An Easy Colorimetric Assay for Glycosyltransferases
Rui Shen, Shuai Wang, Xiaofeng Ma, Junyang Xian, Jing Li, Lianwen Zhang*, and Peng Wang*
College of Pharmacy, Nankai University, Weijin Road 94, Tianjin 300071, P. R. China; fax: +86(022)2350-7880; E-mail: lianwen@nankai.edu.cn; pwang@nankai.edu.cn; shenr05@126.com* To whom correspondence should be addressed.
Received March 31, 2010; Revision received May 27, 2010
Glycosyltransferases are involved in biosynthesis of both protein-bound and non-bound glycans that have multiple and important biological functions in all species. A variety of methods for assaying glycosyltransferase activity have been developed driven by the specific interests and type of information required by researchers. In this work, a novel colorimetric assay for the glycosyltransferase-catalyzed reaction was established. Compared with measuring the newly formed product, which might not exhibit visible absorption, the unreacted acceptor could be readily detected by measuring the visible absorption of the hydrolysis product. In the assay, 4-nitrophenyl-β-D-glycoside (glycosyl-β-pNP) is used as the glycosyl acceptor, which can be hydrolyzed by a special exoglycosidase to release the p-nitrophenol before glycosylation reactions. Absorbance change of the p-nitrophenolate corresponds to unreacted glycosyl acceptor that accompanied the glycosyl transfer. The assay is demonstrated to be useful in the initial characterization of recombinant glycosyltransferases for their kinetic parameters, optimal metal cofactor, and pH value. It provides a simple, sensitive, and quantitative method for assessing glycosyltransferase activity and is thus expected to have broad applications including automated high-throughput screening.
KEY WORDS: glycosyltransferase, colorimetric assay, high-throughput screeningDOI: 10.1134/S0006297910070187
Abbreviations: BSA, bovine serum albumin; ESI-MS, electrospray ionization mass spectrometry; Gal-β-pNP, 4-nitrophenyl-β-D-galactopyranoside; GlcNAc-β-pNP, 4-nitrophenyl-N-acetyl-β-D-glucosamine; HPLC, high performance liquid chromatography; IPTG, isopropyl-1-thio-β-D-galactopyranoside; NMR, nuclear magnetic resonance; NTA, nitrilotriacetic acid; pNP, para-nitrophenol; TLC, thin layer chromatography; UDP-Gal, uridine diphosphate galactose; UDP-GlcNAc, uridine diphosphate-N-acetyl-glucosamine.
Glycosyltransferases are involved in the biosynthesis of
oligosaccharides, which are vital for all living systems. They are an
enormous and diverse family of enzymes catalyzing the transfer of a
monosaccharide unit from a nucleotide or lipid sugar donor to a
specific acceptor molecule, including glycolipids, glycoproteins,
saccharides, natural products, etc., thereby forming glycosidic bonds.
This family of enzymes plays an important role in cell physiology and
has been associated with embryonic maturation, cell surface
recognition, cell development, intracellular migration, and
sperm–egg binding [1-3].
Therefore, the change of glycosyltransferase expression and activity is
relevant to many diseases and can serve as a diagnostic marker for
certain diseases. For examples, α-1,3-galactosyltransferase
activity in the body can lead to autoimmune response or organ allograft
rejection [4-7]. The changes of
N-acetyl-glucosaminetransferase or fucosetransferase activity are
considered an important indicator of tumor and a significant reason for
deterioration or migration [8-9]. The study of glycosyltransferase activity is
intimately associated with the development of new therapeutic agents
[10-12]. The lack of a rapid
and simple method for continuously monitoring of glycosyltransferase
activity can prevent the efficient discovery of potent drug
candidates.
