Characterization of a Monomeric Heat-Labile Classical Alkaline Phosphatase from Anabaena sp. PCC7120
Ming Luo1,2, Yong-Chao Guo1, Jiao-Yu Deng1, Hong-Ping Wei1, Zhi-Ping Zhang1, Yan Leng1,2, Dong Men1,2, Li-Rong Song3, Xian-En Zhang1, and Ya-Feng Zhou1*
1State Key Laboratory of Virology, Wuhan Institute of Virology, Chinese Academy of Sciences, Wuhan 430071, China; fax: +86(027)8719-9492; E-mail: zyf@wh.iov.cn; luoming1976@yahoo.cn; guoyc@ust.hk; dengjy@wh.iov.cn; hpwei@wh.iov.cn; zhangzp@wh.iov.cn; x.zhang@wh.iov.cn2Graduate School, Chinese Academy of Science, Beijing 100039, China; audreyyanleng@yahoo.com.cn; Winter_J_Gates@yahoo.com.cn
3State Key Laboratory of Freshwater Ecology and Biotechnology, Institute of Hydrobiology, Chinese Academy of Sciences, Wuhan, 430072, China; lrsong@ihb.ac.cn
* To whom correspondence should be addressed.
Received March 12, 2010; Revision received March 30, 2010
Alkaline phosphatases (APs), known inducible enzymes of the Pho regulon and poorly characterized in cyanobacteria, hydrolyze phosphomonoesters to produce inorganic phosphate (Pi) during Pi starvation. In this study, two predicted alkaline phosphatase genes in the genome of Anabaena sp. PCC 7120, all2843 and alr5291, were apparently induced during Pi starvation. Sequence analysis showed that alr5291 encodes a protein that is an atypical alkaline phosphatase like other cyanobacteria PhoAs, but the protein encoded by all2843 is very similar to the classical PhoAs, such as Escherichia coli alkaline phosphatase (EAP). To date, there have been no reports about classical phoA in cyanobacterial genomes. The alkaline phosphatase APA, coded by all2843, is characterized as a metalloenzyme containing Mg2+ and Zn2+ with molar ratio of 1 : 2. Site-directed mutagenesis analysis indicated that, though the active center of APA is highly conserved in comparison with EAP, differences do exist between APA and EAP in metal ion coordination. Besides, biochemical analysis revealed that APA is a monomeric protein and inactivated rapidly at 50°C. These results suggest that APA is the first monomeric heat-labile classical PhoA found in cyanobacteria.
KEY WORDS: Anabaena sp. PCC7120, phosphorous starvation, alkaline phosphatase, metalloenzyme, site-directed mutagenesisDOI: 10.1134/S0006297910050172
Abbreviations: AP, alkaline phosphatase; EAP, E. coli alkaline phosphatase; ICP-OES, inductively coupled plasma-optical emission spectrometric analysis; pNPP, para-nitrophenyl phosphate; phoA, alkaline phosphatase-encoding gene; PhoA, product of phoA gene; qPCR, quantitative polymerase chain reaction; SBP, streptavidin-binding peptide.
Phosphorous, an essential element for all organisms, is required for the
biosynthesis of nucleotides, proteins, phospholipids and in the
functional regulation of proteins through phosphorylation. However,
phosphorous is deficient in most ecosystems due to the fact that
inorganic phosphate (Pi) is the only form of phosphorous
that is directly used by cells [1-3]. Phosphate starvation conditions induce the
so-called psi (phosphate starvation-inducible) genes belonging
to the Pho regulon, which code for proteins involved in the transport
and assimilation of Pi and phosphorous-containing compounds
[4].
As important enzymes of the Pho regulon, alkaline phosphatases (APs) (orthophosphate monoester phosphohydrolases (alkaline optimum); EC 3.1.3.1) catalyze the hydrolysis of phosphomonoesters to produce inorganic phosphate [5]. Many APs have been widely investigated since the 1960s. Among these APs, E. coli alkaline phosphatase (EAP) (PhoA) has been well characterized, and its 3-D structure has been resolved at 1.75 Å [5-9]. The enzyme is a homodimeric metalloenzyme activated by Zn2+ and Mg2+ [10, 11].
Cyanobacteria, one of the oldest life forms on Earth [12], survive in a broad range of ecological environments from soil and fresh water to diverse open marine areas and play important roles in the global cycling of carbon, nitrogen, and phosphorus [13, 14]. Similar to other bacteria, they have the Pho regulon [15-17] and produce alkaline phosphatases during Pi starvation [18-21]; however, the alkaline phosphatases in cyanobacteria are poorly characterized. There are three alkaline phosphatases termed PhoA in Synechococcus elongates PCC6301 and PCC7942 and PhoV in Synechococcus elongates PCC7942 [18, 20, 22]. Only two of them, PhoA in Synechococcus PCC6301 and PhoV in Synechococcus PCC7942, have been partially characterized. However, genome informatic analysis indicated most of the putative PhoAs in cyanobacteria were atypical alkaline phosphatases due to low sequence identity to classical PhoA such as EAP [23]. Furthermore, biochemical analysis revealed the alkaline phosphatases in cyanobacteria were apparently inhibited by Zn2+ but strongly activated by Ca2+ [24-26]. The PhoV of Synechococcus PCC7942 was indeed activated by Zn2+ or Ca2+ like EAP [22], but the phoV gene is not present in the genome of Synechococcus PCC7942 [27].
