Investigation of Mechanism of p38 MAPK Activation in Renal Epithelial Cell from Distal Tubules Triggered by Cardiotonic Steroids
O. A. Akimova1,2,3*, O. D. Lopina1, A. M. Rubtsov1, P. Hamet3, and S. N. Orlov1,2,3
1Department of Biochemistry, Faculty of Biology, Lomonosov Moscow State University, 119991 Moscow, Russia; fax: (495) 939-5022; E-mail: ol_akimova@hotmail.com2Institute of General Pathology and Pathophysiology, Russian Academy of Medical Sciences, ul. Baltiiskaya 8, 125315 Moscow, Russia
3Centre de Recherche, Centre Hospitalier de l’Universite de Montreal (CHUM) – Technopole-Angus, 2901 Rachel Est, Montreal, Canada, H1W 4A4
* To whom correspondence should be addressed.
Received March 4, 2010; Revision received April 9, 2010
Ouabain and other cardiotonic steroids (CTS) kill renal epithelial cells from distal tubules (C7-MDCK) via interaction with Na,K-ATPase but independently of inhibition of Na,K-ATPase-mediated ion fluxes. Recently, we demonstrated that modest intracellular acidification and inhibition of p38 MAPK suppress death of C7-MDCK cells triggered by ouabain. In the present study we investigate the mechanism of p38 MAPK activation in renal epithelial cell from distal tubules evoked by cardiotonic steroids. Using Na+/K+ ionophores (monensin, nigericin) and media with different content of monovalent cations, we revealed that p38 MAPK phosphorylation in ouabain-treated renal epithelial cells is not caused by Na,K-ATPase inhibition and inversion of the [Na+]i/[K+]i ratio. We also demonstrated that attenuation of pH from 7.45 to 6.75 did not alter the level of p38 MAPK phosphorylation observed in ouabain-treated cells. Inhibitors of PKA, PKC, and PKG as well as protein phosphatases were unable to abolish p38 MAPK activation triggered by ouabain. Using phosphotyrosine antibodies we did not detect any effect of ouabain on activation of tyrosine kinases. Thus, our results show that activation of p38 MAPK and cytotoxic action of CTS are independent of intracellular Na+, K+, and H+ concentrations. The molecular origin of intermediates of death signaling induced by CTS via conformation changes of Na,K-ATPase with following activation of p38 MAPK should be examined further.
KEY WORDS: Na,K-ATPase, ouabain, ionophores, intracellular Na+ and K+, p38 MAPK, kidney, cell deathDOI: 10.1134/S0006297910080043
Abbreviations: CTS, cardiotonic steroids; [K+]i, intracellular concentration of potassium ions; MAPK, mitogen-activated protein kinase; [Na+]i, intracellular concentration of sodium ions.
Na,K-ATPase (Na+,K+-activated,
Mg2+-dependent adenosine triphosphate phosphohydrolase, EC
3.6.1.3) catalyzes the electrogenic exchange of three intracellular
Na+ ions for two extracellular K+ ions using for
this transport energy that is released during the hydrolysis of ATP. In
this way, Na,K-ATPase plays a key role in the regulation of
intracellular Na+ and K+ concentrations and in
the maintenance of electrical membrane potential, cell volume, and
Na+-coupled transport of amino acids, glucose, nucleotides,
and other compounds with low molecular mass [1-3]. Numerous studies that were made recently
demonstrated that Na,K-ATPase is a receptor for cardiotonic steroids
(CTS) that (without Na-pump inhibition) can activate signal cascades
involved in the regulation of gene expression, proliferation and death
of cells, including receptors for inositol-3-phospates, release of
intracellular Ca2+, Ras, phosphoinositol-3 kinase (PI3K),
and mitogen-activated protein kinase (MAPK) Erk1/2 [4-12]. For example, ouabain, a
hydrophilic CTS that is widely used for experiments in vitro,
protected vascular smooth muscle cells from the development of
programmed cell death (apoptosis) induced by the deprivation of growth
factors [13]. This effect of CTS is due to the
inhibition of Na,K-ATPase, increase of [Na+]i and
expression of Na+i-sensitive genes of early
response: c-Fos and c-Jun, that, in turn, regulate the
expression of antiapoptotic genes including mortalin [14-16].