Numerous detection methods for glycosyltransferase activity have been developed. These methods include the routine monitoring of activity during and after enzyme purification and in biocatalysis, detailed mechanistic and inhibition evaluations, and high-throughput screening for novel glycosyltransferase activities or inhibitors [13-20]. Representative assay methods for all of these applications including their advantages and limitations are summarized by Palcic and Sujino [21]. In the last few years, some new, rapid, and real-time assays for glycosyltransferase have been reported. Recent examples include a pH-sensitive assay, which uses a pH indicator for detection of proton release [22, 23], supramolecular method, which uses a fluorescent chemosensor for a multi-substrate enzymatic reaction [24, 25], etc. The new methods are suitable for high-throughput screening with high sensitivity, but make strict requirements on the pH range of reaction buffer or recognition specificity of fluorescent probe.
Previously, Wang and coworkers reported a novel fluorescence-based assay for the transglycosylation activity. They used a coupled chitinase to capture and hydrolyze the fluorogenic intermediate to detect the transglycosylation activity of endo-β-N-acetyl glucosaminidases [26]. In this paper, we report a new colorimetric assay for lipooligosaccharide glycosyltransferase B (LgtB, β-galactosyltransferase) and illustrate the methodology (Fig. 1). The method is based upon the detection of absorbance changes of p-nitrophenolate in response to the digestion of unreacted glycosyl accepter by a special exoglycosidase. This assay is somewhat like the aforementioned fluorescence-based assay, and it is available for the initial characterization of recombinant glycosyltransferases for their kinetic parameters and optimal metal cofactor and pH value.
Fig. 1. A 96-well plate-based colorimetric high-throughput screening method for glycosyltransferase activity. The enzymatic reactions were incubated in triplicate at 37ºC and stopped by adding Na2CO3 to adjust the pH to 9.6. The formation of the p-nitrophenolate was monitored by measuring A405 with a microplate reader. The absorbance of the solution correlated to the amount of unreacted GlcNAc-β-pNP (or Gal-β-pNP) and to the enzyme activity of glycosyltransferase.
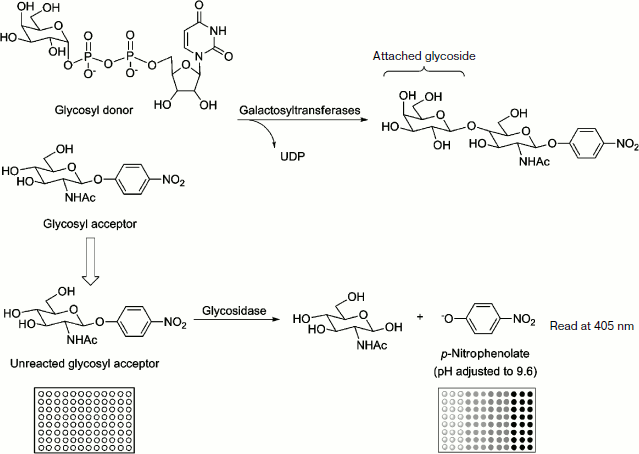
MATERIALS AND METHODS
Materials and equipment. All the reagents including 4-(2-hydroxyethyl)-1-piperazine-ethanesulfonic acid (Hepes), trihydroxymethyl aminomethane (Tris), 4-morpholine-ethanesulfonic acid (Mes), 4-nitrophenyl-N-acetyl-β-D-glucosamine (GlcNAc-β-pNP), 4-nitrophenyl-β-D-galactopyranoside (Gal-β-pNP), uridine diphosphate galactose (UDP-Gal), uridine diphosphate-N-acetyl-glucosamine (UDP-GlcNAc), p-nitrophenol (pNP), isopropyl-1-thio-β-D-galactopyranoside (IPTG), bovine serum albumin (BSA), and ampicillin were purchased from Sigma-Aldrich (USA). Other chemicals were analytical reagent and were used without further purification. Escherichia coli plasmid host strain DH5α was from Gibco-BRL (USA), and E. coli expression host strain BL21(DE3) was purchased from Novagen Inc. (USA). Ni2+-NTA (nitrilotriacetic acid) agarose was purchased from Qiagen (USA). Bio-Gel P2 was purchased from Bio-Rad Laboratories (USA).