Anabaena sp. PCC 7120 is the model species of diazotrophic heterocyst-forming filamentous cyanobacteria. There are no reports on the response of this species to Pi starvation. Two genes, all2843 and alr5291, were predicted to be alkaline phosphatases from genomic analysis [27]. Sequence similarity analysis indicates that alr5291 is probably a homolog of alkaline phosphatase genes from other cyanobacteria [23]. However, all2843 does not show similarity with the predicted PhoAs in cyanobacteria, and no such homolog has been identified even in Anabaena variabilis ATCC 29413, a species very close to Anabaena sp. PCC 7120 [27].
In this study, the expression of psi genes, all2843 and alr5291, in Anabaena sp. PCC 7120 was clearly induced during Pi starvation. Gene all2843 was overexpressed in E. coli and found to encode an alkaline phosphatase termed APA, which is very similar to EAP. The results of site-directed mutagenesis further confirmed that APA has a highly conserved active center as reported for EAP. To date, this is the first time a classic endogenous PhoA gene has been identified in cyanobacteria. Furthermore, unlike most PhoA type alkaline phosphatases, which are dimeric and thermally stable, APA is a monomeric and heat-labile enzyme. This suggests that the protein features of APs are strongly divergent, but the active center is well conserved.
MATERIALS AND METHODS
MnCl2, FeCl3, ZnSO4, CaCl2, CuSO4, CoCl2, MgCl2, and p-nitrophenyl phosphate (pNPP) were obtained from Sigma (USA). The strep-tag purification kit was purchased from IBA (IBA GmbH, Germany).
Strains and plasmids. Escherichia coli DH5α was used as the host cell for cloning purposes. The E. coli ΔphoA strain SM547 (Δ(phoA-proC), phoR tsx: Tn5, Lac, gal K, leu, Strr) used for protein expression was kindly provided by Prof. Kantrowitz (Boston College, USA). The plasmid pASK75-SBP was used to construct vectors for the overexpression of Anabaena sp. PCC 7120 AP.
Bacteria strain and culture conditions. Anabaena sp. strain PCC 7120 was grown to mid-logarithmic phase on a rotary shaker in the light (30 µE·m–2·sec–1). For phosphate deprivation experiments, cells were centrifuged at 5000g for 5 min at room temperature, washed in phosphate-free BG-11 (KH2PO4 was replaced by KCl), and suspended in the same medium at an A730 of 0.5. Escherichia coli DH5α and SM547 were grown in LB medium supplemented with ampicillin (100 µg/ml) when needed.
Construction of APA overexpression vector. The gene all2843 was amplified by PCR from the genomic DNA of Anabaena sp. strain PCC 7120 and ligated into pASK75-SBP. The resulting plasmid pASK75-2843-SBP was confirmed by sequencing.
In vitro mutagenesis. Site-directed mutations were introduced into the selected sites in the all2843 gene by overlap PCR. All fragments were ligated into pASK75-SBP and were subsequently sequenced to confirm the presence of the mutations.
Protein overexpression and purification. The wild-type and mutants of APA were overexpressed and purified using the same procedure. Typically, SM547⁄pASK75-2843-SBP was induced by 200 ng/ml tetracycline (Sigma) at A600 = 0.6 for at least 9 h at 18°C. The cells were harvested by centrifugation. The periplasmic extract was prepared by osmotic shock using polymyxin B, and the recombinant APA (with SBP as fusion partner) was purified using an IBA strep-tag purification kit according to the manufacturer’s guidelines. The purity of the protein solution was estimated by SDS-PAGE, and its concentration was measured by the Quick Start™ Bradford Protein Assay (Bio-Rad, USA).
Measurement of phosphatase activity. Phosphatase activity was quantitatively determined by monitoring the conversion of pNPP to p-nitrophenolate (pNP) at 405 nm at 25°C. For the measurement in cyanobacteria, the cells were suspended in 1 M Tris-HCl (pH 8.0), 150 mM NaCl, and 3.6 mM pNPP (final volume 1 ml) and then incubated at room temperature for 30 min. The reaction was stopped with 100 µl of 5 N NaOH, and the mixture was centrifuged at 6000g for 4 min. The A405 of the supernatant was measured and compared with a standard absorbance curve for p-nitrophenolate (Sigma). For the recombinant AP, the assays were carried out either at pH 8.0 in 1 M Tris-HCl (transphosphorylation activity) or at pH 8.0 in 0.1 M Mops buffer (hydrolysis activity) for 2 min. In each case the ionic strength was maintained constant with 150 mM sodium chloride.
Analytical ultracentrifugation. Molecular mass was measured using an XL-I analytical ultracentrifuge (Beckman Coulter, USA) equipped with a four-cell An-60 Ti rotor. The purified protein (0.4 mg/liter) in 100 mM Tris-HCl buffer (pH 8.0), 150 mM NaCl was centrifuged at 4°C and 262,000g for 4 h, with the same buffer as the control. To determine the molecular mass of the protein, the data were analyzed using the SEDFIT software from http://www.analyticalultracentrifugation.com/download.htm.
Determinations of metal ion content and cation effect studies. The metal ion content in APA was determined using ICP-OES (Vista-MPX; Varian, USA). Purified protein solution (1.6 ml, 1.0 mg/liter) was digested with nitric acid (400 µl) and diluted to 4 ml. The metal ion content in the solution was determined by ICP-OES using metal ion standard solution (GSB 04-1766-2004; General Research Institute for Nonferrous Metals, China). The effects of cations were examined for their influence on enzyme activity. The cations or EDTA were added to the purified APA solution at a final concentration of 1 mM, the mixture was incubated at 4°C overnight, and the residual enzyme activity was measured at pH 8.0 (1 M Tris-HCl).
