REVIEW: 14-3-3 Proteins and Regulation of Cytoskeleton
N. N. Sluchanko and N. B. Gusev*
Department of Biochemistry, Faculty of Biology, Lomonosov Moscow State University, 119991 Moscow, Russia; E-mail: NBGusev@mail.ru* To whom correspondence should be addressed.
Received April 9, 2010
The proteins of the 14-3-3 family are universal adapters participating in multiple processes running in the cell. We describe the structure, isoform composition, and distribution of 14-3-3 proteins in different tissues. Different elements of 14-3-3 structure important for dimer formation and recognition of protein targets are analyzed in detail. Special attention is paid to analysis of posttranslational modifications playing important roles in regulation of 14-3-3 function. The data of the literature concerning participation of 14-3-3 in regulation of intercellular contacts and different elements of cytoskeleton formed by microfilaments are analyzed. We also describe participation of 14-3-3 in regulation of small G-proteins and protein kinases important for proper functioning of cytoskeleton. The data on the interaction of 14-3-3 with different components of microtubules are presented, and the probable role of 14-3-3 in developing of certain neurodegenerative diseases is discussed. The data of the literature concerning the role of 14-3-3 in formation and normal functioning of intermediate filaments are also reviewed. It is concluded that due to its adapter properties 14-3-3 plays an important role in cytoskeleton regulation. The cytoskeletal proteins that are abundant in the cell might compete with the other protein targets of 14-3-3 and therefore can indirectly regulate many intracellular processes that are dependent on 14-3-3.
KEY WORDS: 14-3-3, phosphorylation, cytoskeletonDOI: 10.1134/S0006297910130031
Abbreviations: AANAT, arylalkylamine N-acetyltransferase; GSK3, glycogen synthase kinase 3; MAPKAP, protein kinase activated by MAP-kinase; NES, nuclear export sequence; NLS, nuclear localization sequence; PKA, cAMP-dependent protein kinase (protein kinase A); PKB, protein kinase B (PKB/Akt); PKC, protein kinase C; TPR-domain, tetratricopeptide repeat domain (34-residue domain participating in protein–protein interactions).
Phosphorylation of Ser, Thr, and/or Tyr residues catalyzed by different
protein kinases is one of the ways of posttranslational modification
affecting the structure and properties of many proteins. Transfer of a
phosphate group is accompanied by an increase in negative charge that
might lead to large conformational changes affecting the structure,
properties, and functional activity of phosphorylated proteins. There
are a number of adapter proteins recognizing and specifically
interacting with certain sites of phosphorylated proteins; proteins of
the 14-3-3 family form one of the groups of such adapter proteins.
These proteins were first described about 40 years ago during the
systematic characterization of nervous tissue, where their content
exceeds 1% of the total proteome [1, 2]. At present more than 300 protein targets
interacting with 14-3-3 are described in the literature [3]. The members of the 14-3-3 family are ubiquitous
and participate in regulation of apoptosis, cell cycle, proliferation,
transcription, replication, functioning of ion channels, and
organization of cytoskeleton. Recently published data indicate that
14-3-3 proteins play an important role in neurodegenerative diseases
and carcinogenesis. This leads to growing interest to the investigation
of the structure and properties of 14-3-3 proteins.
This review describes the structure, properties, and mechanism of action of 14-3-3 proteins, analysis of their interaction with protein targets, and their probable involvement in regulation of cytoskeleton.
