Nature of Cation–π Interactions and Their Role in Structural Stability of Immunoglobulin Proteins
I. A. Tayubi and R. Sethumadhavan*
Bioinformatics Division, School of Biosciences and Technology, Vellore Institute of Technology, Vellore 632014, Tamil Nadu, India; fax: 0416-224-3092; E-mail: rsethumadhavan@vit.ac.in; sethurao_2005@yahoo.com; iftikharaslamtayubi@vit.ac.in* To whom correspondence should be addressed.
Received December 8, 2009; Revision received February 8, 2010
Cation–π interactions are known to be important contributors to protein stability and ligand–protein interactions. In this study, we have analyzed the influence of cation–π interactions in single chain immunoglobulin proteins. We observed 87 cation–π interactions in a data set of 33 proteins. These interactions are mainly formed by long-range contacts, and there is preference of Arg over Lys in these interactions. Arg–Tyr interactions are predominant among the various pairs analyzed. Despite the scarcity of interactions involving Trp, the average energy for Trp–cation interactions is quite high. This information suggests that the cation–π interactions involving Trp might be of high relevance to the proteins. Secondary structure analysis reveals that cation–π interactions are formed preferably between residues in which at least one is in β-strand. Proteins having β-strand regions have the highest number of cation–π interaction-forming residues.
KEY WORDS: cation–π, secondary structure, long-range interactions, accessible surface area, stabilization centers, immunoglobulin proteins, structural stabilityDOI: 10.1134/S000629791007014X
Abbreviations: ASA, accessible surface area; LRO, long-range order; SC, stabilizing centers.
The importance of cation–π interaction has been stressed by
several investigators for their role in enhancement of the stability of
thermophilic proteins [1, 2],
folding of polypeptides [3, 4],
and the stability of membrane proteins [5, 6]. Immunoglobulin proteins form a large group of cell
surface and soluble proteins that are involved in the recognition,
binding, or adhesion processes of cells. Immunoglobulin (Ig) was
reported to be the most populous family of proteins in the human genome
[7]. The molecules are categorized as members of
this superfamily based on shared structural features – they all
possess a domain known as an immunoglobulin domain or fold. The
vertebrate immune system has developed into a highly sophisticated form
that gives rapid, measured, and localized response to a vast variety of
pathogens. They are commonly associated with roles in the immune system
[8]. In this study we consider the features of
these proteins, how they interact, and their structural stability.
The prediction of secondary structure is a part of the base of knowledge in all proteins; based on these considerations, our current study might provide understanding of the structurally and energetically significant parameters of immunoglobulin proteins involving cation–π interactions [9]. During the process of protein folding, the cooperative, noncovalent, and long-range interactions between residues provide stability to resist the local tendency for unfolding [10, 11]. Protein structures are stabilized by various noncovalent interactions, including hydrophobic, electrostatic, van der Waals, and hydrogen bonding interactions. These interactions are crucial in many areas of modern chemistry, especially in the field of molecular recognition and for structural stability [12, 13]. As a result, noncovalent interactions of immunoglobulin proteins are recognized to play an important role in the stability and specificity of proteins. In proteins, cation–π interactions occur between the cationic side chain of lysine (K) or arginine (R) and the aromatic side chains of phenylalanine (F), tyrosine (Y), and tryptophan (W) [13]. Clusters of residues in protein structure identified as stabilizing centers (SC) are based on long-range order interactions; most of them are in buried positions [14] and have hydrophobic and aromatic side chains. Gromiha and Selvaraj proposed a parameter, long-range order (LRO) [15], from the knowledge of long-range contacts in protein structure. LRO is a quantitative measure of the number of residue–residue contacts that are close in space (within 8 Å) and far in the sequence (with the minimum separation of 12 residues) normalized by the total number of residues in the protein, as described by Poupon and Mornon [16]. Previous studies on cation–π interactions have focused on various aspects such as their role in ligand recognition [17-19] and protein–drug interactions [20]. There are several instances where cation–π interactions have been shown to play a significant role. For example, the active site of horseradish peroxidase consists of an arginine interacting with an adjacent tyrosine residue to allow aromatic donor binding [21]. Influence of cation–π interactions in protein–DNA complexes has been studied by Gromiha [22]. There are also reports on these kinds of interactions in a set of 62 non-redundant DNA binding proteins by the same author [23]. Recently, our group published work on cation–π interactions in interleukins and tumor necrosis factor (TNF). Noncovalent interactions have been investigated in interleukins and TNF proteins. The results reveal that cation–π interactions are important for understanding stability and functional similarity of proteins [24], and in RNA-binding proteins the energy contribution due to cation–π interaction are increasingly recognized as important noncovalent binding interactions. The influence of cation–π interactions on the stability of RNA binding proteins and its highest occurrence among the aromatic residues have been explored [25].
