REVIEW: Properties of Intraribosomal Part of Nascent Polypeptide
V. A. Kolb
Institute of Protein Research, Russian Academy of Sciences, 142290 Pushchino, Moscow Region, Russia; E-mail: kolb@vega.protres.ru
Received June 30, 2010
This review analyzes the concept according to which the pathway of synthesized peptide from the ribosome peptidyl transferase center to the exit domain goes along the tunnel of the large subparticle. Experimental data on the accessibility of the nascent polypeptide chain to molecules of modifying agents and fluorescence quenchers are considered. Results of localization of the exit site for the nascent peptide on the ribosome surface, possible conformational states of the peptide, and its mobility and folding on the ribosome are analyzed. The analysis is based on the ribosomal tunnel parameters obtained using X-ray crystallography of whole ribosomes and large ribosomal subparticles. Special attention is given to data that do not fit in the concept of the “tunnel for peptide exit” and to results already obtained before the reliable tunnel visualization using X-ray crystallography was achieved.
KEY WORDS: nascent polypeptide, ribosomal tunnel, nascent polypeptide chain exit domain, nascent polypeptide accessibilityDOI: 10.1134/S000629791013002X
Abbreviations: a.a., amino acid residue; NAC, nascent polypeptide-associated complex; PDF, peptide deformylase; PTC, ribosome peptidyl transferase center; SRP, signal recognition particle; TDB, 4-(3-trifluoromethyldiazirino)benzoate; TF, trigger factor.
A polypeptide chain, synthesized by the ribosome, begins in the peptidyl
transferase center (PTC), and as far as elongation proceeds, it follows
to the outlet at the surface of the large subunit along a pathway
hidden inside the ribosome and called the channel for nascent
polypeptide (Fig. 1; see color insert). Attention
of researchers to this element of ribosome structure is due to several
factors. First, the channel serves as the closest environment of the
synthesized polypeptide chain and thus it can impose certain
restrictions on and somewhat contribute to cotranslational formation of
the protein spatial structure. Besides, due to its contacts with the
nascent chain the channel can serve as a sensor generating
conformational “signals” for transduction into the ribosome
functional centers. It is now assumed that the intraribosomal tunnel of
the large subparticle is the channel for the nascent polypeptide (see
review [1]).
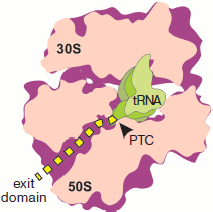
The concept of the channel for the nascent polypeptide in the form of a tunnel in the large ribosome subparticle was first formulated by Lake et al. [2, 3]. It was initially based on two groups of data: results of immunoelectron microscopy concerning localization of the peptide exit site on the ribosome surface, and determination of the length of nascent peptide region protected by the ribosome against the action of proteases.Fig. 1. Location of a synthesized polypeptide in a translating ribosome in accordance with the concept of “tunnel for exit” of a nascent polypeptide chain. The ribosome half with L7/L12 protuberance is removed. The plane of section passes through both subparticles, PTC (arrow), and longitudinal axis of the large subparticle tunnel. The nascent polypeptide is designated by the yellow dotted line. The outline of tRNA localized in the A site is shown in the foreground.
The first attempt to determine the extent of the synthesized peptide shielding against proteolytic effect was made by Malkin and Rich [4]. They treated polysomes isolated from rabbit reticulocytes with different proteases such as pronase, papain, and trypsin mixture with chymotrypsin. The treatment was carried out at 0°C, and the integrity of the ribosomes was checked by their sedimentation properties. The length of the radiolabeled peptide part protected by the ribosome under these conditions was estimated by gel chromatography. The length of the region protected against proteases was 30-35 amino acid residues (a.a.). Unfolding the ribosomes caused by decrease in magnesium concentration in the medium resulted in the complete degradation of the nascent peptide by proteases.
Experiments with “pulse” radiolabeling of nascent peptide and following proteolysis have also shown that just the proximal C-terminal peptide part bound to the ribosome was resistant to protease attack [4].
The authors of this work supposed two possible variants of the synthesized polypeptide chain shielding against proteases: either the nascent chain passes inside rhe ribosome or along its surface but in some groove. Both cases suggest protection of a certain region of polypeptide chain against proteolysis.
Similar results were published in 1970 by Blobel and Sabatini [5]. Polypeptide chains synthesized on rat liver ribosomes underwent limited proteolysis by trypsin and chymotrypsin. The length of the protected part of the nascent chain was approximately 39 a.a. (according to gel chromatography). Ribosome integrity was checked by sedimentation analysis and electrophoresis of ribosomal proteins. It should be noted that proteolysis caused almost complete dissociation of monoribosomes as well as significant changes in mobility of ribosomal proteins.
The shielding of nascent peptide by prokaryotic ribosomes was studied in the group of Davis et al. [6]. Bacillus subtilis ribosomes carrying peptides in vivo labeled with 35S-labeled methionine were treated with pronase. Labeled proteolysis products were analyzed by two methods—gel chromatography and dansylation of N-terminal methionine residue. The latter technique is based on random distribution of methionine residues in in vivo synthesized peptides and on the ability to react with dansyl chloride of methionine with free NH2-group, i.e. localized at the N terminus of a proteolytic fragment. According to the law of probability, just the ratio of total methionine amount to that of N-terminal methionine (determined by amount of dansyl methionine) equals the polypeptide length in a.a. Both approaches gave similar results, i.e. 30 a.a. by gel chromatography and 28 a.a. by dansylation [6].
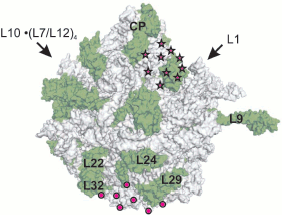
Thus, results of the above-mentioned works [4-6] suggested that under certain conditions the eukaryotic ribosome protects 30-39 a.a. against protease attack, while prokaryotic ribosome protects about 30 a.a. of the C-terminal part of a nascent peptide. However, it remained unclear whether the pathway of synthesized polypeptide chain was inside the ribosome or on its surface; the question concerning conformational state of nascent chain was also open. The site of nascent protein exit on the ribosome surface was directly localized using electron microscopy in order to answer these questions [2, 3]. The use of antibodies to β-galactosidase synthesized by E. coli ribosomes revealed the protein exit site in the large subparticle region remote from the central protuberance and opposed to the subunits interface [2] (Fig. 2; see color insert). This region was called the “exit domain”. According to Lake [3], the exit of ribulose-1,5-diphosphate carboxylase protein at 80S ribosomes of the plant Lemna gibba coincides with the “release domain” of prokaryotic ribosomes.
It is evident that the pathway of the nascent polypeptide chain should begin in the PTC where the next amino acid joins the peptide C terminus. According to data available at that time, PTC were localized at the contacting (concave) surface of the large subparticle next to the central protuberance (head), in the region of the groove separating the subparticle head from its body [7-9].Fig. 2. Exit domain of synthesized polypeptide on the ribosome surface according to electron microscopy data. The outer convex surface of E. coli 50S subparticle is shown in accordance with X-ray data (PDB ID 1VS6). The surface of the RNA is gray, and the surface of proteins is green (designations are given for some proteins). Arrows point to positions of corresponding subparticle protuberances, CP is central protuberance. The domain localization according to Lake [3] is designated by circles, and an alternative localization [28] is shown by asterisks.