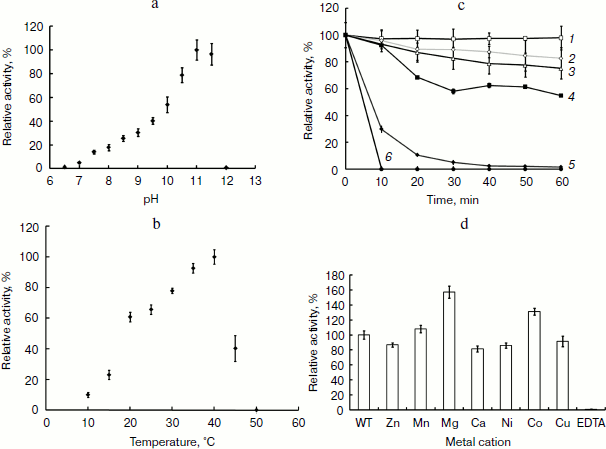
Optimal temperature, optimal pH, and thermal stability. The apparent optimum temperature for transphosphorylation activity of APA was determined by running the standard assay at temperatures ranging from 10 to 50°C in 1 M Tris-HCl buffer (pH 8.0). Purified APA was incubated at each temperature for 10 min prior to the assay of enzyme activity. The apparent optimum pH of the enzyme was determined by running the standard assay using 0.1 M NaOH-glycine in the pH range from 6.5 to 12. Thermostability was measured by incubating the enzyme from 25 to 50°C for 60 min. Samples were taken at 10 min time intervals, and the residue activity was determined by standard assay.
RNA isolation and real-time quantitative PCR. Total RNA from Anabaena sp. PCC 7120 was isolated as described by Ning and Xu [28] and treated three times with RNase-free DNase. For real-time quantitative PCR, a cDNA pool was synthesized using a PrimeScript 1st strand cDNA synthesis kit (Takara Bio Inc., Japan), with 300 ng RNA and 5 µM random 6-mer primer in a reaction volume of 10 µl. Reverse transcription was performed at 37°C for 30 min and then at 85°C for 5 sec to inactivate the reverse transcriptase. For real-time PCR analysis, 1 µl of cDNA samples was used in a 25 µl mixture containing 0.2 µM specific primers (Table 1) and 11.25 µl RealMasterMix (SYBR Green I) (Tiangen, China) using a RT-cycler136 (Capitalbio, China). A control without cDNA was also included. Amplification was initiated at 94°C for 5 min, followed by 40 cycles at 94°C for 30 sec, 60°C for 30 sec, and 72°C for 25 sec. After amplification, melting-curve analysis was performed. The relative amount of transcript was calculated using the RT-Cycler 2.0 Analysis software (Capitalbio) based on the normalized-expression (ΔΔCT, where CT is threshold cycle) method. Transcript levels of target genes were normalized to those of the internal reference gene (rnpB) measured using the same samples. The real-time PCR was performed in triplicate for each sample.
Table 1. Primers used in quantitative
real-time PCR

Phylogenetic analysis. Multiple alignments of the alkaline phosphatase were performed using ClustalW2 [29]. Aligned sequences were analyzed using the maximum likelihood method, assisted with PhyML [30]. Branch support was assessed using 1000 bootstrap replicates [31].
RESULTS
Phylogenic analysis of alr5291 and all2843. The sequence analysis of the proteins encoded by all2843 and alr5291 suggested that alr5291 is an atypical alkaline phosphatase gene like others in cyanobacteria and all2843 belonged to classical phoA like the EAP gene. The phylogenetic tree of the predicted alkaline phosphatases (Fig. 1a), clearly show two clusters of these alkaline phosphatase-like genes: classical phoAs and atypical alkaline phosphatase genes according to the similarities between the coding sequences of these genes. The atypical alkaline phosphatases cluster includes alr5291 and most of the alkaline phosphatases of cyanobacteria. In contrast, all2843 belongs to the classic phoA cluster. Meanwhile, the sequence alignments of APA with four classic phoA type alkaline phosphatases and the PhoV of Synechococcus PCC7942 indicates that they have a conserved active center, further suggesting that APA might be a PhoA homolog protein in cyanobacteria (Fig. 1b).
Fig. 1. Phylogenetic analysis of alkaline phosphatases and conservation of the active site of PhoA. a) Radial tree of alkaline phosphatase genes: all2843 and alr5291 from Anabaena sp. PCC 7120; AVA_2541 from Anabaena variabilis ATCC 29413; sll0654, SYNW2390, and SYNW2391 from Synechocystis sp. PCC 6803 and WH8102; syc0163_d from Synechococcus elongatus PCC strains 6301; Synpcc7942_1392 and PhoV from Synechococcus elongatus PCC strains 7942; VF_A1057 from Vibrio fischeri ES114; Spro_0721 from Serratia proteamaculans; mll4564 and mlr0764 from Mesorhizobium loti; SMc02634 from Sinorhizobium meliloti; gll0893 from Gloeobacter violaceus PCC 7421; PMM0708 and PMN2A_0439 from Prochlorococcus marinus strains CCMP1986 and NATL 2A; EAP from E. coli; TAP from Antarctic bacterium TAB5; PfuAP from Pyrococcus furiosus; PabAP from Pyrococcus abyssi; BsuAP from Bacillus subtilis; BliAP from Bacillus licheniformis; PLAP from human placenta; TNAP from human liver/bone/kidney; DmeAP from Drosophila melanogaster; SgrAP from Streptococcus griseus. Bootstrap values indicated on the figure are from 1000 maximum likelihood trees.