It should be noted that CTS effect on the proliferation and death of cells is tissue specific. For example, long-term treatment of renal epithelial cells from distal tubules, in contrast to vascular smooth muscle cells, triggers a signal cascade that leads to the death of dog renal epithelial cells from distal tubules (MDCK) [17, 18]. This type of death is characterized by mixed signs of necrosis (cell swelling) and apoptosis (activation of caspase-3). It was termed “oncosis” – from the Greek “onkos” that means swelling [19]. It should be noted that, in contrast to the ouabain effect, the inhibition of Na,K-ATPase in the absence of potassium did not affect the viability of MDCK cells but led to an increase of their sensitivity to cytotoxic effect of CTS [20, 21]. Taking into account this observation, we suggested that oncosis is induced by the interaction of CTS with the α-subunit of Na,K-ATPase, but it is not a direct consequence of the inhibition of cations fluxes mediated by this enzyme and the increasing of intracellular [Na+]i/[K+]i ratio [6].
Using pharmacological approaches we did not reveal the participation of Ras and PI3K in the development of MDCK cell death induced by ouabain. Negative data was also obtained as a result of the study of the involvement of [Ca2+]i content, reactive oxygen species, and internalization of Na,K-ATPase in death machinery triggered by ouabain [22, 23]. Further study of the CTS-induced signal cascade have shown that sharp growth of p38 MAPK phosphorylation preceded the development of oncosis in C7-MDCK cells treated by ouabain [24]. Phosphorylation of this form of stress-activated MAPK was found also in other cells that undergo oncosis, whereas the content of P~p38 MAPK did not change in ouabain-resistant cells. Ouabain-induced death of C7-MDCK cells was sharply suppressed in the presence of SB202190, a specific inhibitor of p38 MAPK, but not in the presence of its inactive analog, SB202474, as well as in the presence of inhibitors of Erk and JNK MAPK [24]. We found also that death of C7-MDCK cells was completely eliminated if pH value in the cell is decreased from 7.1 to 6.9 [25]. These data demonstrate that the effect of CTS as oncosis inducers is a consequence of activation of tissue-specific Na+i,K+i-independent pHi-sensitive signal cascade mediated by p38 MAPK. In this work we continued study of involvement of Na+i, K+i, and pHi in phosphorylation of p38 MAPK induced by CTS, as well as a participation in this signal cascade of other protein kinases and phosphatases.
MATERIALS AND METHODS
Reagents. Ouabain was obtained from ICN (USA), 22NaCl and 86RbCl from Amersham (Canada), monensin, valinomycin, and nigericin from Sigma (USA), and Go 6983, Go 6976, H-89, and okadaic acid from Calbiochem (USA). Antibodies for phospho-form of p38-MAPK and phosphotyrosine residues were obtained from Cell Signaling (Canada). Remaining reagents were purchased from Sigma, Gibco BRL (USA), and Anachemia (Canada).
Cell culture. In this work we used Madin–Darby canine kidney epithelial cells (C7-MDCK). The functional characteristics of these cells correspond to that of principal cells from collecting ducts. Cells were seeded in 6-, 12-, and 24-well plates, and also in Petri plates with area 75 cm2 in an incubator (Fisher Scientific) with an atmosphere containing 5% CO2. They were grown in modified Dulbecco’s modified Eagle’s medium (DMEM) containing 5.5 mM glucose, 10% fetal bovine serum, penicillin (100 units/ml), and streptomycin (100 µg/ml). In some experiments cells were incubated in media A, B, and C. Medium A contained (mM): NaCl, 109.4; KCl, 5.4; CaCl2, 1.8; MgSO4, 0.8; NaHCO3, 29.8; NaH2PO4, 0.9; Hepes, 8.4; glucose, 5; and also vitamins and amino acids in concentrations that usually are used in DMEM. Media B and C had the same composition as medium A except the concentration of Na+ and K+: [Na+]o = 145.5 mM, [K+]o = 0.05 mM for medium B and [Na+]o = 30.7 mM, [K+]o = 114.8 mM for medium C. In experiments on pH effect on cell morphology and p38 MAPK activation triggered by ouabain, we used control (pH 7.45) and acidified (pH 6.75) media. Control and acidic media contained (mM): NaCl, 130; KCl, 5; CaCl2, 1; MgCl2, 1; NaH2PO4, 0.9; Hepes, 20; glucose, 5; and also vitamins and amino acids in concentrations that are usually used in DMEM; pH was adjusted to the values 7.45 and 6.75 by the addition of Tris. After experiment cell morphology was evaluated by Diaphot phase contrast microscopy (Nikon, Japan) without preliminary fixation.