STRUCTURE AND PROPERTIES OF 14-3-3 PROTEINS
Historical Overview and Nomenclature
The proteins of the 14-3-3 family were first described in 1967 in the process of systematic classification of nervous tissue proteins [4]. The peculiar name reflects the fraction number enriched in these proteins on ion-exchange chromatography of bovine brain extract and the position of these proteins on starch gel electrophoresis. These proteins are abundant in brain, have low pI value (4.0-4.5), and the molecular mass of the 14-3-3 monomer is close to 30 kDa [5].At first it was supposed that 14-3-3 participates in regulation of neurotransmitter synthesis. In 1987 it was shown that 14-3-3 activates key enzymes of serotonin and catecholamine synthesis, i.e. tryptophan- and tyrosine monooxygenases [6]. Later the heterogeneous sample of brain 14-3-3 was separated by reverse-phase high-pressure liquid chromatography, and several isoforms marked by Greek letters from α to σ were obtained in purified state [7]. This type of separation revealed nine isoforms of 14-3-3, so-called α, β, γ, δ, ε, ζ, η, τ (or θ), and σ. Further investigations revealed that the α isoform is indistinguishable from the β isoform phosphorylated at Ser184, whereas the δ isoform is identical to the ζ isoform also phosphorylated at Ser184 (through this review we use the numbering of amino acid residues corresponding to the ζ isoform of human 14-3-3) [8].
It is worth mentioning that except for the classical designation 14-3-3 with the corresponding Greek letter, the same proteins are sometimes marked as Leonardo (Drosophila 14-3-3ε), Bilardo (Drosophila 14-3-3ζ), Stratifin (14-3-3σ), BAP-1 (14-3-3τ/θ), CBP (Cruciform-Binding Protein), KCIP-1 (protein kinase C inhibitor-1), Exo1 (stimulator of Ca2+-dependent exocytosis), GF14 (plant G-box binding factor), etc. [5].
In the beginning of the 1990s it was shown that 14-3-3 undergoes phosphorylation [9], participates in regulation of certain protein kinases [10], and is widely expressed in different tissues of eukaryotes [11-13]. All these facts attracted attention of many investigators and led to the beginning of intensive investigation of 14-3-3.
The 3D structure of mammalian 14-3-3ζ and τ was described in 1995 [14, 15]. 14-3-3 form dimers, and the monomers of the dimers are easily exchangeable. This leads to formation of both homo- and heterodimers of 14-3-3 [16]. The dimeric structure is necessary for normal functioning of 14-3-3, and destabilization of the dimer decreases the interaction of 14-3-3 with different target proteins (i.e. Raf kinase and p53) [17, 18].
Isoforms and Phylogeny of the 14-3-3 Family
14-3-3 proteins are ubiquitous and are detected in practically all eukaryotes. Moreover, as a rule each species contains more than two different isoforms of this protein. The rare exclusions are the fungus Candida albicans and the protozoan Dictyostelium discoideum, which contain a single isoform of 14-3-3. Although at the beginning only one isoform of 14-3-3 was detected in Drosophila [19], later investigations revealed at least two (ε and ζ) isoforms of 14-3-3 in this organism [20].The members of 14-3-3 family are rather conservative. For instance, the homology of the frog Xenopus tropicalis 14-3-3ζ and the corresponding human protein determined by the BLAST program (http://www.ncbi.nlm.nih.gov/blast/Blast.cgi) is close to 90%. Homology of the primary structure of human 14-3-3ζ with the corresponding proteins of Bombyx mori and Drosophila melanogaster are close to 81-83% [21]. The identity of plant (Arabidopsis thaliana) 14-3-3ω and its nearest human analog is close to 75%. Homology of 14-3-3 isoforms isolated from the same species is even higher, and this complicates obtaining antibodies specific to particular isoforms of 14-3-3 [19].
As already mentioned, human tissues contain seven 14-3-3 isoforms, i.e. β, γ, ε, η, σ, τ (which is also designated as θ), and ζ [7]. Earlier described α and δ isoforms are indistinguishable from phosphorylated β and ζ isoforms, respectively [8]. The isoforms of 14-3-3 differ in the structure of short variable stretches of the primary structure. However, they are not the products of alternative splicing and each isoform is coded by separated gene [19].