This means that in order to appear in the “exit domain”, the N terminus of a nascent peptide has to cover the distance from the PTC to this domain which, according to Lake et al., extends along the straight line approximately 140 Å for prokaryotic ribosomes [2, 3] and 160 Å for eukaryotic ribosomes [3]. If it is supposed that the nascent peptide is in the fully extended chain conformation, when about 3.6 Å corresponds to 1 a.a., then peptides of 33-45 and 37-51 a.a. (for pro- and eukaryotic ribosomes, respectively) will reach the ribosome surface. The assessed values coincide with the length of the chain region protected by the ribosome against proteolysis [4-6]. This led to a conclusion concerning the existence of a tunnel between the PTC and “exit domain” inside the large subparticle. The synthesized peptide, localized in this tunnel, is inaccessible for proteases.
Thus, the concept of ribosomal channel for nascent polypeptide was formulated using the following interrelated conditions:
a) the ribosome-synthesized polypeptide appears at the ribosome surface in the “exit domain” of the large subparticle;
b) nascent polypeptide passes from PTC to the “exit domain” along the tunnel through the large subparticle;
c) within the tunnel the polypeptide is in the conformation of unfolded and fully extended chain.
In this review the concept is analyzed in the light of experimental data concerning the accessibility of nascent polypeptide chain to molecules of modifying agents and fluorescence quenchers, results of the localization of exit site for the nascent peptide on the ribosome surface, possible conformational state of nascent polypeptide, and data on its folding on the ribosome. Special attention is given to data that do not fit in the concept of “the tunnel for peptide exit” and to the results obtained when there was no reliable tunnel visualization by X-ray crystallography. The analysis is based on the ribosomal tunnel properties revealed by X-ray crystallographic structures of 70S ribosomes and 50S subparticles.
Cryoelectron microscopy of translating ribosomes
The first attempts to visualize a ribosomal tunnel were made using three-dimensional reconstruction of electron-microscopic images of flat two-dimensional crystals of 70S ribosomes and 50S subparticles isolated and purified from the thermophilic microorganism Bacillus stearothermophilus [10, 11]. Regions of low electron density in large ribosomal subunit can be seen on the obtained images. The authors believe that these regions within 50S subparticles correspond to hollows or cavities 100 Å in length and 25 Å in diameter. Something resembling a ribosomal tunnel was also detected in eukaryotic ribosomes by three-dimensional reconstruction of electron microscopic images [12]. Unfortunately, resolution of the technique used at that time was within 30-47 Å, which is obviously insufficient for reliable visualization of a tunnel of 25 Å in diameter. The fact that this structural element is not always well seen on three-dimensional reconstruction of electron microscopic images [10-14] was explained by the authors by the possibility of its filling with the nascent peptide [15].
Along with development of electron microscopy technique and improvement of the mathematical apparatus of image recognition and analysis, it became possible to obtain high quality cryoelectron microscopic images of ribosomes and their complexes. In works of van Heel’s [16, 17] and Frank’s [18, 19] groups not simply a tunnel, but rather a network of tunnels penetrating the large subunit of bacterial ribosome were visualized along with quite a set of intraribosomal hollows, voids, and gaps [20]. The authors of work [18] also supposed, on the basis of observed tunnel branching, a possible multiplicity of the nascent polypeptide exit sites on the ribosome surface. Resolution achieved in these works was from 20-23 [16, 17] to 25 Å [18, 19], which, unfortunately, did not allow reliable determination of the morphological characteristics of a tunnel. The method was also applied to eukaryotic ribosomes, plant [21] and yeast [22], and at the resolution of 35-38 Å; their significant morphological similarity with bacterial ribosomes along with the presence of the variable diameter tunnel, penetrating 60S subunit and ending with a narrow outlet, were found [22]. Comparison of more detailed (at resolution of 24 Å) results of three-dimensional reconstruction of cryoelectron-microscopic images of yeast and rabbit ribosomes resulted in similar conclusions [23]. The authors attribute to intraribosomal tunnel a length about 100 Å and the averaged diameter of approximately 20 Å. Similar data were also obtained for the tunnel in rat liver ribosomes [24].
In recent works of Beckman’s group on cryoelectron microscopy of a homogeneous preparation of ribosomes carrying defined length polypeptide chains, it has been reported that the nascent polypeptide is visualized in the large subparticle tunnel [25-27]. In fact, authors of these works managed to achieve the resolution record for this method of 5.8 Å [25] and to detect in this case electron density islets within the tunnel. The electron-dense objects found in the tunnel were considered as the nascent chain regions. Of course, even such high resolution is not enough for identification of individual amino acids in a nascent polypeptide. The fact that the nascent chain in this case is not continuous was explained by different mobility of its different regions. Besides, the authors attribute to the synthesized polypeptide a set of certain interactions with the tunnel walls; changes in these interactions accompany each elongation step. The existence of the polypeptide chain multicenter binding to the tunnel walls should make significantly difficult the translocation of synthesized polypeptide along the ribosome, and it would certainly slow down the nascent chain release from the ribosome after termination or reaction with puromycin. However, no description of such retardation was found in the literature. Besides, multicenter binding should significantly decrease the nascent polypeptide mobility within the tunnel and thus to convey electron density close to that of low-mobility regions of ribosome structure. Electron density of the object, considered by the authors of work [25] as the nascent polypeptide chain, is much lower.
Localization of NASCENT polypeptide exit domain on THE ribosome
Localization of the nascent polypeptide exit domain carried out in Lake’s group using immunoelectron microscopy can be not completely precise due to the large size of the synthesized protein. In fact, not the place of peptide exit from the ribosome, but rather the site of surface localization of antigen determinants of a large completed protein was determined [2, 3], which is not the same thing. That is why experiments on refinement of “exit domain” localization were undertaken with ribosomes charged with short peptides carrying at the N terminus dinitrophenyl hapten, which interacted with corresponding antibodies [28]. The cell-free translation system where certain amino acids were excluded was used to obtain short peptides on E. coli ribosomes. The system included only amino acids incorporated in the 42 a.a.-long N-terminal region of MS2 phage coat protein. As a result, two sites of localization of the nascent peptide N terminus were revealed: the first coincided with Lake’s “exit domain”, while the other was localized between the base of the central protuberance and the L1 ridge. The authors supposed [28] that the N terminus of the nascent peptide after leaving the PTC moves further along a groove on the surface of 50S subparticle and finishes at the “exit domain” according to Lake.