Intracellular content of exchangeable Na+ and K+ was measured as the steady-state distribution of 86Rb and 22Na that was established after 5 h of incubation [20, 21]. With this aim, C7-MDCK cells in 24-well plates were washed by phosphate-buffered saline (PBS, pH 7.4). After that 1 ml of medium containing 0.5 µCi/ml 86RbCl or 4 µCi/ml 22NaCl was added (complete composition of the medium is given above) and cells were incubated for 5 h at 37°C. Then cells were transferred on ice, washed by cold solution containing 100 mM MgCl2 and 10 mM Hepes (pH 7.4), and lysed by solution with 1% SDS and 4 mM EDTA. Contents of intracellular Na+ and K+ were calculated as A/am, where A is radioactivity of lysate (cpm) obtained from cells containing m mg of protein, and a is specific radioactivity of the incubation medium (cpm/nmol).
Western blot analysis. After completing the experiment, cells were lysed by buffer containing 150 mM NaCl, 1% Triton X-100, 0.1% SDS, 2 mM EDTA, 2 mM EGTA, 25 mM Hepes (pH 7.5), 10% glycerin, 1 mM NaF, 200 µM Na3VO4, and proteinase inhibitors (1 µg/ml leupeptin, 1 µg/ml aprotinin, and 1 mM PMSF). After incubation for 15 min, the lysate was centrifuged at 20,000g for 10 min. SDS sample buffer was added to samples, and they were kept 5 min at 95°C. Proteins in samples were separated by using PAGE according to the Laemmli procedure in the presence of SDS with 4% stacking and 10% running gels. Proteins separated by PAGE were transferred to nitrocellulose membrane (Mini Trans-Blot; Bio-Rad, USA) by transblotting during 1.5 h at 400 mA in buffer containing 25 mM Tris/192 mM glycine (pH 8.3), 20% methanol. To check quality of protein transfer, the membrane was stained by Ponceau S die (0.02%), and then blocked at room temperature by 5% fat-free dry milk dissolved in PBS during 1 h under stirring. After that the membrane was incubated overnight with primary antibodies against phospho-MAPK or phosphotyrosine residues at 4°C under constant stirring. Then the membrane was washed (3 times for 15 min with PBS containing 0.05% Tween 20) and incubated with secondary antibodies. Proteins were visualized using a kit for chemiluminescence (ECL; Amersham) and photo film (Kodak, USA) with subsequent development (Kodak X-Omat 2000A).
RESULTS
Role of Na+i and K+i in CTS-induced phosphorylation of p38 MAPK. Previously we have shown that ouabain induced maximal increase of p38 MAPK phosphorylation in C7-MDCK cells after 4-6 h of incubation [24]. It was noted also that p38 MAPK phosphorylation preceded the death of cells that started from 8-10 h and completed by 24 h after the ouabain addition. In contrast to the ouabain effect, the inhibition of Na,K-ATPase that took place in K+-free medium was not accompanied by the growth of the content of p38 MAPK phospho-form [24]. These findings indicate that the effect of CTS on the phosphorylation of this MAPK is not a direct consequence of inhibition of Na,K-ATPase and the increase of [Na+]i/[K+]i ratio. However, because of the leakage of intracellular K+, the extracellular concentration of K+, [K+]o, is increased in the initially K+-free medium up to 30-50 µM [21], which might be enough for the moderate activation of Na,K-ATPase [1, 2]. In this connection we compared effects of ouabain on the content of intracellular Na+ and K+ and the phosphorylation of p38 MAPK in control medium A and in medium B that contain 5.4 and 0.05 mM KCl, respectively.
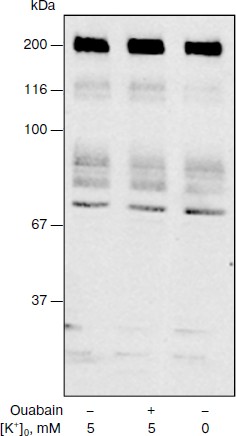
As can be seen from the Table 1, incubation of the cells in medium B with low K+ concentration for 1.5 h resulted in 8-fold increase of [Na+]i, which was approximately the same as in the case of the inhibition of Na+-pump by ouabain in control medium A. Despite the sharp growth of content of Na+i, medium B did not induce the increase of p38 MAPK phosphorylation. The addition of ouabain to the medium B was accompanied by 15-fold increase of p38 MAPK phosphorylation, although the enhancement in the content of Na+i was insignificant (<20%) (Fig. 1 and Table 1).