ESI-MS was recorded on a Finnigan LCQ-Advantage spectrometer and 1H-NMR spectra were recorded on a Bruker 400 M NMR spectrometer. The products of the LgtB-catalyzed glycosylation reaction and the kinetics were analyzed on a Shimadzu series HPLC instrument. The absorbance at 405 nm of p-nitrophenolate was recorded on a Thermo full wavelength microplate reader.
Cloning, expression, and purification of recombinant enzymes. The preparations of the recombinant plasmid DNA constructs of pET15b-LgtA, pET15b-LgtB, and pET15b-LgtC were reported previously [27-29]. The constructs were transformed into E. coli DH5α for amplification. Positive clones were identified by restriction enzyme digestion and sequencing. The correct sequences were subsequently inserted into the expression host, E. coli BL21(DE3).
For protein expression, E. coli BL21(DE3) harboring the recombinant plasmid was incubated in 4 ml Luria–Bertani (LB) medium (10 g tryptone, 5 g yeast extract, 10 g NaCl per liter) containing 0.1 mg/ml ampicillin with rapid shaking (220 rpm) at 37°C overnight. Then 4 ml of the overnight culture was transferred into 200 ml LB with 0.1 mg/ml ampicillin and incubated at 37ºC. When A600 reached 0.6-0.8, IPTG was added to a final concentration of 1 mM (LgtA) or 0.5 mM (LgtB and LgtC) and protein expression was allowed to proceed at 16ºC (160 rpm) overnight (LgtA and LgtC) or 25ºC (250 rpm) for 4 h (LgtB). All the cells were harvested by centrifugation (8000 rpm, 10 min, 4ºC), resuspended with 20 mM PBS buffer (pH 7.8, 0.5 M NaCl), and then broken by ultrasonication. After centrifugation (12,000 rpm, 30 min, 4ºC) of the cell lysate, the supernatant was filtered (0.45 µm) and applied onto a Ni2+-NTA affinity column which bound to the N-terminal 6×His tag sequence in the recombinant enzyme. All procedures were performed in a cold room (4-7ºC). The Ni2+-NTA column was washed with six column volumes of binding buffer (5 mM imidazole, 20 mM PBS, pH 7.8, 0.5 M NaCl), followed by six volumes of washing buffer (20 mM imidazole, 20 mM PBS, pH 7.8, 0.5 M NaCl). The overexpressed protein was eluted with eight volumes of elution buffer (200 mM imidazole, 20 mM PBS, pH 7.8, 0.5 M NaCl). Fractions containing the purified enzyme were combined and then concentrated by ultrafiltration at 4ºC three times and dialyzed against 20 mM PBS (pH 7.5, 0.15 M NaCl) buffer. Glycerol was added to a final concentration of 50%, and the enzyme solution was then stored at –20ºC in a freezer.
HPLC assay for glycosylation products and kinetics of LgtB. To identify the products synthesized by the purified glycosyltransferase and verify the accuracy of colorimetric assay, HPLC assays were performed. The reaction mixture contained 50 mM Hepes buffer (pH 7.2), UDP-Gal, GlcNAc-β-pNP, MnCl2, appropriate purified LgtB, and 0.1 mg/ml BSA. A control assay was prepared under the same conditions but without LgtB. Reactions were incubated at 37ºC for 30 min and quenched by adding ice-cold 12% acetonitrile. The samples were then kept on ice until aliquots of 10 µl were injected and analyzed on a Shimadzu LC-10A system equipped with a temperature control unit and an ultraviolet detector. A reverse-phase Premier C18 column (250 × 4.6 mm, 5 µm particle size; Shimadzu, Japan) protected with a C18 guard column cartridge was used. The mobile phase was 12% acetonitrile, and the absorbance at 254 nm was measured. Apparent kinetic parameters were obtained by varying the UDP-Gal concentration from 0.01 to 0.4 mM (0.01, 0.025, 0.05, 0.1, 0.2, and 0.4 mM) and a fixed concentration of GlcNAc-β-pNP (0.1 mM). Lineweaver–Burk plots were obtained from the average values of duplicate assay results.
