RCs of the single mutant GM203D Rba. sphaeroides [20], in which the decay of P* stimulated emission is slowed by ~3-fold in comparison with native RCs [21]. In deuterated Rba. sphaeroides R-26 RCs, an isotopic drop of the fundamental mode of the oscillations at 32 cm–1 by 1.3-1.6-fold takes place in the kinetics of charge separated states P+BA– and P+HA– [14, 17, 19, 22]. In dry films of pheophytin-modified RCs of Rba. sphaeroides R-26, from the structure of which the HOH55 is assumed to be excluded, a significant decrease of oscillation amplitudes at 32 and 120 cm–1 is observed in the kinetics of the BA– absorption band at 1020 nm together with a slowing of P+BA– accumulation [14, 17]. In dry films of the RCs of YM210W and YM210L Rba. sphaeroides mutants, the BA– absorption band at 1020 nm is not registered, and a marked decrease of oscillation amplitude is observed in the kinetics of P* stimulated emission at 940 nm [19].
Combinations of different mutations activating electron transfer in the B-branch and hindering it in the A-branch are used for studying the electron transfer processes in the B-branch of cofactors, which is almost inactive in native RCs. The LM212H mutation in Rba. capsulatus RCs, by which the native leucine M212 is replaced by histidine liganding the Mg atom, changes the molecule of BPheo HA by the BChl molecule denoted as β, which leads to significant increase of the free energy level of the P+β– state in comparison with P+HA–. By combining this mutation with the GM201D mutation that replaces the native glycine M201 by aspartic acid near ring V of the BA molecule and raises the P+BA– free energy level, it is possible to slow and to destabilize the electron transfer in the A-branch by several-fold [23]. As a result, a developing of the P+HB– state with a 15% quantum yield as well as strong acceleration of recombination processes in the A-branch is observed in the double mutant GM201D/LM212H Rba. capsulatus RCs. An adding of the third mutation SL178K replacing serine L178 by lysine near ring V of the BB molecule and decreasing the energy level of the P+BB– state leads to increase of a quantum yield of electron transfer in the B-branch up to 23% [23]. A similar result is achieved by FL121D mutation, which does not alter the pigment composition of the RC but raises the P+HA– energy level considerably. In the triple mutant FL97V/FL121D/LM212H Rba. capsulatus, the P+HB– state is formed with quantum yield of 12%, and adding of the forth mutation GM201D increases it up to 18% [24]. In the recently obtained mutant FL181Y/YM208F/LM212H Rba. capsulatus, the quantum yield of the P* → P+HB– reaction reaches 30% in photochemically active fraction of RCs (about 50% of total amount), and a further electron transfer to QB is registered with 13% total quantum yield of the P* → P+HB– → P+QB– process [25, 26]. In photochemically active fraction of mutant DLL-FYLFM Rba. capsulatus RCs, the 80% record quantum yield of P* → P+HB– reaction is registered, and the process of electron transfer in the B-branch of this mutant as well as in the A-branch is close to an activation-less process [27]. Electron transfer to HB in the B-branch is observed in the heterodimer Rba. capsulatus RCs containing BChl and BPheo in the structure of primary electron donor and the supplementary mutation LM212H [28]. Coherent B-branch electron transfer has been found in mutant HM182L Rba. sphaeroides RCs, in which the monomer BChl BB molecule is substituted by BPheo ΦB molecule, by registration of ΦB absorption band bleaching at 785 nm [29]. The reversible B-branch electron transfer occurs in RCs of this mutant at 40 fsec delay after excitation of P, which is earlier by 80 fsec than the analogous transfer in the A-branch detected by the BA– absorption band appearance at 1020 nm. In C. aurantiacus RCs, the reversible B-branch electron transfer begins even at smaller 10-20 fsec delay after excitation of P and is detected by the oscillations in the kinetics of the ΦB absorption band at 785 nm [30].
In the present work, coherent processes accompanying electron transfer were studied in the triple mutant SL178K/GM203D/LM214H Rba. sphaeroides RCs by difference (light – dark) absorption spectroscopy with 18 fsec resolution. The applied set of the mutations was equivalent with the triple mutant SL178K/GM201D/LM212H Rba. capsulatus mutations and was used for B-branch activation on the background of electron transfer braking in the A-branch [23]. The difference absorption spectra were analyzed in the 940-1060 nm range (stimulated emission of dimer P* and absorption of BA– and β– anions) and in the 735-775 nm range (B-branch HB absorption band bleaching) in the delay range of –0.1 to 4 psec from photoexcitation of P at 870 nm at 293 and 90 K. A weak absorption band was found at 1018 nm which is formed ~100 fsec later than excitation of P and reflected the A-branch electron transfer with creation of the P+β– and/or the P+BA– states. The oscillations in the kinetics of this band reflect a coherent component of A-branch electron transfer. These oscillations do not include the 32 cm–1 mode characteristic for RCs containing the HOH55 water. An aperiodic oscillation with a maximum at ~50 fsec delay caused by B-branch coherent electron transfer was found in the ΔA kinetics of the HB absorption band at 751 nm. The results are analyzed in terms of an influence of nuclear wave packet motion in the P* state upon the electron transfer, of relative positions of the energy levels of the charge separated states, and of an influence of the HOH55 water upon the kinetics of primary charge separation.
