REVIEW: Ribosomal Tunnel and Translation Regulation
A. A. Bogdanov*, N. V. Sumbatyan, A. V. Shishkina, V. V. Karpenko, and G. A. Korshunova
Belozersky Institute of Physico-Chemical Biology and Chemical Faculty, Lomonosov Moscow State University, 119991 Moscow, Russia; E-mail: bogdanov@belozersky.msu.ru* To whom correspondence should be addressed.
Received July 20, 2010
This review describes the results of recent studies of the ribosomal tunnel (RT), the major function of which is to allow the smooth passage of nascent polypeptides with different sequences from the peptidyl transferase center of the ribosome to the tunnel exit, where the folding of protein molecules begins. The features of structural organization of RT and their role in modulation and stabilization of the nascent chain conformation are discussed. Structural features of macrolide binding sites as well as application of macrolide antibiotics and their derivatives as tools to investigate ligand–tunnel wall interactions are also considered. Several examples of strong and specific interactions of regulatory polypeptides with nucleotide and amino acid residues of RT that lead to ribosome stalling and translational arrest are described in detail. The role of these events in regulation of expression of certain genes is discussed on the basis of recent high-resolution structural studies of nascent chains in the RT.
KEY WORDS: ribosome, ribosomal tunnel, growing polypeptide chain, macrolides, leader peptides, translation regulation, cation–π interactionsDOI: 10.1134/S0006297910130018
Abbreviations: cryo-EM, cryoelectron microscopy; FRET, Förster (fluorescence) resonance energy transfer; PTC, ribosome peptidyl transferase center; RT, ribosomal tunnel.
The polypeptide chain of newly formed protein molecules remains bound to
the ribosome during translation of genetic information up to completion
of its synthesis. In this case, beginning from the first steps of the
translation process, part of the synthesized polypeptide chain is
situated in the ribosomal tunnel (RT). This important structural
element of the ribosome is located in its large subunit; its beginning
overlaps with the ribosome peptidyl transferase center (PTC), and at
the end of the RT ribosome-associated proteins involved in
cotranslational folding and modification of the protein molecule are
localized. Thus, the main function of the RT is to provide for the
unobstructed release of the newly synthesized polypeptide chain from
the ribosome and its delivery to the place of formation of a
functionally useful protein molecule.
The main stages, leading participants, and basic principles of protein biosynthesis on ribosomes were established by the beginning of 1970s thanks to rapid progress of molecular biology during the first 15 years of its existence. The ribosomal tunnel was not then an exception. At the end of 1960s and beginning of 1970s, first Malkin and Rich [1] and then Blobel and Sabatini [2] found that 30-40 amino acid residues at the C-terminal part of the protein synthesized by rabbit reticulocytes and rat liver, respectively, are not cleaved by proteolytic enzymes. They concluded that in the ribosome there is a channel or tunnel within which the newly-synthesized polypeptide chains are transferred. However, only 12 years later, Lake et al. returned to the idea that there is a tunnel within the ribosome that transports the synthesized protein [3, 4]. They used immunoelectron microscopy and found that already practically completed newly formed protein molecule is located at the ribosome site directly opposite to the site of the beginning of its synthesis. Several years later, Milligan and Unwin [5] and then Yonath et al. [6] discovered the tunnel in the ribosome large subunit by cryoelectron microscopy (cryo-EM) and also supposed the concept that the polypeptide chains synthesized by the ribosome are transposed along the RT. Interpretation of these results met quite reasonable for that time criticism of other authors who believed that the polypeptide synthesized by the ribosome is transposed along its surface [7, 8]. More detailed arrangement and parameters of the RT were much later described by Frank et al. [9] who significantly increased the cryo-EM resolution in studies of ribosomes and their functional complexes. However, finally the question concerning RT as a structural element of large ribosomal subunit serving as a guide for newly synthesized polypeptides could be considered as solved only after publication of a work by Choi and Brimacombe in 1998 [10]. These authors studied covalent cross-linking of N-termini of E. coli peptides of different length synthesized by ribosomes with 23S rRNA. The results were analyzed within the framework of the model of spatial structure of 50S subunit of E. coli ribosome elaborated by Brimacombe, van Heel, et al. on the basis of data obtained by high-resolution cryo-EM and numerous results of chemical, biochemical, and genetic analyses. The model described with high precision the RT arrangement in the body of 50S subunit [11], and after publication of atomic models of the large subparticle of archaebacterial [12, 13] and eubacterial [14] ribosome obtained by X-ray analysis, it appeared that nucleotide residues of 23S rRNA detected in experiments on covalent cross-linking are really arranged in a proper order on the RT walls [15, 16].
The existence of sufficiently high resolution X-ray data and corresponding atomic models of the ribosome, its subunits, and a number of ribosomal functional complexes with substrates and protein translation factors during the past decade had an enormous effect on development of investigations in the field of protein biosynthesis (for review see [18]). The present-day concepts of RT structure and functioning are also significantly based on these founding works either directly or indirectly via interpretation of biochemical and genetic data in the light of existing atomic structures. Just they constitute the basis of this review. The reader interested in results of previous works can find them in excellent review by Hardesty and Kramer [19].
There are several reasons why RT attracts intent attention of researchers. First, it is still poorly studied element of the ribosome. Its detailed study began less than 10 years ago. Second, the RT differs in principle from now known channels in membrane structures used for transposition of proteins and peptides. Third, binding sites of many (including clinically important) antibiotics are localized in the RT, owing to which modification of RT walls results in bacterial (and pathogenic among them) resistance to antibacterial preparations. Finally, the RT walls take part in monitoring of amino acid sequence of the polypeptide moving along it. In some cases polypeptides become involved in strong interactions with the RT walls, which results in translation arrest. This event is the key moment in regulation of transcription/translation of some genes. Although now not so many systems are described in which RT are involved in regulation of expression of genetic information, it is already clear that they greatly differ in their functional importance and the regulatory mechanisms themselves are rather variable (for example, see [20]).
STRUCTURE OF THE RIBOSOMAL TUNNEL
As already mentioned, the ribosomal tunnel is unique: all presently known cell channels, tunnels, and pores (with only one interesting exception [21, 22]) through which ions, water molecules, and low and high molecular weight compounds (including proteins and nucleic acids) as well as macromolecular complexes are transported are built of protein molecules. However, the RT walls and their nearest environment consist mainly of nucleotide residues of the large ribosomal subunit rRNA and only of two or three small segments of ribosomal proteins (Fig. 1). Nucleotide residues of five out of six domains that are usually distinguished in the secondary structure of this rRNA are involved in the structural organization of the RT [11]. Already this single fact shows that the ribosomal tunnel is formed due to a complex system of tertiary contacts between rRNA regions remote in its secondary structure. Thus, RT resembles all the other functional centers of the ribosome built mainly of rRNA nucleotide residues (while PTC is entirely built from them). The overwhelming majority of RT-forming nucleotides are rather conservative [13].
The length of the ribosomal tunnel is directly related to the size of the large ribosomal subunit and its rRNA: in bacterial ribosomes it is approximately 90 Å [13, 14], in eukaryotic ribosomes about 100 Å [23], and in mitochondrial ribosomes about 60 Å [24, 25]. At the same time, the RT diameter in all ribosomes, independently of their source, is the same. It is approximately 15 Å in the upper (adjacent to PTC) third of the RT, in the middle part the tunnel narrows down 10 Å diameter, and then the tunnel expands again and a funnel-shaped structure with maximal diameter of approximately 25 Å is formed at its outlet. In the narrowed central part of the tunnel its walls are mainly formed by amino acid residues of β loops of the large ribosomal subunit proteins L4 and L22 (in eukaryotes L17) [13]. The β-loop of L23 protein (L32a in archaeans, Rpl25 in yeasts, L39 in other eukaryotes) is also involved in the RT wall structure organization close to its outlet [13, 14]. The outlet itself is framed by a globular part of L22 protein (Rpl17 in yeasts) as well as by proteins L24 (Rpl24 in yeasts), L29 (Rpl29 in yeasts), and L32 [23].Fig. 1. Scheme of arrangement of the ribosomal tunnel in the ribosome 50S subunit (cross-section along long axis of the RT). The macrolide binding site and proteins involved in the RT wall formation are shown (the upper part of the RT is shown in more detail in Fig. 9). Adapted with permission from [86].
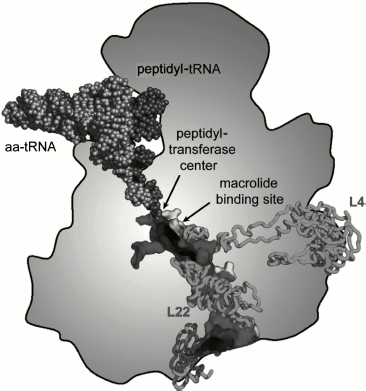
The ensemble of proteins localized at the outlet from the RT fulfils an extremely important function: it forms a platform for binding a group of protein factors and enzymes carrying out early stages of cotranslational processing of newly formed proteins as well as transport to receptors of proteins on cellular membranes, the proteins carrying in their N-terminal part secretion signal sequences [26]. A site for TF chaperon (so-called trigger factor) and associated polypeptide deformylase (that removes formyl group from N-terminal methionine residue) binding to bacterial ribosome is now clearly detected among these proteins [27]. This complex also includes methionine aminopeptidases (MAP) removing N-terminal methionine residue (both in pro- and eukaryotes), peptidyl-prolyl-isomerases, and N-acetylases [28]. At the same place at the outlet from the RT a small fraction of ribosomes is associated with SRP (signal-recognizing particles) intended for transfer of a ribosome, synthesizing membrane, or secreted protein to a translocon localized in the endoplasmic reticulum [26].