Hence, we have analyzed the cation–π interaction in immunoglobulin proteins. The energetic contribution of cation–π interactions is revealed for each of the 33 proteins, and all six pairs of residues (Arg–Phe, Arg–Tyr, Arg–Trp, Lys–Phe, Lys–Tyr, and Lys–Trp) involved in such interactions are investigated.
MATERIALS AND METHODS
Searching of PDB for immunoglobulin proteins. The protocol we have used for our current study of immunoglobulin proteins in terms of PDB IDs available in the literature [26] and cross validation with the SCOP database [27] is that they come under the category of immunoglobulin-like β-sandwich fold. The set was obtained with the following conditions: (i) the three dimensional structures of these proteins have been solved with ≤3.0 Å resolution, (ii) the similarity search using PSI-BLAST yielded e-value of less than 0.001, and (iii) sequence identity is less than 80%. The complexes, whose proteins were homologous but recognized different nucleotide sequences, were included in the PDB IDs.
The PDB tags of the proteins are: 1igm, 1wz1, 1cfv, 1a4j, 1a4k, 1fl3, 1nbv, 1ktr, 1mpa, 1cbv, 1ap2, 1cgs, 1vpo, 1sbs, 1bbd, 32c2, 1afv, 1a6w, 1l7t, 1um5, 1jpt, 1ken, 1mci, 1a8j, 2mpa, 1e6o, 1mfb, 1mck, 1mcp, 1l6x, 1k6q, 1h3u, and 2gj7.
Computation of amino acid composition. The amino acid composition for each amino acid residue that is involved in cation–π interactions (Lys, Arg, Phe, Trp, and Tyr) was computed using the standard formula:
comp(i) = n(i)/N,
where n(i) is the number of amino acids of type “i” and N is the total number of amino acids in the protein.
Energetic contribution due to cation–π interactions. To provide an energetic evaluation of all potential cation–π interactions in immunoglobulin proteins, their structures are identified and evaluated using the energy-based program CAPTURE (http://capture.caltech.edu/) developed by Gallivan and Dougherty [27]. The percentage composition of a specific amino acid residue contributing to cation–π interactions is obtained from the equation:
compcat–π(i) = ncat–π(i) × [100/n(i)],
where “i” stands for the five residues (Lys, Arg, Phe, Trp, and Tyr), ncat–π is the number of residues involved in cation–π interactions, and n(i) is the number of residues of type “i” in the considered protein structures.
We have computed the energetic contribution of cation–π interactions for each enzyme in the data set and for all possible pairs of positively charged and aromatic amino acids. The total cation–π interaction energy (Ecat–π) was divided into electrostatic (Ees) and van der Waals energy (Evw) and computed using the program CAPTURE, which implemented a subset of OPLS [28] force field to calculate the energies. The electrostatic energy (Ees) is calculated using the equation:
Eel = Σqiqje2/rij,
where qi and qj are the charges for the atoms i and j, respectively, and rij is the distance between them. The van der Waals energy is given by:
Evw = 4εij[(σij12/rij12) – (σij6/rij6)],
where σij = (σiiσjj)1/2 and εij = (εiiεjj)1/2; σ and ε are the van der Waals radius and well depth, respectively.