MATERIALS AND METHODS
Genetic manipulations and obtaining of mutant RCs were carried out according to [31-33]. RCs of mutant SL178K/GM203D/LM214H Rba. sphaeroides were isolated by treatment of chromatophores with N,N-dimethyldodecylamine-N-oxide (LDAO) detergent followed by purification using DEAE cellulose [29, 34]. RCs were suspended in 10 mM Tris-HCl buffer (pH 8.0) with 0.05% Triton X-100. Low temperature measurements at 90 K were performed on samples containing 65% glycerol. Optical density of the samples before adding the glycerol was 0.5 at 860 nm in a 1-mm optical pathlength cuvette at room temperature. To keep the state PBAHAQA–, 5 mM of sodium dithionite was added to RC samples. Absorption spectra of non-excited samples were measured on a Shimadzu UV-1601 PC (Japan) spectrophotometer.
Femtosecond measurements of absorption changes (light – dark) were performed using a laser spectrometer based on a Tsunami Ti-sapphire femtosecond mode locked laser continuously pumped by a Millennia laser (both from Spectra Physics, USA). Laser pulses were amplified in a Ti-sapphire 8-passed amplifier and were used for generation of a continuum in water. A minor part of the continuum (~4%) was used for probing of the samples. For excitation of the sample, a band centered at 870 nm was cut from the continuum by filters. The duration of pump and probe pulse was 18 fsec. An optical multichannel analyzer (Oriel, France) connected with polychromator was used for measuring of absorption spectral changes at 15 Hz frequency. The delay between exciting and probing pulses was set with 1-fsec accuracy and was varied with a variable 15-100 fsec step. After additional compensation the temporal dispersion does not exceed 30 fsec in the range of 940-1060 nm and 15 fsec in the range of 735-755 nm. Difference absorption spectra are averaged over 7000-10,000 measurements at each delay. The sensitivity of the spectrometer was (1-3)·10–5 OD units. The kinetics of absorption changes (ΔA) at 1018 and 751 nm were plotted using the absorption band amplitudes measured at their maximum with an additional subtraction of broadband background (see further Figs. 3 and 6). Polynomial non-oscillating curves, which were found mathematically, were used for approximation of the experimental kinetics. In study of the early processes of charge separation, which are adiabatic to a marked degree, this approach is more useful than an ordinary exponential approximation, which was used only for evaluation of time constants of different processes. Oscillatory parts of the kinetics were found as a result of the subtraction of the approximating curves from the initial kinetics and were analyzed by Fourier transformation to obtain a frequency spectrum of the oscillations. Non-oscillatory curves were corresponded to a minimal noise of Fourier spectra and to minimal amplitude of the oscillations.
RESULTS
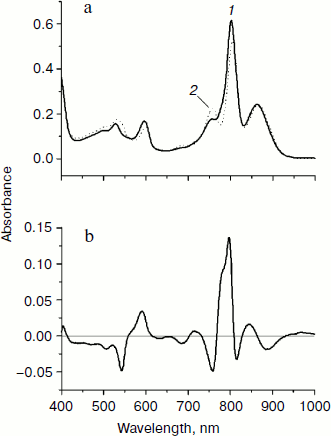
In Fig. 2, normalized absorption spectra measured in the triple mutant SL178K/GM203D/LM214H Rba. sphaeroides RCs (solid line) and in the native Rba. sphaeroides RCs (dotted line) at 293 K (Fig. 2a) are shown together with their difference (Fig. 2b). The main changes in the mutant spectrum deal with the LM214H mutation, which alters the HA molecule by the BChl molecule denoted as β that is proved by pigment extraction in the LM214H Rba. sphaeroides mutant [35]. A disappearance of the HA absorption band is observed at ~760 nm (Qy transition) and at 543 nm (Qx transition), and also an appearance of β absorption band at ~778 nm (Qy transition) and 590 nm (Qx transition) is observed. The β absorption bands are overlapped with the absorption bands of other BChl molecules at ~600 and ~800 nm. The HB molecule has absorption bands at ~755 (Qy transition) and ~530 nm (Qx transition). The dimer P absorption band of the triple mutant shifted slightly to the short wavelength side is at 862 nm (866 nm in the Rba. sphaeroides RCs).
In Fig. 3, the difference (light – dark) absorption spectra of the mutant SL178K/GM203D/LM214H Rba. sphaeroides RCs measured at 90 K (a) and 293 K (b) at various temporal delays from the 18 fsec pulse excitation at 870 nm are shown for the 975-1055 nm range. A weak absorption band is observed at 1018 nm, which appears on the background of long wavelength slope of the P* stimulated emission band at ~100 fsec delay. An oscillatory increase and decrease of this band as a whole with a period of ~200 fsec is observed during the first 0.5 psec at 90 K (for details see the kinetics in Fig. 5). These initial oscillations are almost absent at 293 K. A monotonous increase of the amplitude of the 1018 nm band is observed at longer delays. The bandwidth of the 1018 nm band is ~30 nm both at 90 and 293 K, that is somewhat more than in native Rba. sphaeroides RCs at 1020 nm (~20 nm at 90 K) [14]. The 1018 nm band might belong to the radical-anion β– or BA–.Fig. 2. Absorption spectra (a) and their difference (b) of the triple mutant SL178K/GM203D/LM214H Rba. sphaeroides RCs (solid line, curve 1) and of the native Rba. sphaeroides RCs (dotted line, curve 2) at 293 K. The absorption spectra are normalized by the long wavelength P band at 862-866 nm.