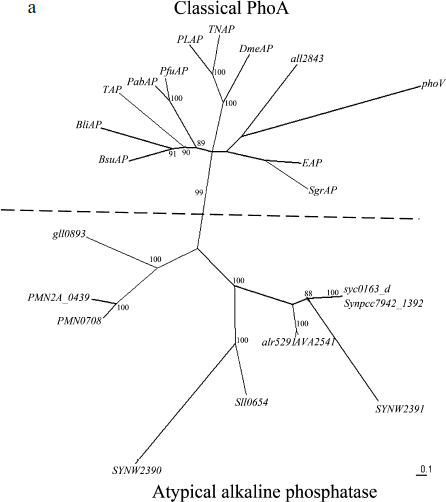
Alkaline phosphatase was induced after deprivation of phosphate. Anabaena sp. PCC 7120 in phosphate-free medium had increased alkaline phosphatase activity as measured in suspensions of intact cells that were initially cultured in BG-11 medium containing 175 µM phosphate (Fig. 2a). After nine days of phosphate deprivation, the total alkaline phosphatase activity of the cultures was elevated 2.5-fold, whereas no obvious increase in phosphatase activity was detected in the BG-11 culture.Fig. 1. Phylogenetic analysis of alkaline phosphatases and conservation of the active site of PhoA. b) Conservation of the active site of PhoA. Shown is the peptide sequence alignment of APA with other members of PhoAs. Abbreviations: APA, Anabaena sp. PCC 7120 APA; BsuAP, Bacillus subtilis AP IV; EAP, E. coli AP; PLAP, Homo sapiens AP placental type; TAP, Antarctic bacterium TAB5 AP; PhoV, PhoV from Synechococcus elongatus PCC strains 7942. Strictly conserved residues, residues conserved more than 80 and 60% are highlighted in black, dark gray, and light gray, respectively. Symbols above the sequences indicate the conserved residues that are involved in the active site: solid arrows, coordinating directly to the metal ions; open arrows, forming the salt-link important for phosphate binding; solid circle, phosphoseryl intermediate formation; open circle, phosphate coordination; solid triangle, stabilizing active site.

The qPCR method was used to measure the transcript levels of all2843 and alr5291. The mRNA levels were analyzed on days 0, 0.5, 1, 2, 3, 5, 7, and 9 after the cells were grown under the phosphate-free conditions. The results indicated the transcription of both all2843 and alr5291 were upregulated during phosphate starving. However, their expression patterns over time and their mRNA levels were different from one another. The transcription of all2843 was always upregulated when the cells were starved of phosphate, with an increase of almost 20-fold on the 5th day of phosphate depletion (Fig. 2b, shaded bars) while the transcription level of all5291 was downregulated to 0.6-fold during the initial 12 h of phosphate deprivation, followed by an increase to 3.5-fold on day 5 (Fig. 2b, open bars). Interestingly, the RNA expression level in alr5291 was almost 30 times higher than the level expressed in all2843 in the same phosphate-containing media (Fig. 2c).Fig. 2. Induction of alkaline phosphatases of Anabaena sp. PCC 7120. a) Alkaline phosphatase activity of Anabaena sp. PCC 7120 after the transfer of cells to phosphate-free medium. Absorbance of the culture was measured at 730 nm. Cells were resuspended in fresh phosphate-free BG-11 medium at A730 of 0.5, and alkaline phosphatase activity was assayed at 0, 0.5, 1, 2, 3, 5, 7, and 9 days after the transfer. b) Relative abundance of transcripts determined by real-time quantitative PCR. Relative abundance of transcripts of all2843 (shaded bars) and alr5291 (open bars) were normalized to those of rnpB as an internal standard. Amount of the transcripts of each gene measured at 0 h after the deprivation of phosphorous was set at 1, and all other data were calculated relative to this value. c) Different expression levels of all2843 and alr5291 in phosphate-containing medium. Lines: 1) rnpB; 2) alr5291; 3) all2843.
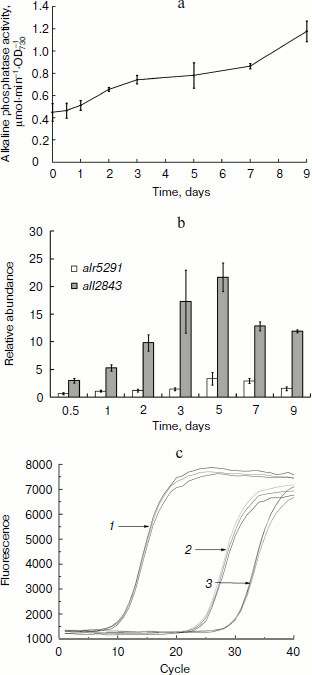
APA is a monomeric protein. The recombinant APA was overexpressed in E. coli and purified by strep-tag affinity chromatography. A single protein band was observed near 66 kDa by SDS-PAGE (Fig. 3a). Gel filtration and analytical ultracentrifugation indicated that the molecular mass of the recombinant APA was 69 kDa (Fig. 3b) and 70 kDa (Fig. 3c), respectively. According to theoretical molecular mass of 73.43 kDa, it was concluded that APA is a monomeric protein.