Most nucleotide residues of rRNA forming the RT walls are exposed to the tunnel space by their heterocyclic bases [13]. For a protein polypeptide chain located in an RT, this creates the possibility of establishing hydrogen bonds and hydrophobic contacts (see in more detail section “Involvement of Ribosomal Tunnel in Regulation of Gene Expression”). The presence of residues of the rRNA sugar-phosphate skeleton near tunnel walls creates negative potential within the tunnel, which is unevenly distributed along the RT [29, 30]. Actually, if short blocks of arginine or lysine residues are introduced into the growing polypeptide chain using gene-engineering techniques, its movement along the tunnel will be difficult and restriction in moving forward depends on the actual arrangement of such blocks in the RT [31]. However, there are no large hydrophobic or hydrophilic regions on the tunnel walls: their presence would make the tunnel impassable for many amino acid sequences present in proteins. The RT is highly hydrated; moreover, it is connected with the surface of the large subunit by numerous “microchannels” through which water molecules and hydrated ions (but not polypeptide chains) can pass rather easily [32].
The ribosome is a molecular machine characterized by pronounced conformational mobility [33, 34]. During the elongation cycle there is both movement of ribosomal subunits relative each other [34] and periodic structural changes in ribosomal subunits and translation factors coordinated with formation of each new peptide bond [35-37]. Therefore, it is not surprising that the problem of RT conformational mobility is often discussed in the literature. The extreme point of view was put forward by Frank et al. [38] who compared tunnel parameters in wild-type E. coli ribosomes and in ribosome mutants resistant to the antibiotic erythromycin and containing amino acid substitutions in the regions of proteins L4 and L22 exposed inside the tunnel. In their interpretation of the results obtained using cryo-EM, they concluded that mutations in proteins involved in formation of the RT walls result in almost doubling of its diameter. It was supposed that in the much narrowed RT part (i.e. in the region formed by proteins L4 and L22) the tunnel pushes the polypeptide chain to the RT outlet, working as peristaltic pump, i.e. it periodically narrows and widens [38]. Another group of authors, also on the basis of cryo-EM data of relatively low resolution, supposed that the RT dimensions may as much increase as is necessary for polypeptide chain folding into “rudimentary” globular structures [39]. However, probably these suppositions will remain fascinating hypotheses: detailed analysis of X-ray data has shown that such large change in RT parameters is impossible because it requires global change of the whole structure of the large ribosomal subunit, especially rigid at the RT region [32]. The maximal diameter of the protein molecule structural element that can be disposed in the RT corresponds to α-helix. Nevertheless, as will be shown below, local conformational changes in nucleotide and amino acid residues, lining the RT walls are not only possible, but they occur during functioning of the ribosomal tunnel. It is supposed that these conformational transitions are involved in the signal cascade transduction along the RT walls (for example, see [40]), which well corresponds to current concepts of the relationship between the ribosome functional centers [41, 42].
CONFORMATION OF THE GROWING POLYPEPTIDE CHAIN IN THE RIBOSOMAL
TUNNEL
There are several experimental approaches in the literature allowing isolation of ribosome preparation together with newly synthesized polypeptide. In some cases they are based on the use in a cell-free system of protein synthesis of a template, a segment of natural mRNA free of stop codons (so-called “truncated” or shortened mRNA [43, 44]). In other cases sequences encoding a polypeptide able to stop translation (usually called “stop-peptide”) were included into mRNA constructs, and as a rule, a special “anchor” peptide was added at the N-terminus of the chain for subsequent affinity chromatography [45]. Complexes obtained by these methods have been recently studied by Beckmann et al. using cryo-EM with record high resolution of 5.5-7.0 Å [46, 47]. It should be noted so far the study of ribosome complex with fixed growing polypeptide has not been achieved by X-ray analysis, although systematic attempts have been made.
As we have already emphasized the growing polypeptide can be located in the ribosomal tunnel either in the form of α-helix or as unfolded chain. Knowing the length of the RT (90 Å) and the distance per single amino acid residue in any polypeptide secondary structure, one can easily estimate that the part of a completely α-helical polypeptide localized in the RT will consist of (90 : 1.5) or approximately 60 amino acid residues; for completely unfolded polypeptide (90 : 3.5) this value would be about 25. However, it has been long known from biochemical data that the ribosome protects against cleavage by proteases the newly synthesized polypeptide up to 40 amino acid residues in length [1, 2]. So it can be supposed that within the RT a part of the polypeptide chain is in unfolded conformation, whereas another part acquires the shape of α-helix. All presently available structural data support this hypothesis.
Johnson et al. [48] first showed using the FRET technique that at least part of transmembrane segments of membrane proteins synthesized by a ribosome without leaving the RT acquire compact, most likely α-helical conformation. They suggested that it is formed in the upper part of the RT (i.e. in the region of the PTC) and is preserved during transfer of the segment along the tunnel. The latter conclusion was based on data on the chemical cross-linking of the growing polypeptide with the RT walls, according to which a transmembrane segment moved along the tunnel, retaining its compaction, first cross-linked to protein L4, then to L7 (L22), and finally to L39 [48]. Further Deutsch et al. in their broad study on model [30] and natural [49] systems using a set of chemical methods also showed that segments of polypeptide localized in the RT of eukaryotic ribosome are able to form α-helices. This happens only when this type of secondary structure is that preferred for the amino acid sequence of the segment. In the first case, oligo-alanine blocks of 5, 10, and 15 amino acid residues, for which α-helical conformation is known to be preferable, were introduced into different positions of polypeptide chain of a protein domain fragment forming potassium channel. Using truncated mRNA, the peptide was “frozen” in the RT. In this case the peptide contained a single cysteine residue localized at a distance of 38 amino acid residues from its C-end (i.e. from the PTC). If the peptide was in unfolded conformation, then the cysteine residue was outside the RT and it could be detected using chemical modification by N-polyethylene glycol maleimide. The reagent had molecular mass 5 kDa and could not penetrate into the RT. However, if oligo-alanine segments acquired α-helical conformation, then the cysteine residue was inside the RT and did not undergo chemical modification. They found that in some regions the growing peptide is able to fold into sufficiently stable α-helices. Even more convincing results were recently obtained by the same research group during investigation of conformation in the RT of natural protein segments containing more than one cysteine residue [49, 50]. Using reagents penetrating into the RT and catalyzing formation of S–S bonds, they estimated distances between cysteine residues in the polypeptide chain and concluded that the RT plays an active role in generation of α-helical conformation of the newly synthesized protein. In this case the outlet-adjacent third of the RT, named “alpha-zone”, exhibits the most pronounced ability to modulate helix formation. Just during transfer into this zone transition of the polypeptide secondary structure from unfolded to α-helical occurred. Since these helices were preserved upon translocation into membrane structures, a conclusion was drawn concerning the importance of the growing polypeptide chain within the tunnel for folding in protein biogenesis.
It is necessary to note here that already in the middle of the 1980s Lim and Spirin made detailed theoretical conformational analysis of the peptidyl transferase reaction and concluded that already at early stages of protein synthesis the polypeptide chain folds to a regular α-helical structure. They supposed that formation of such structure is important for following correct folding of the newly synthesized protein macromolecule [51].
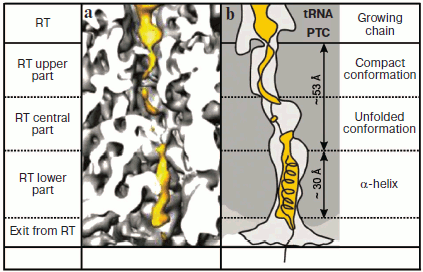
Recently Beckman et al. directly identified α-helical segments of the protein polypeptide chain growing in the ribosomal tunnel [52]. As already mentioned, in this laboratory such high resolution was achieved for cryo-EM analysis of the ribosome that electron density corresponding to protein α-helices could be clearly observed with the background of different structural elements of the ribosome. They included a 25-membered polypeptide into two different regions of the type II dipeptidyl peptidase B segment synthesized by 80S ribosome of wheat germs. It was encoded in truncated mRNA (without stop-codon) of 90-nucleotide-long. The polypeptide insert was the five-fold repeat of EAAAK sequence that in solution [53] and within one of fish antifreeze proteins [54] exists in the form of classical α-helix due to the presence of alanine blocks and formation of salt bridges between glutamic acid and lysine residues. When after translation arrest this segment was localized in the last third of the RT, it acquired α-helical conformation clearly distinguished by cryo-EM (Fig. 2; see color insert). However, if it was placed in the RT central part the polypeptide chain remained unfolded, although as noted above, its amino acid sequence in solution prefers α-helical conformation. They identified some polypeptide contacts with tunnel walls and concluded that L39 protein is the most important component of the RT “α-zone”. Interestingly, in both cases formation of compact structure was observed in polypeptide regions (free of inserted segments) localized in the upper part of the RT near the PTC. However, these structures were not folded into canonical α-helical conformation [52].
Thus, together the above-mentioned data show that, first, segments of protein polypeptide chain with particular amino acid sequences growing in the RT are able to acquire α-helical conformation, and second, transition into this conformation (as well as in the unfolded chain conformation) is modulated by the interaction of the polypeptide with the tunnel walls in its specific zones. The last fact, namely, the active role of the RT in organization and stabilization of secondary structure of polypeptide chain synthesized on the ribosome is especially important because it is known that secondary structure of disordered region of a protein molecule fluctuates at enormous rates (exceeding by many orders of magnitude the rate of protein synthesis) between conformations specific of α-helix, β-structure, and polyproline type II helix [55].Fig. 2. Growing polypeptide chain in the RT of eukaryotic ribosome. a) Distribution in the RT of the electron density of the polypeptide and its environment according to CEM data. The tRNA and polypeptide are shown in yellow. b) Schematic interpretation of electron-microscopic image shown in Fig. 2a. The figures were created on the basis of data presented in [52].
INTERACTION OF THE RIBOSOMAL TUNNEL WITH ANTIBIOTICS
Bacterial ribosomes serve as target for almost half of clinically used antibiotics [56]. Therefore, determining the atomic structure of their complexes with different classes of antibiotics (see reviews [57, 58]) can be considered as one of main achievements of ribosome crystallography. Although these studies are first of all important for design of new antibacterial drugs and elucidation of mechanisms of resistance of protein synthesis to antibiotics, they brought and continue to bring extremely important information concerning potential sites of interaction between the growing polypeptide chain and the RT walls during protein synthesis.
