Molecular Genetic Characterization of the Thermostable L-Lactate Dehydrogenase Gene (ldhL) of Thermoanaerobacter ethanolicus JW200 and Biochemical Characterization of the Enzyme
Q. Zhou* and W.-L. Shao
Jiangsu Key Laboratory for Biodiversity and Bio-resources, College of Life Sciences, Nanjing Normal University, Nanjing 210046, China; E-mail: qqzhouqing@hotmail.com* To whom correspondence should be addressed.
Received October 26, 2009; Revision received November 10, 2009
The structural gene ldhL for a thermostable L-(+)-lactate dehydrogenase was cloned from Thermoanaerobacter ethanolicus JW200. The nucleotide sequence of the ldhL gene was determined and shown to have the capacity to encode a protein of 311 amino acids (33.5 kDa). By 5′-RACE analysis, the ldhL transcription start point was confirmed to be derived from the –10 region closest to the initiation codon. The enzyme was overexpressed in Escherichia coli and purified to homogeneity by nickel-affinity chromatography. It was shown to be allosteric in the presence of fructose-1,6-bisphosphate. The optimum pH and temperature for the enzyme were 5.8 and 60°С in the pyruvate reduction and 7.0 and 70°С in the lactate oxidation reaction, respectively. The kinetic parameters Km,app and kcat,app for pyruvate were 0.18 mM and 520 U/mg, respectively, and in the absence of fructose-1,6-bisphosphate, a 1.2-fold increase in Km,app and a 16-fold decrease in kcat,app were determined. The Km,app and kcat,app values for lactate were 60 mM and 0.58 U/mg, respectively, and they were not affected by fructose-1,6-bisphosphate. The enzyme was greatly inhibited by Zn2+, Ag+, Cu2+, Fe3+, and Pb3+. The extreme thermostability of the enzyme was reflected in its unaltered activity over 5 h at 70°C.
KEY WORDS: L-(+)-lactate dehydrogenase, Thermoanaerobacter ethanolicus JW200, allosteric, thermostable, transcription start pointDOI: 10.1134/S0006297910040188
Abbreviations: DTT, dithiothreitol; FBP, fructose-1,6-bisphosphate; L-LDH, L-(+)-lactate dehydrogenase; TeLdhL, L-LDH of T. ethanolicus JW200.
Thermoanaerobacter ethanolicus JW200, isolated originally from
thermal springs in Yellowstone National Park, USA [1], is an extremely thermophilic, non-spore-forming
anaerobic bacterium. This bacterium uses the Embden–Meyerhof
pathway to convert glucose to pyruvate. Under stress conditions, such
as high glucose concentration (>10 g/liter), pyruvate is converted
to lactate as a major fermentation product [2].
Recently, more and more uses of lactic acid have focused on the production of the non-chlorinated solvent ethyl lactate and the biodegradable plastic polylactic acid with an eye to ecological interest. However, L-(+)-lactic acid is mostly the isomer preferred [3]. L-(+)-lactic acid is produced from pyruvate by L-(+)-lactate dehydrogenase (L-LDH) (EC 1.1.1.27) with the concomitant oxidation of NADH. L-LDHs from only several thermophiles have been studied in the past [4-6]. But the strains with high growth temperature, such as Lactobacillus helveticus [6], do not always produce thermostable L-LDH.
Understanding of the molecular genetics of T. ethanolicus JW200 is not far advanced. Only a few of the genes of the bacterium concerned with fermentative metabolism have been cloned and expressed in E. coli for possible uses in industrial biotransformations [7, 8]. To our knowledge, the L-LDH of T. ethanolicus JW200 (TeLdhL) has received no reported attention, though it is a very important enzyme. In this work, we report the cloning, DNA sequencing, and promoter analysis of the ldhL gene from T. ethanolicus JW200. Moreover, the expression, purification, and biochemical characterizations of TeLdhL are described.