Table 1. Effect of different concentrations of extracellular Na+, K+, and ouabain on intracellular concentration of Na+ and K+ and p38 MAPK phosphorylationFig. 1. Results of Western-blot analyses demonstrating the effect of ouabain (3 µM) on the phosphorylation of p38 MAPK in C7-MDCK cells after 5 h incubation in control medium A ([Na+]o = 140.1 mM; [K+]o = 5.4 mM), in medium B with low K+ concentration ([Na+]o = 145.5 mM; [K+]o = 0.05 mM), and in medium C with high K+ concentration and decreased Na+ concentration ([Na+]o = 30.7 mM; [K+]o = 114.8 mM). Complete composition of medium A is given in “Material and Methods”.

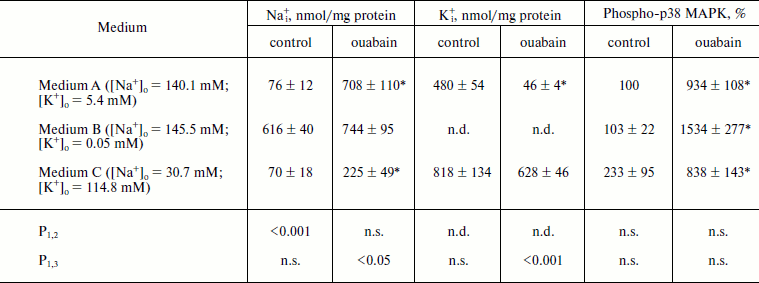
Note: C7-MDCK cells were incubated during 5 h in different media A, B, and C ± 3 µM ouabain. Complete compositions of the used media are given in “Materials and Methods”. Content of phospho-p38 MAPK in medium A without ouabain was taken as 100%. The mean values ± S.E. from experiments performed in quadruplicate (for Na+ and K+) and in triplicate (for phospho-MAPK) are given in the table; n.d., not determined; n.s., not significant.
* p < 0.05 in comparison with values obtained without ouabain.
To investigate further the role of Na+ and K+ in the phosphorylation of p38 MAPK, we investigated the action of ouabain under conditions that eliminated transmembrane K+ gradient using medium C with ([Na+]o = 30.7 mM, [K+]o = 114.8 mM), and also in the presence of Na+ and K+ ionophores. As one can expect, the addition of ouabain to medium C definitely did not affect [K+]i and significantly reduced the growth of [Na+]i in comparison with the control medium (Table 1). We found that medium C itself did not effect the increase of the content of P~p38 MAPK in the presence of ouabain in comparison with control medium A (Fig. 1 and Table 1).
The Na+ ionophore monensin induced 4-fold elevation of [Na+]i but it did not alter the content of [K+]i, whereas nonselective Na+/K+-ionophore nigericin led to 10-fold growth of [Na+]i and 3-fold decrease of [K+]i (Table 2). We did not find significant effect of selective K+-ionophore valinomycin on the intracellular content of Na+ and K+, which appears to be due to the initially high permeability of the membrane of MDCK cells for K+ and the values of electrical membrane potential that is close to steady-state potential for K+. Monensin did not alter the content of phospho-form of p38 MAPK, whereas it was increased in the presence of nigericin and valinomycin by 3-4 times. In control medium as well as in the presence of monensin, ouabain increased the content of P~p38 MAPK by 10-15-fold; in the presence of nigericin and valinomycin this parameter increased by 3-4 times (Table 2).
Table 2. Effect of ouabain and ionophores on
intracellular concentrations of Na+ and K+ and
p38 MAPK phosphorylation
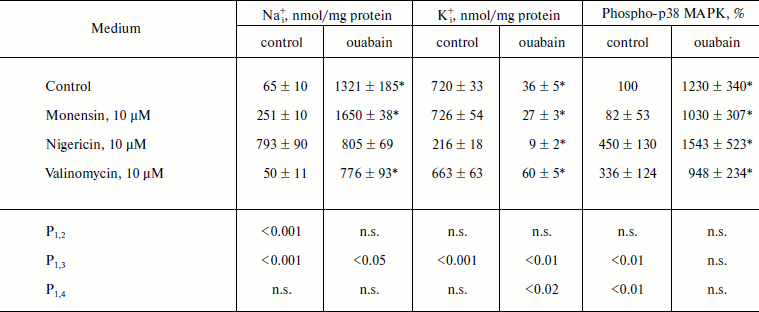
Note: C7-MDCK cells were incubated during 5 h in DMEM in the
presence of ionophores and 3 µM ouabain. Content of
phospho-p38 MAPK without ionophores and ouabain was taken as 100%. The
mean values ± S.E. from experiments performed in
quadruplicate (for Na+ and K+) and in triplicate
(for phospho-p38 MAKP) are given in the table; n.s., not
significant.