As already mentioned, the “macrolide-binding site” (MBS) is localized in the RT not far from the PTC. It occupies approximately the upper third of the RT (see also Fig. 1). Functional groups of some macrolides and related ketolides also reach the RT walls in its narrowed central part [59, 60]. It is assumed that after binding to MBS, antibiotics prevent the growth of the polypeptide chain on the ribosome.
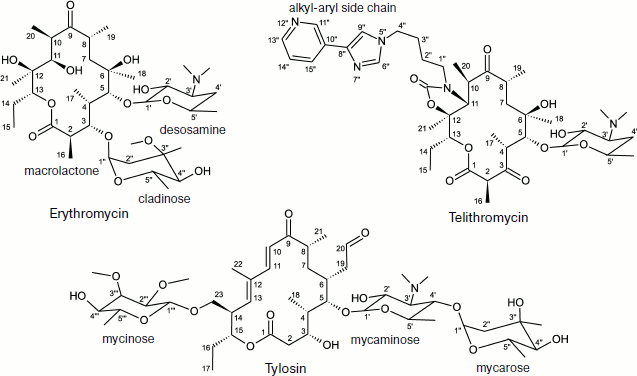
Macrolides comprise a family of natural and semisynthetic antibiotics widely used in clinical and veterinary science designed on the basis of 12-16-membered lactones to which carbohydrate substituents are attached (Fig. 3) [61]. In particular, all macrolides contain one or more carbohydrate residues bound by glycoside bonds to position C5 of the lactone ring and conventionally directed “upstream” along the RT, while some of them, including a mycinose residue in position C14 of the lactone ring, orient along the RT walls in the opposite direction, conventionally “downstream” along the RT [59, 60, 62]. The binding sites of different macrolides are distinct, although they overlap significantly [63].
The arrangement of the MBS in the large ribosomal subunit was initially detected using biochemical and genetic methods of investigation. It was found that elements of the central loop of domain V and helix 35 of domain II of 23S rRNA are involved in MBS formation [64]. It was shown by mutagenesis that all macrolides form at least the same “point” contact, namely, with nucleotide A2058 (here and everywhere in the text, if it is not especially specified, numbering of nucleotide residues is given for E. coli 23S rRNA) located in the central loop of domain V of E. coli 23S rRNA [64, 65]. Methylation of the amino group of adenine residue in position 2058 of rRNA by Erm type methyl transferases [66, 67] as well as substitution of this base for G, C, or U result in significant decrease of ribosome affinity to macrolides [68] and emergence of bacterial resistance to these antibiotics. Most likely, methylation of A2058 sterically prevents macrolide binding in the MBS. Mutations of A2058-adjacent nucleotides also result in bacterial resistance to antibiotics [69-71]. Besides, it was found that the mutant with G2032A substitution is more sensitive to erythromycin, while deletions in rRNA domain II decrease growth rate of bacterial cells in the presence of antibiotic [68]. The use of site-directed mutagenesis has shown that residues C2611, G2057 (domain V), G748 (hairpin 35 of domain II) [65], A752 (hairpin 35 of domain II) [64], Lys90 (L4), Gly64 (L22) [38, 72] are directly or indirectly involved in formation of the MBS.Fig. 3. Chemical structure of representatives of macrolide antibiotics (erythromycin, tylosin) and ketolides (telithromycin).
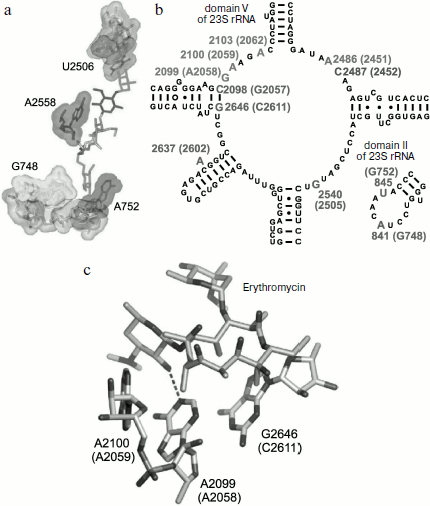
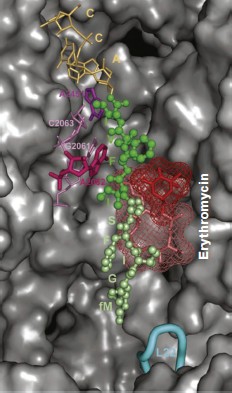
Biochemical studies, in particular chemical probing, confirmed the data of mutagenesis [62, 64, 71, 73-75]. All macrolides protect A2058 and A2059 residues of the central part of domain V from chemical modification. Tylosin, desmycosin, carbomycin, and spiramycin also prevent modification of the A2062 residue [30]. Besides, according to data of chemical probing, A2572 and U2609 also interact with antibiotics in the RT. Macrolides having two sugar residues in position 5 and a carbohydrate residue in position 23 of the lactone cycle (tylosin, carbomycin, and spiramycin) bind to nucleotides U2506 and A752 of hairpin 35 of domain II. Figure 4 shows the organization in the 23S rRNA spatial and secondary structures of its nucleotide residues involved in MBS formation.
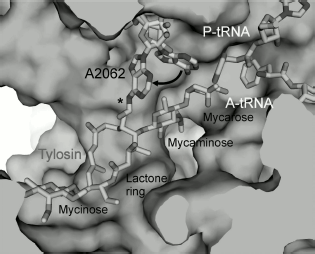
The detailed pattern of macrolide binding (carbomycin A, spiramycin, tylosin, azithromycin, erythromycin, etc.) to the RT was obtained using X-ray analysis with crystals of their complexes with large ribosomal subunit of Haloarcula marismortui and Deinococcus radiodurans [59, 60]. Although on the whole the X-ray data agree with earlier results of biochemical and genetic experiments, only they made it possible to detect the exact arrangement of lactone rings and carbohydrate residues of antibiotics in the RT [76, 77]. The X-ray analysis has also shown that in the case of the tylosin group of macrolides there is reversible formation of a covalent bond between the antibiotic aldehyde group in the lactone ring position C6 and N6 amino group of the 23S rRNA residue A2062, after which the lactone ring fills the RT clearance (Fig. 5). It became clear that macrolide binding in the RT is mainly due to hydrogen bonds and hydrophobic interactions. Carbohydrate residues play an important role in formation of contacts between the antibiotic molecule and the RT walls, their fraction being 50-60% of the total surface of the molecule involved in the interaction. Accordingly, the more sugar residues bound to lactone ring, the stronger is the interaction between the antibiotic molecule and the RT walls, and the higher is its antibiotic activity. Each carbohydrate residue forms at least one hydrogen bond with rRNA nucleotides. Thus, the OH group of a desosamine residue (14-15-membered macrolides) and mycaminose (16-membered macrolides) (O2A) forms a hydrogen bond with N1 of A2058 (Fig. 4c). Base pair C2611 and G2057 can also be involved in hydrogen bonding with desosamine [59] or (Fig. 4b) stabilize hydrophobic interactions of the lactone [60]. Besides, a desosamine residue is potentially able to interact with the oxygen atom from a phosphate residue of G2505. Mycarose establishes hydrogen bond with the G2505 residue. The tylosin mycinose also interacts with amino acid residues of ribosomal protein L22, whereas furosamine of spiramycin interacts with the L4 protein. As for lactone rings of macrolides, they have such conformation in the RT that nonpolar substituents are positioned at the same side of the ring plane, forming a nonpolar surface; polar substituents are turned to the opposite side, thus forming the hydrophilic surface of the lactone. It also became clear from data obtained in this cycle of works that in the region of macrolide binding a hydrophobic surface is formed on the RT walls because the aromatic ring of the C2611 residue protrudes into the RT clearance, while the A2058 residue is involved in stacking interactions with G2057. There is such conformation of this 23S rRNA region that the heterocyclic bases in nucleotides A2059 and A2058 cannot be parallel [78]. A hydrophobic groove is formed in the RT wall with which the lactone ring of macrolides interacts in positions C4-C7 [78]. According to X-ray data, stacking interactions should also be established between reoriented A2062 and the hydrophobic surface of the lactone.Fig. 4. Macrolide antibiotics in ribosomal tunnel. a) Tylosin disposition relative to most important 23S rRNA nucleotide residues forming the macrolide binding site. b) Position of 23S rRNA nucleotide residues forming the macrolide binding site in secondary structure of domains II and V. Numbering as in H. marismortui 23S rRNA (numbers of corresponding nucleotide residues in E. coli 23S rRNA are shown in parentheses). c) Arrangement of erythromycin molecule relative to neighboring H. marismortui 23S rRNA nucleotide residues (numbers of corresponding nucleotide residues in E. coli 23S rRNA are shown in parentheses). The model is based on X-ray data (PDB index 1YI2).
It is discussed in the literature whether macrolides really completely block the RT passage, thus stopping the movement of the growing polypeptide chain, or macrolide in the tunnel is only an obstacle resulting in tunnel narrowing in the MBS region, through the remained space of which the growing polypeptide chain is still able to squeeze. Data published in [79, 80] indicate that the ability of macrolides to block passage of growing polypeptide chain along the tunnel is not absolute. However, in some cases antibiotics still hinder the movement of the growing polypeptide, thus activating different mechanisms of protein translation arrest (see section “Involvement of Ribosomal Tunnel in Regulation of Gene Expression”) and it is especially important that in this way they activate dissociation of peptidyl-tRNA complex from the ribosome [80].Fig. 5. Detailed model of arrangement of tylosin in the RT. The asterisk indicates the covalent bond formed by the tylosin aldehyde group and the amino group of A2062. The arrow points to transition of A2062 from “closed” to “open” conformation accompanying the formation of this bond. Adapted with permission from [91].