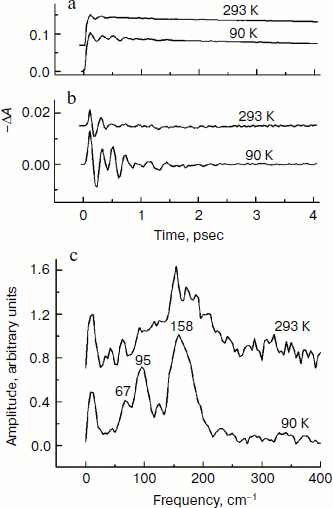
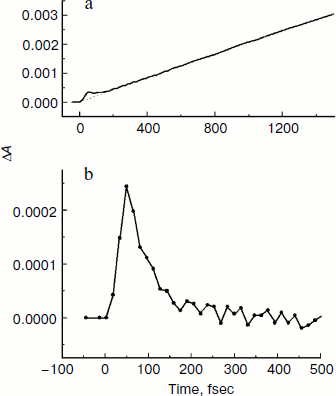
In Fig. 4, the ΔA kinetics of P* stimulated emission at 940 nm (a), their oscillatory components (b), and spectra of Fourier transformation of the oscillatory components (c) are shown for the mutant SL178K/GM203D/LM214H Rba. sphaeroides RCs excited by 18 fsec pulses at 870 nm at 90 and 293 K. The P* emission appears at 940 nm at ~100 fsec delay after excitation of P and then starts to decay in an oscillatory manner because of charge separation. The non-oscillatory part of the 90 K kinetics is well approximated by a sum of two exponents with time constants of ~14 psec (67%) and ~100 psec (33%). At 293 K, the best approximation is the sum of exponents with time constants of ~18 psec (81%) and ~150 psec (19%). For more precise determination of the time constants, it is necessary to widen the time window of the measurements, which is beyond a frame of the present work. At 90 K, the oscillations have a main frequency at 158 cm–1 and minor components with 95 and 67 cm–1 frequencies and decay during 1 psec after excitation of P (Fig. 4, b and c). At 293 K, the oscillations have ~2-fold smaller amplitude and decay much faster during a time of less than 0.5 psec. The Fourier spectrum of the oscillations at 293 K looks like a broad band centered at 154 cm–1 with a complicated form.Fig. 3. Difference (light – dark) absorption spectra of mutant SL178K/GM203D/LM214H Rba. sphaeroides RCs in the 975-1055 nm range at 90 (a) and 293 K (b) at various temporal delays from the moment of excitation by 18 fsec pulses at 870 nm. Numbers are delays in fsec. Amplitude of the absorption band in the maximum at 1018 nm (shown by double arrows) from which a broadband background was subtracted was used for plotting of the kinetics in Fig. 5. The curves are shifted vertically for clearness.
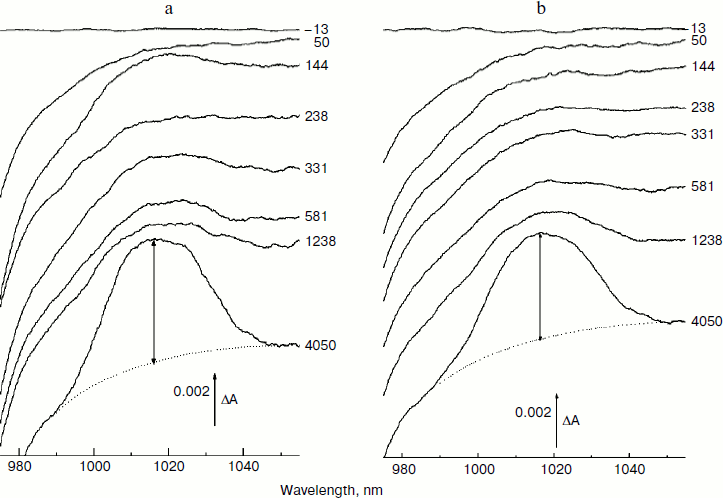
In Fig. 5, the ΔA kinetics at 1018 nm (a), its oscillatory components (b), and spectra of Fourier transformation of the oscillatory components (c) are shown for the mutant SL178K/GM203D/LM214H Rba. sphaeroides RCs excited by 18 fsec pulses at 870 nm at 90 and 293 K. The ΔA kinetics reflects an accumulation of the charge-separated states with absorption band at 1018 nm being accompanied by distinct oscillations. At 90 K, the initial oscillations with a ~200 fsec period decayed during ~0.5 psec and lower frequency oscillations with complicated form decayed much later at ~2.5 psec are observed in the kinetics. The Fourier spectrum of these oscillations includes a broad band at 138 cm–1 and narrower bands at 15, 45, and 66 cm–1. At 293 K, the oscillations are different: the component with ~200 fsec period is almost absent in it, but lower frequency components decaying at ~2 psec are present. The Fourier analysis of the oscillations at 293 K reveals a dominant band at 40 cm–1 and minor bands at 12, 57, and 100 cm–1.Fig. 4. Kinetics of ΔA at 940 nm (a), its oscillatory components (b), and spectra of Fourier transformation of the oscillatory components (c) of mutant SL178K/GM203D/LM214H Rba. sphaeroides RCs excited by 18 fsec pulses at 870 nm at 90 and 293 K. Numbers in Fig. 4c show the characteristic frequencies of the Fourier spectra. The curves are shifted vertically for clearness.