MATERIALS AND METHODS
Bacterial strains and plasmids. Thermoanaerobacter ethanolicus JW200 was cultivated as described previously [1]. Escherichia coli JM109 (Promega, USA) was used as host for gene cloning in plasmid pMD19T (Takara, Japan), and E. coli BL21(DE3) (Promega) was used as host for gene expression in the plasmid pET20b (Novagen, USA).
Cloning and sequencing of ldhL. The genomic DNA of T. ethanolicus JW200 was prepared by the standard method [9]. The fragment containing gene ldhL was amplified from genomic DNA by using Ex-taq polymerase (Takara) with a primer pair (5′-TCACTTTTTATGAGTTCTTCCAT-3′ and 5′-TTATATATCAAGCTCTTGTA-3′) designed on the basis of putative ldhL gene of Thermoanaerobacter pseudethanolicus 39E, and then ligated into pMD19T for sequencing. The ldhL gene of T. ethanolicus JW200 was synthesized from genomic DNA by using Pyrobest DNA polymerase (Takara) with a specific primer pair (5′-CCCCATATGAGCAAAATATCTGTAAT-3′, with a NdeI site boldfaced, and 5′-CCCCTCGAGTATATCAAGCTCTTGTATTA-3′, with a XhoI site boldfaced). The 0.95 kb PCR product was cloned into NdeI-XhoI sites of pET20b to yield pET-ldhL.
RNA isolation and 5′-RACE analysis for determining the transcription start site. Total RNA was isolated and purified from T. ethanolicus JW200 cells by using PureLinkTM Micro-to-Midi Total RNA purification kit (Invitrogen, USA) according to the manufacturer’s protocol. The quantity and quality of purified RNA were determined spectrophotometrically and by formaldehyde agarose gel electrophoresis. First-strand cDNA was synthesized by using SuperscriptTM III first-strand synthesis system (Invitrogen) according to the manufacturer’s protocol with a gene-specific primer A (5′-TTATATATCAAGCTCTTGTA-3′). Then the first-strand cDNA was treated with RNase H and precipitated by adding three volumes of ethanol/3 M sodium acetate, pH 5.2 (30 : 1), dissolved in water, and subsequently self-ligated by using T4 RNA ligase (NEB, USA). One microliter of the ligation reaction mixture was used as template for the following PCR. The gene-specific primer B (5′-ATTACAGATATTTTGCTCAT-3′) and primer C (5′-TCTGGCACAGTTCTTGAC-3′) were applied. The resulting products were cloned into pMD19T and sequenced to determine the transcription start site.
Expression and purification of the TeLdhL protein. TeLdhL fused with 6×His-tag at C-terminus was expressed in E. coli BL21(DE3) harboring pET-ldhL and purified by nickel-affinity chromatography (Novagen) according to manufacturer’s protocol (pET His Taq System; Novagen), except that cell-free extracts were obtained by centrifugation after a heat treatment at 65°C for 15 min and then loaded onto the column. The eluted protein was further transferred into 20 mM Tris buffer (pH 7.5) containing 1 mM dithiothreitol (DTT) and 10% (v/v) glycerol by dialysis. The purity and integrity of 6×His-tagged TeLdhL were checked by SDS-PAGE on a 12% gel.
Enzyme assays. The standard assay of TeLdhL activity for pyruvate reduction was performed spectrophotometrically (A320) at 60°С in 50 mM potassium hydrogen phthalate-imidazole buffer (pH 5.8) containing 1 mM pyruvate, 0.2 mM NADH, and 0.2 mM fructose-1,6-bisphosphate (FBP). The standard assay for L-lactate oxidation was performed with 100 mM L-lactate and 1 mM NAD+ in potassium hydrogen phthalate-imidazole buffer (pH 7.0) at 70°С. One unit of enzyme activity was defined as the quantity of enzyme that catalyzed the oxidation of 1 µmol of NADH or reduction of 1 µmol of NAD+ per minute under the above assay conditions. The protein concentration was determined by the Bradford method using BSA as a standard.