The X-ray analysis of ribosome complexes with macrolides elucidated in many respects the molecular reasons for emergence in bacteria of resistance to these antibiotics. It was mentioned previously that one of the reasons of bacterial resistance to antibiotics is the result of methylation of 23S rRNA (most often mono- and dimethylation of the N6 amino group of A2508 nucleotide as well as methylation of different purine nucleotides of domains II and V of 23S rRNA). About 20 erm (erythromycin ribosome methylation) genes encoding methyl transferase are known that are associated with transposons and can be localized both on plasmids and chromosomes. These methylases are widespread among aerobic and anaerobic Gram positive and Gram negative bacteria [70, 81, 82]. Methylation of macrolide targets defines the high level of their resistance to these antibiotics (see, for example, [83]). Mono- and dimethylation of A2058 and G748 produces resistance to actually all macrolides because these residues are involved in formation of opposite regions of the RT walls, and simultaneous modifications at both residues disturb formation of hydrogen bonds between nucleotides and the antibiotic molecule [65, 84]. Lowering the sensitivity to macrolides in strains S. pneumoniae, S. pyogenes, and S. oralis also causes mutations in genes of ribosomal proteins L4 and L22. Most often C substitutes A2062, and less common are replacements by other nucleotides without a free amino group which is necessary for antibiotic binding. In particular, in the case of tylosin group macrolides this results in the loss of ability to form a covalent bond at the aldehyde group. Replacement of the A2058 residue by G [85] makes less hydrophobic the contact surface of the RT nucleotides in the macrolide-binding site, so that interaction with the hydrophobic surface of the lactone ring is distorted.
Although there are still no answers to many questions concerning mechanisms of translation inhibition by macrolides [86], elucidation of structure of their complexes with the RT has already brought appreciable practical results: it became possible to design on the basis of this knowledge principally new inhibitors of bacterial protein synthesis, able to overcome the resistance of pathogens to antibiotics presently used in clinics (see review [58]). Data from these works are also extremely important for identification of the RT regions and particular functional rRNA and protein groups able to interact with growing peptide via modulation and stabilization of its conformation within the tunnel and its involvement in translation arrest. Here we should first of all distinguish practically all nucleotide residues of rRNA of the large ribosomal subunit domain V, covering the walls of the upper third of the RT; nucleotide sequences of the loop of helix 35 (domain II), localized near the narrowing of the RT, and a number of amino acid residues of β-loop apexes of proteins L4 and L22 forming this narrowing. Unfortunately, so far only one X-ray study has been published for a complex of ribosome with antibiotic (negamycin) that binds to the RT region localized not far from the tunnel outlet [87]. In principle, this is of great interest because negamycin can be considered as an analog of growing peptide chain. However, this complex is still studied at insufficiently high resolution.
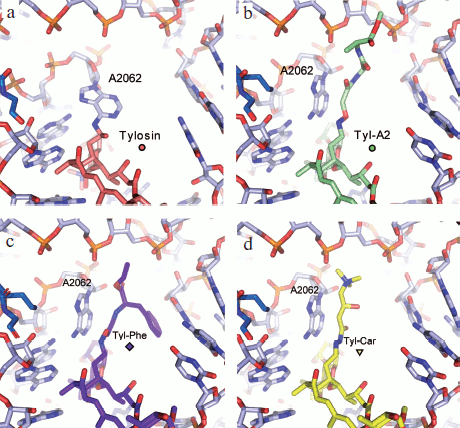
Based on information from X-ray analysis of antibiotic complexes with ribosome, we synthesized amino acid and peptide macrolide derivatives that appeared to be useful tools for modeling the behavior of a growing peptide within the RT. The main idea of these investigations was to use the tylosin group macrolides as “anchoring” molecules carrying amino acids or short peptides (Fig. 6) [88]. In this case the lactone rings and carbohydrate residues of the antibiotics were modified so that the amino acid or peptide could directly contact potential sites of interaction of the growing peptide with the RT [89, 90]. Binding of these compounds to ribosome and their ability to inhibit protein synthesis in a cell-free system were studied. Besides, the structure of the alanylalanine derivative of tylosin at position C20 was studied by X-ray analysis (Fig. 7b; see color insert) [91]. It was found that the RT cavity formed upon transition of nucleotide residue A2062 from open conformation to the closed one (for more details of this conformational transition see next section) can interact with hydrophobic (Fig. 7c) and positively charged (Fig. 7d) residues of the growing protein chain. It was also shown that the ensemble formed by the A752 residue and its environment, on one side, and the Arg92 residue of L22 protein, on the other, can serve as the binding site for side chains of aromatic amino acids of the growing chain (A. V. Shishkina, unpublished data).
Fig. 6. Chemical formula of tylosin. Arrows point to positions of tylosin modification by amino acid and peptide moieties (see details in text).
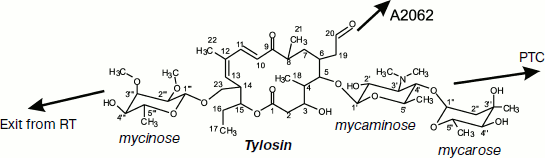
Fig. 7. Molecular models of tylosin and its derivatives at C20 in the RT “adenine pocket”) [91]. a) Tylosin covalently bound to A2062. Model based on X-ray data (PDB index 1K9M). b) C20-alanylalanyl–tylosin. Model based on X-ray data [57, 91]. c) C20-phenylalanyl–tylosin. The aromatic ring of phenylalanine is involved in stacking interaction with C2586. d) C20-carnityl–tylosin. The positively charged amino acid residue forms an ion pair with a phosphate group of sugar-phosphate backbone of 23S rRNA.
INVOLVEMENT OF THE RIBOSOMAL TUNNEL IN REGULATION OF GENE EXPRESSION
As mentioned above, the RT should provide for the transfer of the growing protein chain in the ribosome body. In early works there even emerged a not so fortunate image of the RT as a “passive Teflon-like tunnel”. However, before it was proved that the RT is the guide for the ribosome-synthesized protein chains, it was known that in a number of special cases the newly formed polypeptide chain has strong interactions with the ribosome that results in ribosome stalling [92-94]. Later it was found that translation arrest is the basis of mechanisms of expression regulation of some genes, including those responsible for secretion [95], amino acid metabolism [96], and antibiotic resistance [97]. Usually translation arrest occurs at the stage of elongation or termination and covers only ribosomes already involved in protein synthesis. As a rule, only so-called leader polypeptides encoded in the 5′-terminal operon region act as “stop peptides”. Accordingly, translation arrest in the RT regulates expression of genes localized downstream from the leader sequence.
In genes whose expression is regulated via translation arrest, there are encoded mRNA exhibiting unique secondary structure that is rearranged in the way required by the classical mechanism of transcription attenuation. In each case the ribosome binding site for the sequence encoding drug resistance is localized within one of two inverse repetitive sequences. This means that in transcripts this site will be included in a stable hairpin structure that prevents translation. The leader sequence encoding a short regulatory peptide is localized upstream from this structure [92].
In most known cases “stop peptides” inhibit translation in combination with specific low molecular weight coeffectors. Thus, the level of synthesis of fungal carbamoyl phosphate synthetase is regulated by a short “stop peptide” encoded in the 5′-terminal region of its gene. In this case, translation arrest and decrease of the enzyme synthesis level occurs upon increase in intracellular arginine concentration [98]. A similar situation is observed for animal S-adenosylmethionine decarboxylase. In this case spermidine acts as the effector [99]. Below we shall describe the most important examples of regulation of expression of genetic information with direct involvement of the RT, and we shall consider the possible nature of recognition of the growing regulatory polypeptide amino acid sequence.
Regulation of cat and clmA Gene Expression
Discovery of the fact that in eubacteria two inducible genes of chloramphenicol resistance are regulated by the classical mechanism of transcription attenuation were among first indications that expression of some genes is controlled by specific peptides. Lovett et al. showed that the main step of transcription attenuation, temporary ribosome stalling, is caused by the inhibition of peptidyl transferase activity by short peptides (5-8 amino acid residues) that are N-terminal parts of polypeptides encoded in the leading sequence of controlled genes. Short peptides influence PTC activity in cooperation with inducer chloramphenicol and are active exclusively in cis [92, 100]. Inhibition of peptidyl transferase activity depends on the primary structure of the peptide. Based on these facts, the authors supposed that cis-acting regulatory peptides should specifically bind to some PTC-associated ribosomal element. Since the growing peptides studied by the authors competed for an erythromycin binding site [101], there is no doubt that their target was localized in the RT.
It is known that genes cat and clmA are responsible for inducible chloramphenicol resistance in Gram-positive bacteria. In both systems the ribosome-binding site is masked in the RNA secondary structure, and initiation of translation is practically impossible. Chloramphenicol induces cat and clmA expression by ribosome retardation in a particular site of translated leading sequence preceding this secondary structure element.
In the case of cat genes, noticeable induction occurs only when the ribosome is suspended at the sixth codon of the leading sequence. In the case of clmA translation activation occurs due to suspension of the ribosome at the ninth codon. Chloramphenicol, an inducer of cat and clmA gene expression, inhibits translation elongation at randomly chosen sites and is not able to provide for site-specific ribosome retardation. Specificity of translation arrest is then provided by the leading sequence codons preceding the ribosome stopping site and encoding a pentapeptide in the case of the cat gene and an octapeptide in the case of clmA, inhibiting PTC activity [102].
Regulation of ermC Gene Expression
The Erm type methyl transferase is encoded by the ermC gene. This enzyme methylates amino group of A2058 in 23S rRNA, which, as shown above, results in the resistance of the ribosome to several classes of antibiotics including macrolides [103]. Expression of this gene in Staphylococcus aureus is regulated by a “stop peptide” encoded in the leader sequence (19 codons) located at the distance of 65 nucleotide residues ahead of the beginning of ermC. In the absence of antibiotics constitutive translation of leader sequence occurs. In this case, mRNA of the ErmC protein is not translated because the ribosome-binding site is shielded in its secondary structure. In the presence of antibiotic the synthesis of leader peptide stops at the ninth codon, and the mRNA secondary structure rearranges in such a way that the site of ErmC protein translation initiation becomes accessible for the ribosome and its synthesis proceeds freely [104].