* p < 0.05 in comparison with values obtained without
ouabain.
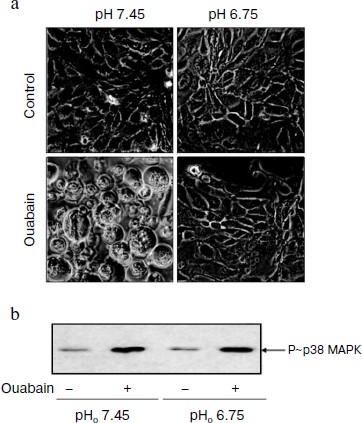
Role of pHi in ouabain-induced phosphorylation of p38 MAPK. We have shown earlier that moderate acidification of the incubation medium as well as of the cytoplasm after the addition of inhibitors of Na+/H+ exchanger led to the suppression of ouabain-triggered death of C7-MDCK cells [25]. In this work we compared the effect of ouabain on cell viability and on activation of p38 MAPK under control conditions (pH 7.45) and after weak acidification of the medium (pH 6.75). In accordance with the data obtained in the previous study, the addition of ouabain (3 µM) led to the loss of intercellular contacts. As a result, the cells lost the connection with support and became ball-shaped (Fig. 2a), whereas ouabain did not influence the cell morphology in the acidic medium (Fig. 2a), their connection with the support and viability that was determined as the level of chromatin fragmentation and the activity of caspase-3 (data not presented). In contract to the cytotoxic effect of ouabain, the acidification of the medium did not effect the phosphorylation of p38 MAPK either in the absence or in the presence of ouabain (Fig. 2b).
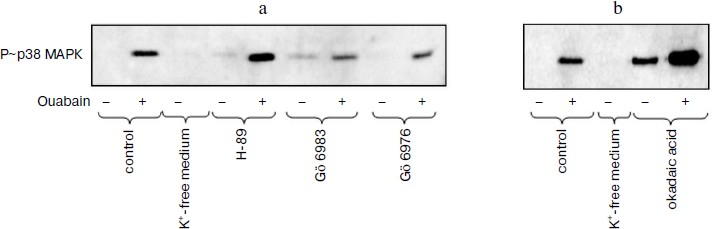
Role of protein kinases and protein phosphatases in transduction of CTS-induced signal of cell death. Numerous studies present evidence that in the cells from a number of tissues the phosphorylation of MAPK as a result of the effect of growth factors and other stimuli is due to the activation of serine-threonine protein kinases including protein kinases A, C, and G [26, 27]. In accordance with data of the Calbiochem catalog 2008/2009, compound Go 6983 inhibits all isoforms of PKC except PKC-µ (IC50 10-60 nM), whereas compound Go 6976 specifically inhibits PKC-α and PKC-β1 (IC50 2 and 6 nM, respectively). As can be seen from Fig. 3a, these compounds at concentration 5 µM did not alter the ouabain-induced phosphorylation of p38 MAPK. It was shown using purified enzyme preparations that H-89 suppressed the activity of protein kinases A, C, and G with IC50 ~ 50 nM, 14 µM, and 350 nM, respectively. When higher concentrations were used, the inhibition of activities of PKB, GSK, RS6K, AMP-activated protein kinase, Rho-dependent protein kinase ROCK-II, and kinase CHK1 was also observed. H-89 at concentration 10 µM did not affect the ouabain-induced phosphorylation of p38 MAPK (Fig. 3a).Fig. 2. Effect of pH on cell morphology and activation of p38 MAPK induced by ouabain. a) Phase contrast microscopy of C7-MDCK cells (200-fold magnification) after 18 h incubation in media with pHo 7.45 and 6.75 in the presence of ouabain (3 µM) and without it. b) Immunoblotting demonstrating effect of 6 h incubation without ouabain and in the presence of ouabain (3 µM) on p38 MAPK phosphorylation. Complete compositions of the media are given in “Materials and Methods”.