Effect of pH and temperature on enzyme activity. The enzyme activity was measured in the pH range from 3.8 to 8.2 for both the reduction of pyruvate to lactate and the oxidation of lactate to pyruvate at 65°С. Buffers used were 50 mM potassium hydrogen phthalate-imidazole buffer (pH 3.8-8.6). To test the effect of temperature on enzyme activity, reactions were performed at various temperatures (30-95°C) in the direction of pyruvate reduction at pH 5.8 and in the direction of lactate oxidation at pH 7.0. The reaction temperatures were adjusted in a temperature-controlled water circulation bath.
Kinetics parameters. For analysis of enzyme kinetics of TeLdhL, varying concentrations of pyruvate between 0.01 and 7.5 mM were used while the NADH was kept at 0.2 mM; varying concentrations of NADH between 0.025 and 1 mM were used while the pyruvate was kept at 1 mM; varying concentrations of lactate between 40 and 150 mM were used while the NAD+ was kept at 1 mM; varying concentrations of NAD+ between 0.05 and 1.5 mM were used while the lactate was kept at 100 mM. The results were analyzed by double reciprocal Lineweaver–Burk plots.
Substrate specificity of TeLdhL. Various combinations (1 mM) of 2-ketobutyrate, phenylpyruvate, or α-ketoglutaric acid for pyruvic acid; 100 mM succinic acid or malic acid for lactate; 0.2 mM NADPH for NADH were tested for their ability to act as substrates of the TeLdhL. The tests were run under the standard assay conditions. Results with tested combinations were compared with the standard reaction.
Inhibition studies. The TeLdhL was incubated in the presence of potential inhibitors in 50 mM potassium hydrogen phthalate-imidazole buffer (pH 5.8) at room temperature for 30 min. The activities for pyruvate reduction were compared with that of the untreated control, which was taken as 100%, by the standard assay.
Stability of TeLdhL. The pH stability of TeLdhL was determined by preincubating the enzyme (20 mg/ml) in 50 mM potassium hydrogen phthalate-imidazole buffer (pH 3.8-8.2) at 70°С for 1 h. The kinetics of thermal deactivation were determined by incubating the same TeLdhL preparations at 60, 70, and 80°С in 50 mM potassium hydrogen phthalate-imidazole buffer (pH 6.2) for different times. The residual activities by standard assay were respectively compared with that of the untreated control, which was taken as 100%.
RESULTS
Sequence analysis. The nucleotide sequence of the 1141-bp fragment containing the ldhL gene and its flanking sequence from T. ethanolicus JW200 genomic DNA was obtained from GenBank (accession No. EU421945). The molar GC content of ldhL was 37.5%. The deduced primary structure of the encoded TeLdhL was composed of 311 amino acids, with a predicted molecular mass of 33.5 kDa. The amino acid sequence of TeLdhL was found to be by 96% identical to the putative L-LDH of T. pseudethanolicus 39E and by 73% identical to that of Thermoanaerobacterium saccharolyticum. TeLdhL contained a G-X-G-X-X-G motif, which was common to most NAD-linked dehydrogenases [10], close to the N-terminus. Signal peptide was predicted by SignalP V2.0 HMM (Signal peptide probability 0.836) with cleavage site probability 0.431 between residue 26 and 27, and deletion of the putative signal peptide sequence led to non-expression of TeLdhL.
Determination of the ldhL transcription start point. Total RNA was isolated from an 8-h anaerobic culture of T. ethanolicus JW200, reverse transcribed, and subjected to 5′-RACE analysis. The transcription start point was located 36 nucleotides upstream of the ATG initiation codon (Fig. 1). A typical prokaryotic ribosome binding site, AGGAGA [11], was identified at 7 nucleotides upstream of the initiation codon. One dyad symmetry region (22 bp) was located in the –35 region.