Another way of the “exit domain” detection is localization on the ribosome of molecules interacting with relatively short nascent polypeptides. The trigger factor (TF), the signal recognition particle (SRP), peptide deformylase (PDF), as well as membrane complex for protein translocation (translocon) can serve as such molecules. For proteins translocated cotranslationally into endoplasmic reticulum, exit of their N termini on the ribosome surface is coupled with the obligatory association of the nascent chain with the translocon. An analog of such associate, consisting of vacant yeast ribosomes and heterotrimeric complex Sec61 (without nascent polypeptide, membrane, and other translocon components), was obtained and studied using cryoelectron microscopy [29]. It appeared that the intraribosomal tunnel is not only clearly seen on three-dimensional reconstruction of electron microscopic images but is exactly coaxial to trimeric complex Sec61. In other words, the ribosomal tunnel opened directly into the internal, intersubunit hole of Sec61 trimer, which was supported by the author’s conclusion concerning intraribosomal tunnel identity to the channel for nascent polypeptide chain [29]. However, when translocon structure was detected by X-ray analysis, it became clear that only the Sec61α or SecY subunit rather than trimer can serve as the peptide- conducting channel of the translocon [30]. This means that most likely the tunnel should open into an internal cavity of this subunit rather than into intersubunit space of the translocon. Interestingly, cryoelectron microscopy of translating ribosomes, following the early electron microscopic work on the nascent peptide N terminus localization on the ribosome [28], revealed two sites of translocon binding. Together with Lake’s “exit domain”, a site near L1 protuberance was also able to bind translocon [31]. The authors of the above-mentioned work from Frank’s group claim that the observed translocon interaction with the site of L1 protein is caused by translocon affinity to mRNA rather than to nascent peptide, and so the detected complex is not related to the peptide exit to the ribosome surface. However, the translocon affinity to mRNA was never shown before. In order to find the existence of complete conformity between data obtained in works [28] and [31], one has to suppose that antibodies to dinitrophenyl hapten also exhibited affinity to 5′-proximal site of mRNA, which left the decoding region of the ribosome and appeared at its surface near the L1 protuberance.
However, localization of sites for binding proteins and complexes interacting with nascent polypeptides, namely, SRP [32] delivering translating ribosomes to membranes for cotranslational transmembrane translocation of synthesized polypeptides, PDF [33] removing formyl group of N-terminal formylmethionine, and TF [34, 35] exhibiting chaperone activity, leads to the same site near the exit from the tunnel. The authors of work [36] suppose that the site for binding of methionine amino peptidase (MetAP), removing N-terminal methionine from many nascent polypeptides, is localized in the same place. However, it is obvious that simultaneous binding of all these macromolecules on a relatively small region of the ribosome surface is impossible. Besides, their interaction with the N terminus of a nascent chain should happen in turn (so, MetAP interacts with polypeptide only after removal of formyl group by PDF [37, 38]). Thus, the existence of overlapping or identical binding sites makes no sense. It should also be noted that in fact sites for binding small PDF and TF fragments rather than full-sized proteins were determined using X-ray analysis. For this aim grown crystals of ribosomes were soaked in solutions of appropriate protein fragments. It is clear that crystalline package and inter-ribosomal contacts within it do not allow interaction of macromolecules with all regions of the ribosome surface — contacts with only sterically available ones are possible. This means that the used experimental approach could restrict the range of search and exclude the detection of the real binding sites.
The above-mentioned objections concerning the exactness of the exit domain localization become more important in the light of results of electron microscopy of platinum replicas of intact fragments of rough endoplasmic reticulum [39]. The authors found that the lumen of intact translocon in a complex with the ribosome is coaxial to the intersubunit space of the ribosome rather than to intraribosomal tunnel. According to the data, both ribosome subunits contact translocon proteins, and the peptide exit site is localized immediately at the intersubunit fissure [39]. Such discrepancy from results of Frank’s group [31] can be explained both by differences in experimental technique and in extent of intactness of the experimental object.
In either event, we could not find indisputable data indicative of the nascent polypeptide moving along the intraribosomal tunnel, although some data point to just such localization of the nascent polypeptide chain.
INTRARibosomal tunnel visualization by X-ray analysis
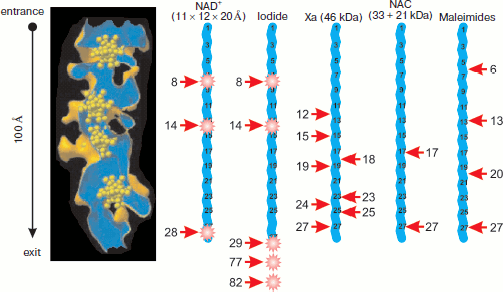
The first successful resolution of the 50S ribosome subunit structure from the archaeon Haloarcula marismortui with 9 Å resolution revealed an intraribosomal tunnel passing from PTC to the “exit domain” [40]. Improvement of resolution to 5 Å [41] made it possible to characterize the tunnel morphology: its length is somewhat more than 100 Å, and its midline is almost straight. The tunnel walls are not smooth — they have a developed surface of complex morphology. The diameter of the tunnel is about 20 Å. The subunit crystals for X-ray analysis and phasing by heavy metal substitutions were kept in solution of heteropolytungstate, each molecule of which contained 11 tungsten atoms. It appeared that four heavy metal clusters were localized within the intraribosomal tunnel [41] (Fig. 3; see color insert).
Further improvement of resolution to 2.4 Å and obtaining highly ordered subunit crystals made it possible to determine that the tunnel walls consist mainly of rRNA and to identify ribosomal proteins that are localized near the tunnel or comprise a part of its walls [42]. It was found that tunnel constriction and a small bend near the PTC are formed by L4 and L22 proteins that enter the tunnel directly by their non-globular regions at opposite sides [42, 43]. The bend is localized at a distance of 20-35 Å from the PTC. The tunnel diameter (mean value 15 Å) varies from 20 Å in the most ample place to 10 Å in two constrictions, one near the PTC and the other at a distance of 28 Å from the tunnel exit. A region of the L22 protein makes up the most part of the protein component of the tunnel surface; the contribution of L39e protein, obviously non-structurized in free form, is also pronounced [42, 43]. All five domains of 23S rRNA are involved in formation of tunnel surface: 28 sites forming tunnel walls are localized along the whole length of the RNA molecule and usually belong to loops in its secondary structure. The beginning of the tunnel is formed by the V domain of 23S rRNA, and the next 20 Å of tunnel length are formed together by regions of domains II and IV and by proteins L22 and L4. The distal tunnel half is covered by regions of domains I and III, protein L39e, and a region of the globular part of L22 protein [43]. However, on the whole the tunnel surface is free of extended charged or hydrophobic regions, which according to authors of work [43] makes possible movement of unfolded polypeptide chain within the tunnel without its binding and fixing to the walls. The constriction formed by L22 and L4 proteins is supposedly able to function as a valve opening in response to a signal from translocon binding or, on the contrary, signal from the valve may prepare binding to translocon because the globular part of L22 protein is localized near the exit domain (together with proteins L19, L23, L24, L29, and L31e) [43].Fig. 3. Accessibility of synthesized peptide for interaction with fluorescence quenchers and modifying agents. The ribosomal tunnel is shown in longitudinal section on the left. Heteropolytungstate clusters (shown in yellow, see [41]) are present in the tunnel. Nascent 28-membered polypeptide chains (blue broken line) in fully extended conformation are shown on the same scale with tunnel (the chain and tunnel length is 100 Å). Arrows point to accessible amino acid residues, the agent names being shown above corresponding nascent chain. The residue ordinal number is given near the arrow (the C-terminal residue is considered as first). Fluorophore positions within chains are shown by asterisks. See explanations and references in the text.