Okadaic acid is known to inhibit the serine-threonine protein phosphatases except protein phosphatases PP-1C and PP-2B. Phosphatases PP-1 and PP-2A are suppressed by this compound with IC50 ~ 10-15 and 0.1 nM, respectively. As should be expected, treatment of C7-MDCK cells by okadaic acid increased basal level of phosphorylation of serine and threonine residues that was recorded using anti-P~p38 MAPK antibodies (Fig. 3b). Nevertheless, the increase of ouabain-induced p38 MAPK phosphorylation was retained in the presence of this phosphatase inhibitor (Fig. 3b).Fig. 3. Effect of inhibitors of serine-threonine protein kinases (a) and protein phosphatases (b) on p38 MAPK phosphorylation in control and in cells treated by ouabain. Cells were incubated during 6 h in the DMEM ± 3 µM ouabain, 10 µM H-89, 5 µM Go 6983, 5 µM Go 6976, and 1 µM okadaic acid.
It is known that signals for proliferation involving phosphorylation of p38 and other MAPK are due to the activation of tyrosine protein kinases [26, 27]. We cannot find significantly different change of the level of phosphorylation of tyrosine residues of proteins with molecular masses from ~200 to 20 kDa in C7-MDCK cells that were treated by ouabain (3 µM) for 5 h (Fig. 4). A slight increase of tyrosine phosphorylation of a protein with molecular mass 195 kDa that was found by Contreras and coworkers in MDCK cells after their incubation with 10 µM ouabain during 8 h [28] was not found in our experiments.
Fig. 4. Results of Western-blot analyses demonstrating the effect of ouabain (3 µM) on the content of proteins with phosphorylated tyrosine residues in C7-MDCK cells. Cells were incubated during 5 h in control medium or in K+-free medium A. Composition of medium A is given in “Materials and Methods“.
DISCUSSION
In contrast to smooth muscle cells [28] and cells of nervous tissue [29, 30], incubation of endothelial cells of vessels [31] and renal epithelial cells from distal tubules (MDCK cells) with CTS during 24 h [17, 18, 21] leads to cell death. Similar to ouabain, the removal of potassium ions from the medium for MDCK cultivation induced inhibition of Na,K-ATPase and sharp growth of [Na+]i/[K+]i ratio but did not cause cytotoxic effect [20, 32]. Data obtained in experiments with 3H-labeled ouabain binding indicated that only one ouabain binding site exists in MDCK cells, and the affinity of this site to ouabain sharply increased when extracellular concentration of potassium ions is decreased [21], which is in agreement with numerous data obtained with purified preparations of Na,K-ATPase [1, 2, 33, 34]. These results suggest that CTS induce death of MDCK via their interaction with Na,K-ATPase α-subunit but independently of inhibition of ion fluxes mediated by this enzyme.
Investigation of the signal cascade inducing cell death demonstrated that ouabain activated p38 MAPK in MDCK cells, but it did not affect the activity of this protein kinase in smooth muscles cells or in other cells that are resistant to CTS. It was shown also that ouabain-triggered death was prevented as result of inhibition of p38, but not JNK and Erk, which indicated the involvement of this MAPK in the transduction of toxic signal [24]. Transfer of MDCK cells to K+-free medium led to 10-fold decrease of the range of ouabain concentrations that induced increase of p38 MAPK phosphorylation [24]. This indicates that the signal cascade is induced by the interaction of this CTS with Na,K-ATPase α-subunit. In the present work, we carried out further investigation of the mechanism of activation of p38 MAPK by CTS. We obtained the following principal results.