Kinetics by colorimetric assay. In a 50 mM Hepes buffer (pH 7.2) containing different concentrations of p-nitrophenol, 0.5 M Na2CO3 solution was added to adjust final pH to 9.6; the A405 was recorded, according to which the calibration curve was established.
The enzymatic assays were carried out in Hepes buffer (50 mM, pH 7.2) containing GlcNAc-β-pNP (or Gal-β-pNP), MnCl2, and appropriate purified glycosyltransferase. The reactions were started by adding UDP-Gal (or UDP-GlcNAc) and incubated at 37ºC for 30 min. The donor was omitted in the control reaction. Then the glycosylation reactions were terminated by the addition of an excess of exoglycosidase. The hydrolysis reactions were continued to incubate at 37ºC for 30 min and quenched by adding Na2CO3 to adjust the pH to 9.6. The formation of the p-nitrophenolate was monitored by measuring A405 with the microplate reader. The absorbance of the solution correlated to the amount of unreacted GlcNAc-β-pNP (or Gal-β-pNP) and to the glycosyltransferases activity. All measurements were performed in triplicate and values averaged. For reaction catalyzed by glycosyltransferase, 0.1 mg/ml BSA was added. Apparent kinetic parameters were obtained by varying the UDP-Gal (or UDP-GlcNAc) concentration from 5 to 100 μM (5, 10, 15, …, 90, 95, and 100 μM) and a fixed concentration of GlcNAc-β-pNP (or Gal-β-pNP) (25 μM). The kinetic parameters were calculated from the Lineweaver–Burk plots that were obtained from the average values of triplicate assay results.
To evaluate the reliability of the method, LgtA (β-N-acetyl-glucosaminyltransferase) and LgtC (α-galactosyltransferase) activities were determined under the same conditions as described above.
Effect of pH value and cofactor by colorimetric assay. Glycosyltransferase activity was determined as described above with the following modifications. Reactions under different pH conditions were performed in different buffer systems (Mes buffer in a pH range of 6.0-7.0; Hepes buffer in a pH range of 6.8-8.2; Tris-HCl buffer in a pH range of 8.0-9.0) at the concentration of 50 mM. For the examination of metal ion dependency the following metals were used in form of 2.5 μM MeCl2: Mg, Ca, Cu, Zn, Co, Ni, and Mn. For a control reaction without metal ion influence, 25 μM EDTA was added to the assay.
Preparation of glycosylation products with LgtB. The synthesis of Gal-β1,4-GlcNAc-β-pNP was performed in 50 mM Hepes buffer (pH 7.2) containing 10 mM GlcNAc-β-pNP, 12 mM UDP-Gal, 1 mM MnCl2, appropriate purified LgtB, and 0.1 mg/ml BSA. The reaction was carried out at 37°C and monitored by TLC as previously described [27]. When the acceptor had disappeared or no further formation of the product was observed, the reaction was stopped by adding an equal volume of ice-cold ethanol. After the precipitated protein was removed by centrifugation (8000 rpm, 20 min), the supernatant was concentrated and purified using a Bio-Gel P2 size exclusion chromatography column. Fractions containing the purified products were collected and lyophilized to give Gal-β1,4-GlcNAc-β-pNP as a white powder. Yield 80.2%; (ESI-MS): 527.14 [M+Na]+; 1H-NMR (400 MHz, D2O): δ8.16 (d, 2H), 7.12 (d, 2H), 5.27 (d, J = 8.5 Hz, 1H), 4.38 (d, J = 7.6 Hz, 1H), 4.06 (dd, 1H), 3.85-3.46 (m, 11H), 1.92 (s, 3H). The result indicated that there was β-configuration of the galactose residue, which was transferred to GlcNAc-β-pNP.