Classification of long-range orders. The short-, medium-, and long-range interactions for immunoglobulins was evaluated computationally, i.e. contacts between two residues that are close to the residues coming within a sphere of 8 Å was computed as described in [29]. For a given residue, the comparison of the surrounding residue is analyzed in terms of the location at the sequence level. The contribution from <±4 are treated as short-range contacts, >±4 to <±20 as medium-range contacts, and >20 are treated as long-range contacts. This classification enables us to evaluate the contribution of long-range contacts in the formation of cation–π interactions.
Computation analysis of solvent accessibility and secondary structure analysis in cation–π interaction-forming residues in immunoglobulin proteins. We investigated the structural significance of secondary structure and solvent accessibility, which are the two major intermediate steps in understanding the structure and function of proteins. We systematically analyzed the preference for each of the cation–π interaction-forming residues based on their location in different secondary structures of immunoglobulin proteins and their solvent accessibility. Solvent accessibility is the ratio between the solvent accessible surface area of a residue in a 3D structure and in an extended tripeptide conformation. We obtained the solvent accessible surface area (ASA) information using Net ASA view (http://www.netasa.org/) [30]. The entire implementation of ASA View for all PDB proteins as a whole or for an individual chain can be accessed at http://www.netasa.org/asaview/. Requirements for the uses are simply the PDB code or the coordinate file. Solvent accessibility was divided into three classes, buried, partially buried, and exposed indicating, respectively, low, moderate, and high accessibility of the amino acid residues to the solvent. We used the DSSP program (http://swift.cmbi.kun.nl/gv/dssp/) [31] to obtain information about secondary structure. The secondary structures were classified into α-helix, β-strand, and random coil as suggested by Heringa and Argos [32]. Solvent accessibility was divided into three classes, i.e. 0-20, 20-50, and >50% indicating the least, moderate, and high accessibility of the amino acid residues, respectively.
Computation of stabilization center. Stabilization centers are clusters of residues that are involved in medium- or long-range interactions. Residues can be considered part of stabilization centers if they are involved in medium- or long-range interactions and if two supporting residues can be selected from both of their flanking tetra peptides, which together with the central residues form at least seven out of the nine possible contacts. We used the server available at http://www.enzim.hu/scide [33] for this purpose.
Conservation score. The Consurf program (Consurf server http://consurf.tau.ac.il/) was used to analyze conservation score of cation–π interacting amino acid residues in each immunoglobulin protein [35]. The server computes the conservation based on comparison of the sequence of a PDB chain with the proteins deposited in Swiss-Prot [36] and finds the ones that are homologous to the PDB sequence. The number of PSI-BLAST iterations and the E-value cutoff used in all similarity searches were 1 and 0.001, respectively. All the sequences that are evolutionarily related with each one of the proteins in the data set were used in subsequent multiple alignments. Based on these protein sequence alignments, the residues are classified into nine categories from highly variable to highly conserved. Residues with a score of 1 are considered highly variable, and residues with a score of 9 are considered highly conserved.