The resolution of the Thermus thermophilus 70S ribosome structure at 7.8 [44] and 5.5 Å [45] also revealed intraribosomal tunnel with similar parameters in the 50S subunit. The exit domain on the T. thermophilus ribosome is surrounded by proteins L22, L24, and L29. A tunnel was also found in the 50S subunit of eubacterial mesophile Deinococcus radiodurans, whose structure was obtained at resolution of 3.1 Å [46]. Differences in tunnel morphology and exit domain environment for archaean and eubacterial microorganisms are that proteins L23 and L31e from H. marismortui are fused in the single polypeptide L23 from D. radiodurans.
Obviously, the well documented fact of intraribosomal tunnel existence does not prove that the nascent polypeptide chain during translation passes along it to the exit from the ribosome, although a number of indirect data point to such “route” of the nascent peptide [47]. Unfortunately, results of X-ray analysis of ribosomes or 50S ribosomal subunits containing peptidyl-tRNA with fairly lengthy polypeptide chain are not yet available.
SIZE OF NASCENT POLYPEPTIDE SEGMENT HIDDEN IN THE RIBOSOME
The hypothesis concerning fully extended conformation of the nascent chain within ribosome and tunnel length of 100 Å (120 Å for eukaryotes) suggests that at least 28 a.a. of synthesized polypeptide (33 residues for eukaryotic ribosomes) should be hidden in the intraribosomal tunnel. These estimations well agree with results of controlled proteolysis [4-6], although Malkin and Rich reasonably noted that the peptide region of 30-40 a.a. might be protected against protease attack due to steric hindrance to protease access to the peptide, even localized on the ribosome surface, in the groove but not within the tunnel [4]. The same arguments could be put forward to discuss results of works [48, 49], a coauthor of which is the author of this review. We showed earlier that biosynthesis of globular proteins is accompanied by folding of the nascent polypeptide chain (cotranslational folding) which results in release from the ribosome of folded protein with fully formed spatial structure [50, 51]. The nascent polypeptide folding into biologically active protein is also possible without its release from the ribosome in the case of removal of natural termination codons from the reading frame and elongation of the C-terminal region by additional amino acid sequence which is not encoded in the wild-type gene. Obviously, the additional C-terminal segment holds the folding protein away from the PTC of the ribosome, thus allowing the natural C terminus of the chain to occupy the necessary position in the folded protein structure. It appeared that in the case of the eukaryotic ribosome the length of this additional segment should be no less that 26 a.a. [48, 49]. Taking into consideration the fact that several C-terminal residues (no more than 12) can be removed from luciferase structure without significantly lowering its activity, the obtained value corresponds to estimation of the length of fully extended polypeptide within the tunnel of the eukaryotic ribosome. It should be noted that estimation of the length of the nascent chain intraribosomal segment in works [48, 49] was carried out on intact ribosomes not damaged by either proteolysis or covalent cross-links. Such sparing experimental conditions were used in the group of Rodnina, who found that SRP is capable to form complex with ribosomes bearing short nascent polypeptide chains. The chains contained N-terminal signal peptides and were completely hidden within the ribosome [52]. To explain the observed interaction, the authors used the hypothesis of conformational signal transduction from the tunnel with signal peptide inside it to the particle-binding site on the ribosome surface. It is possible that a simpler explanation will be suitable, which suggests the signal peptide or SRP localization on the ribosome provides for the access of the particle even to short nascent chains.
The discovery of NAC complex associated with nascent peptides [53] called in question the hypothesis concerning the intraribosomal tunnel as the channel for nascent peptide. It was shown that a cytoplasmic factor, an αβ-heterodimer with 33 and 21 kDa subunits and abundant in the cytoplasm, interacts with nascent polypeptide chains immediately upon their appearance from the ribosome [53]. The NAC factor function consists in prevention of cotranslational translocation through membrane of peptides that are not designated for this. NAC reversibly associates with all nascent peptide chains, and in this case several factor molecules interact with each chain. NAC is easily displaced from such complex by SRP, but only if the nascent chain contains an N-terminal signal peptide. The ribosomal complex with SRP then takes part in translocation, while NAC-containing ribosomal complexes do not interact with the translocon [53, 54]. Apparently NAC dissociates from such complexes upon cotranslational folding of the nascent polypeptide. Identification of region of NAC binding to nascent polypeptide resulted in an unexpected result: it appeared that in the absence of NAC, Xa factor hydrolyzes the nascent peptide at the recognition site localized at a distance of only 12 a.a. from the PTC [55]. Taking into consideration the tunnel diameter and dimensions of Xa factor (46 kDa protein), one can definitely state that the factor cannot get into the tunnel. The authors introduced the protease Xa recognition site into nascent peptide at different distance, namely at 12, 15, 18, 19, 23, 24, 25, 27, 33, 35, 36, 37, 38, 39, 40, 41, 42, 44, 47, 58, and over 100 residues from the C terminus and observed Xa-specific hydrolysis in each of these variants in the absence of NAC (Fig. 3). In the presence of NAC, hydrolysis was observed only at sites remote from the PTC by over 33 a.a. No hydrolysis was observed at sites closer to the C terminus, namely 12, 15, 18, 19, 23, 24, 25, and 27 a.a. from the PTC [55]. Most likely NAC association with the nascent polypeptide sites remote from the PTC by more than 33 a.a. is less stable and does not prevent interaction with Xa factor.
It was found in the same work that NAC cross-link with nascent chain lysines carrying photoactivated group TDB occurs at the distance of 17, 27, 30, 33, 34, 35, 36, 38, 43, 44, 46, 47, 49, 53, 55, 58, and 100 a.a. from the PTC (Fig. 3). In this case covalent cross-link with NAC does not prevent either further elongation of the synthesized polypeptide or successful termination of its synthesis and release from the ribosome [55].
The authors conclude that the nascent polypeptide chain is protected against proteolysis by associated NAC factor rather than by the ribosomal tunnel. In the absence of NAC the synthesized polypeptide is attacked by protease at the distance of only 12 a.a. from the PTC, which is incompatible with the nascent chain localization in the intraribosomal tunnel.
CONFORMATION OF NASCENT POLYPEPTIDE INTRARIBOSOMAL SEGMENT
The assumption concerning fully extended peptide conformation within the ribosome is one of main points underlying localization of the nascent polypeptide inside the ribosomal tunnel. An evident disadvantage of this assumption was noted by Lim and Spirin in 1986 [56]. Obviously, the fully extended chain should willingly interact with its RNA–protein environment by hydrogen bonds formed in this case between polypeptide and its nearest milieu rather than between the polypeptide amino acid residues. As said above, this could result in fixation of the nascent peptide and difficulty in its further elongation. It is also true that this possibility becomes less probable in the absence of extended charged (polar) or hydrophobic regions on the internal surface of the ribosomal tunnel [43]. Besides, the fully extended conformation, when there is 3.6 Å per 1 a.a., is sterically strained. To achieve this conformation, it is necessary to apply to the peptide a mechanical traction hardly provided by the ribosome during synthesis.