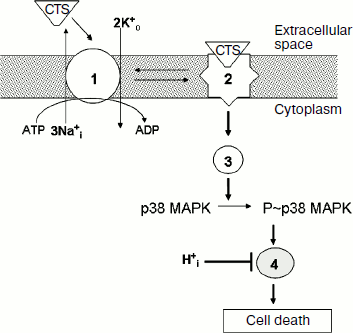
First, activation of p38 MAPK does not connect with the inhibition of ion fluxes mediated by Na,K-ATPase and with the increase of [Na+]i/[K+]i ratio. Indeed, sharp inhibition of Na,K-ATPase in medium B with low concentration of K+ that did not contain ouabain did not induce p38 MAPK phosphorylation, whereas ouabain increased phosphorylation of this MAPK in medium C with high concentration of K+ and low concentration of Na+, which are conditions under which this CTS did not significantly affect the [Na+]i/[K+]i ratio (Table 1). Moreover, the Na+-ionophore monensin induced 4-fold increase of [Na+]i but did not alter the content of [K+]i, whereas the nonselective Na+/K+ ionophore nigericin led to 10-fold elevation of [Na+]i and 3-fold decrease of [K+]i (Table 2). Despite the sharp increase of [Na+]i, monensin did not influence the content of P~p38 MAPK. Ouabain increased the content of this phosphoprotein in the presence of monensin as well as in the presence of nigericin (Table 2). These results are in agreement with a hypothesis suggesting the participation of p38 MAPK in the transduction of tissue-specific Na+i,K+i-independent cytotoxic signal produced by CTS through the conformational change of Na,K-ATPase α-subunit (Fig. 5).
Second, the cytotoxic effect of ouabain is completely eliminated when extracellular pHo is decreased from 7.45 to 6.75, but pH change did not affect the increase of p38 MAPK phosphorylation induced by this CTS (Fig. 2). These data show that activation of p38 MAPK precedes activation of the pH-sensitive mediator that involved in the transduction of cytotoxic signal (Fig. 5).Fig. 5. Working model illustrating the mechanism of the effect of CTS and pHi on the death of renal epithelial cells from distal tubules. 1, 2) Na,K-ATPase in active and inactive state, respectively; 3) mediators of signal cascade resulting in activation of p38 MAPK as a consequence of conformational change of Na,K-ATPase α-subunit induced by CTS binding; 4) unidentified pH-sensitive element of signal cascade triggered as a result of p38 MAPK phosphorylation.
Third, we did not reveal participation of serine-threonine protein kinase A, C, and G in the activation of p38 MAPK (Fig. 3) and also the influence of ouabain on the activity of tyrosine protein kinases (Fig. 4). These negative results are in agreement with the lack of significant influence of the inhibitors of serine-threonine and tyrosine protein kinases on the cytotoxic effect of ouabain that was noted in our previous studies [22].
Thus, our data demonstrate a Na+i-, K+i-, and H+i-independent mechanism of p38 MAPK activation by ouabain and its involvement in the cytotoxic effect of CTS. Further studies should identify mediators of this signal cascade induced by CTS via the change of Na,K-ATPase conformation and resulting activation of p38 MAPK, as well as pHi-sensitive mediator in the chain of transduction of cytotoxic signal that due to activation of p38 MAPK plays a key role in the transduction of this signal (Fig. 5).
This study was done with the financial support of grants from the Canadian Institute of Medical Research and the Russian Foundation for Basic Research (Nos. 09-04-00646, 08-04-01321, 10-04-00348) (SNO, ODL, OAA) and also a scholarship of the Canadian Fund of Kidney Investigation (OAA).
REFERENCES
1.Blanco, G., and Mercer, R. W. (1998) Am. J.
Physiol., 275, F633-F650.
2.Mobasheri, A., Avila, J., Cozar-Castellano, I.,
Brownleader, M. D., Trevan, M., Francis, M. J. O., Lamb, J. F., and
Martin-Vassalo, P. (2000) Biosci. Rep., 20, 51-91.
3.Mongin, A. A., and Orlov, S. N. (2001)
Pathophysiology, 8, 77-88.
4.Orlov, S. N., Akimova, O. A., and Hamet, P. (2005)
Curr. Hypertens. Rev., 1, 243-257.
5.Aperia, A. (2007) J. Intern. Med.,
261, 44-52.
6.Orlov, S. N., and Hamet, P. (2006) Acta Physiol.
(Oxford), 187, 231-240.
7.Orlov, S. N., and Hamet, P. (2006) J. Membr.
Biol., 210, 161-172.
8.Dvela, M., Rosen, H., Feldmann, T., Nesher, M., and
Lichtstein, D. (2007) Pathophysiology, 14, 159-166.
9.Schoner, W., and Scheiner-Bobis, G. (2007) Am.
J. Physiol. Cell Physiol., 293, C509-C536.
10.Tian, J., and Xie, Z.-J. (2008)
Physiology, 23, 205-211.
11.Bagrov, A. Y., Shapiro, J. I., and Fedorova, O.