The genes of 14-3-3 isoforms are usually located on different chromosomes. For instance, the genes of human η and β isoforms are located on the 22nd and 20th chromosomes [22]. These data agree well with the results obtained with the Human Genome Database. According to Aitken, the genes coding β, η, σ, τ/θ, γ, and ε isoforms are located on chromosomes 20, 22, 1, 2, 7, and 17, respectively. The main gene encoding the full-size 14-3-3ζ is located on the chromosome 8 [19]. In addition to the so-called working genes, the human genome contains a large number of pseudogenes that do not participate in expression of functionally active proteins [5]. For instance, pseudogenes of the ε isoform were detected on chromosomes 2 and 7, whereas pseudogenes of the ζ isoform were detected on chromosomes 5, 6, 9, 11, and 15. Several sequences corresponding to σ-isoform were also detected in the human genome [19]. Our search for the main working genes of different 14-3-3 isoforms using the BLAT program of the Human Genome Browser (http://genome.ucsc.edu/cgi-bin/hgBlat?command=start) confirms the presence of a number of 14-3-3 pseudogenes. It is probable that these pseudogenes were formed by inserting into chromosomes of DNA fragments formed by retroviral reverse transcriptase from mRNA of different isoforms of intracellular 14-3-3.
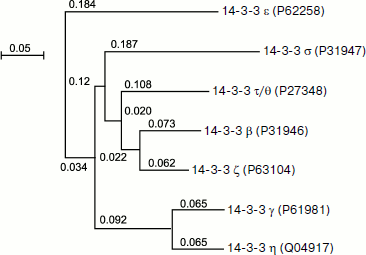
Figure 1 represents the phylogenetic tree depicting homology of the primary structure of different human isoforms of 14-3-3. The tree was built by multiple alignment of the primary structure using the ClustalW v.1.83 program (http://www.ebi.ac.uk/Tools/clustalw/index.html). Alignment was performed on the basis of Gonnet and Blosum matrices with parameters gap open = 10 and extension gap = 0.05 and provides similar results for both matrices, which are presented as a tree using the NJplot program (http://pbil.univ-lyon1.fr/software/njplot.html). The data of Fig. 1 indicate that the primary structure of certain pairs of 14-3-3 isoforms, namely η and γ, β and ζ, as well as σ and τ/θ are especially similar to each other. The data presented might indicate the presence of a common precursor of the six above-mentioned isoforms of 14-3-3. At the same time, the ε isoform undergoes the largest changes in the course of evolution and is significantly different from the other six isoforms of 14-3-3 [23].
Fig. 1. Phylogenetic tree of seven 14-3-3 isoforms constructed using ClustalW. The identification numbers of the corresponding isoforms in the UniprotKB database are indicated in parentheses. The scale corresponds to 0.05 replacements in the ancestral polypeptide per amino acid residue.
Distribution of 14-3-3 in Human Tissues
14-3-3 proteins are ubiquitously expressed in human tissues. Data of radioimmunoassay indicate high content of 14-3-3 in brain, testis, and intestine where its content is higher than 0.5% of all soluble proteins. In the case of brain the level of 14-3-3 is close to 1.3% of soluble protein, which is equal to about 40 µM calculated per 14-3-3 monomer [1]. At the same time kidney and hemolysate contain low quantities of 14-3-3, which are close to 0.008% of soluble protein [1]. Immunofluorescence and Western blotting data confirm the presence of all seven isoforms of 14-3-3 in cornea and eye mucosa as well as the presence of ζ and γ isoforms in human tear fluid [24].At present 14-3-3 is considered as a perspective marker of different neurodegenerative diseases and carcinogenesis [25, 26]. Indeed, it has been found that the concentration of 14-3-3 in cerebrospinal fluid is significantly increased in the case of different forms of dementia, encephalopathies, and tumors of the central nervous system. Increased concentration of 14-3-3 in the liquor was also detected in the case of Creutzfeldt–Jakob disease [23, 27]. In the case of tuberculous meningitis and multiple sclerosis, the level of 14-3-3 in liquor is increased up to 100-130 ng/ml [1]. Moreover, the level of 14-3-3 is increased in different forms of cancer [28-30]. All these data make 14-3-3 a perspective marker of different human diseases.