RESULTS AND DISCUSSION
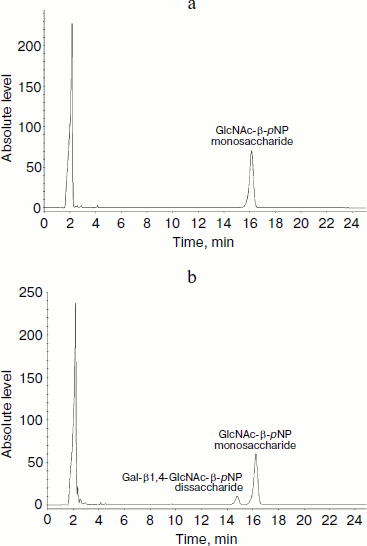
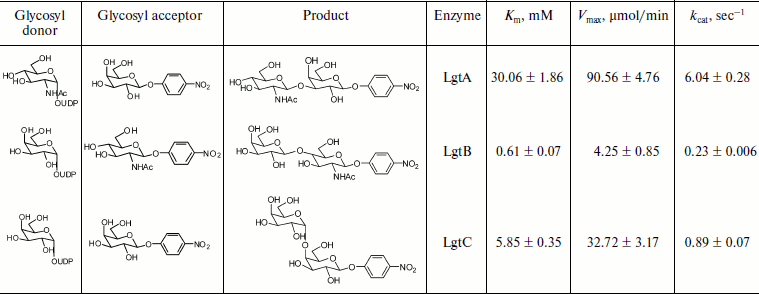
HPLC assay for glycosylation products and kinetics of LgtB. LgtB is a galactosyltransferase, which catalyzes the transfer of galactose from UDP-Gal to N-acetylglucosamine or its glycosides, forming a β-1,4 linkage. The optimized chromatographic conditions for the separation were used to analyze the appearance of the glycosyltransferase reaction products (Fig. 2). Resolution and selectivity were satisfactory. In the control assay, the acceptor (GlcNAc-β-pNP) showed an obvious absorption peak at 16.2 min. After the glycosylation reaction, the peak at 16.2 min decreased and a new peak at 15.1 min concomitantly appeared. This suggested that galactose was transferred to GlcNAc-β-pNP, forming Gal-β1,4-GlcNAc-β-pNP. When the mole number of reaction substrate was the same, there was approximate 18% GlcNAc-β-pNP converted to Gal-β1,4-GlcNAc-β-pNP over the 30 min reaction period. The LgtB apparent Km values for UDP-Gal obtained by HPLC assay was 0.43 ± 0.09 mM, and other kinetic parameters are listed in Table 1.
Table 1. Apparent kinetic parameters for LgtB activity by HPLC assays*Fig. 2. HPLC analysis of LgtB glycosylation reaction product using GlcNAc-β-pNP as an acceptor substrate (b). The reaction was performed at 37ºC for 30 min and was stopped by adding ice-cold 12% acetonitrile. A control assay was prepared under the same conditions but without LgtB (a). The appearance of the products was detected by using a reverse-phase Premier C18 column and 12% acetonitrile as the mobile phase at a flow rate of 1 ml/min. The absorbance at 254 nm was measured.
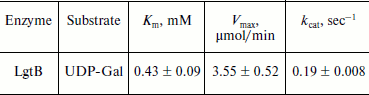
*Assays were performed in triplicate in 50 mM Hepes buffer (pH 7.2) with varied concentrations of UDP-Gal (0.01, 0.025, 0.05, 0.1, 0.2, and 0.4 mM) and a fixed concentration of GlcNAc-β-pNP (0.1 mM). LgtB was used at the same quantity for these assays.