The polypeptide chain in α-helical conformation is devoid of the above-mentioned disadvantages. The advantage of α-helical conformation is also its steric acceptability for any amino acid sequence: even the presence of proline residues only slightly distorts its geometric parameters [56]. The diameter of α-helix slightly exceeds the diameter of any extended structure with side amino acid residue chains; therefore, if the ribosomal channel accommodates extended polypeptide, it can accommodate the α-helical polypeptide as well.
There is 1.5 Å per 1 a.a. in α-helical conformation. This means that 67-80 a.a. can be localized in a 100-120 Å long tunnel, which contradicts both results of limited proteolysis [4-6] and data on the size of additional C-terminal luciferase segment [48, 49]. The conclusion following from this contradiction is that either the conformation of the nascent polypeptide chain is not α-helical, or that the nascent chain does not pass through the ribosomal tunnel. Authors of recent works [25-27] on cryoelectron microscopy of ribosomes carrying the defined length polypeptide chains did not find in the ribosome a nascent polypeptide in fully α-helical conformation. Even if the achieved resolution is worse than the claimed 5.8 Å [25], it is doubtful that an α-helix of over 40 a.a. would remain undetectable. Most likely, the whole intraribosomal region of the nascent polypeptide is not in α-helical conformation, although formation of small α-helical sites resulting in chain compaction and enlargement of the region protected by the ribosome against modifications was shown [57, 58].
ACCESSIBILITY OF INTRARIBOSOMAL PART OF SYNTHESIZED POLYPEPTIDE
CHAIN
Conformational state, polarity of the environment, and accessibility of nascent polyphenylalanine and polylysine peptide chains to small molecules were studied using fluorescent labels attached to the N-terminal amino acid residue. Results obtained in laboratory of Hardesty [59-61] revealed significant differences in characteristics of nascent polyphenylalanine and polylysine peptides. Thus, the N terminus of polylysine during the whole synthesis was localized in hydrophobic polar environment, while fluorescence anisotropy sharply decreased at the very beginning of elongation and remained low during the whole translation process. Low values of fluorescence anisotropy are indicative of molecule mobility, which agrees poorly with its localization in such a massive particle as the ribosome [59, 60]. The N terminus of nascent polyphenylalanine, on the contrary, detected both the presence of hydrophobic environment and shielding against solvent, as well as sufficiently high fluorescence anisotropy, slowly increasing during elongation after low but with a sharp fall at the very beginning of translation [59-61]. The authors of the above-mentioned works concluded that exit from PTC causes some increase in mobility of the nascent polyphenylalanine N terminus (fall of anisotropy), but as elongation proceeds the peptide forms a non-structured hydrophobic mass, which also contradicts its localization within the tunnel.
Investigations of accessibility of fluorescent labels within nascent peptides to molecules of different fluorescence quenchers are also inconsistent with localization of the ribosome-synthesized peptide in the intraribosomal tunnel. Thus, N-terminal fluorophores of nascent polyalanine, polyserine, and polylysine chains, independently of the chain length, were practically equally available for sufficiently large molecules of the quencher methyl viologen (molecular mass 257, dimensions are comparable with those of tryptophan) [62]. Evidently, short nascent chains, if they were in the tunnel, would be less accessible to methyl viologen due to significant reduction of solid angle at which the fluorophore can be attacked by the quencher. However, this was not observed.
In a series of methodologically similar works of Johnson’s group [63-65], lifetimes of the fluorophore excited state and Stern–Volmer constant were measured to determine quantitatively quenching efficiency (that is the fluorophore accessibility for collisions with quencher molecules). The fluorophore was incorporated into nascent preprolactin (of both wild-type and mutant) at the distance of 8, 14, 29, 77, and 82 a.a. from the PTC [63] (Fig. 3). Efficiency of fluorophores’ quenching by iodide ion was practically equal independently of their localization in the synthesized preprolactin sequence, which points to equal accessibility of the nascent polypeptide to collisions with iodide ion. Evidently, the ability of quencher to collide with polypeptide in a narrow tunnel should be significantly deteriorated compared to a peptide lying on a plane. However, in fact values of Stern–Volmer constant for ribosome-bound fluorescence-labeled peptides is only 1.5-2 times lower than for free peptides [63] in solution. This result being expected upon solid angle halving due to fluorophore attachment to the plane from volume rather by fitting them in a tunnel of 15 Å diameter. Only joining of translating ribosomes to the membrane isolates the nascent peptide from cytoplasmic molecules, and fluorescence quenching by iodide becomes impossible both from the cytoplasm [63] and from a pore in the reticulum lumen (for peptides not exceeding 70 a.a.) [64].
The complete shielding of nascent chain against the cytoplasm [63, 64] in the membrane-associated ribosome led the authors to suggest that the chain passes through the intraribosomal tunnel. Their own results on fluorophore quenching near PTC of free ribosomes remained undiscussed.
The technique of nascent polypeptide fluorescence labeling was also applied to determination of the translocon pore diameter [65], which gave an unexpected result. Molecules of different size were used for quenching fluorescence upon collision with the fluorophore, in particular nicotinamide adenine dinucleotide (NAD+) having dimensions 11 × 12 × 20 Å in the anhydrous state. Fluorophores were localized at different distances from the C terminus of the peptide. Three variants of distances are important for this study: a) 28 a.a.; b) 8 and 14 a.a. in a single chain; c) 1, 22, and 28 a.a. in a single chain (Fig. 3). It is surprising, but practically equal efficiency of fluorescence quenching by NAD+ was registered for all three variants of fluorophore localization in preprolactin peptides on free ribosomes. The free fluorophore accessibility to quenching was only three times higher than that of ribosome-bound fluorophore, which was indicative of almost unhindered diffusion of such a large molecule as NAD+ near the nascent polypeptide [65]. Apparently, a tunnel with mean diameter of 15 Å and also filled with the nascent chain had to exclude diffusion into the pore of a molecule with dimensions 11 × 12 × 20 Å, and thus the values of quenching efficiency are incompatible with the nascent polypeptide chain localization in the tunnel of the large ribosomal subunit.
Anomalously high accessibility of the intraribosomal part of the nascent polypeptide was also found by Lu et al. [66], who measured electrostatic potential around the nascent chain (“in the ribosomal exit tunnel”). In this work the authors measured the rate at which a cysteine residue in an assigned position in the nascent chain (and hence at the assigned distance from the ribosome PTC) reacted with compounds that modified the sulfhydryl group. This made possible, in addition to electrostatic potential calculation, to estimate the residue accessibility to interaction with substituted methane thiosulfonates and maleimides. It appeared that cysteine residues located at a distance of 6, 13, 20, and 27 a.a. from the PTC (Fig. 3) react with maleimides at relatively high rate, approximately 1/10 of the maximal rate characteristic of cysteine residues outside of the ribosome (at a distance of 67 and 74 a.a. from the PTC) [66]. Surprisingly, the reaction rate and therefore the cysteine group accessibility were practically independent of the reacting residue position in the range from 6 to 27 residues from the C terminus [66]. This means that diffusion of relatively large reagent molecules around most of the nascent peptide proceeded at equal efficiency, which is poorly consistent with the nascent chain arrangement in the tunnel, which allows for molecules entering the tunnel from the surrounding solution only from one side, from the exit domain. These results seems even less understandable if the already mentioned data of cryoelectron microscopy [27] are considered, according to which α-helical conformation of nascent peptide can emerge at the entrance into the tunnel from the cytoplasm. Obviously, helical regions should serve as reliable “stoppers” preventing contacts of cytoplasmic molecules with the nascent chain regions localized between the helical region and the PTC.