In Fig. 6, the difference absorption spectra of the mutant SL178K/GM203D/LM214H Rba. sphaeroides RCs measured in the 735-775 nm range at 90 K are shown for various temporal delays from the excitation by 18 fsec pulses at 870 nm. The bleaching of the HB absorption band at ~751 nm reflects the B-branch electron transfer from P* to HB with the formation of the P+HB– state. This bleaching becomes noticeable at 34 fsec delay and develops on the background of a broadband pedestal. A small bleaching is observed at ~755 nm at 19 fsec delay on the background of a pedestal reflecting mainly the P* state. The kinetics of HB absorption band bleaching at ~751 nm plotted on the basis of the measured ΔA spectra are shown in Fig. 7 in the delay range of 0 to 1.5 psec. The bleaching at ~751 nm appears at the small ~20 fsec delay after the excitation moment and quickly increases up to a local maximum at ~50 fsec delay. Then an approximate constancy of the bleaching amplitude is observed in the 50-150 fsec delay range followed by its permanent growth. The result of subtraction of a monotonous approximation from the ΔA kinetics shown in Fig. 7b reflects their variable part, which has the form of a single maximum at ~50 fsec delay. Note that oscillations of other types are not revealed in the 751 nm kinetics on the background of the noise with an amplitude of ~ (1-3)·10–5 OD units.Fig. 5. Kinetics ΔA at 1018 nm (a), its oscillatory components (b), and spectra of Fourier transformation of the oscillatory components (c) of mutant SL178K/GM203D/LM214H Rba. sphaeroides RCs excited by 18 fsec pulses at 870 nm at 90 and 293 K. Numbers in Fig. 5c show the characteristic frequencies of the Fourier spectra. The curves are shifted vertically for clearness.
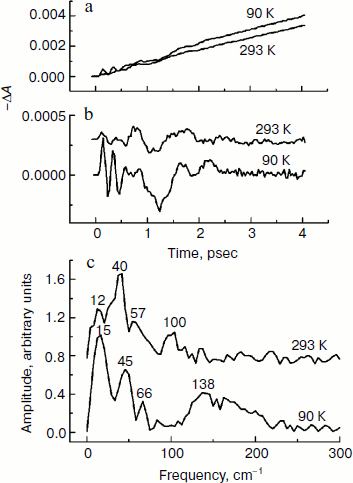
Fig. 6. Difference (light – dark) absorption spectra of mutant SL178K/GM203D/LM214H Rba. sphaeroides RCs in the 735-775 nm range at 90 K at various temporal delays from the moment of excitation by 18 fsec pulses at 870 nm. The spectra in the delay range of –13 to 144 fsec multiplied by 10 are shown in Fig. 6a. The spectra in the delay range of 503 to 4050 fsec are shown in Fig. 6b. Numbers are delays in fsec. Amplitude of the bleaching band in the maximum at ~751 nm (shown by double arrows in one of the spectra) from which a broadband background was subtracted was used for plotting of the kinetics in Fig. 7. The curves in Fig. 6a are shifted vertically for clearness.
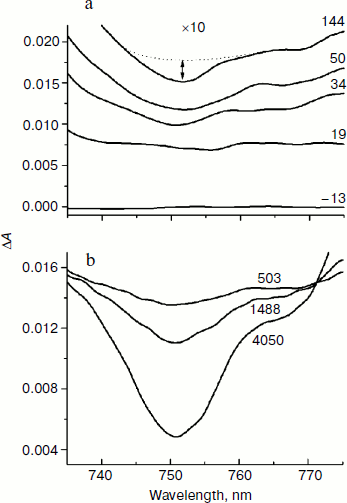
Fig. 7. Kinetics of ΔA at 751 nm (a) of mutant SL178K/GM203D/LM214H Rba. sphaeroides RCs excited by 18 fsec pulses at 870 nm at 90 K. An approximating curve is shown by dots. The difference of the kinetics and the approximating curve is shown in Fig. 7b in the delay range of –50 to 500 fsec.
DISCUSSION
In the present work, the coherent processes accompanying electron transfer in the A- and B-branch of cofactors are investigated in the triple mutant SL178K/GM203D/LM214H Rba. sphaeroides RCs by difference absorption spectroscopy with 18 fsec resolution. Studies of noncoherent electron transfer in the analogous triple mutant of Rba. capsulatus showed that the primary electron acceptor in the A-branch is, probably, the BChl β molecule that substituted the native HA molecule because of the LM212H mutation [23]. The data set from [23] suggest that the P+β– free energy level of this mutant is slightly lower than the P* level, and P+BA– level is slightly above the P* level, which does not exclude participation of the BA molecule in electron transfer by a superexchange mechanism. A different situation is observed in the LM214H Rba. sphaeroides mutant, where the P+BA– level is, most probably, lower than the P+β– level, and both of the levels are close to the P* level, which leads to primary electron transfer to BA [36, 37]. Adding of the GM203D mutation raises the P+BA– level significantly, which can put this level even closer to the P+β– and P* levels in the double mutant GM203D/LM214H Rba. sphaeroides. Calculations show that the P+BB– level is higher than the P* level in native Rba. sphaeroides RCs, and that the BB molecule can not participate directly in the B-branch electron transfer [38]. Adding of the third mutation SL178K lowers the P+BB– level of the triple mutant SL178K/GM201D/LM212H Rba. capsulatus somewhat, but it is still slightly above the P* level [23]. One can assume that the P+BB– level might be somewhat higher than the P* level in the analogous mutant of Rba. sphaeroides also.