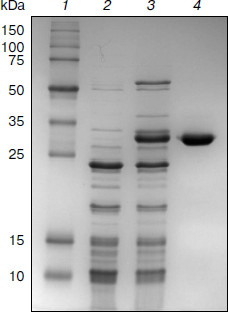
Purification of TeLdhL. TeLdhL samples from the different treatment steps were analyzed by 12% SDS-PAGE. One major protein band corresponding to the expected 33.5-kDa 6×His-tagged TeLdhL was detected in the elution fraction (Fig. 2).Fig. 1. Thermoanaerobacter ethanolicus JW200 ldhL promoter elements. The ATG start codon is indicated in italic type. The –35 and –10 sites are in bold letters, and the Shine–Dalgarno sequences are underscored. The ldhL transcription start point is marked by an asterisk. The region of dyad symmetry is indicated with opposing arrows.

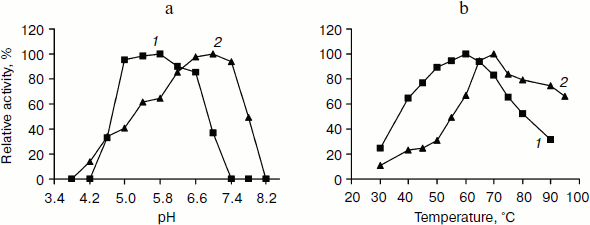
pH and temperature dependences on TeLdhL activity. Figure 3 shows pH and temperature profiles for TeLdhL activity. In the pyruvate reduction reaction, TeLdhL had a pH optimum between 5.0 and 5.8, and the temperature optimum was at 60°C. In the lactate oxidation reaction, TeLdhL had a pH optimum between 6.6 and 7.4, and the temperature optimum was at 70°C.Fig. 2. SDS-PAGE analysis of TeLdhL expression and purification in E. coli. Lanes: 1) molecular mass marker; 2) crude extracts of IPTG-induced E. coli BL21(DE3) harboring plasmid pET20b after heat treatment (65°C, 15 min) as negative control; 3) crude extracts of IPTG-induced E. coli BL21(DE3) harboring plasmid pET-ldhL after heat treatment (65°C, 15 min); 4) the purified recombinant TeLdhL protein after nickel-affinity chromatography. Gel electrophoresis was performed in a 12% SDS-polyacrylamide gel and was followed by staining with Coomassie brilliant blue R-250.
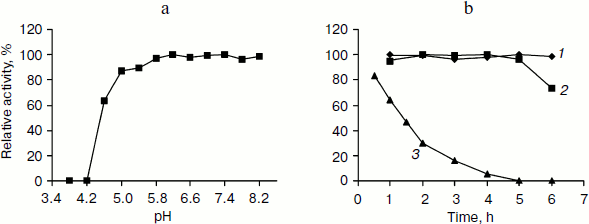
Incubation of TeLdhL at lower pH resulted in a rapid inactivation of the enzyme (Fig. 4a). After incubation in 50 mM potassium hydrogen phthalate-imidazole buffer (pH 5.8-8.2) at 70°C for 1 h, TeLdhL still retained all its original activity. And TeLdhL showed high thermostability with a 1.5-h half-life at 80°C, and it did not lose activity even when incubated at 70°С for 5 h (Fig. 4b).Fig. 3. Effects of pH (a) and temperature (b) on TeLdhL activity. The catalytic direction of pyruvate reduction and lactate oxidation are designated as 1 and 2, respectively.