RESULTS
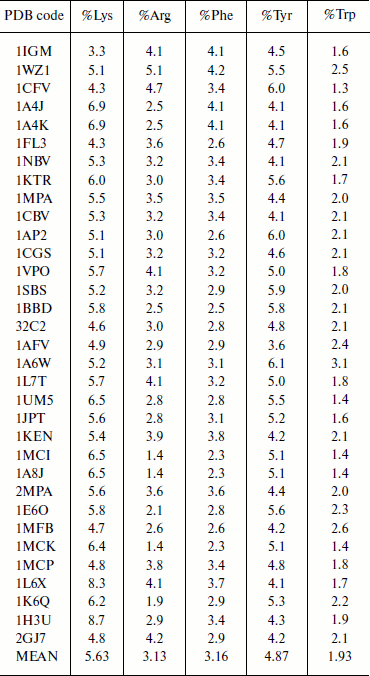
Composition of aromatic and positively charged amino acids in immunoglobulin proteins. The composition of amino acid residues that are involved in cation–π interactions was analyzed and the results are presented in Table 1. We observed that Lys has higher occurrence than Arg in immunoglobulin proteins [6, 8]. Tyr has higher occurrence than Phe among the aromatic residues, and Trp has the lowest occurrence. Generally the composition of cation–π interaction forming residues is similar to other globular proteins [6, 8].
Table 1. Composition of cation–π
forming residues in immunoglobulin proteins
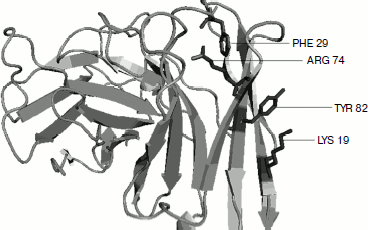
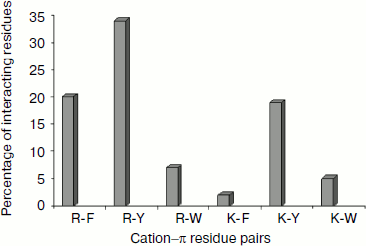
Cation–π residue pairs involved in immunoglobulin proteins. In this work we have studied 33 proteins with β-structure. The number of cation–π interactions ranged within 1-6. Our study shows that 5, 14, and 20% of the proteins had 1, 2, and more than 2 interactions, respectively. The energetically significant cation–π interacting residues are Arg–Phe, Arg–Tyr, Arg–Trp, Lys–Phe, Lys–Tyr, and Lys–Trp pairs. The PyMOL view of Arg–Phe and Lys–Trp interacting pairs for the protein with PDB ID 1wz1 is shown in Fig. 1. It was found that among the cation–π interactions involving Arg residues Arg–Tyr interactions were more often found than Arg–Phe and Arg–Trp interactions. Among the cation–π interactions involving Lys residues Lys–Tyr interaction was higher than Lys–Phe and Lys–Trp interactions. These results are shown in Fig. 2. Individually Arg and Tyr were more often observed in cation–π interactions, and also the Arg–Tyr and Lys–Tyr pairs were more common than the other four pairs. Hence, Arg–Tyr and Lys–Tyr interactions may play the main role in the stability of immunoglobulin proteins.
Fig. 1. PyMOL view of Arg–Phe and Lys–Tyr interacting pairs in 1wz1.
Energetic contribution of cation–π interactions in immunoglobulin proteins. The 33 immunoglobulin proteins were investigated, and we found 87 energetically significant cation–π interactions, the total cation–π energy ranging from –2.45 (PDB ID 1mck) to maximum –30.92 (PDB ID 1cfv) kcal/mol. The energetic contribution of each cationic–aromatic pair of amino acids was computed, and the results are presented in Table 1 in the supplement available on the journal site (http://protein.bio.msu.ru/biokhimiya). The pairwise cation–π interaction energy between the cationic and aromatic residues shows that Arg–Trp (–6.41) energy is the strongest and Arg–Phe is the lowest (–3.43) among the six possible pairs as shown in Fig. 3. In the 33 proteins it was found that 55% showed a cation–π energy less than –10 kcal/mol, 21% from –10 to –20 kcal/mol, and 24% of them showed a cation–π interaction energy greater than –20 kcal/mol. Most of the cation–π interactions have energy in the range of –3 to –6 kcal/mol.Fig. 2. Cation–π interacting residue pairs in immunoglobulin proteins.