Structural Organization of 14-3-3
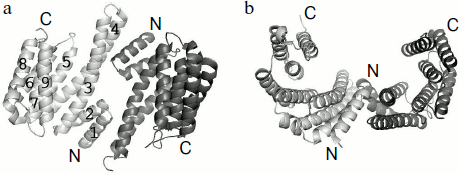
The 3D structure of all seven isoforms of human 14-3-3 is described in the literature [31, 32]. The data of X-ray crystallography indicate that the 14-3-3 subunits form a dimer that is similar to a horseshoe with an inner channel depth and width of about 20 and 35 Å, respectively (Fig. 2) [14, 15]. The linear size of this dimeric structure calculated by the PyMOL v.1.1 program using the X-ray crystallography data obtained on human 14-3-3ζ (PDB identification number 1QJB) is close to 55-60 Å in width, about 75-80 Å in length, and about 35-40 Å in height. α-Helices are the main structural elements of 14-3-3. Theoretical predictions performed by Psipred and Sable2 programs indicate that about 65-75% of 14-3-3 structure consists of α-helices. These predictions correlate well with the data of CD spectroscopy [33, 34] and X-ray crystallography (PDB ID 1QJB) showing that about 70% of 14-3-3 is indeed α-helical.
Each monomer of 14-3-3 consists of nine α-helices indicated by Arabic numbers starting from the N-terminal α1 up to the C-terminal α9 (Fig. 2). Helices α5-α9 form the side walls of a cup-like structure. Amino acid residues of helices α1-α4 form the bottom of the dimeric structure where two subunits interact with each other. At this site the α1 and α2 helices of one monomer interact with the α3 and α4 helices of the second subunit, so the subunits of 14-3-3 are oriented antiparallel to each other. The α3 and α4 helices are the longest (about 55 Å), whereas the α1 and α2 helices are the shortest (about 25 Å in length) (Fig. 2).Fig. 2. α-Helical structure of 14-3-3 dimer. The PyMOL program was used for presentation of X-ray crystallography data obtained on 14-3-3ζ (PDB ID 1QJB): a) top view; b) side view of 14-3-3 dimer. Two subunits are shown in light and dark gray; numbers correspond to the number of α-helices in the 14-3-3 monomer.
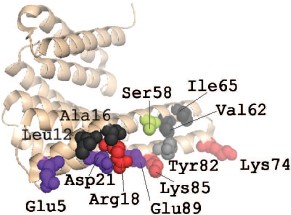
As already mentioned, the dimeric structure is important for proper functioning of 14-3-3. The dimeric structure of 14-3-3ζ is probably stabilized by three salt bridges formed by Arg18-Glu89, Glu5-Lys74, and Asp21-Lys85 (there are six such bridges in the dimer) and by a number of weakly polar and hydrophobic interactions with participation of Leu12, Ala16, Ser58, Val62, Ile65, and Tyr82 (Fig. 3; see color insert) [32]. The first salt bridge, Arg18-Glu89, and all the above mentioned weak contacts are found in homodimers formed by all human 14-3-3 isoforms. The second salt bridge, Glu5-Lys74, is absent in the dimers of the σ, η, ε, and γ isoforms. The third salt bridge, Asp21-Lys85, participates in formation of dimers of all 14-3-3 isoforms except that of ε. In the case of the σ isoform an alternative salt bridge, Lys9-Glu83, is postulated in the literature [32]. The X-ray crystallographic data were confirmed by means of site directed mutagenesis. Indeed, mutation of the residues 5 and 12 in the α1-helix, as well as of residues 82, 85, and 87 in the α4-helix of 14-3-3ζ, lead to destabilization of intersubunit contacts [35]. Similar results were obtained with the σ isoform. Mutation of residues in position 5 of the α1-helix, position 20 of the α2-helix, as well as position 55 of the α3- and position 80 in α4-helices were accompanied by changes in the intersubunit contacts of this isoform of 14-3-3 and to changes in the interaction of σ isoform with other isoforms of this protein [35]. It is supposed that Ser58 also plays an important role in dimer formation (Fig. 3). This residue is located in the α3-helix of the ζ, β, ε, γ, and η isoforms of 14-3-3 (isoforms σ and τ contain Ala in this position). The data in the literature indicate that phosphorylation of this residue might significantly affect the state of the 14-3-3 dimer [18, 36, 37].