V. (2009) Pharmacol. Rev., 61, 9-38.
12.Taurin, S., Hamet, P., and Orlov, S. N. (2003)
Mol. Biol., 37, 371-381.
13.Orlov, S. N., Thorin-Trescases, N., Kotelevtsev,
S. V., Tremblay, J., and Hamet, P. (1999) J. Biol. Chem.,
274, 16545-16552.
14.Taurin, S., Dulin, N. O., Pchejetski, D.,
Grygorczyk, R., Tremblay, J., Hamet, P., and Orlov, S. N. (2002) J.
Physiol., 543, 835-847.
15.Taurin, S., Seyrantepe, V., Orlov, S. N.,
Tremblay, T.-L., Thibaut, P., Bennett, M. R., Hamet, P., and
Pshezhetsky, A. V. (2002) Circ. Res., 91, 915-922.
16.Haloui, M., Taurin, S., Akimova, O. A., Guo,
D.-F., Tremblay, J., Dulin, N. O., Hamet, P., and Orlov, S. N. (2007)
FEBS J., 274, 3257-3267.
17.Ledbetter, M. L., Young, G. J., and Wright, E. R.
(1986) Am. J. Physiol., 250, C306-C313.
18.Bolivar, J. J., Lazaro, A., Fernandez, S.,
Stefani, E., Pena-Cruz, V., Lechene, C., and Cereijido, M. (1987)
Am. J. Physiol., 253, C151-C161.
19.Orlov, S. N., and Hamet, P. (2004) Adv. Exp.
Med. Biol., 559, 219-233.
20.Pchejetski, D., Taurin, S., der Sarkissian, S.,
Lopina, O. D., Pshezhetsky, A. V., Tremblay, J., DeBlois, D., Hamet,
P., and Orlov, S. N. (2003) Biochem. Biophys. Res. Commun.,
301, 735-744.
21.Akimova, O. A., Bagrov, A. Y., Lopina, O. D.,
Kamernitsky, A. V., Tremblay, J., Hamet, P., and Orlov, S. N. (2005)
J. Biol. Chem., 280, 832-839.
22.Akimova, O. A., Lopina, O. D., Hamet, P., and
Orlov, S. N. (2005) Pathophysiology, 12, 125-135.
23.Akimova, O. A., Hamet, P., and Orlov, S. N.
(2008) Pflugers Archiv-Eur. J. Physiol., 455,
711-719.
24.Akimova, O. A., Lopina, O. D., Rubtsov, A. M.,
Gekle, M., Tremblay, J., Hamet, P., and Orlov, S. N. (2009)
Apoptosis, 14, 1266-1273.
25.Akimova, O. A., Pchejetski, D., Hamet, P., and
Orlov, S. N. (2006) Pflugers Arch., 451, 569-578.
26.Kyriakis, J. M., and Avruch, J. (2001)
Physiol. Rev., 81, 807-869.
27.Pearson, G., Robinson, F., Beers, G. T., Xu, B.
E., Karandikar, M., Berman, K., and Cobb, M. H. (2001) Endocrin.
Rev., 22, 153-183.
28.Contreras, R. G., Shoshani, L., Flores-Maldonado,
C., Lazaro, A., and Cereijido, M. (1999) J. Cell Sci.,
112, 4223-4232.
29.Stel’mashuk, E. V., Isaev, N. K., Andreeva,
N. A., and Viktorov, I. V. (1996) Bull. Exp. Biol. Med.,
122, 163-166.
30.Akimova, O. A., Mongin, A. A., Hamet, P., and
Orlov, S. N. (2006) Cell. Mol. Biol., 52, 71-77.
31.Orlov, S. N., Thorin-Trescases, N., Pchejetski,
D., Taurin, S., Farhat, N., Tremblay, J., Thorin, E., and Hamet, P.
(2004) Pflugers Arch., 448, 335-345.
32.Contreras, R. G., Flores-Maldonado, C., Lazaro,
A., Shoshani, L., Flores-Benitez, D., Larre, I., and Cereijido, M.
(2004) J. Membr. Biol., 198, 147-158.
33.Wallick, E. T., and Schwartz, A. (1988) Meth.
Enzymol., 156, 201-213.
34.Lingrel, J. B., Croyle, M. L., Woo, A. L., and
Arguello, J. M. (1998) Acta Physiol. Scand., 163 (Suppl.
643), 69-77