Effects of various reagents on TeLdhL activity. At a concentration of 1 mM EDTA, ATP and the metal ions Mg2+, Co2+, Ca2+, Mn2+, Ni2+ changed TeLdhL activity by less than 6%, Fe2+ inhibited by 74%, whereas Zn2+, Ag+, Cu2+, Fe3+ and Pb3+ completely inhibited the enzyme. DTT and 2-meraptoethanol (1 mM) were slightly inhibitory (19 and 10% inhibition, respectively). SDS (0.1%) inhibited by 100%, whereas 0.1% Triton X-100 and urea stimulated activity by 23 and 9%, respectively. NAD+ (0.5 mM) inhibited TeLdhL activity by 7%.Fig. 4. Stability of the purified TeLdhL. a) pH stability of the purified TeLdhL. The enzyme was incubated in 50 mM potassium hydrogen phthalate-imidazole buffer of various pH values at 70°С for 1 h. b) Kinetics of thermal deactivation of the purified TeLdhL at different temperatures (°C): 1) 60; 2) 70; 3) 80. The enzyme was incubated in 50 mM potassium hydrogen phthalate-imidazole buffer (pH 6.2) and the activity was measured at different times.
Substrate specificity. The relative activities for 2-ketobutyrate, α-ketoglutaric acid, and phenylpyruvate at 1 mM were 35, 13, and 0%, when compared to that against pyruvate taken as 100%. In the reverse reaction, succinic and malic acids at 100 mM were equally effective substrates as lactate. TeLdhL was found to be NADH-dependent. When NADH was replaced by NADPH as coenzyme, no activity was observed.
Determination of the catalytic parameters Km,app and kcat,app of TeLdhL. The purified recombinant TeLdhL is an allosteric enzyme that is activated by FBP only in the direction of pyruvate reduction. It had Km,app and kcat,app values of 0.18 mM and 520 U/mg, respectively, for pyruvate in the presence of FBP. In the absence of FBP, a 1.2-fold increase in Km,app and a 16-fold decrease in kcat,app for pyruvate were recorded. The Km,app and kcat,app values of the enzyme for lactate were 60 mM and 0.58 U/mg, respectively. As for the coenzyme, the Km,app and kcat,app were unaffected by FBP. NADH had Km,app and kcat,app values of 0.09 mM and 0.30 U/mg, while NAD+ had Km,app and kcat,app values of 0.45 mM and 269 U/mg, respectively.
DISCUSSION
This first report of the cloning and expression of a thermostable TeLdhL from the thermophile T. ethanolicus JW200 provides evidence on the molecular bases for promoter sequence, allosteric properties, and biochemical characterizations of the enzyme.
FBP functions as a positive allosteric effector for the recombinant purified TeLdhL, just as for some gram-positive bacterial LDHs [12]. Even in the absence of FBP, the catalytic efficiency (kcat,app/Km,app) of TeLdhL was much higher in the direction of pyruvate reduction than in the direction of lactate oxidation. Therefore, TeLdhL probably catalyzes pyruvate reduction rather than lactate oxidation in vivo. The enzyme was found to be quite thermostable. These properties suggest that the enzyme might be of great potential for future applications.
Few transcriptional analyses of bacterial LDH genes have been reported so far. The only examples are analyses of ldhLs from Lactococcus lactis [13], Bifidobacterium longum [14], and Pediococcus acidilactici [15]. A small number of Thermoanaerobacter promoter sequences have been identified [16-19], most by homology to known promoters in E. coli. We have identified the transcription start point from the 5′ end of the T. ethanolicus ldhL transcript, and a promoter sequence has been defined at a location close to the translation start. The –10 sequence showed relatively low A+T content and might weaken the transcription initiation. One dyad symmetry region (22 bp) located in the –35 region might be involved in the transcriptional regulation of ldhL. These findings might account for the malfunction of the promoter of ldhL in E. coli (data not shown).
The data on substrate specificity of TeLdhL showed that the relative activities of these 2-oxocarboxylic acids were pyruvic acid > 2-ketobutyrate > α-ketoglutaric acid > phenylpyruvate. This might be because the longer aliphatic chain and the complex benzene ring influence the binding and orientation of the substrate in the enzyme active site. Although most enzymes of lactate dehydrogenase/malate dehydrogenase family have high substrate specificity for either lactate or malate, the TeLdhL displayed low specificity, like the corresponding protein of Mycoplasma genitalium [20].