APA is a heat-labile alkaline phosphatase. To further characterize the biochemical feature of this AP, the heat resistance, pH, and temperature profiles were investigated. The optimal pH of the enzyme was shown to be 11.0, but it was fully inactive above pH 12.0 (Fig. 4a). The optimal temperature for APA was clearly shown to be 40°C, and its activity dropped drastically at higher temperatures (Fig. 4b). The enzyme was shown to be very stable at lower temperatures as 25 and 30°C but denatured quickly at 45°C, losing almost 70% of its activity, and was fully inactive at 50°C within 10 min (Fig. 4c). Taken together, these results showed that APA is a heat-labile alkaline phosphatase with optimal activity at pH 11.0, which is stable from 25 to 30°C but is denatured quickly beyond 45°C.Fig. 3. Molecular mass determination of APA. a) SDS-PAGE analysis of purified APA. Lanes: 1) protein marker; 2) purified APA protein. b) Gel filtration. Molecular mass markers: thyroglobulin (669 kDa, No. 1), apoferritin (443 kDa, No. 2), β-amylase (200 kDa, No. 3), alcohol dehydrogenase (150 kDa, No. 4), bovine serum albumin (66 kDa, No. 5) and carbonic anhydrase (29 kDa, No. 6). c) Analytical ultracentrifugation.
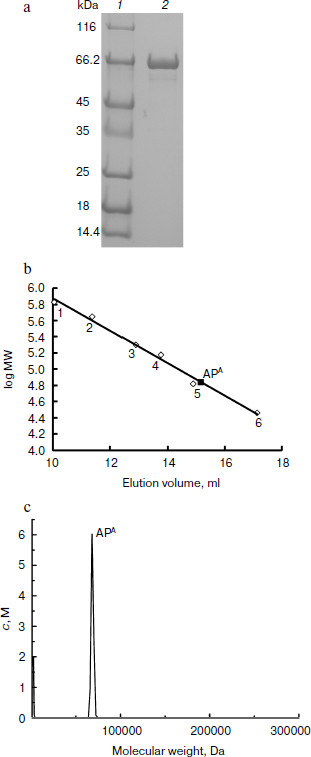
To date, research indicates that APs are classic metalloenzymes [5], including most of PhoAs such as EAP, which contains one Mg2+ and two Zn2+ ions per molecule. In our study the effect of divalent cations and the metal content of APA were examined. Mg2+, Mn2+, Ca2+, Cu2+, Zn2+, Ni2+, and EDTA were added to the purified enzyme solution at a final concentration of 1 mM. As shown in Fig. 4d, the enzyme activity was strongly inhibited in the presence of 1 mM EDTA by chelation of the metal ion from the enzyme active center, whereas the enzyme exhibited 1.6- and 1.4-fold activation in the presence of 1 mM Mg2+ and Co2+, respectively. These results indicated that the APA activity is dependent on the presence of divalent cations. The presence of metal ions in APA was further confirmed using inductively coupled plasma-optical emission spectrometry (ICP-OES). The metal ion content was calculated using a calibration curve obtained for each metal ion with subtraction of the background signal. ICP-OES showed that APA contained magnesium and zinc with an average molar ratio of 0.81 ± 0.15 and 1.55 ± 0.19, respectively, suggesting that the APA protein is a metalloenzyme containing one Mg2+ and two Zn2+ ions per molecule, which is in agreement with studies of EAP.Fig. 4. Effect of pH, temperature, and metal ions on the activity of APA and thermal stability of APA. a) pH profile of APA (0.1 M NaOH-glycine for the pH range 6.5 to 12.0). b) Temperature profile of APA. Tris-HCl buffer (1 M, pH 8.0) was used as the solvent. c) Thermal stability of APA. Lines: 1) 25°C; 2) 30°C; 3) 35°C; 4) 40°C; 5) 45°C; 6) 50°C. d) Effect of metal ions on APA. Various metal ions were added to APA at a final concentration of 1 mM and incubated at 4°C. The enzyme activity was measured in 1 M Tris-HCl (pH 8.0), 150 mM sodium chloride at 25°C. Activity is expressed as specific activity relative to a control incubated in the absence of any metal ions.
Steady state kinetics of APA. Alkaline phosphatase hydrolyses phosphomonoesters to inorganic phosphates and alcohol or transphosphorylates phosphomonoesters to new phosphoesters in the presence of alcohol [6]. The enzymatic dynamics and the kinetic parameters of APA at pH 8.0 are summarized. In the presence of phosphate acceptor (1 M Tris-HCl buffer), the specific activity, Km, and kcat were 16.3 ± 2.4 units/mg (one unit is defined as the amount of enzyme which hydrolyzes 1 µmol substrate pNPP in 1 min), 14.5 ± 0.1 µM, and 29.9 ± 1.1 sec–1, respectively. Deprivation of the phosphate acceptor resulted in much lower catalytic activity (6.0 ± 0.6 units/mg) with a Km of 3.6 ± 0.3 µM. The product inorganic phosphate is also a competitive inhibitor of the AP: the Ki of phosphate was 47.2 ± 1.5 µM in 0.1 M Mops buffer (pH 8.0). However, the Km was much lower than those reported for alkaline phosphatases of other cyanobacteria [18].
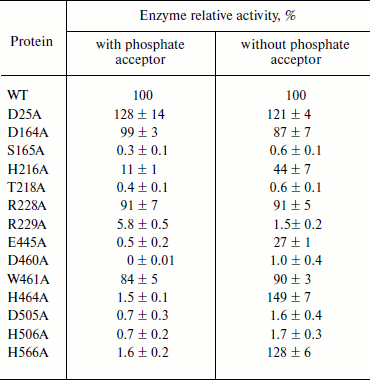
The active site of PhoA is highly conserved in APA. The alignment of APA with other phoA alkaline phosphatases showed that the residues corresponding to the active site of PhoA are highly conserved in APA (Fig. 1b). All these residues were chosen for further study to confirm that they are involved in the enzyme active site. Alanine was introduced into APA at these selected sites by site-directed mutagenesis, and Table 2 shows the specific activities of these mutant enzymes. The activity of mutants S165A, H216A, T218A, R229A, E445A, D460A, D505A, and H506A were significantly decreased in the presence or absence of a phosphate acceptor. H464A and H566A were almost inactive in 1 M Tris buffer, but had an increased activity in 0.1 M Mops buffer. The mutants D164A, R228A, and W461A showed a slight loss of activity, and D25A exhibited a slight increase in activity under each reaction condition.