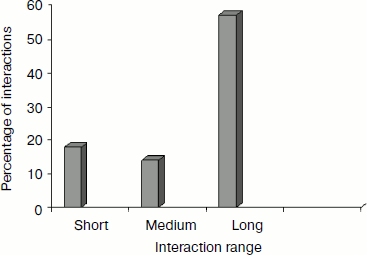
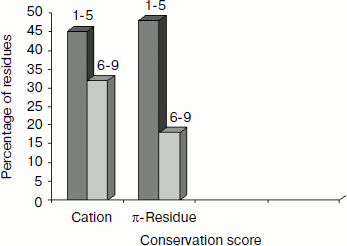
Separation in primary sequence and conservation score. The distance in the primary sequence was calculated between the cationic and the aromatic residues for each cation–π interaction, and the results are shown in Fig. 4. Those from <±4 are treated as short-range contacts, >±4 to <±20 as medium-range contacts, and >20 are as long-range contacts. In our study group 20, 15, and 60% of immunoglobulin proteins exhibited short-, medium-, and long-range interactions, respectively. This result revealed that majority of the cation–π interactions in immunoglobulin proteins are long-range interactions. This result reflects the importance of long-range interactions to the stability of immunoglobulin proteins. We used the Consurf program to calculate the conservation score for cation–π interaction forming residues for proteins 1ktr and 1um5 whose conservation scores are not available. We found that 34% of cationic residues and 17% of π residues have conservation score from 6-9 (these residues are most conserved) and 45% of cationic residues and 47% π residues have conservation score from 1-5 (these residues are more variable) (see Fig. 5).Fig. 3. Average cation–π interaction energy for the interacting residue pairs.
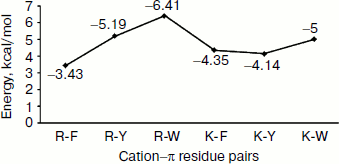
Fig. 4. Separation in primary sequence of cation–π interacting residues.
Solvent accessibility of cation–π interaction-forming residues. We estimated the solvent accessibility of all residues that are involved in cation–π interaction using DSSP [31]. We analyzed the percentage of cation–π interaction forming residues at various ranges of solvent accessibility defined as: 0-20% (buried), 20-50% (partially buried), and >50% (surface exposed). The average solvent accessibility of the Arg, Lys, Phe, Tyr, and Trp residues involved in cation–π interactions is 56, 24, 21, 49, and 10%, respectively (Fig. 6). The solvent accessibility of Arg and Tyr residues is significantly higher than that of other cation–π forming residues [37, 38]. From this classification, we observed that Arg and Tyr residues were mostly in the exposed region, Lys and Phe were mostly in the partially buried region, while Trp was mostly be in the fully buried regions.Fig. 5. Cation–π interacting residues and conservation score.
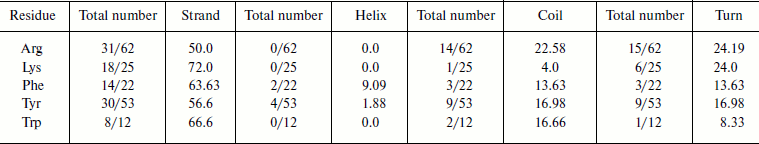
Cation–π interaction-forming residues in different secondary structures. We calculated the occurrence of cation–π interaction-forming residues and in different secondary structures of immunoglobulin proteins (Table 2). We found that in the immunoglobulin proteins cation–π interaction-forming Lys was predominantly in strand, while Arg was dominantly in strands and also to some extent in random coil and turn regions. Most of the aromatic cation–π interaction-forming residues are in strand.Fig. 6. Cation–π interaction residues in different accessible surface area (ASA) ranges.