Formation of homo- and heterodimers is thought to be one of the factors affecting the specificity of 14-3-3 towards different protein targets [16, 35]. It is worthwhile to mention that only certain isoforms of 14-3-3 are able to form heterodimers. For instance, there are non-conservative replacements in the site of interaction of 14-3-3σ. The non-conservative residues Ser5, Glu20, Phe25, Gln55, and Glu80 of this isoform provide for formation of homodimers, but prevent formation of heterodimers. This leads to unique properties of 14-3-3σ [35, 38].Fig. 3. Site of interaction of 14-3-3 monomers. The PyMOL program was used for presentation of X-ray crystallography data obtained on 14-3-3ζ (PDB ID 1QJB). The structure of 14-3-3 monomer is shown as beige helices. Uncharged residues involved in dimerization are shown in black and gray. Ser58 participating in regulation of dimerization is shown in green. The residues involved in formation of salt bridges between 14-3-3 monomers are shown in red (positively charged) and violet (negatively charged).
Formation of heterodimers containing different isoforms increases the possibilities of 14-3-3. Heterodimers composed of ε and ζ or of τ and ζ isoforms might be useful for the binding of certain substrates or provide for interaction of two different protein targets that in the isolated state do not interact with each other. For instance, it has been shown that Nedd4-2, having several sites of 14-3-3 binding and involved in aldosterone-dependent regulation of sodium channels, interacts with heterodimer composed of β and ε isoforms of 14-3-3 [39]. Formation of 14-3-3 heterodimers is also necessary for the binding of cruciform structures of DNA, which are often located at replication origin and might determine the level of replication [40].
Analysis of the primary structure and the domain search performed using the InterProScan program (http://www.ebi.ac.uk/InterProScan) did not reveal any similarities between 14-3-3 family proteins and the domains of other proteins present in the rather wide database of InterPro. However, Ostrerova et al. supposed that members of the 14-3-3 family are to some extent homologous to synuclein, which is highly expressed in brain. Analysis of the primary structure of 14-3-3 and synuclein revealed two short peptides having 40% homology [41]. Moreover, it has been shown that synuclein and 14-3-3 interact with each other and are able to bind the same protein targets, such as protein kinase C and Bad. Therefore, it was concluded that synuclein and 14-3-3 have similar functions [41].
It is worthwhile mentioning that Korean scientists [42] also detected the similarity of 14-3-3 and synuclein. However, the similarity was detected in sites that were completely different from the sites postulated by Ostrerova et al. [41]. The primary structure of the similar sites detected by the Korean group in 14-3-3 and synuclein resembles that of the crystallin domain of small heat shock proteins. Probably this is the reason why both synuclein and 14-3-3 have chaperone-like activity.
Recently published data indicate that plant 14-3-3 GF14ω (general factor 14ω), which is homologous to human 14-3-3ζ, contains a sequence similar to that of the canonical EF-hand involved in the binding of divalent cations (Ca2+ and Mg2+) [43]. Comparing the structure of plant GF14ω and human 14-3-3ζ using the BLAST program, we detected in the human protein a sequence identical to that of its plan counterpart, namely ELDTLSEESYKD. This sequence is remotely similar to the loop of the EF-hand. However, the experimental data of Aitken’s group failed to confirm the Ca2+-binding properties of mammalian 14-3-3 [33].