Table 2. Relative activity of APA
mutants. Activity is expressed relative to wild type APA.
The assays were carried out either in 1 M Tris-HCl, pH 8.0
(with phosphate acceptor) or in 0.1 M MOPS, pH 8.0 (without
phosphate acceptor). In each case the ionic strength was maintained
with 150 mM NaCl
DISCUSSION
Alkaline phosphatases exist in all organisms and have been particularly well studied in E. coli [5-9]. Although not well studied, several APs in cyanobacteria have been identified, but they did not show similarity with EAP [18, 20]. Only one PhoA homolog, PhoV, has been reported in Synechococcus elongates PCC7942 and was found to be similar to EAP. However, the gene phoV is not present in its sequenced genome. The reason is still unclear, but perhaps a plasmid might carry its coding sequence.
Here we identified two endogenous genes, all2843 (gene location, 3463675 to 3465558 in Anabaena sp. PCC 7120 genome) and alr5291 (gene location, 6310388 to 6312055 in Anabaena sp. PCC 7120 genome), encoding two putative AP orthologs. The phylogenic analysis concluded that the gene alr5291 encodes an atypical alkaline phosphatase like many others in cyanobacteria [32]. Instead, the gene all2843 encoded a classical phoA alkaline phosphatase. Both of the two genes might be involved in phosphorus assimilation because their expressions are induced under conditions of Pi starvation, which is consistent with other cyanobacteria [21]. The gene alr5291 was likely constitutively expressed in phosphorous sufficient medium, and its transcription level was higher than all2843 by almost 30-fold. Moreover, only the AP encoded by alr5291 was found to have a signal peptide, which drives the AP secretion to the periplasmic space [33], suggesting that the AP activity outside of the cell mainly relies on the protein encoded by alr5219. This is also consistent with the correlation of upregulation of the alr5219 mRNA level (3.5-fold) and the enhancement of AP activity (2.5-fold), rather than the 20-fold upregulation of all2843 mRNA. The exact functions of these two APs in cyanobacteria need to be further investigated.
The metal content test suggested that there are two zinc ions and one magnesium ion in APA, the same as for EAP. However, APA is activated by Co2+ (not by Zn2+ as for EAP). This difference might have resulted from changes in the amino acids required for ion binding. Similar to the EAP double mutant D153H/K328W, which is also activated by Co instead of Zn [34], the corresponding residues in APA are histidine and tryptophan, exactly the same as in the EAP mutant D153H/K328W.
Sequence alignment of APA with other PhoAs indicated that residues corresponding to the active site of PhoA are highly conserved in APA. As previous studies on EAP and PLAP had shown, the residues responsible for the phosphoseryl intermediate formation [35], phosphate coordination [36], and metal coordination [37-42] were critical for retaining the enzymatic activity, and replacement of any of them with alanine resulted in the loss of activity. According to the alignment result, in APA the phosphoseryl intermediate formation site is Ser165; the phosphate coordination site is Arg229; the metal coordination sites are D25, Thr218, Glu445, Asp460, His464, Asp505, His506, and His566. Our mutagenesis test also indicated that mutations of these residues led to drastic decreases in enzymatic activity except for the D25A mutation. Very interestingly, the D25A mutant had an even higher catalytic activity than the wild-type enzyme, whereas the corresponding EAP mutant D51A was almost inactive [43]. The discrepancy between the APA mutant D25A and the EAP mutant D51A might result from variations in structural features between APA and EAP. In EAP, Asp101 stabilizes the active site, and the mutant D101A showed no change in activity [44]. In our study, the corresponding APA mutant D164A also exhibited nearly full wild-type enzyme activity. The other important residues in EAP active site, Asp153 and Lys328, are responsible for the forming a salt link, which is important for phosphate binding and indirectly coordinated with the Mg ion through water-mediated hydrogen bands [45, 46]. Both the mutants D153A and K328A showed increased activity [45, 46]. Our results showed that the corresponding mutants in APA, H216A and W461A, exhibit decreased activity. As the residues Asp25, His216, and Trp461 should all interact with the magnesium ion directly or indirectly, it indicates that the environment of magnesium ion in APA might be somewhat different from that in EAP. Further investigations will be necessary to determine the fine mechanisms for this interesting observation. Besides, replacement of Arg228, the residue adjacent to the predicted phosphate coordination site Arg229, led to a slight decrease in activity. This might be due to lower Km value. A similar observation was previously made with EAP in our lab: substitution of Lys167, which is adjacent to Arg166, resulted in a more positively charged environment around the substrate binding pocket, which could stabilize the transition state complex and accelerate the rate of hydrolysis of the covalent E–P intermediate [47]. These results clearly show that the active sites of PhoA are highly conserved in APA and further support the conclusion that APA is a classical PhoA type alkaline phosphatase, though difference do exist between this enzyme and those from other species.
Based on the result of bioinformatic, biochemical, and mutagenesis analysis, APA was confirmed to be a classical PhoA. This is the first time a classical PhoA has been identified in cyanobacteria.