In the present work, an absorption band at 1018 nm appearing at ~100 fsec delays after P excitation has been found in the triple mutant SL178K/GM203D/LM214H Rba. sphaeroides for the first time (Fig. 3). This band is shifted by ~2 nm to the short wavelength spectral area in comparison with the BA– absorption band of native Rba. sphaeroides RCs at 1020 nm, and it has somewhat larger bandwidth and somewhat different form (more plane apex) than those of native Rba. sphaeroides RCs [14]. Generally speaking, this absorption band might belong to BA– as well as to β– or to their sum in some proportion, which can be varied by the delay variation. Since the Qy absorption band of the β molecule (~780 nm) is different from the analogous BA band (~800 nm) (Fig. 2), one can assume that anion bands of these molecules would be different too. However, the data in Fig. 3 indicates that: i) either the absorption band being detected at 1018 nm belongs to one of the two molecules (BA– or β–); ii) or the sum of two spectrally non-distinguishable absorption bands of BA– and β– is registered. The spectral indistinguishability of the BA– and β– absorption band might mean that a common mixed state P+β–/P+BA– exists that is formed when the energy levels of the two states are close enough to each other and the two molecules BA– and β– are close in space to each other too. The nature of the mixing can be either classical (thermal) or quantum. Intense mixing of the P+β–/P+BA– states is supposed to occur in FL121D and LM212H Rba. capsulatus mutants [23, 37].
The presence of the oscillations in the 1018 nm band similar to the oscillations in the BA– absorption band of the native Rba. sphaeroides RCs at 1020 nm [14] reflects coherent modulation at early stages of charge separation. The fact that the 1018 nm absorption band does not change its form and spectral position over the whole range of delays in the experiment indicates the absence of the coherent modulation inside the state being formed as a result of electron transfer process (P+β– and/or P+BA–). Then the oscillations in the kinetics at 1018 nm (Fig. 5) reflect, probably, a coherent component of the reaction P* → P+β–/P+BA–. A synchronism of the initial oscillations with a ~150 cm–1 frequency in the 1018 nm band (Fig. 5) with the analogous oscillations in the P* stimulated emission band at 940 nm (Fig. 4) having mostly vibrational nature supports this assumption too. A general picture of the oscillations in the emission band at 940 nm and in the absorption band at 1018 nm of the triple mutant SL178K/GM203D/LM214H Rba. sphaeroides has been proved to be quite similar with the analogous oscillations of the native Rba. sphaeroides RCs studied by us in [13, 14]. This picture reflects a domination of the oscillation mode at ~150 cm–1 in the P* stimulated emission and the presence of distinct low frequency oscillation modes at 12-70 cm–1 in the kinetics of the absorption band at 1018 nm. At 90 K, the oscillations in the 1018 nm band are similar to the oscillations in the kinetics of the BA– absorption band at 1020 nm of the native Rba. sphaeroides RCs [14]. At 293 K, the oscillations in the 1018 nm band includes mostly the low frequency modes that makes them more similar to the oscillations in the HA band bleaching at 760 nm observed during the accumulation of the P+HA– state in the native Rba. sphaeroides RCs [13].
It is necessary to consider the role of the GM203D mutation for further discussion. Initially, the GM203D mutation was introduced to RC structure in order to add a hydrogen bond to the 131-ketocarbonyl group of the BA molecule by replacing Gly by Asp, which was assumed to cause a decrease in the free energy level of the P+BA– state [21]. However, results of the studies of various Rba. capsulatus RCs with the GM201D mutation (the analog of the GM203D mutation in Rba. sphaeroides) are more consistent with the P+BA– free energy level being slightly above the P* level [6, 23]. According to resonance Raman scattering spectroscopy, Asp M201 can be deprotonated and negatively charged at room temperature [39]. Therefore, it was concluded that introducing Asp M201 is accompanied by lowering of the midpoint redox potential (Em) of the BA/BA– pair that leads to an increase in the P+BA– free energy level and, as a consequence, to the lengthening of P* lifetime [39]. The effect of Asp M201 deprotonation is decreased significantly at low temperatures [39]. Under these conditions the P+BA– level might stay below the P* level in the GM201D mutant, which might lead to decrease in the P* lifetime at low temperature. As a matter of fact, lowering temperature from 285 to 77 K leads to the increase of the P* lifetime from 7.6 to 10 psec in the GM201D Rba. capsulatus mutant and to the decrease of the P* lifetime from 15 to 13 psec in the GM201D/LM212H Rba. capsulatus double mutant [39]. The percentages of these changes are much less than the 2-3-fold acceleration of the primary charge separation reaction in the native Rba. sphaeroides and Rba. capsulatus RCs under the same temperature lowering [13, 14, 39].
It is shown in the present work that the P* lifetime of the triple mutant SL178K/GM203D/LM214H Rba. sphaeroides is decreased from ~18 to ~14 psec on lowering the temperature from 293 to 90 K. Simultaneously, a considerable growth of the 150 cm–1 oscillation mode is observed in the kinetics of the state absorbing at 1018 nm on lowering the temperature from 293 to 90 K (Fig. 5). Taken together, these two facts might mean that the P+BA– level is higher than the P* level in this triple mutant at 293 K, and that the β molecule is a primary electron acceptor in the A-branch. At 90 K, the P+BA– level is lowered below the P* level and the BA molecule begins to participate directly in the A-branch electron transfer of the triple mutant SL178K/GM203D/LM214H Rba. sphaeroides. The specific mechanism of this participation can be different: parallel P* → P+BA– and P* → P+β– transfers, two-step transfer P* → P+BA– → P+β–, or participation in the mixed state P+BA–/P+β–, which has quantum rather than thermal nature at low temperatures.