Steric restrictions applied by the tunnel also make improbable passing through it for nascent polypeptide with a massive group covalently attached to the side group of one of the amino acid residues. Nevertheless, attachment of a large eosin fluorophore (11 × 20 Å) to the N-terminal methionine did not prevent synthesis of chloramphenicol acetyltransferase and rhodanese, although it significantly slowed translation initiation down (elongation was decelerated to a lesser extent) [67]. The fact of successful synthesis of enzymatically active proteins modified by eosin at their N termini agrees well with results of covalent cross-linking the nascent polypeptide with NAC, which does not prevent either further elongation of the synthesized polypeptide or its termination and release from the ribosome [55]. Thus, both of these facts cause serious doubts about the nascent peptide chain localization in the ribosomal tunnel.
Experiments on the cotranslational folding of globin also contradict the hypothesis concerning the pathway of the nascent polypeptide in the tunnel of the large subunit [68, 69]. The authors tested formation of native structure of α-globin by the heme-binding ability of nascent polypeptide chain and found that the specific binding of the ligand occurs with nascent peptides of 140, 100, and 86 a.a. and is not observed with shorter chains of 75, 65, and 34 residues [69]. This suggested formation by the nascent 86-membered polypeptide of spatial structure close to native α-globin structure.
It is known that histidine residues in positions 58 and 87 are important for heme binding. Despite the absence of histidine-87 from the 86-membered globin fragment, the heme-binding pocket was formed. As follows from the native α-globin structure, C-terminal residues of the synthesized 86-membered peptide form contacts necessary for heme binding and retention. Localization of these residues in the ribosome tunnel far from the forming protein globule would prevent their participation in ligand binding. The assumption concerning folding within the ribosome tunnel and, as a result, concerning localization within it of 86-membered globin fragment is excluded due to steric limitations (the possibility of the synthesized protein folding in the ribosome tunnel was analyzed in [70]).
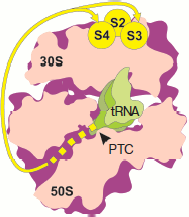
Experiments on mapping the nearest environment of nascent polypeptide chain as its elongation on the ribosome proceeded was carried out using the TDB photo-activating group covalently bound to the N terminus of synthesized polypeptides of different length and sequence [71-74]. The N-terminal fragments of the following proteins were used in these works: tetracycline resistance protein, E. coli ompA, and protein encoded by gene 60 of T4 bacteriophage (subunit of DNA topoisomerase). As elongation proceeded, peptides consecutively produced cross-links with nucleotides of domains V, II, III, and I of 23S rRNA [71-73]. Taking into consideration that the tunnel beginning is formed by domain V of 23S rRNA (PTC), its middle part is formed by regions of domains II and IV, while the distant region is formed by regions of domains I and III [43], it can be concluded that the nascent peptide moves along the tunnel to exit from the ribosome. At the same time, it appeared that all studied peptides (from 1 to 50 a.a.) retained the ability to cross-link with nucleotides of domain V. Paradoxically, topoisomerase peptides (gene 60) of 31, 47, and 50 a.a. produced cross-links both with domain I, some regions of which are localized at the exit from the tunnel, and with domain V near the entrance to the tunnel [73]. Moreover, beginning with the length of 6 a.a., ompA peptides cross-linked with both 50S and 30S subunits [74]. Exactly like ompA peptides, N-terminal topoisomerase fragments of 30, 46, and 49 residues gave cross-links with the small ribosome subunit and with yield identical to that with 50S subunit [74]. The authors identified regions of the small subunit interacting with nascent chains. They were proteins S1, S2, S4, and S3 (to a lesser extent) localized (except S1) near joining of the subunit “head” and “body”, at the side turned to the cytoplasm [75]. No cross-links with 16S rRNA were identified due to their low efficiency [74]. Evidently so short nascent chains are not able to “reach” proteins of 30S subunit and cross-link with them if they were to pass through the tunnel (Fig. 4; see color insert). This means that synthesized peptides do not pass through the tunnel.
On the basis of their results, the authors of works [71-74] drew the conclusion concerning unusual flexibility of nascent polypeptide chains, exhibiting such mobility during cotranslational folding, that they were able to contact both with ribosomal tunnel and exit from it as well as with PTC. They believe that the folding of the nascent polypeptide takes place within the tunnel. However, detection of morphological characteristics of the tunnel using X-ray analysis of ribosome and subunit crystals clearly shows that this is hardly possible because the tunnel lumen is too narrow. Folding (and, in general, three-dimensional mobility of the chain N terminus) in so limited space is unlikely [70].Fig. 4. Mapping the nascent polypeptide chain contacts with the ribosome using N-terminal photo-activating cross-links (according to [74]). Designations as in Fig. 1. The intraribosomal region of nascent chain (30-40 a.a.) is shown by the yellow dotted line. Yellow lines show the pathway of the peptide N terminus from the tunnel exit to the small subparticle proteins producing cross-links (yellow circles).
CONCLUSION
The resolution of the ribosome structure by X-ray analysis unambiguously solved the question concerning the existence of the tunnel in the large subunit and detected its morphological characteristics. At the same time, there is no reliable experimental support for the idea that the intraribosomal tunnel serves for the exit of the nascent polypeptide chain from the ribosome. On the contrary, some of the above-mentioned data give rise to doubts in this point of view. Summarizing the arguments, one can say the following:
a) the site of nascent polypeptide appearance on the ribosome surface is detected ambiguously, and there are two different results concerning the exit domain localization;
b) it is unlikely that the nascent polypeptide passes through the ribosome being in the conformation of fully extended chain;
c) the intraribosomal tunnel does not shield the nascent polypeptide against interactions with a number of proteins and against collisions with large molecules of fluorescence quenchers;
d) the intraribosomal tunnel diameter is too small to explain high mobility of the nascent chain, its cotranslational folding within the tunnel, and the ability of its N terminus to interact with the small ribosomal subunit and the PTC at the length of 30-50 a.a.
One can surmise that the tunnel diameter can change during translation and the tunnel in translating ribosome is more spacious than that in a crystal. At the present time there are no experimental confirmations of this. On the contrary, it was confirmed that the 50S subunits in a crystal are active at least in the transpeptidation reaction, but authors do not report about changes in tunnel morphology before and after reaction [76].
The supposition that not all synthesized peptides pass through the ribosomal tunnel and that the tunnel is a regulatory element involved in sorting [77] or defining the rate of translation [77, 78] is more probable, but also needs experimental checking. The available data on cross-links of peptides of different sequences with ribosome components rather point to the similarity than the differences in the nearest environment of the nascent polypeptide chains with N-terminal signal peptide and without it [73, 74].