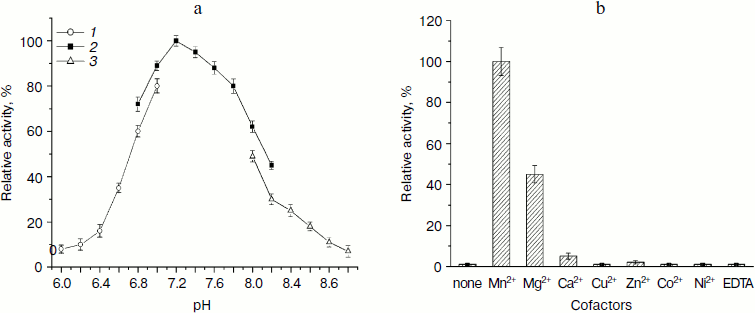
Effect of pH value and cofactor by colorimetric assay. The activity assay of LgtB under different pH conditions demonstrated that the enzyme catalyzes the galactosyl transferring reaction under neutral condition (pH 7.2-7.8) with optimal activity at pH 7.2 (Fig. 3a). This pH value is similar to reported pH optima for other β1,4-galactosyltransferase [30, 31]. More than 90% of the activity was observed at pH 7.4 or 7.6. There was a sharp decline in enzymatic activity below pH 6.8 or above pH 7.8. The activity dropped to about 35 and 40% when the pH was at 6.6 and 8.2, respectively. In addition, the enzyme presented lower activity in Mes buffer or Tris-HCl buffer compared with that in Hepes buffer at the same pH value, respectively. LgtB showed preference for certain buffer systems, with the Hepes buffer system being the most efficient.
Then the effects of various metal ions and the chelating agent on the enzyme activity of LgtB were examined. As shown in Fig. 3b, the activity of LgtB was found to be absolutely dependent on specific divalent metal ions, and selectivity towards metal ions appeared to be more obvious. MnCl2 significantly supported its activity at the concentration of 2.5 μM, and MgCl2 also showed certain activity, which was about 45% of that with MnCl2. None of the other divalent metal cations (Ca2+, Cu2+, Zn2+, Co2+, and Ni2+) were efficient cofactors for this enzyme. In addition, the enzyme showed no activity in the absence of metal ion or in the presence of EDTA. The dependence of a metal ion for its activity was similar to previous reports [32].Fig. 3. Effects of pH and divalent metal ions on β1,4-galactosyltransferase activity of LgtB. a) pH profile (Mes (1), Hepes (2), Tris-HCl (3)). b) Effects of divalent metal ions.
Kinetic characterization of glycosyltransferases with acceptor GlcNAc-β-pNP (or Gal-β-pNP). The apparent kinetic parameters were estimated by colorimetric assay with varied donor concentration along with fixed acceptor concentration. As shown in Fig. 4a, the enzymatic reactions were under initial rate conditions [33]. The average values from triplicate assays were used to obtain Lineweaver–Burk plots (Fig. 4b). The apparent Km, Vmax, and kcat values were calculated accordingly and are summarized in Table 2. The apparent Km value of LgtB for UDP-Gal was 0.61 ± 0.07 mM under conditions as the section of kinetics by colorimetric assay described above. It agreed with the results of the kinetics by HPLC assay. The apparent Km values of LgtA and LgtC for UDP-GlcNAc and UDP-Gal were 30.06 ± 1.86 and 5.85 ± 0.35 mM, respectively (a plot of reaction velocity versus substrate concentration and Lineweaver–Burk plot of LgtA or LgtC are not shown).
Table 2. Apparent kinetic parameters for glycosyltransferase activity of LgtA, LgtB, and LgtC by colorimetric assays*Fig. 4. Kinetic curve of LgtB. a) Plot of reaction velocity versus substrate concentration for LgtB. b) Lineweaver–Burk plot of LgtB for calculation of Km.