Table 2. Frequency of occurrence of
cation–π interaction-forming residues in different secondary
structures
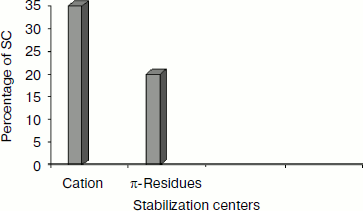
Stabilization centers of cation–π interaction-forming residues in immunoglobulin proteins. We computed the stabilization center for all cation–π interaction forming residues of immunoglobulin proteins using the program SCide (Fig. 7). It was found that 35% of cationic residues and 20% of π residues had one or more stabilization centers. Cationic residues were found to have more stabilization centers than π residues. This trend was different with an earlier report on RNA binding proteins [25]. It is interesting to note that all the five residues found in cation–π interactions are important in locating one or more stabilization centers. These observations strongly reveal that these residues can contribute significantly to the structural stability of these proteins in addition to participating in cation–π interactions.
Fig. 7. Stabilization centers in immunoglobulin protein.
DISCUSSION
The percentage of specific amino acid residues contributing to cation–π interactions was calculated for immunoglobulin proteins. Further, the characteristic features of residues involved in cation–π interactions have been evaluated in terms of secondary structure, solvent accessibility, conservation score, and stabilizing centers (see Table 2 in supplement available on the journal site http://protein.bio.msu.ru/biokhimiya). We observed that the cation–π interaction energy for pairs with Arg is stronger than with Lys. Most of the cation–π interactions in immunoglobulin proteins are of residues distant from each other in the primary sequence, i.e. long-range interactions. Arg–Trp and Arg–Tyr have the strongest cation–π interaction energy among the six possible residue pairs. Secondary structure and solvent accessibility of residues in immunoglobulin proteins reveal that cation–π interaction-forming Lys residues are mostly in strand, while Arg is mainly in strand but also sometimes in coil and turn regions. Most of the cation–π interaction-forming aromatic residues are in strand. While Arg residues are usually solvent exposed, Lys is often partially buried and the cation–π interaction-forming aromatic amino acids Tyr and Phe are partially buried and Trp residues are buried. In this comprehensive approach our study articulates significant cation–π interactions and gives deeper understanding about proficiency, specificity, and stability of immunoglobulin proteins. Overall we report here that all proteins with β structure have long-range cation–π interactions, Arg–Trp and Arg–Tyr pairs have the strongest cation–π interaction energies, and Lys and Arg are primarily in strand. We believe that this report will help to understand structural stability and will promote the use of immunoglobulin proteins as peptide based drugs.
The authors thank the Management of Vellore Institute of Technology for providing the facilities to carry out this work.
REFERENCES
1.Gromiha, M. M., Thomas, S., and Santhosh, C. (2002)
Prep. Biochem. Biotech., 32, 355-362.
2.Chakravarty, S., and Varadarajan, R. (2000)
Biochemistry, 41, 8152-8161.
3.Shi, Z., Olson, C. A., and Kallenbach, N. R. (2002)
J. Am. Chem. Soc., 124, 3284-3291.
4.Burghardt, T. P., Juranic, N., Macura, S., and
Ajtai, K. (2002) Biopolymers, 63, 261-272.
5.Mulhern, T. D., Lopez, A. F., D’Andrea, R.
J., Gaunt, C., Vandeleur, L., Vadas, M. A., Booker, G. W., and Bagley,
C. J. (2000) J. Mol. Biol., 297, 989-1001.
6.Gromiha, M. M. (2003) Biophys. Chem.,
103, 251-258.
7.Harpaz, Y., and Chothia, C. (1994) J. Mol.
Biol., 238, 528-539.
8.Barclay, A. (2003) Semin. Immunol.,
15, 215-223.
9.Hunter, C. A., and Sanders, J. K. M. (1990) J.
Am. Chem. Soc., 112, 5525-5534.
10.Dill, K. A. (1990) Biochemistry,
29, 7133-7155.
11.Rose, G. D., and Wolfenden, R. (1993) Biophys.
Biomol. Struct., 22, 381-415.