A role of the tunnel in translation rate regulation was proposed when the mutations were analyzed that eliminate the elongation blocking during synthesis of SecM protein. The blocking, according to authors’ hypothesis [78], was caused by tunnel constriction. Mutations indeed were mapped in the narrowest part of the tunnel, in L22 protein, but they consisted of glycine-91 and alanine-93 replacement by residues with larger side chains. In other words, the tunnel lumen had to be reduced after the mutations. Later it became clear that blocking the nascent peptide elongation by the arresting sequence of SecM is not due to steric hindrances interfering in the chain moving along the tunnel, but is stimulated by events in the ribosome PTC: prolyl tRNA in the A site is not able to accept the peptide bound to glycyl tRNA located in the P site [79].
The role of the intraribosomal tunnel is still unclear and poorly studied. The localization of the nascent chain on the ribosome will probably be finally determined by X-ray crystallography. Direct answer to questions concerning function of this part of ribosome structure also depends on the results of X-ray crystallography of translating ribosomes.
The author is deeply indebted to his colleagues M. S. Svetlov and A. Kommer for critical reading of the manuscript and constructive advice for its improvement.
This work was supported by the Russian Foundation for Basic Research (grant 09-04-01447a), program of Presidium of Russian Academy of Sciences “Molecular and Cell Biology”, and Program for support of leading research schools of the Russian Federation (grant NSh-8488.2010.4).
REFERENCES
1.Selmer, M., and Liljas, A. (2008) Structure, 16, 498-500.2.Bernabeu, C., and Lake, J. A. (1982) Proc. Natl. Acad. Sci. USA, 79, 3111-3115.
3.Bernabeu, C., Tobin, E. M., Fowler, A., Zabin, I., and Lake, J. A. (1983) J. Cell Biol., 96, 1471-1474.
4.Malkin, L. I., and Rich, A. (1967) J. Mol. Biol., 26, 329-346.
5.Blobel, G., and Sabatini, D. D. (1970) J. Cell Biol., 45, 130-145.
6.Smith, W. P., Tai, P. C., and Davis, B. D. (1978) Proc. Natl. Acad. Sci. USA, 75, 5922-5925.
7.Luhrmann, R., Bald, R., Stoffler-Meilicke, M., and Stoffler, G. (1981) Proc. Natl. Acad. Sci. USA, 78, 7276-7280.
8.Olson, H. M., Grant, P. G., Cooperman, B. S., and Glitz, D. G. (1982) J. Biol. Chem., 257, 2649-2656.
9.Spirin, A. S., and Vasiliev, V. D. (1989) Biol. Cell, 66, 215-223.
10.Arad, T., Piefke, J., Weinstein, S., Gewitz, H. S., Yonath, A., and Wittman, H. G. (1987) Biochimie, 69, 1001-1005.
11.Yonath, A., Leonard, K. R., and Wittmann, H. G. (1987) Science, 236, 813-816.
12.Milligan, R. A., and Unwin, P. N. T. (1986) Nature, 319, 693-696.
13.Radermacher, M., Wagenknecht, T., Verschoor, A., and Frank, J. (1987) EMBO J., 6, 1107-1114.
14.Eisenstein, M., Sharon, R., Berkovitch-Yellin, Z., Gewitz, H. S., Weinstein, S., Pebay-Peyroula, E., Roth, M., and Yonath, A. (1991) Biochimie, 73, 879-886.
15.Yonath, A., and Wittmann, H. G. (1989) Trends Biochem. Sci., 14, 329-335.
16.Stark, H., Mueller, F., Orlova, E. V., Schatz, M., Dube, P., Erdemir, T., Zemlin, F., Brimacombe, R., and van Heel, M. (1995) Structure, 3, 815-821.
17.Stark, H., Orlova, E. V., Rinke-Appel, J., Junke, N., Mueller, F., Rodnina, M., Wintermeyer, W., Brimacombe, R., and van Heel, M. (1997) Cell, 88, 19-28.
18.Frank, J., Zhu, J., Penczek, P., Li, Y., Srivastava, S., Verschoor, A., Radermacher, M., Grassucci, R., Lata, R. K., and Agrawal, R. K. (1995) Nature, 376, 441-444.
19.Frank, J., Verschoor, A., Li, Y., Zhu, J., Lata, R. K., Radermacher, M., Penczek, P., Grassucci, R., Agrawal, R. K., and Srivastava, S. (1995) Biochem. Cell Biol., 73, 757-765.
20.Yonath, A., and Berkovitch-Yellin, Z. (1993) Curr. Opin. Struct. Biol., 3, 175-181.
21.Verschoor, A., Srivastava, S., Grassucci, R., and Frank, J. (1996) J. Cell Biol., 133, 495-505.
22.Verschoor, A., Warner, J. R., Srivastava, S., Grassucci, R., and Frank, J. (1998) Nucleic Acids Res., 26, 655-661.
23.Morgan, D. G., Menetret, J.-F., Radermacher, M., Neuhof, A., Akey, I. V., Rapoport, T. A., and Akey, C. W. (2000) J. Mol. Biol., 301, 301-321.
24.Dube, P., Weiske, M., Stark, H., Schatz, M., Stahl, J., Zemlin, F., Lutsch, G., and van Heel, M. (1998) Structure, 6, 389-399.
25.Seidelt, B., Innis, C. A., Wilson, D. N., Gartmann, M., Armache, J.-P., Villa, E., Trabuco, L. G., Becker, T., Mielke, T., Schulten, K., Steitz, T. A., and Beckmann, R. (2009) Science, 326, 1412-1415.
26.Becker, T., Bhushan, S., Jarasch, A., Armache, J.-P., Funes, S., Jossinet, F., Gumbart, J., Mielke, T., Berninghausen, O., Schulten, K., Westhof, E., Gilmore, R., Mandon, E. C., and Beckmann, R. (2009) Science, 326, 1369-1373.
27.Bhushan, S., Gartmann, M., Halic, M., Armache, J.-P., Jarasch, A., Mielke, T., Berninghausen, O., Wilson, D., and Beckmann, R. (2010) Nat. Struct. Mol. Biol., 17, 313-317.
28.Ryabova, L. A., Selivanova, O. M., Baranov, V. I., Vasiliev, V. D., and Spirin, A. S. (1988) FEBS Lett., 226, 255-260.
29.Beckmann, R., Bubeck, D., Grassucci, R., Penczek, P., Verschoor, A., Blobel, G., and Frank, J. (1997) Science, 278, 2123-2126.
30.Van den Berg, B., Clemons, W., Jr., Collinson, I., Modis, Y., Hartmann, E., Harrison, S., and Rapoport, T. (2004) Nature, 427, 36-44.
31.Mitra, K., Schaffitzel, C., Shaikh, T., Tama, F., Jenni, S., Brooks, C. L. III, Ban, N., and Frank, J. (2005) Nature, 438, 318-324.
32.Schaffitzel, C., Oswald, M., Berger, I., Ishikawa, T., Abrahams, J. P., Koerten, H., Koning, R., and Ban, N. (2006) Nature, 444, 503-506.