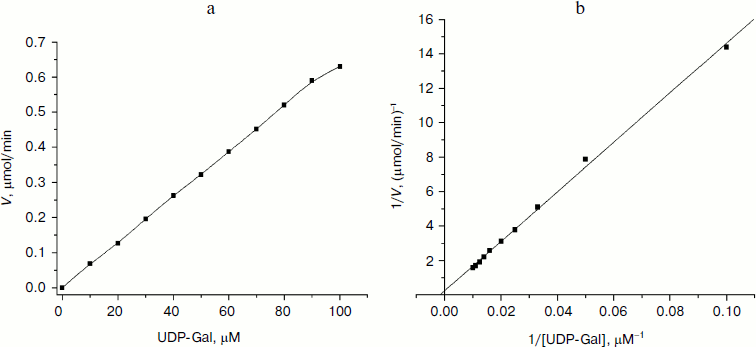
*Assays were performed in Hepes buffer (50 mM, pH 7.2) with either varied concentrations of UDP-Gal (or UDP-GlcNAc) (5, 10, 15, …, 90, 95, and 100 μM) and a fixed concentration of GlcNAc-β-pNP (or Gal-β-pNP) (25 μM). The measurements were done in triplicate and the data shown are the average ± standard deviation.
Because the extinction coefficient for p-nitrophenolate is relatively large, the color change is obvious. The colorimetric assay, which is based on the detection of absorbance changes of the p-nitrophenolate, has many advantages such as high sensitivity, low detectable limit, and simple operation. However, crude extracts that contain substrate hydrolases cannot be used for coupled assays due to interference for quantitative analysis. The subtle differences in substrate specificity for a single enzyme might not be convenient to discern with this colorimetric method because they require different glycosidase.
We have adopted a new colorimetric assay for three glycosyltransferases. The assay is useful in the initial characterization of recombinant glycosyltransferase, including determination of optimal metal cofactor and pH value, calculation of kinetic parameter, and screening of potential inhibitors or effectors of the enzymes. It offers a simple and sensitive method for glycosyltransferase assay and is expected to offer further impetus for discovering new and potent therapeutic agents that are directed at glycosyltransferase.
We are grateful for the financial support of the China Postdoctoral Science Foundation (20090450086), National Program on Key Basic Research Project (2007CB914403), and National Natural Science Foundation of China (20872068).
REFERENCES
1.Holemann, A., and Seeberger, P. H. (2001) Curr.
Opin. Biotechnol., 15, 615-622.
2.Bertozzi, C. R., and Kiessling, L. L. (2001)
Science, 291, 2357-2364.
3.Weijers, C. A., Franssen, M. C., and Visser, G. M.
(2008) Biotechnol. Adv., 26, 436-456.
4.Fang, J. W., Li, J., Chen, X., Zhang, Y. N., Wang,
J. Q., Guo, Z. M., Zhang, W., Yu, L. B., Brew, K., and Wang, P. G.
(1998) J. Am. Chem. Soc., 120, 6635-6638.
5.Chen, X., Andreana, P. R., and Wang, P. G. (1999)
Curr. Opin. Chem. Biol., 3, 650-658.
6.Posekany, K. J., Pittman, H. K., Bradfield, J. F.,
Haisch, C. E., and Verbanac, K. M. (2002) Infect. Immun.,
70, 6215-6222.
7.Chong, A. S.-F., Blinder, L., Ma, L., Yin, D. P.,
Shen, J. K., Williams, J. W., Byrne, G., Schwarz, A., Diamond, L. S.,
and Logan, J. E. (2000) Transpl. Immunol., 8, 129-137.
8.Bao, X. F., Kobayashi, M., Hatakeyama, S., Angata,
K., Gullberg, D., Nakayama, J., Fukuda, M. N., and Fukuda, M. (2009)
PNAS, 106, 12109-12114.
9.Yamada, N., Chung, Y. S., Takatsukal, S., Arimoto,
Y., Sawadal, T., Dohi, T., and Sowal, M. (1997) Brit. J. Cancer,
76, 582-587.