It is found in the present work that the P* lifetime of the triple mutant SL178K/GM203D/LM214H Rba. sphaeroides at 293 K (~18 psec) exceeds that of the native RCs (4.3 psec) by more than 4-fold, and by more than 10-fold at 90 K (~14 and 1.2 psec, respectively) (Fig. 4 and [13, 14]). It is possible to suggest at least two possible reasons of this slowing. The first is an alteration in the energetics of the primary electron transfer reaction in the A-branch caused by strong rise of the P+BA– and P+β– energy levels. In the analogous triple mutant SL178K/GM203D/LM214H Rba. capsulatus, the rise of the P+β– level relative to the P+HA– level is estimated as 150-200 meV, and the rise of the P+BA– level is estimated as 100-150 meV [24]. The second reason is a possible exclusion of the HOH55 water from the RC structure of the triple mutant SL178K/GM203D/LM214H Rba. sphaeroides by analogy with the GM203D and FM197R/GM203D Rba. sphaeroides mutants [19, 20], though the data on the three-dimensional structure of this mutant is not yet available. X-Ray analysis of the crystal structure of the mutant GM203L and FM197R/GM203D Rba. sphaeroides RCs showed an absence of the HOH55 water in the RC structure [19, 20]. This water molecule is present in the structure of the native Rba. sphaeroides RCs (Fig. 1). The same structural alteration is supposed also in RCs of the single mutant GM203D [20]. Because of neutrality of Leu, the GM203L mutation does not create any influence on the M203 residue on the primary electron transfer reaction, unlike the GM203D mutation that simplified analysis of the results. It was found that more than 4-fold slowing of the P* → P+BA– reaction takes place in the mutant GM203L Rba. sphaeroides in comparison with the native Rba. sphaeroides RCs [19]. Besides, the 32 cm–1 spectral component and its overtones, which are clearly observed in the oscillations of the native Rba. sphaeroides RCs, is completely absent in the analogous oscillations of the kinetics of the P* stimulated emission and BA– absorption in this mutant [19]. A series of experiments with deuterated Rba. sphaeroides RCs and with dry films of Rba. sphaeroides RCs suggests that the 32 cm–1 oscillation mode reflects a rotation of the HOH55 water appearing at femtosecond excitation [14, 17, 19, 22]. The absence of the 32 cm–1 rotation mode and of its overtones in the oscillations of the kinetics at 940 nm (P* emission) and at 1018 nm (BA– and β– absorption) might indicate the absence of HOH55 in RCs of the triple mutant SL178K/GM203D/LM214H Rba. sphaeroides (Figs. 4 and 5). Identification of the low frequency spectral components observed in the mentioned oscillations requires further study.
The absence of the HOH55 water in the triple mutant SL178K/GM203D/LM214H Rba. sphaeroides RCs can have two consequences. First, a rather strong hydrogen bond (4.1 kcal/mol) is registered between the 131-ketocarbonyl group of the BA molecule and the HOH55 molecule in oxidized native RCs (Fig. 1) that can stabilized the BA– [20]. Thus, the absence of this hydrogen bond can lead to the rising of the P+BA– level and to the slowing of the P* → P+BA– reaction. However, it is not clear whether the formation of the hydrogen bond between the 131-ketocarbonyl group of BA and HOH55 occurs within the P+BA– lifetime (~1 psec in native Rba. sphaeroides RCs at 293 K [13]). Second, the position of the HOH55 water molecule permits this molecule to be a part of the electron transfer chain consisting from the following polar atoms: N-Mg(PB)-N-C-N(HisM202)-HOH55-O=(BA) (Fig. 1). From this point of view, lengthening of the P* lifetime in the triple mutant in comparison with the native Rba. sphaeroides might mean damage to intactness of this polar group chain by the exclusion of the water HOH55.
In the present work, the coherent single peak-like oscillation accompanying the B-branch electron transfer is found in the triple mutant SL178K/GM203D/LM214H Rba. sphaeroides RCs for the first time (Fig. 7). This oscillation reaches its maximum at the ~50 fsec delay that is more than 50 fsec earlier than the first maximum of the synchronous oscillations at 940 and 1018 nm reflecting the A-branch electron transfer (Figs. 4 and 5). A similar result was obtained by us in the mutant HM182L Rba. sphaeroides RCs, in which the native BB molecule is replaced by the BPheo ΦB molecule. In this mutant, the coherent oscillations in the kinetics of the ΦB absorption band bleaching looks like a single peak with maximum at ~40 fsec, whereas the first maximum of the BA– absorption band oscillations appears noticeably later at 120 fsec [29]. The coherent electron transfer in the B-branch forestalling those in the A-branch was found by us in C. aurantiacus RCs [30]. In all of the mentioned cases the B-branch electron transfer starts almost immediately after excitation, whereas the A-branch electron transfer is delayed by a time approximately equal to the half oscillation period.