33.Bingel-Erlenmeyer, R., Kohler, R., Kramer, G., Sandikci, A., Antolic, S., Maier, T., Schaffitzel, C., Wiedmann, B., Bukau, B., and Ban, N. (2008) Nature, 452, 108-113.
34.Ferbitz, L., Maier, T., Patzelt, H., Bukau, B., Deuerling, E., and Ban, N. (2004) Nature, 431, 590-596.
35.Baram, D., Pyetan, E., Sittner, A., Auerbach-Nevo, T., Bashan, A., and Yonath, A. (2005) Proc. Natl. Acad. Sci. USA, 102, 12017-12022.
36.Addlagatta, A., Quillin, M. L., Omotoso, O., Liu, J. O., and Matthews, B. W. (2005) Biochemistry, 44, 7166-7174.
37.Adams, J. M. (1968) J. Mol. Biol., 33, 571-589.
38.Takeda, M., and Webster, R. E. (1968) Proc. Natl. Acad. Sci. USA, 60, 1487-1494.
39.Miyaguchi, K., and Reese, T. S. (1996) J. Struct. Biol., 116, 413-417.
40.Ban, N., Freeborn, B., Nissen, P., Penczek, P., Grassucci, R. A., Sweet, R., Frank, J., Moore, P. B., and Steitz, T. A. (1998) Cell, 93, 1105-1115.
41.Ban, N., Nissen, P., Hansen, J., Capel, M., Moore, P. B., and Steitz, T. A. (1999) Nature, 400, 841-847.
42.Ban, N., Nissen, P., Hansen, J., Moore, P. B., and Steitz, T. A. (2000) Science, 289, 905-920.
43.Nissen, P., Hansen, J., Ban, N., Moore, P. B., and Steitz, T. A. (2000) Science, 289, 920-930.
44.Cate, J., Yusupov, M. M., Yusupova, G. Zh., Earnest, T. N., and Noller, H. F. (1999) Science, 285, 2095-2104.
45.Yusupov, M. M., Yusupova, G. Zh., Baucom, A., Lieberman, K., Earnest, T. N., Cate, J. H. D., and Noller, H. F. (2001) Science, 292, 883-896.
46.Harms, J., Schluenzen, F., Zarivach, R., Bashan, A., Gat, S., Agmon, I., Bartels, H., Franceschi, F., and Yonath, A. (2001) Cell, 107, 679-688.
47.Eisenstein, M., Hardesty, B., Odom, O. W., Kudlicki, W., Kramer, G., Arad, T., Franceschi, F., and Yonath, A. (1994) in Biophysical Methods in Molecular Biology (Pifat, G., ed.) Balaban Press, Rehovot, Izrael, pp. 213-246.
48.Makeyev, E., Kolb, V., and Spirin, A. (1996) FEBS Lett., 378, 166-170.
49.Kolb, V. A., Makeyev, E. V., Kommer, A., and Spirin, A. S. (1995) Biochem. Cell Biol., 73, 1217-1220.
50.Kolb, V., Makeyev, E., and Spirin, A. (1994) EMBO J., 13, 3631-3637.
51.Kolb, V., Makeyev, E., and Spirin, A. (2000) J. Biol. Chem., 275, 16597-16601.
52.Bornemann, T., Jockel, J., Rodnina, M., and Wintermeyer, W. (2008) Nat. Struct. Mol. Biol., 15, 494-499.
53.Wiedmann, B., Sakai, H., Davis, T. A., and Wiedmann, M. (1994) Nature, 370, 434-440.
54.Moller, I., Beatrix, B., Kreibich, G., Sakai, H., Lauring, B., and Wiedmann, M. (1998) FEBS Lett., 441, 1-5.
55.Wang, S., Sakai, H., and Wiedmann, M. (1995) J. Cell Biol., 130, 519-528.
56.Lim, V. I., and Spirin, A. S. (1986) J. Mol. Biol., 188, 565-574.
57.Lu, J., and Deutsch, C. (2005) Biochemistry, 44, 8230-8243.
58.Lu, J., and Deutsch, C. (2005) Nat. Struct. Mol. Biol., 12, 1123-1129.
59.Hardesty, B., Picking, W. D., and Odom, O. W. (1990) Biochim. Biophys. Acta, 1050, 197-202.
60.Picking, W. D., Odom, O. W., Tsalkova, T., Serdyuk, I., and Hardesty, B. (1991) J. Biol. Chem., 266, 1534-1542.
61.Odom, O. W., Picking, W. D., Tsalkova, T., and Hardesty, B. (1991) Eur. J. Biochem., 198, 713-722.
62.Picking, W. D., Picking, W. L., Odom, O. W., and Hardesty, B. (1992) Biochemistry, 31, 2368-2375.
63.Crowley, K. S., Reinhart, G. D., and Johnson, A. E. (1993) Cell, 73, 1101-1115.
64.Crowley, K. S., Liao, S., Worrell, V. E., Reinhart, G. D., and Johnson, A. E. (1994) Cell, 78, 461-471.
65.Hamman, B. D., Chen, J.-C., Johnson, E. E., and Johnson, A. E. (1997) Cell, 89, 535-544.
66.Lu, J., Kobertz, W., and Deutsch, C. (2007) J. Mol. Biol., 371, 1378-1391.
67.Ramachandiran, V., Willms, C., Kramer, G., and Hardesty, B. (2000) J. Biol. Chem., 275, 1781-1786.
68.Komar, A. A., Kommer, A., Krasheninnikov, I. A., and Spirin, A. S. (1997) FEBS Lett., 326, 261-263.
69.Komar, A. A., Kommer, A., Krasheninnikov, I. A., and Spirin, A. S. (1997) J. Biol. Chem., 272, 10646-10651.
70.Voss, N. R., Gerstein, M., Steitz, T. A, and Moore, P. B. (2006) J. Mol. Biol., 360, 893-906.
71.Stade, K., Riens, S., Bochkariov, D., and Brimacombe, R. (1994) Nucleic Acids Res., 22, 1394-1399.
72.Stade, K., Junke, N., and Brimacombe, R. (1995) Nucleic Acids Res., 23, 2371-2380.
73.Choi, K. M., and Brimacombe, R. (1998) Nucleic Acids Res., 26, 887-895.
74.Choi, K. M., Atkins, J. F., Gesteland, R. F., and Brimacombe, R. (1998) Eur. J. Biochem., 255, 409-413.
75.Brodersen, D. E., Clemons, W. M., Jr., Carter, A. P., Wimberly, B. T., and Ramakrishnan, V. (2002) J. Mol. Biol., 316, 725-768.
76.Schmeing, T. M., Seila, A. C., Hansen, J., Freeborn, B., Soukup, J. K., Scaringe, S. A., Strobel, S. A., Moore, P. B., and Steitz, T. A. (2002) Nature Struct. Biol., 9, 225-230.
77.Tenson, T., and Ehrenberg, M. (2002) Cell, 108, 591-594.
78.Nakatogava, H., and Ito, K. (2002) Cell, 108, 629-636.
79.Muto, H., Nakatogawa, H., and Ito, K. (2006) Mol. Cell, 22, 545-552.