The molecular mechanism of induction of ermC gene expression by the antibiotic erythromycin was studied in detail by Mankin et al. [97]. They showed that upon ribosome arrest, 9-membered “stop peptide” is bound to tRNA localized in the ribosome P-site; the C-terminal tetrapeptide sequence IFVI of the “stop peptide” is critical for efficient induction, and replacement of Ile9, localized in PTC, even by the structurally similar Val residue eliminates it; the arrested ribosome contains both “stop peptide” and erythromycin; nucleotide residue A2062 is critical for the induction process: in mutants with its replacement by C or U practically no ermC induction by erythromycin is observed; and finally, combined arrangement of the “stop peptide” and erythromycin in the RT results in inactivation of the PTC (although erythromycin itself does not inhibit the peptidyl transferase reaction) [97]. The supposed structure of the complex is shown in Fig. 8 (see color insert). The authors suggested that such complex formation is accompanied by transition of A2062 from “open” conformation (when the heterocyclic base “looks” out at the RT open space) into the “close” one (when the adenine ring is oriented along the RT wall). A similar conformational transition was observed upon binding in the RT of amino acid and peptide tylosin derivatives (see section “Interaction of the Ribosomal Tunnel with Antibiotics”) [91]. Mankin et al. supposed that the change in A2062 conformation modulates structural transformations in the ribosome PTC, resulting in inactivation of the latter [97].
Actually, in the crystalline structure of the 50S ribosomal subunit not bound to substrates the triad of nucleotide residues G2061-A2062-C2063 forms a very unusual structure: G2061 is practically intercalated between A2062 and C2063; G2061 and C2063, flanking A2062, are focused directly on the active PTC site. Residue G2061 forms a hydrogen bond with A2451, one of key elements of the PTC. Residue C2063 is also hydrogen-bonded with PTC elements, namely, with A2450 [12]. Mutations of any of these residues (G2061, C2063, A2450, or A2451) are critical for ribosome functioning, making it absolutely inactive [105]. Thus, residue A2062 is bound to the PTC active site via a system of internucleotide interactions; therefore, movement of this residue probably really results in structural changes in the PTC that directly affects ribosome functioning. It is quite probable that residue A2062 is involved in the growing peptide chain monitoring and signal transduction from the RT to the PTC resulting in change in translation level [97].Fig. 8. Interaction of erythromycin with the ermC “stop peptide”. Computer model. The C-terminal model of ermC that is absolutely necessary for translation arrest is shown in dark-green. Adapted from [79] with permission.
SecM “Stop Peptide” and Regulation of Expression of Gene secA
Protein SecA plays an important role in E. coli protein secretion and transport through membranes [94, 106]. Expression of the secA gene in E. coli is regulated by its leader region that contains an open reading frame (ORF) encoding protein secM (170 codons). When the ribosome-binding site on leader mRNA is available, a similar site on the secA region is involved in formation of mRNA secondary structure, which prevents initiation of SecA synthesis. Translation initiation and polymerization of the first 165 amino acids proceed until Gly-tRNA, corresponding to codon Gly165, at which the ribosome stops enters the P-site. The SecM polypeptide chain contains a signal sequence at the N terminus, and in the case of normal work of intracellular protein synthesis apparatus the N-terminal part of the SecM peptide will be removed from the tunnel, which will result in the release of the ribosome from arrested state using SRP. In this case no activation of gene secA expression is observed. The key moment in the work of this system is that ribosome arrest takes place upon SecA deficiency in the cell, i.e. under conditions of disturbed protein secretion. The ribosome stops at secM codon 165, that results in rearrangement of mRNA secondary structure, and another ribosome translates part of mRNA in which SecA protein is encoded [94].
Nakatogawa and Ito showed using mutation analysis [106] that ribosome stalling at the secM ORF is caused by the sequence of C-terminal region of the growing polypeptide F150XXXXWIXXXXGIRAGP166, where X means positions in polypeptide chains which can be replaced by alanines without disturbance of SecM “stop function” [94]. Critical for stopping are residues Phe150, Trp155, and Ile156 along with six C-terminal residues GIRAGP166. The distance between these important residues is also essential [107]. The fact that translation arrest is due to entry into the RT of just the above-mentioned sequence was proved by incorporation of encoding segments of the secM gene into genes of different proteins, which resulted in significant decrease in their expression level [104]. As already mentioned, in the arrested complex the growing peptide is joined to tRNAGly localized in the ribosome P-site [104]. It is necessary to note that a proline residue delivered by aminoacyl-tRNA into the A-site in accordance with codon 166 is also absolutely necessary for translation arrest. In other words, in this system Pro-tRNA plays the role of effector of translation arrest. Simultaneous presence of peptidyl-tRNA in the P-site and of aminoacyl-tRNA in the A-site of arrested ribosome indicates that translation arrest is caused by the inability of the ribosome to catalyze formation of a peptide bond between Gly165 and Pro166. At least one reason of this event is that the proline residue (due to its unique structure among protein amino acids and low nucleophilicity) is a bad substrate both for ribosome A- and P-sites). In the case of A-site this can be illustrated by the fact that the proline-acetylated phenylalanine-tRNA reacts with fMet-tRNA at a rate several orders of magnitude lower than for Phe-tRNA [108]. In the case of the P-site it is known that peptidyl-tRNA located in this site, with a peptide residue carrying proline at the C terminus, poorly reacts with puromycin [109, 110].
A special role of a proline residue located at the C-terminus of the growing peptide, which due to its arrangement in the RT causes translation arrest, was studied sufficiently comprehensively for YbeL protein that is also able to cause translation arrest [111]. Systemic mutation analysis of the YbeL C-terminal part showed that replacement of the C-terminal Pro by different types of amino acids results in normal translation termination. The amino acid residue in the next to C-terminal proline position (–2) appears to be also important for transfer-messenger RNA (tmRNA) binding efficiency. For example, substitution of Arg, Asn, Leu, Trp, Cys, His, Thr, or Met for Glu (–2) in the wild-type YbeL protein greatly increases the translation arrest, and the efficiency of translation arrest is also influenced by some substitutions in –3 position. Thus, the presence of Pro at the C-terminus of the growing peptide is necessary but not enough for inhibition for peptidyl-tRNA cleavage. Some amino acid residues of the growing peptide located inside the RT also contribute to this process [112].
It is interesting that although no noticeable similarity is yet found in amino acid sequence of presently known “stop peptides”, their C-terminus is most often occupied by proline. This has been especially clearly shown recently by Buskirk et al. [113] who elaborated a clever scheme for “stop peptide” sequence screening in a peptide bank containing a huge set of short peptides. Their scheme for selection of such peptides is based on use of a natural bacterial cell system and its key component, tmRNA, with the help of which the latter overcomes translation arrest and eliminates unfinished (defective) proteins [114]. The tmRNA template region was changed so that it provided for completion of a protein that defined the bacterial cell survival in the presence of the antibiotic kanamycin. The peptide library was incorporated into the C terminus of the unfinished protein. Just library components that provided for translation arrest made it finally possible to select kanamycin-resistant cells. The authors identified three classes of “stop peptides”: 1) with diverse sequences but with obligatory C-terminal proline; 2) similar to SecM C-terminal region; 3) a new type of peptide with FxxYxIWPP sequence, where x is any amino acid residue [113].
There is one more factor important for efficient translation arrest by the stop peptide of SecM protein; it is conformation of the latter within the RT. When a ribosome synthesizes the SecM sequence necessary for translation arrest, some of its regions already during synthesis acquire conformation more compact than completely unfolded (in some parts it may be α-helical) [115]. This fact was detected by Johnson et al. using FRET. They showed that the absence of such compaction in the “stop sequence” C-terminal region, directly adjacent to the PTC (the extent of compaction can be regulated by changing certain amino acid residues in SecM) makes translation arrest less efficient or completely eliminates it [115]. They suggest that the special conformation is a required spatial co-location of key SecM amino acid residues with RT sensor elements.
Recently Yap and Bernstein performed detailed genetic analysis of SecM and obtained convincing confirmation of this supposition [116]. Studying mutations in SecM that suppress its ability to arrest translation and comparing amino acid sequences of this protein encoded in genomes of numerous different eubacteria, they concluded that only R163 and Pro166 residues are absolutely necessary for arrest of elongation of this protein. Different amino acid residues revealed previously in SecM [94, 107] important for fulfilling this function create such conformation of its polypeptide chain in RT that is necessary for fixing R163 opposite its specific site on the tunnel wall. They do not discuss the question of which nucleotide site of 23S rRNA this could be.
In this connection the following should be noted. In our previous analysis of atomic structure of complexes of Haloarcula marismortui 50S ribosome subunit with analogs of aminoacyl- and peptidyl-tRNA [117, 118] we paid attention, in particular, to the fact that the oxygen atom in the carbonyl group of the peptide bond between the C-terminal and next to it amino acid residues in posttranslocational complex (analog of peptidyl-tRNA is localized in P-site) forms a hydrogen bond with exocyclic group of G2061 [119]. It was supposed that this bond stabilizes the initial conformation of the growing polypeptide chain in the RT. If this supposition is correct, then in the case of translation arrest using SecM, G2061 will interact with the peptide backbone of the Ala164-Gly165 sequence. In this case it is quite probable that positively charged side chain of key residue R163 will occupy the place in the “adenine pocket” formed by A2062 and its environment. Formation of a similar complex was observed in the case of a carnitine derivative of tylosin (see section “Interaction of Ribosomal Tunnel with Antibiotics” and Fig. 7d). Another possibility is formation of the so-called cation–π complex (see in more detail about this type of complexes in the next section) between a positively charged arginine group and the heterocyclic base in A2062. The fact that mutations at A2062 deprive SecM of the ability to arrest translation favors both suppositions (N. Vazquez-Laslop and A. S. Mankin, personal communication). However, the choice between the two alternative mechanisms is evidently possible only using X-ray analysis of 50S ribosomal subunit complex with SecM in the ribosomal tunnel.