The totality of these results allows us to describe in details the process of the formation and of the coherent motion of the photoinduced nuclear wave packet in the triple mutant SL178K/GM203D/LM214H Rba. sphaeroides. In Fig. 8, a simplified one-dimensional scheme is shown for the potential energy surfaces of the ground and excited P states and for the charge separated states P+BA–/P+β– and P+HB–. This scheme has illustrative purpose, and therefore the scale of the energy and coordinate axes is not kept and the form of the curves is arbitrary. With the purpose of simplifying, the P+BA– and P+β– states are shown by one curve, and both the reactions P* → P+BA–/P+β– and P* → P+HB– are considered to be in the same plane. A splitting of the potential surfaces can occur in the area of their intersection in proportion to a value of electron coupling V (not shown for simplicity).
Excitation of P by 18 fsec pulses with a broad spectrum creates the nuclear wave packet which begins an oscillatory motion along the P* surface. Right after formation the wave packet is on the left slope of the P* surface and emits light at 895 nm [3, 10]. The fact that the oscillation in the HB absorption band appears immediately after excitation indicates proximity of the intersection area of the P* and P+HB– surfaces to the area of the nuclear wave packet formation. At the ~150 fsec delay the oscillation at 751 nm has disappeared completely, which means a complete reversibility of the B-branch electron transfer induced by the nuclear wave packet. At the ~100 fsec delay the wave packet arrives in the intersection area of the P* and P+BA–/P+β– surfaces on the right slope of the P* surface. The wave packet emits at ~930-940 nm in this area, and the phase of this emission is opposite to the phase of the 895 nm emission [10]. The wave packet appearance in the intersection area of the P* and P+BA–/P+β– surfaces leads to the appearance of the oscillation in the absorption band at 1018 nm that is in phase with the oscillation in the P* emission at 940 nm (Figs. 4 and 5). The absorption at 1018 nm is strongly decreased after reflection of the wave packet in the area of surface intersection and its going away in the opposite direction, which indicates the reversibility of the P* → P+BA–/P+β– coherent reaction and the absence of a stabilization of separated charges at this delay. Thus, the coherent components of the electron transfer reactions in the A- and B-branches are separated in time, and also the B-branch transfer starts earlier by ~50 fsec than those in the A-branch. At the ~200 fsec delay, the wave packet appears again on the left slope of the P* surface near the intersection area of the P* and the P+HB– surfaces, but it does not exert any influence on the transfer reaction (Fig. 7). Probably, the wave packet energy was already decreased at this time and became insufficient for the electron to get over a potential barrier in this area. At the ~300 fsec delay, the wave packet appears again on the right slope of the P* surface near the intersection area of the P* and the P+BA–/P+β– surfaces that leads to the appearance of the second maximum in the oscillations of the absorption at 1018 nm and of the P* emission at 940 nm. The wave packet is diffused during its motion because of vibrational relaxation and scattering on the phonons of surroundings that leads to the registration of the few oscillation periods only. Note that the coherent electron transfer in both branches occurs on the background of the ordinary, noncoherent transfer.Fig. 8. A simplified illustrative scheme of mutual arrangement of potential energy surfaces of the ground and excited P states and of the primary charge separated states P+BA–/P+β– and P+HB–.
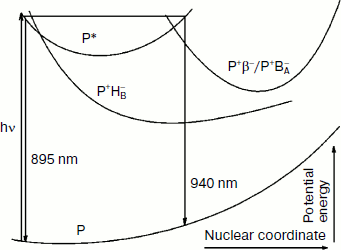
Thus, coherent processes of charge separation are found in both branches of cofactors in the triple mutant SL178K/GM203D/LM214H Rba. sphaeroides RCs by the method of femtosecond difference absorption spectroscopy. These processes are initiated by the nuclear wave packet motion along the potential energy surface of the P* state and are registered by the reversible formation of the charge separated states P+BA–/P+β– in the A-branch and P+HB– in the B-branch. The P+HB– state is formed earlier than the P+BA–/P+β– state by ~50 fsec, which might be connected with a difference in the nuclear configurations that are optimal for electron transfer along the two branches.
This work was done with financial support of the Presidium of the Russian Academy of Sciences (program “Molecular and Cell Biology”) and the Russian Foundation for Basic Research (grant No. 08-04-00888).
REFERENCES
1.Deisenhofer, J., Epp, O., Miki, K., Huber, R., and
Michel, H. (1984) J. Mol. Biol., 180, 385-398.
2.Allen, J. P., Feher, G., Yeates, T. O., Komiya, H.,
and Rees, D. C. (1987) Proc. Natl. Acad. Sci. USA,
84, 5730-5734.
3.Shuvalov, V. A. (2000) Transformation of Solar
Energy in Primary Act of Charge Separation in Reaction Centers of
Photosynthesis [in Russian], Nauka, Moscow.
4.Kennis, J. T. M., Shkuropatov, A. Ya, van Stokkum,
I. H. M., Gast, P., Hoff, A. J., Shuvalov, V. A., and Aartsma, T. J.
(1997) Biochemistry, 36, 16231-16238.
5.Carter, B., Boxer, S. G., Holten, D., and Kirmaier,
C. (2009) Biochemistry, 48, 2571-2573.
6.Heller, B. A., Holten, D., and Kirmaier, C. (1995)
Science, 269, 940-945.
7.Marcus, R. A. (1988) in The Photosynthetic
Bacterial Reaction Center: Structure and Dynamics (Breton, J., and
Vermeglio, A., eds.) Plenum Press, New York-London, pp. 389-398.
8.Larsson, S., and Ivashin, N. V. (1999) J. Appl.