TeLdhL activity was inhibited by the reducing agents DTT and 2-mercaptoethanol, suggesting that intact disulfide groups are essential for the enzyme activity. NAD+ slightly inhibited TeLdhL in the direction of pyruvate reduction due to higher Km,app value for NAD+ and lower Km,app value for NADH. This finding might suggest that the enzyme reaction is not sensitive to the balance of NADH and NAD+. For many LDHs, ATP was found to be the most potent inhibitor [21]. But the fact that ATP had no effect on the TeLdhL might indicate that the enzyme is not sensitive to the intracellular pool of ATP.
REFERENCES
1.Weigel, J., and Ljungdahl, L. G. (1981) Arch.
Microbiol., 128, 343-348.
2.Lacis, L. S., and Lawford, H. G. (1991) Appl.
Environ. Microbiol., 57, 579-585.
3.Skory, C. D. (2000) Appl. Environ.
Microbiol., 66, 2343-2348.
4.Taguchi, H., Yamashita, M., Matsuzawa, H., and
Ohta, T. (1982) J. Biochem., 91, 1343-1348.
5.Ostendorp, R., Auerbach, G., and Jaenicke, R.
(1996) Protein Sci., 5, 862-873.
6.Savijoki, K., and Palva, A. (1997) Appl.
Environ. Microbiol., 63, 2850-2856.
7.Holt, P. J., Williams, R. E., Jordan, K. N., Lowe,
C. R., and Bruce, N. C. (2000) FEMS Microbiol. Lett.,
190, 57-62.
8.Mai, V., Wiegel, J., and Lorenz, W. W. (2000)
Gene,' 247, 137-143.
9.Ausubel, F. M., Brent, R., Kingston, R. E., Moore,
D. D., Seidman, J. G., Smith, J. A., and Struhl, K. (1998) Current
Protocols in Molecular Biology, John Wiley and Sons, New York, pp.
2.4.1-2.4.5.
10.Branden, C., and Tooze, J. (1991) Introduction
to Protein Structure, Garland Publishing, New York-London, pp.
141-159.
11.Shine, J., and Dalgarno, L. (1975) Nature,
254, 34-38.
12.Garvie, E. I. (1980) Microbiol. Rev.,
44, 106-139.
13.Llanos, R. M., Hillier, A. J., and Davidson, B.
E. (1992) J. Bacteriol., 174, 6956-6964.
14.Minowa, T., Iwata, S., Sakai, H., Masaki, H., and
Ohta, T. (1989) Gene, 85, 161-168.
15.Garmyn, D., Ferain, T., Bernard, N., Hols, P.,
and Delcour, J. (1995) Appl. Environ. Microbiol., 61,
266-272.
16.Erbeznik, M., Dawson, K. A., and Strobel, H. J.
(1998) J. Bacteriol., 180, 1103-1109.
17.Jørgensen, S. T., Tangney, M., Staines, R.
L., Amemiya, K., and Jørgensen, P. L. (1997) Biotechnol.
Lett., 19, 1027-1031.
18.Erbeznik, M., Strobel, H. J., Dawson, K. A., and
Jones, C. R. (1998) J. Bacteriol., 180, 3570-3577.
19.Holt, P. J., Williams, R. E., Jordan, K. N.,
Lowe, C. R., and Bruce, N. C. (2000) FEMS Microbiol. Lett.,
190, 57-62.
20.Wu, G., Fiser, A., Ter Kuile, B., Sali, A., and
Muller, M. (1999) Proc. Natl. Acad. Sci. USA, 96,
6285-6290.
21.Brown, A. T., and Wittenberger, C. L. (1972)
J. Bacteriol., 110, 604-615.