Participation of Leader Peptide TnaC in Regulation of the Tryptophanase Operon
Many bacteria are able to cleave tryptophan and use its degradation products for their own vital activity. For example, in E. coli indol formed from tryptophan serves as a signal component in the so-called “quorum sensing” processes, biofilm formation, and regulation of expression of some genes [120-122]. Tryptophanase is the main enzyme of tryptophan catabolism in Gram-negative bacteria; it cleaves tryptophan to indol, pyruvate, and ammonia, making tryptophan an intracellular source of carbon and nitrogen [123]. The tryptophanase (tna) operon of E. coli consists of transcribed leading regulatory sequence (320 bp) followed by two structural genes tnaA and tnaB. A leader sequence encodes a 24-membered regulatory “stop peptide” TnaC; tnaA encodes tryptophanase, and tnaB encodes tryptophan permease. Transcription of structural genes begins in the case of transcription termination with involvement of Rho factor at the transcription arrest sites positioned immediately after tnaC. The Rho factor effect is prevented by high tryptophan concentrations. Induction by tryptophan requires synthesis of TnaC containing a necessary tryptophan residue in position 12 [124]. It was shown that induction is possible due to inhibition by tryptophan of RF2 termination factor activity at stop codon of tnaC [125].
Three regions essential for its expression regulation by free tryptophan are distinguished in leader transcript of the tna operon: the TnaC encoding sequence, binding site of Rho termination factor, and RNA sites necessary for Rho-dependent transcription termination. These sites precede the two structural genes of the operon—tnaA and tnaB. It is supposed that leader peptide TnaC regulates Rho activity as follows: during translation it transduces to the ribosome the signal for tryptophan binding, which results in inhibition of TnaC-tRNAPro splitting off, which in turn causes ribosome arrest at the tnaC stop codon. Such arrested ribosome blocks the Rho factor binding site (rut-site) and thus prevents Rho factor binding to transcript and transcription termination. When it is impossible for Rho to exhibit its activity, RNA polymerase, stopped next to the leader region, restores the transcription process by displacement onto the operon structural genes [126].
If intracellular tryptophan concentration is below that necessary for induction, the ribosome completes the TnaC peptide synthesis, TnaC-peptidyl-tRNA is cleaved, and the ribosome is released. As a result, the rut-site in leader RNA is able to bind Rho factor, which in this case forms a contact with RNA polymerase stopped at the RNA leader region, and causes transcription termination. The regulatory region of the E. coli tna operon is rather similar to regulatory regions of this operon in some other bacterial species [126]. When cells are grown in a medium deprived of catabolite repressor, CAP protein is inactivated and initiation of tna operon transcription occurs [127].
For a long time, investigations in the laboratory of Yanofsky were aimed at answering the question of how RT walls recognize the Trp12 residue of the growing chain and how this recognition results in formation of a free tryptophan binding site at the ribosome. The approximate arrangement of the most important TnaC-tRNAPro Trp12 in the 50S ribosome subunit was determined in experiments showing that Lys11 in TnaC-tRNAPro can be covalently cross-linked to A750 of 23S rRNA, and that Trp12 in TnaC-tRNAPro “protects” against methylation of A788 base in 23S rRNA [127]. Also, it was found that some nucleotides of 23S rRNA and one residue of L22 ribosomal protein are necessary for induction by tryptophan and are involved in formation of the regulatory region in the ribosomal tunnel, the Trp12 binding site in TnaC-tRNAPro. Supposedly, these important residues are U2609 and A752 in 23S rRNA and K90 of L22 protein. They concluded that the site for free tryptophan binding is localized in the A-site region of PTC, and this interaction inhibits hydrolysis of the aminoacyl bond in TnaC-tRNAPro catalyzed by termination factor [124]. It was found in experiments with the wild-type ribosomes containing TnaC-tRNAPro that free tryptophan inhibits puromycin and sparsomycin binding in the A-site. However, in the presence of mutations at one of the three above-mentioned essential 23S rRNA bases, free tryptophan inhibits neither the puromycin reaction nor sparsomycin binding [128]. The above-mentioned facts suggest that after binding to the PTC A-site, free tryptophan induces structural alterations in this functional center of the ribosome [129].
The tryptophan residue in TnaC position 12 is absolutely necessary for induction of the tna operon by free tryptophan. No induction takes place upon replacement of Trp12 by arginine, removal of Trp12 from peptide chain by deletion of the corresponding codon, or by incorporation of amino acid residues between Trp12 and the C-terminal Pro (Pro24). However, replacement of some amino acid residues between Trp12 and Pro24 and deletions and insertions in the N-terminal region of TnaC have practically no effect on induction level. Pro24 is also absolutely necessary, first, for TnaC functioning as “stop peptide”, and second, so that free tryptophan can induce the tryptophanase operon [130].
So, this indicates that bacterial ribosomes contain a specific site for interaction with a tryptophan residue of the growing peptide. The distance from this site to the end of the tunnel should be 35-40 Å, because Trp12 is the 13th amino acid residue from the C terminus of TnaC peptide. So it was supposed that the tryptophan binding site is localized in the middle part of the RT, in the region of its narrowing. It was also supposed that Trp12 binding to its specific center on the RT wall serves as a signal for formation of an inducer (free tryptophan) binding site in the PTC A-site region and that inducer binding to this site causes changes in PTC preventing cleavage of TnaC-peptidyl-tRNAPro [130, 131].
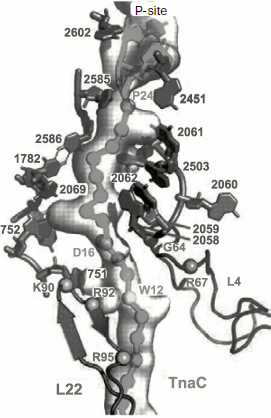
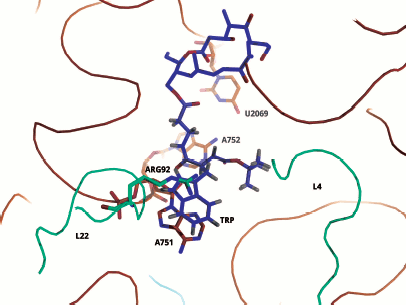
Most of these suppositions were confirmed when it became possible to study the E. coli ribosome complex with TnaC by high resolution cryo-EM [46]. It appeared that this “stop peptide” exists in the RT in unfolded but quite definite conformation because it forms numerous contacts with the tunnel walls. In this case Trp12 is really localized in the narrowed part of the tunnel and opposite to the tunnel wall there is Arg92 residue of L22 protein, while nucleotide residue A751 is near it (Fig. 9). We used results of inhibition of the cell-free translation system by the above-described peptide derivatives of macrolides (including tryptophan-containing ones) and computer modeling, and we found that the aromatic ring of a tryptophan residue in TnaC can intercalate between the positively charged side group of Arg92 and adenine residue A751 (A. V. Golovin and A. A. Bogdanov, unpublished data) (Fig. 10; see color insert). This complex is stabilized both by well-known hydrophobic stacking interactions and by cation–π interaction of Arg with Trp. The last type of intermolecular interactions in proteins and their complexes with nucleic acids has been intensely studied in recent times (see, for example, [132]). The possibility of contact of Arg92 with Trp12 in TnaC due to cation–π interactions has been recently demonstrated by Trabuco et al. [133] by quantum-mechanical calculations (molecular dynamics). They also described another important contact between TnaC and the RT walls, namely ion pair formation between the K90 residue in L22 protein and Asp16 in TnaC. The high probability of such contact is based not only on biochemical and genetic data [130] but also on the fact that the last amino acid residue is completely conservative in all bacterial TnaC analogs [126].
Fig. 9. The TnaC leader peptide in the E. coli ribosomal tunnel. The model is based on the high-resolution cryo-EM data [46]. Circles indicate amino acid residues playing the key role in translation arrest. Numbers of nucleotide and amino acid residues whose mutations disturb translation arrest are shown in bold.
It is necessary to note that cryo-EM analysis of TnaC complex with ribosome gave an additional very important result: it was found that the binding of this “stop peptide” to RT causes major conformational changes exactly in the part of the PTC responsible for translation termination [46].Fig. 10. Computer model illustrating possible binding of Trp12 residue of the TnaC leader peptide to the tryptophan binding center of the ribosome 50S subunit. For model plotting, the tylosin group macrolide derivative (OMT) was used in which the tryptophan residue was bound to the OMT lactone ring by the corresponding length “bridge”. The lactone ring position in the RT exactly corresponds to that in the tylosin complex with 50S subunit of H. marismortui (PDB index 1K9M). It is seen that the tryptophan aromatic ring is intercalated between a positively charged arginine group and the adenine ring in A751. The model was designed by A. V. Golovin and is provided through his courtesy.
In conclusion of this section, it is impossible to ignore in comparison of SecM and TnaC binding to RT that, despite absence of similarity in their amino acid sequences, mechanisms of translation arrest by these peptides are based on the same principles. First, a proline residue should be present in the PTC A- or P-site. Second, only one or two strong contacts with the RT walls, depending on the peptide secondary structure, are enough for arrest of the polypeptide chain movement along the RT. Third, information on these contacts should be somehow delivered into the PTC where the effector (inducer) binding site is formed.
From the intensive studies of the past decade, the ribosomal tunnel was transformed from a sufficiently illusory structural element of the ribosome into an object of intense attention of molecular biologists, although not everybody believed in its existence. Investigation of its complexes with antibiotics significantly accelerated the creation of new generations of antibacterial preparations. Analysis of RT complexes with peptides capable of translation arrest resulted in discovery of new regulatory mechanisms of expression of genetic information. At the same time, it is obvious that the answer to the main question of how the growing polypeptide chain overcomes such a long (within protein molecule dimensions) path remains unanswered. In fact, “stop peptides” that received much attention in this article are only an exception from the rule. The large majority of protein polypeptide chains synthesized by the ribosome should freely travel through the ribosomal tunnel. But what is the driving force of their transfer along the RT? No doubt this is one of main problems of the dynamics of ribosome functioning without resolution of which there will remain a serious gap in our concepts of mechanisms of protein biosynthesis.