12.Ponnuswamy, P. K., and Gromiha, M. M. (1994)
J. Theor. Biol., 166, 63-74.
13.Pace, C. N. (1995) Meth. Enzymol.,
259, 538-554.
14.Dosztanyi, Z., Fiser, A., and Simon, I. (1997)
J. Mol. Biol., 272, 597-612.
15.Gromiha, M. M., and Selvaraj, S. (2001) J.
Mol. Biol., 310, 27-32.
16.Poupon, A., and Mornon, J. P. (1999) FEBS
Lett., 452, 283-289.
17.Zacharias, N., and Dougherty, D. A. (2002)
Trends Pharmacol. Sci., 23, 281-287.
18.Zhong, W., Gallivan, J. P., Zhang, Y., Li, L.,
Lester, H. A., and Dougherty, D. A. (1998) Proc. Natl. Acad.
Sci. USA, 95, 12088-12093.
19.Scrutton, N. S., and Raine, A. R. C. (2000)
Biochem. J., 319, 1-8.
20.Liu, R., Pidikiti, R., Petersen, C. E., Bhagavan,
N. V., and Eckenhoff, R. G. (2002) J. Biol. Chem., 277,
36373-36379.
21.Ma, J. C., and Dougherty, D. A. (1997) Chem.
Rev., 97, 1303-1324.
22.Gromiha, M. M., Santhosh, C., and Suwa, M. (2004)
Polymer, 45, 633-639.
23.Gromiha, M. M. (2005) Polymer, 46,
983-990.
24.Anand, A., Sudha, A., Lazar Mathew, and
Sethumadhavan, R. (2006) Cytokine, 35, 263-269.
25.Anand, A., Sudha, A., Lazar Mathew, and
Sethumadhavan, R. (2007) Int. J. Biol. Macromol., 40,
479-483.
26.Berman, H. M., Westbrook, J., Feng, Z., et
al. (2000) Nucleic Acids Res., 28, 235-242.
27.Murzin, A. G., and Brenner, S. E., Hubbard, T.,
and Chothia, C. (1995) J. Mol. Biol., 247, 536-540.
28.Gallivan, J. P., and Dougherty, D. A. (1999)
Proc. Natl. Acad. Sci. USA, 96, 9459-9464.
29.Jorgensen, W. L., Maxwell, D. S., and Rives, J.
T. (1996) J. Am. Chem. Soc., 118, 11225-11236.
30.Gromiha, M. M., and Selvaraj, S. (2004)
Biophys. Mol. Biol., 86, 235-277.
31.Kabsch, W., and Sander, C. (1983)
Biopolymers, 22, 2577-2637.
32.Heringa, J., and Argos, P. (1989)
Proteins, 37, 30-43.
33.Dosztanyi, Z. S., Magyar, C. S., Tusnady, E., and
Simon, I. (2003) Bioinformatics, 19, 899-900.
34.Gromiha, M. M., Pujadas, G., Magyar, C.,
Selvaraj, S., and Simon, I. (2004) Proteins, 55,
316-329.
35.Glaser, F., Pupko, T., Paz, I., Bell, R. E.,
Bechor, D., Martz, E., and Ben-Tal, N. (2003) Bioinformatics,
19, 163-164.
36.Boeckman, B., Bairoch, A., Apweiler, R., Blatter,
M. C., Estreicher, A., Gasteiger, E., Martin, M. J., Michoud, K.,
O’Donovan, C., Phan, I., Pilbout, S., and Schneider, M. (2003)
Nucleic Acids Res., 31, 365-370.
37.Gromiha, M. M., Oobatake, M., Kono, H.,
Uedaira, H., and Sarai, A. (1999) Protein Eng., 12,
549-555.
38.Gilis, D., and Rooman, M. (1997) J. Mol.
Biol., 272, 276-290.
Supplementary TABLE 1 (MS Word)
Supplementary TABLE 2 (MS Word)