10.Compain, P., and Martin, O. R. (2001) Bioorg.
Med. Chem., 9, 3077-3092.
11.Lee, L. V., Mitchell, M. L., Huang, S. J., Fokin,
V. V., Sharpless, K. B., and Wong, C. H. (2003) J. Am. Chem.
Soc., 125, 9588-9589.
12.Pasquarello, C., Picasso, S., Demange, R.,
Malissard, M., Berger, E. G., and Vogel, P. (2000) J. Org.
Chem., 65, 4251-4260.
13.Khraltsova, L. S., Sablina, M. A., Melikhova, T.
D., Joziasse, D. H., Kaltner, H., Gabius, H.-J., and Bovin, N. V.
(2000) Anal. Biochem., 280, 250-257.
14.Yan, L. Y., Smith, F., and Cummings, R. D. (1994)
Anal. Biochem., 223, 111-118.
15.Gosselin, S., Alhussaini, M., Streiff, M. B.,
Takabayashi, K., and Palcic, M. M. (1994) Anal. Biochem.,
220, 92-97.
16.Pace, M., Agnellini, D., Gardana, C., Mauri, P.
L., and Pietta, P. G. (1995) J. Chromatogr. A., 691,
331-336.
17.Monegal, A., Pinyol, R., and Planas, A. (2005)
Anal. Biochem., 346, 115-123.
18.Zhang, C. S., Albermann, C., Xun, F., and
Thorson, J. S. (2006) J. Am. Chem. Soc., 128,
16420-16421.
19.Gao, H., and Leary, J. A. (2003) J. Am. Soc.
Mass. Spectrom., 14, 173-181.
20.Yang, M., Brazier, M., Edwards, R., and Davis, B.
G. (2005) ChemBioChem., 6, 346-357.
21.Palcic, M. M., and Sujino, K. (2001) Trends
Glycosci. Glyc., 13, 361-370.
22.Deng, C. H., and Chen, R. R. (2004) Anal.
Biochem., 330, 219-226.
23.Persson, M., and Palcic, M. M. (2008) Anal.
Biochem., 378, 1-7.
24.Wongkongkatep, J., Miyahara, Y., Ojida, A., and
Hamachi, I. (2006) Angew. Chem. Int. Ed., 45,
665-668.
25.Chen, X. Q., Jou, M. J., and Yoon, J. Y. (2009)
Org. Lett., 11, 2181-2184.
26.Hauser, S., Song, H. J., Li, H. G., and Wang,
L.-X. (2005) Biochem. Biophys. Res. Commun., 328,
580-585.
27.Liu, Z. Y., Lu, Y. Q., Zhang, J. B., Pardee, K.,
and Wang, P. G. (2003) Appl. Environ. Microbiol., 69,
2110-2115.
28.Zhang, J. B., Kowal, P., Fang, J. W., Andreana,
P., and Wang, P. G. (2002) Carbohydr. Res., 337,
969-976.
29.Liu, X. W., Xia, C. F., Li, L., Guan, W. Y.,
Pettit, N., Zhang, H. C., Chen, M., and Wang, P. G. (2009) Bioorg.
Med. Chem., 17, 4910-4915.
30.Sauerzapfe, B., Namdjou, D.-J., Schumacher, T.,
Linden, N., Krenek, K., Kren, V., and Elling, L. (2008) J. Mol.
Catal. B. Enzym., 50, 128-140.
31.Namdjou, D. J., Chen, H. M., Vinogradov, E.,
Brochu, D., Withers, S. G., and Wakarchuk, W. W. (2008)
ChemBioChem., 9, 1632-1640.
32.Witsell, D. L., Casey, C. E., and Neville, M. C.
(1990) J. Biol. Chem., 265, 15731-15737.
33.Wang, J. Y., Zhu, S. G., and Xu, C. F. (2002)
Biochemistry, 3rd Edn., Higher Education Press, Beijing, pp.
351-368.