The authors are grateful to A. S. Mankin and A. V. Golovin for presented unpublished materials.
This work was supported by the Russian Foundation for Basic Research (grants 04-04-49480-a, 07-04-00902-a, 10-04-01187-a, 09-04-12064-ofi_m) and grants of President of Russian Federation for State Support of Leading Scientific Schools NSh-1284.2008.4 and NSh-3314.2010.4.
REFERENCES
1.Malkin, L. I., and Rich, A. (1967) J. Mol.
Biol., 26, 329-346.
2.Blobel, G., and Sabatini, D. D. (1970) J. Cell.
Biol., 45, 130-145.
3.Bernabeu, C., and Lake, J. A. (1982) Proc. Natl.
Acad. Sci. USA, 79, 3111-3115.
4.Bernabeu, C., Tobin, E., Fowier, A., Zabin, I., and Lake, J. A.
(1983) J. Cell. Biol., 96, 1471-1474.
5.Milligan, R. A., and Unwin, P. N. T. (1986) Nature,
319, 693-695.
6.Yonath, A., Leonard, K. R., and Wittmann, H. G.
(1987) Science, 236, 813-816.
7.Ryabova, L. A., Selivanova, O. M., Baranov, V. I.,
Vasiliev, V. D., and Spirin, A. S. (1988) FEBS Lett.,
226, 255-260.
8.Wang, S., Sakai, H., and Wiedmann, M. (1995) J.
Cell. Biol., 130, 519-528.
9.Frank, J., Zhu, J., Penczek, P., Li, Y.,
Srivastava, S., Verschoor, A., Radermacher, M., Grassucci, R., Lata, R.
K., and Agrawal, R. K. (1995) Nature, 376, 441-444.
10.Choi, K. M., and Brimacombe, R. (1998) Nucleic
Acid Res., 15, 887-895.
11.Mueller, F., Sommer, I., Baranov, P., Matadeen,
R., Stoldt, M., Woehnert, J., Goerlach, M., van Heel, M., and
Brimacombe, R. (2000) J. Mol. Biol., 298, 35-59.
12.Ban, N., Nissen, P., Hansen, J., Moore, P. B.,
and Steitz, T. A. (2000) Science, 289, 905-920.
13.Nissen, P., Hansen, J., Ban, N., Moore, P. B.,
and Steitz, T. A. (2000) Science, 289, 920-930.
14.Harms, J., Schluenzen, F., Zarivach, R., Bashan,
A., Gat, S., Agmon, I., Bartels, H., Franceschi, F., and Yonath, A.
(2001) Cell, 107, 679-688.
15.Brimacombe, R. (2000) Structure, 8,
R195-R200.
16.Sergiev, P., Leonov, A., Dokudovskaya, S.,
Shpanchenko, O., Dontsova, O., Bogdanov, A., Rinke-Appel, J., Mueller,
F., Osswald, M., von Knoblauch, K., and Brimacombe, R. (2001) Cold
Spring Harb. Symp. Quant. Biol., 66, 87-100.
17.Sergiev, P. V., Dontsova, O. V., and Bogdanov, A.
A. (2001) Mol. Biol. (Moscow), 35, 559-583.
18.Schmeing, T. M., and Ramakrishnan, V. (2009)
Nature, 461, 1234-1242.
19.Hardesty, B., and Kramer, G. (2001) Progr.
Nucleic Acid Res. Mol. Biol., 66, 41-66.
20.Ramu, H., Mankin, A., and Vazquez-Laslop, N.
(2008) Mol. Microbiol., 71, 811-824.
21.Reusch, R. N., and Sadoff, H. L. (1988) Proc.
Natl. Acad. Sci. USA, 85, 4176-4180.
22.Reusch, R. N. (2000) Biochemistry
(Moscow), 65, 280-295.
23.Rospert, S. (2004) Curr. Biol., 14,
R386-R388.
24.Mears, J. A., Cannone, J. J., Stegg, S. M.,
Guttell, R. R., Agrawal, R. K., and Harvey, S. C. (2002) J. Mol.
Biol., 321, 215-234.
25.Sharma, M. R., Booth, T. M., Simpson, L., Maslov,
D. A., and Agrawal, R. K. (2009) Proc. Natl. Acad. Sci. USA,
106, 9637-9642.
26.Kovalskaya, O. M., Sergiev, P. V., Bogdanov, A.
A., and Dontsova, O. A. (2007) Uspekhi Biol. Khim., 47,
129-188.
27.Kramer, G., Boehringer, D., Ban, N., and Bukau,
B. (2009) Nat. Struct. Mol. Biol., 16, 589-597.
28.Giglione, C., Filulaine, S., and Meinnel, T.
(2009) Trends Biochem. Sci., 34, 417-426.
29.Lu, J., Kobertz, W. R., and Deutsch, C. (2007)
J. Mol. Biol., 371, 1378-1391.
30.Lu, J., and Deutsch, C. (2005) Nat. Struct. Biol.,
12, 1123-1129.
31.Lu, J., and Deutsch, C. (2008) J. Mol.
Biol., 384, 73-86.
32.Voss, N. R., Gerstein, M., Steitz, T. M., and
Moore, P. B. (2006) J. Mol. Biol., 360, 893-906.
33.Steitz, T. A. (2008) Nat. Rev. Mol. Cell
Biol., 9, 242-253.
34.Spirin, A. S. (2009) J. Biol. Chem., 284,
21103-21119.
35.Valle, M., Zavialov, A., Sengupta, J., Rawat, U.,
Ehrenberg, M., and Frank, J. (2003) Cell, 114,
123-134.
36.Schmeing, T. M., Voorhees, R. M., Kelly, A. C.,
Gao, Y. G., Murphy, F. V., 4th, Weir, J. R., and Ramakrishnan, V.
(2009) Science, 326, 688-694.
37.Gao, Y. G., Selmer, M., Dunham, C. M.,
Weixbaumer, A., Kelly, A. C., and Ramakrishnan, V. (2009)
Science, 326, 694-699.
38.Gabashvili, I. S., Grgory, S. T., Valle, M.,
Grassucci, R., Worbs, M., Wahl, M. C., Dahlberg, A. T., and Frank, J.
(2001) Mol. Cell, 8, 181-188.
39.Gilbert, R. J., Fucini, P., Connel, S., Fuller,
S. D., Nierhaus, K. H., Robinson, C. V., Dobson, C. M., and Stuart, D.
I. (2004) Mol. Cell, 14, 57-66.
40.Fulle, S., and Gohlke, H. (2009) J. Mol.
Biol., 387, 502-517.
41.Sergiev, P. V., Bogdanov, A. A., and Dontsova, O.
A. (2005) FEBS Lett., 579, 5439-5442.
42.Kiparisov, S. V., Sergiev, P. V., Bogdanov, A. A., and
Dontsova, O. A. (2006) Mol. Biol. (Moscow), 40,
755-768.
43.Halic, M., Becker, T., Pool, M. R., Spahn, C. M.,
Grassucci, R. A., Frank, J., and Beckmann, R. (2004) Nature,
427, 808-814.
44.Halic, M., Becker, T., Mielke, T., Pool, M. R.,
Wild, K., Sinning, I., and Beckmann, R. (2006) Nature,
444, 507-511.
45.Schaffitzel, C., and Ban, N. (2007) J. Struct.
Biol., 158, 463-471.
46.Seidelt, B., Innis, C. F., Wilson, D. N.,
Gartmann, M., Armache, J.-P., Villa, E., Trabuco, L. G., Becker, T.,
Mielke, T., Schulten, K., Steitz, T. A., and Beckmann, R. (2009)
Science, 326, 1412-1415.
47.Becker, T., Bhushan, S., Jarasch, A., Armache, J.-P., Funes,
S., Jossinet, F., Gumbart, J., Mielke, T., Berninghausen, O., Schulten,
K., Westhof, E., Gilmore, R., Mandon, E. C., and Beckmann, R.
(2009) Science, 326, 1369-1373.
48.Woolhead, C. A., McCormick, P. J., and Johnson,
A. E. (2004) Cell, 116, 725-736.
49.Kosolapov, A., and Deutsch, C. (2009) Nat.
Struct. Biol., 16, 405-411.
50.Tu, L., and Deutsch, C. (2010) J. Mol.
Biol., 396, 1346-1350.
51.Lim, V. I., and Spirin, A. S. (1986) J. Mol.
Biol., 188, 565-577.
52.Bhushan, S., Gartmann, M., Halic, M., Armache,
J.-P., Jarasch, A., Mielke, T., Berninghausen, O., Wilson, D. N., and
Beckmann, R. (2010) Nat. Struct. Biol., 17, 313-317.
53.Marqusee, S., and Baldwin, R. L. (1987) Proc.
Natl. Acad. Sci. USA, 84, 8898-8902.
54.Sicheri, F., and Yang, D. S. (1995)
Nature, 375, 427-431.
55.Barron, L. D., Hecht, L., and Wilson, G. (1997)
Biochemistry, 36, 13143-13147.
56.Mankin, A. (2001) Mol. Biol. (Moscow),
35, 597-609.
57.Wilson, D. N., Harms, J. M., Nierhaus, K. H.,
Schluenzen, F., and Fucini, P. (2005) Biol. Chem., 386,
1239-1252.
58.Wimberly, B. T. (2009) Curr. Opin. Investig.
Drugs, 10, 750-765.
59.Schlunzen, F., Zarivach, R., Harms, J., Bashan,
A., Tocilj, A., Albrecht, R., Yonath, A., and Franceschi, F. (2001)
Nature, 413, 814-821.
60.Hansen, J., Ippolito, J. A., Ban, N., Nissen, P.,
Moore, P. B., and Steitz, T. A. (2002) Mol. Cell, 10,
117-128.
61.Omura, S. (2002) Macrolide Antibiotics:
Chemistry, Biology and Practice, 2nd Edn., Academic
Press.
62.Hansen, L. H., Mauvais, P., and Douthwaite, S.