Spectrosc., 66, 539-543.
9.Stowell, M. H. B., McPhillips, T. M., Rees, D. C.,
Soltis, S. M., Abresch, E., and Feher, G. (1997) Science,
276, 812-816.
10.Vos, M. H., Rappaport, F., Lambry, J.-C., Breton,
J., and Martin, J.-L. (1993) Nature, 363, 320-325.
11.Stanley, R. J., and Boxer, S. G. (1995) J.
Phys. Chem., 99, 859-863.
12.Yakovlev, A. G., Shkuropatov, A. Ya., and
Shuvalov, V. A. (2000) FEBS Lett., 466, 209-212.
13.Yakovlev, A. G., Shkuropatov, A. Ya., and
Shuvalov, V. A. (2002) Biochemistry, 41,
2667-2674.
14.Yakovlev, A. G., Shkuropatov, A. Ya., and
Shuvalov, V. A. (2002) Biochemistry, 41, 14019-14027.
15.Streltsov, A. M., Vulto, S. I. E., Shkuropatov,
A. Ya., Hoff, A. J., Aartsma, T. J., and Shuvalov, V. A. (1998) J.
Phys. Chem. B, 102, 7293-7298.
16.Vos, M. H., Rischel, C., Jones, M. R., and
Martin, J.-L. (2000) Biochemistry, 39,
8353-8361.
17.Yakovlev, A. G., Vasilieva, L. G., Shkuropatov,
A. Ya., Bolgarina, T. I., Shkuropatova, V. A., and Shuvalov, V. A.
(2003) J. Phys. Chem. A, 107, 8330-8338.
18.Potter, J. A., Fyfe, P. K., Frolov, D., Wakeham,
M. C., van Grondelle, R., Robert, B., and Jones, M. R. (2005) J.
Biol. Chem., 280, 27155-27164.
19.Yakovlev, A. G., Jones, M. R., Potter, J. A.,
Vasilieva, L. G., Shkuropatov, A. Y., and Shuvalov, V. A. (2005)
Chem. Phys., 319, 297-307.
20.Fyfe, P. K., Ridge, J. P., McAuley, K. E.,
Cogdell, R. J., Isaacs, N. W., and Jones, M. R. (2000)
Biochemistry, 39, 5953-5960.
21.Williams, J. C., Alden, R. G., Murchison, H. A.,
Peloquin, J. M., Woodbury, N. W., and Allen, J. P. (1992)
Biochemistry, 31, 11029-11037.
22.Shuvalov, V. A., and Yakovlev, A. G. (2003)
FEBS Lett., 540, 26-34.
23.Kirmaier, C., Weems, D., and Holten, D. (1999)
Biochemistry, 38, 11516-11530.
24.Roberts, J. A., Holten, D., and Kirmaier, C.
(2001) J. Phys. Chem. B, 105, 5575-5584.
25.Kirmaier, C., and Holten, D. (2009) J. Phys.
Chem. B, 113, 1132-1142.
26.Kee, H. L., Laible, P. D., Bautista, J. A.,
Hanson, D. K., Holten, D., and Kirmaier, C. (2006) Biochemistry,
45, 7314-7322.
27.Chuang, J. I., Boxer, S. G., Holten, D., and
Kirmaier, C. (2008) J. Phys. Chem. B, 112,
5487-5499.
28.Kirmaier, C., Bautista, J. A., Laible, P. D.,
Hanson, D. K., and Holten, D. (2005) J. Phys. Chem. B,
109, 24160-24172.
29.Yakovlev, A. G., Shkuropatova, T. A., Vasilieva,
L. G., Shkuropatov, A. Y., Gast, P., and Shuvalov, V. A. (2006)
Biochim. Biophys. Acta, 1757, 369-379.
30.Yakovlev, A. G., Shkuropatova, T. A., Vasilieva,
L. G., Shkuropatov, A. Y., and Shuvalov, V. A. (2008) J. Bioinform.
Comput. Biol., 6, 643-666.
31.Paddock, M. L., Rongey, S. H., Feher, G., and
Okamura, M. Y. (1989) Proc. Natl. Acad. Sci. USA,
86, 6602-6606.
32.De Boer, A. L., Neerken, S., de Wijn, R.,
Permentier, H. P., Gast, P., Vijgenboom, E., and Hoff, A. J. (2002)
Photosynth. Res., 71, 221-239.
33.Goldsmith, J. O., and Boxer, S. G. (1996)
Biochim. Biophys. Acta, 1276, 171-175.
34.Shuvalov, V. A., Shkuropatov, A. Ya., Kulakova,
S. M., Ismailov, M. A., and Shkuropatova, V. A. (1986) Biochim.
Biophys. Acta, 849, 337-348.
35.Heller, B. A., Holten, D., and Kirmaier, C.
(1995) Biochemistry, 34, 5294-5302.
36.Kirmaier, C., Laporte, L., Schenck, C. C., and
Holten, D. (1995) J. Phys. Chem., 99, 8910-8917.
37.Heller, B. A., Holten, D., and Kirmaier, C.
(1996) Biochemistry, 35, 15418-15427.
38.Alden, R. G., Parson, W. W., Chu, Z., and
Warshel, A. (1995) J. Am. Chem. Soc., 117,
12284-12302.
39.Czarnecki, K., Kirmaier, C., Holten, D., and
Bocian, D. (1999) J. Phys. Chem. A, 103, 2235-2246.