APA also showed some apparent differences from EAP. For example, it was characterized to be a monomeric heat-labile protein, while most other PhoAs are dimeric [48-50] and heat-resistant enzymes [51, 52]. Previous work on EAP showed that the introduction of amino acids with large, bulky, and charged side chains into the interface of EAP disturbs the formation of the dimeric structure. The replacement of Thr59 with Arg resulted in the formation of a monomeric EAP mutant with little activity, and in B. subtilis APIV, a monomeric PhoA, the corresponding residue is also Arg [53]. Amino acid alignments showed that the residue in APA, which corresponds to Thr59 in EAP, is also an Arg. APA is likely a monomer because it lacks the interactions necessary for dimerization. In EAP, the substitution of Thr to Arg at residue 59 also impacted the stability of the enzyme. The Tm for the T59R mutant was 43°C, significantly lower than the 97°C observed for the wild-type enzyme [53]. Therefore, the differences in residues between APA and EAP are likely one of the factors responsible for the thermal instability of APA. Computational analysis has indicated that the decrease in content of charged residues causes protein destabilization [54]. It is commonly recognized that charged residues are usually located at the surface of proteins and stabilize the proteins by electrostatic interactions. The content of the charged residues (Lys, Arg, Glu, and Asp) of APA (16.6%) is significantly lower than that of EAP (20.7%). The decrease in electrostatic interactions might be another factor responsible for the thermal instability of APA. In addition, there are four cysteines that form two disulfide bonds in EAP [6], and only two cysteines exist in APA. The decrease in the number of disulfide bonds might also contribute to the cold-adaptation of APA [55].
Taken together, in this report, APA coded by all2843 was identified to be a classical PhoA and to be induced during phosphate starvation. Biochemical analysis indicated that it is a monomeric heat-labile enzyme, which distinguishes it from most alkaline phosphatases. Data of site-direct mutagenesis showed that, though the active sites of PhoA are highly conserved in APA, metal ion coordination of APA might be different from that of EAP to some extent. This study broadens our understanding of cyanobacterial alkaline phosphatases.
We are grateful to Professor Xu-Dong Xu for helpful advice, Dr. Yuan Gao for RNA operation, Mr. Yi-Jun Zhang for technical assistance in ICP-OES, and Professor Simon Rayner and Dr. Hai-zhou Liu for bioinformatics analysis.
Li-Rong Song and Ya-Feng Zhou were supported by the National Basic Research Program of China (973 program No. 2008CB418000). Others were supported by the Chinese Academy of Sciences and State Key Laboratory Funds.
REFERENCES
1.Wu, J., Sunda, W., Boyle, E. A., and Karl, D. M.
(2000) Science, 289, 759-762.
2.Sundareshwar, P. V., Morris, J. T., Koepfler, E.
K., and Fornwalt, B. (2003) Science, 299, 563-565.
3.Thingstad, T. F., Krom, M. D., Mantoura, R. F.,
Flaten, G. A., Groom, S., Herut, B., Kress, N., Law, C. S., Pasternak,
A., Pitta, P., Psarra, S., Rassoulzadegan, F., Tanaka, T., Tselepides,
A., Wassmann, P., Woodward, E. M., Riser, C. W., Zodiatis, G., and
Zohary, T. (2005) Science, 309, 1068-1071.
4.Vershinina, O. A., and Znamenskaia, L. V. (2002)
Mikrobiologiya, 71, 581-595.
5.Coleman, J. E., and Gettins, P. (1983) Adv.
Enzymol. Relat. Areas Mol. Biol., 55, 381-452.
6.Kim, E. E., and Wyckoff, H. W. (1991) J. Mol
Biol., 218, 449-464.
7.Stec, B., Holtz, K. M., and Kantrowitz, E. R.
(2000) J. Mol. Biol., 299, 1303-1311.
8.Orhanovic, S., and Pavela-Vrancic, M. (2003)
Eur. J. Biochem., 270, 4356-4364.
9.Derman, A. I., and Beckwith, J. (1991) J.
Bacteriol., 173, 7719-7722.
10.Coleman, J. E., Nakamura, K., and Chlebowski, J.
F. (1983) J. Biol. Chem., 258, 386-395.
11.Sowadski, J. M., Handschumacher, M. D., Murthy,
H. M., Foster, B. A., and Wyckoff, H. W. (1985) J. Mol. Biol.,
186, 417-433.
12.Schopf, J. W. (1993) Science, 260,
640-646.
13.Field, C. B., Behrenfeld, M. J., Randerson, J.
T., and Falkowski, P. (1998) Science, 281, 237-240.
14.Sanudo-Wilhelmy, S. A., Kustka, A. B., Gobler, C.
J., Hutchins, D. A., Yang, M., Lwiza, K., Burns, J., Capone, D. G.,
Raven, J. A., and Carpenter, E. J. (2001) Nature, 411,
66-69.
15.Watson, G. M., Scanlan, D. J., and Mann, N. H.
(1996) FEMS Microbiol. Lett., 142, 105-109.
16.Hirani, T. A., Suzuki, I., Murata, N., Hayashi,
H., and Eaton-Rye, J. J. (2001) Plant Mol. Biol., 45,
133-144.
17.Suzuki, S., Ferjani, A., Suzuki, I., and Murata,
N. (2004) J. Biol. Chem., 279, 13234-13240.
18.Block, M. A., and Grossman, A. R. (1988) Plant
Physiol., 86, 1179-1184.
19.Bone, D. H. (1971) Arch. Microbiol.,
80, 147-153.