(1999) Mol. Microbiol., 31, 623-631.
63.Moazed, D., and Noller, H. F. (1987)
Biochemie, 69, 879-884.
64.Poulsen, S. M., Kofoed, C., and Vester, B. (2000)
J. Mol. Biol., 304, 471-481.
65.Liu, M., and Douthwaite, S. (2002) Proc. Natl.
Acad. Sci. USA, 99, 14658-14663.
66.Lai, C. J., and Weisblum, B. (1971) Proc.
Natl. Acad. Sci. USA, 68, 856-860.
67.Skinner, R., Cundliffe, E., and Schmidt, F. J.
(1983) J. Biol. Chem., 258, 12702-12706.
68.Douthwaite, S. (1992) J. Bacteriol.,
174, 1333-1338.
69.Sigmund, C. D., and Morgan, E. A. (1982) Proc.
Natl. Acad. Sci. USA, 79, 5602-5606.
70.Weisblum, B. (1995) Antimicrob. Agents
Chemother., 39, 577-585.
71.Vester, B., and Douthwaite, S. (2001)
Antimicrob. Agents Chemother., 45, 1-12.
72.Gregory, S. T., and Dahlberg, A. E. (1999) J.
Mol. Biol., 289, 827-834.
73.Garza-Ramos, G., Xiong, L., Zhong, P., and
Mankin, A. (2001) J. Bacteriol., 183, 6898-6907.
74.Petropoulos, A. D., Kouvela, E. C., Dinos, G. P.,
and Kalpaxis, D. L. (2008) J. Biol. Chem., 283,
4756-4765.
75.Rodriguez-Fonseca, C., Amils, R., and Garrett, R.
A. (1995) J. Mol. Biol., 247, 224-235.
76.Auerbach, T., Bashan, A., and Yonath, A. (2004)
Trends Biotechnol., 22, 570-576.
77.Steitz, T. A. (2005) FEBS Lett.,
579, 955-958.
78.Tu, D., Blaha, G., Moore, P. B., and Steitz, T.
A. (2005) Cell, 121, 257-270.
79.Vazquez-Laslop, N., Thum, C., and Mankin, A. S.
(2008) Mol. Cell, 30, 190-202.
80.Tenson, T., Lovmar, M., and Ehrenberg, M. (2003)
J. Mol. Biol., 330, 1005-1014.
81.Matsuoka, M., Inoue, M., Endo, Y., and Nakajima,
Y. (2003) FEMS Microbiol. Lett., 220, 287-293.
82.Furneri, P. M., Rapazzo, G., Musumarra, M.,
Pietro, P., Catania, L. S., and Roccasalva, L. S. (2001)
Antibiot. Chemother., 45, 2958-2960.
83.Sutcliffe, J. A. (2004) Features,
70, 513-519.
84.Douthwaite, S., Crain, P. F., Liu, M., and
Poehlsgaard, J. (2004) J. Mol. Biol., 337, 1073-1077.
85.Pfister, P., Corti, N., Hobbie, S., Bruell, C.,
Zarivach, R., Yonath, A., and Bottger, E. C. (2005) Proc. Natl.
Acad. Sci. USA, 102, 5180-5185.
86.Mankin, A. S. (2008) Curr. Opin.
Microbiol., 11, 414-421.
87.Schroeder, S. J., Blaha, G., and Moore, P. B.
(2007) Antimicrob. Agents Chemother., 51, 4462-4465.
88.Sumbatyan, N. V., Korshunova, G. A., and
Bogdanov, A. A. (2003) Biochemistry (Moscow), 68,
1156-1158.
89.Korshunova, G. A., Sumbatyan, N. V., Fedorova, N.
V., Kuznetsova, I. V., Shishkina, A. V., and Bogdanov, A. A. (2007)
Bioorg. Khim., 33, 235-244.
90.Sumbatyan, N. V., Kuznetsova, I. V., Karpenko, V.
V., Fedorova, N. V., Chertkov, V. A., Korshunova, G. A., and Bogdanov,
A. A. (2010) Bioorg. Khim., 36, 265-276.
91.Starosta, A. L., Karpenko, V. V., Shishkina, A.
V., Mikolajka, A., Sumbatyan, N. V., Schluenzen, F., Korshunova, G. A.,
Bogdanov, A. A., and Wilson, D. N. (2010) Chem. Biol.,
17, 504-514.
92.Lovett, P. S., and Rogers, E. J. (1996)
Microbiol. Rev., 60, 366-385.
93.Morris, D. R., and Geballe, A. P. (2000) Mol.
Cell Biol., 20, 8635-8642.
94.Tenson, T., and Ehrenberg, M. (2002) Cell,
108, 591-594.
95.Nakatogawa, H., and Ito, K. (2002) Cell,
108, 629-636.
96.Gong, F., and Yanofsky, C. (2002) Science, 297,
1864-1867.
97.Vazquez-Laslop, N., Thum, C., and Mankin, A. S.
(2008) Mol. Cell, 30, 190-202.
98.Wang, Z., Fang, P., and Sachs, M. (1997) Mol.
Cell Biol., 18, 7528-7536.
99.Raney, A., Law, G. L., Mize, G. J., and Morris,
D. R. (2002) J. Biol. Chem., 277, 5988-5994.
100.Lovett, P. S. (1994) J.
Bacteriol., 176, 6415-6417.
101.Gu, Z., Harrod, R., Rogers, E. J., and Lovett,
P. S. (1994) Proc. Natl. Acad. Sci. USA, 91,
5612-5616.
102.Rogers, E. J., and Lovett, P. S. (1994) Mol.
Microbiol., 12, 181-186.
103.Weisblum, B. (1995) Antimicrob. Agents
Chemother., 39, 797-805.
104.Ramu, H., Mankin, A., and Vasquez-Laslop, N.
(2008) Mol. Microbiol., 71, 811-824.
105.Bayfield, M. A., Thompson, J., and Dahlberg, A.
E. (2004) Nucleic Acids Res., 32, 5512-5518.
106.Nakatogawa, H., and Ito, K. (2004)
ChemBioChem, 5, 48-51.
107.Muto, H., Nakatogawa, H., and Ito, K. (2006)
Mol. Cell, 22, 545-552.
108.Pavlov, M. Y., Watts, R. E., Tan, Z., Cornish,
V. W., Ehrenberg, M., and Forster, A. C. (2009) Proc. Natl. Acad.
Sci. USA, 106, 50-54.
109.Muto, H., and Ito, K. (2008) Biochem.
Biophys. Res. Commun., 366, 1043-1047.
110.Wohlgemuth, I., Brenner, S., Beringer, M., and
Rodnina, M. V. (2008) J. Biol. Chem., 283,
32229-32235.
111.Suhonara, T., Abo, T., Inada, T., and Aiba, H.
(2002) RNA, 8, 1416-1427.
112.Roche, E. D., and Sauer, R. T. (2001) J.
Biol. Chem., 276, 28509-28515.
113.Tanner, D. R., Carillo, D. A., Woolstenhulme,
C. J., Broadbent, M. A., and Buskirk, A. R. (2009) J. Biol.
Chem., 284, 34809-34818.
114.Shpanchenko, O. V., Ivanov, P. V., Zvereva, M.
E., Bogdanov, A. A., and Dontsova, O. A. (2004) Mol. Biol.
(Moscow), 38, 914-925.
115.Woolhead, C. A., Johnson, A. E., and Bernstein,
H. D. (2006) Mol. Cell, 22, 587-598.
116.Yap, M.-N., and Bernstein, H. D. (2009) Mol.
Cell, 34, 201-211.
117.Schmeing, T. M., Seila, A. C., Hansen, J. L.,
Freeborn, B., Soukup, J. K., Scaringe, S. A., Strobel, S. A., Moore, P.
B., and Steitz, T. A. (2002) Nat. Struct. Biol., 9,
225-230.
118.Hansen, J. L., Schmeing, T. M., Moore, P. B.,
and Steitz, T. A. (2002) Proc. Natl. Acad. Sci. USA, 99,
11670-11675.
119.Bogdanov, A. A. (2003) Mol. Biol.
(Moscow), 37, 436-439.
120. Wang, D., Ding, X., and Rather, P. N. (2001) J.
Bacteriol., 183, 4210-4216.
121.Ren, D., Bedzyk, L. A., Ye, R. W., Thomas, S.
M., and Wood, T. K. (2004) Appl. Environ. Microbiol., 70,
2038-2043.
122.Hirakawa, H., Inazumi, Y., Masaki, T., Hirata,
T., and Yamaguchi, A. (2005) Mol. Microbiol., 55,
1113-1126.
123.Kazarinoff, M. N., and Snell, E. E. (1977)
J. Biol. Chem., 252, 7598-7602.
124.Gollnicky, P., and Yanofsky, C. J. (1990) J.
Bacteriol., 172, 3100-3107.
125.Gong, F., and Yanofsky, C. (2001) J. Biol. Chem.,
276, 1974-1983.
126.Yanofsky, C. (2007) RNA, 13,
1141-1154.
127.Gong, F., Ito, K., Nakamura, Y., and Yanofsky,
C. (2001) Proc. Natl. Acad. Sci. USA, 98, 8997-9001.
128.Gong, M., Cruz-Vera, L. R., and Yanofsky, C.
(2007) J. Bacteriol., 189, 3147-3155.
129.Cruz-Vera, L. R., Rajagopal, S., Squires, C.,
and Yanofsky, C. (2005) Mol. Cell, 19, 333-343.
130.Cruz-Vera, L. R., New, A., Squires, C., and Yanofsky, C.
(2007) J. Bacteriol., 189, 3140-3146.
131.Mankin, A. S. (2006) Trends Biochem.
Sci., 31, 11-13.
132.Torrice, M. M., Bower, K. S., Lester, H. A.,
and Dougherty, D. A. (2009) Proc. Natl. Acad. Sci. USA,
106, 11919-11924.
133.Trabuco, L. G., Harrison, C. B., Schreiner, E.,
and Schulten, K. (2010) Structure, 18, 627-637.