20.Ray, J. M., Bhaya, D., Block, M. A., and
Grossman, A. R. (1991) J. Bacteriol., 173, 4297-4309.
21.Doonan, B. B., and Jensen, T. E. (1980)
Microbios, 29, 185-207.
22.Wagner, K. U., Masepohl, B., and Pistorius, E. K.
(1995) Microbiology, 141 (Pt. 12), 3049-3058.
23.Su, Z. C., Olman, V., and Xu, Y. (2007) Bmc
Genomics, 8, 156-167.
24.Ihlenfeldt, M. J. A., and Gibson, J. (1975)
Arch. Microbiol., 102, 23-28.
25.Grainger, S. L., Peat, A., Tiwari, D. N., and
Whitton, B. A. (1989) Microbios, 59, 7-17.
26.Kumar, A., Singh, S., and Tiwari, D. N. (1992)
World J. Microbiol. Biotechnol., 8, 585-588.
27.Kaneko, T., Nakamura, Y., Wolk, C. P., Kuritz,
T., Sasamoto, S., Watanabe, A., Iriguchi, M., Ishikawa, A., Kawashima,
K., Kimura, T., Kishida, Y., Kohara, M., Matsumoto, M., Matsuno, A.,
Muraki, A., Nakazaki, N., Shimpo, S., Sugimoto, M., Takazawa, M.,
Yamada, M., Yasuda, M., and Tabata, S. (2001) DNA Res.,
8, 205-213, 227-253.
28.Ning, D., and Xu, X. (2004) Microbiology,
150, 447-453.
29.Larkin, M. A., Blackshields, G., Brown, N. P.,
Chenna, R., McGettigan, P. A., McWilliam, H., Valentin, F., Wallace, I.
M., Wilm, A., Lopez, R., Thompson, J. D., Gibson, T. J., and Higgins,
D. G. (2007) Bioinformatics, 23, 2947-2948.
30.Guindon, S., and Gascuel, O. (2003) Syst.
Biol., 52, 696-704.
31.Felsenstein, J. (1985) Evolution,
39, 783-791.
32.Sebastian, M., and Ammerman, J. W. (2009) ISME
J., 3, 563-572.
33.Gierasch, L. M. (1989) Biochemistry,
28, 923-930.
34.Wojciechowski, C. L., and Kantrowitz, E. R.
(2002) J. Biol. Chem., 277, 50476-50481.
35.Stec, B., Hehir, M. J., Brennan, C., Nolte, M.,
and Kantrowitz, E. R. (1998) J. Mol. Biol., 277,
647-662.
36.Chaidaroglou, A., Brezinski, D. J., Middleton, S.
A., and Kantrowitz, E. R. (1988) Biochemistry, 27,
8338-8343.
37.Xu, X., and Kantrowitz, E. R. (1993)
Biochemistry, 32, 10683-10691.
38.Xu, X., and Kantrowitz, E. R. (1992) J. Biol.
Chem., 267, 16244-16251.
39.Tibbitts, T. T., Xu, X., and Kantrowitz, E. R.
(1994) Protein Sci., 3, 2005-2014.
40.Martin, D. C., Pastra-Landis, S. C., and
Kantrowitz, E. R. (1999) Protein Sci., 8, 1152-1159.
41.Ma, L., and Kantrowitz, E. R. (1994) J. Biol.
Chem., 269, 31614-31619.
42.Kozlenkov, A., Manes, T., Hoylaerts, M. F., and
Millan, J. L. (2002) J. Biol. Chem., 277,
22992-22999.
43.Tibbitts, T. T., Murphy, J. E., and Kantrowitz,
E. R. (1996) J. Mol. Biol., 257, 700-715.
44.Chaidaroglou, A., and Kantrowitz, E. R. (1989)
Protein Eng., 3, 127-132.
45.Matlin, A. R., Kendall, D. A., Carano, K. S.,
Banzon, J. A., Klecka, S. B., and Solomon, N. M. (1992)
Biochemistry, 31, 8196-8200.
46.Xu, X., and Kantrowitz, E. R. (1991)
Biochemistry, 30, 7789-7796.
47.Xu, H., Zhang, X., Zhang, Z., Zhang, Y., and
Cass, A. E. G. (2003) Biocatal. Biotransform., 21,
41-47.
48.Bradshaw, R. A., Cancedda, F., Ericsson, L. H.,
Neumann, P. A., Piccoli, S. P., Schlesinger, M. J., Shriefer, K., and
Walsh, K. A. (1981) Proc. Natl. Acad. Sci. USA, 78,
3473-3477.
49.Le Du, M. H., Stigbrand, T., Taussig, M. J.,
Menez, A., and Stura, E. A. (2001) J. Biol. Chem., 276,
9158-9165.
50.Helland, R., Larsen, R. L., and Asgeirsson, B.
(2009) Biochim. Biophys. Acta, 1794, 297-308.
51.Janeway, C. M., Xu, X., Murphy, J. E.,
Chaidaroglou, A., and Kantrowitz, E. R. (1993) Biochemistry,
32, 1601-1609.
52.Zappa, S., Rolland, J. L., Flament, D., Gueguen,
Y., Boudrant, J., and Dietrich, J. (2001) Appl. Environ.
Microbiol., 67, 4504-4511.
53.Boulanger, R. R., Jr., and Kantrowitz, E. R.
(2003) J. Biol. Chem., 278, 23497-23501.
54.Nakashima, H., Fukuchi, S., and Nishikawa, K.
(2003) J. Biochem., 133, 507-513.
55.Thornton, J. M. (1981) J. Mol. Biol.,
151, 261-287.