Analysis of the structure of 14-3-3, p53, and IκBα revealed that the α9-helix, located on the very C-terminus of 14-3-3, contains a 13-residue peptide having primary structure corresponding to the canonical nuclear export sequence (NES) providing for the export of the protein from the nucleus [44]. It was supposed that this sequence is important for intracellular location of 14-3-3 and its interaction with cytosolic proteins [44, 45]. However, later investigations did not confirm the presence of NES in the structure of 14-3-3. The intracellular distribution of 14-3-3 seems to be dependent on the ability of 14-3-3 to mask or unmask the nuclear export sequence located in the primary structure of the target proteins [32, 46]. All attempts to determine nuclear location sequence (NLS) inside 14-3-3 were unsuccessful [47]. It is supposed that the ability to enter nucleus is due to formation of tight complexes between 14-3-3 and certain target proteins carrying in their primary structure a typical NLS. However, it is difficult to exclude the possibility that 14-3-3 contains an unusual signal sequence that remains undetermined and which is responsible for transportation of 14-3-3 from cytosol to nucleus [48].
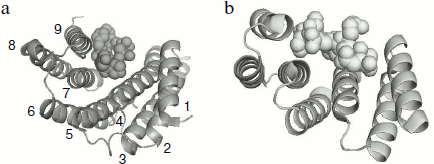
A tetratricopeptide repeat domain (TPR-domain) was detected in the C-terminal part of 14-3-3 [49]. This domain having similar primary, secondary, and tertiary structure was earlier detected in the structure of different proteins. This domain is usually composed of 34-residue repeats formed by a pair of antiparallel α-helices separated by a turn. The presence of a number of these repeats can lead to formation of a bundle of α-helices rotated from each other by a certain angle. Figure 4 presents the comparison of the structure of monomer of 14-3-3 with the TPR-domain of p60 (or Hop), which provide for interaction of heat shock proteins Hsp70 and Hsp90.
These elements of tertiary structure are ideal for the binding of different ligands and are detected in the primary structure of more than 800 proteins performing different functions (http://pawsonlab.mshri.on.ca/index.php?option=com_content&task=view&Itemid=64&id=183). It is probable that the TPR-domain formed by the C-terminal α-helices of interacting subunits are important for positioning of the target proteins, which are bound in the internal channel of 14-3-3 (see below).Fig. 4. Comparison of the tertiary structure of 14-3-3 monomer binding phosphopeptide (a) and the TPR-domain of Hop protein interacting with a fragment of Hsp70 (b). The PyMOL program was used for depicting data of X-ray crystallography of 14-3-3 (PDB ID 1QJB) and Hop (PDB ID 1ELW). Ribbon models are used for showing 14-3-3 α-helices (numbered from 1 to 9) and Hop structure. Space filling models are used for showing the structure of ligands.
Regulation of 14-3-3 by Phosphorylation
The functioning of 14-3-3 can be regulated in a number of different ways. First, regulation can be achieved by changes in 14-3-3 content. With cell models it was shown that overexpression of 14-3-3 is accompanied by increase in total activity of Raf-1 and decrease in protein kinase C activity [50]. As already mentioned, suppression of 14-3-3 expression (or at least decrease in expression of σ isoform) can lead to development of oncological diseases. At the same time, serious damages of DNA are accompanied by p53-dependent induction of 14-3-3σ synthesis [50]. Second, certain isoforms of 14-3-3 are caspase substrates. Limited proteolysis of certain isoforms of 14-3-3 leads to liberation of proapoptotic proteins (Bad and Bax), resulting in initiation of apoptosis [51, 52]. Third, 14-3-3 undergoes different posttranslational modifications. For instance, acetylation of Lys residues of 14-3-3 involved in substrate binding might significantly affect protein–protein interactions [53]. Moreover, 14-3-3 proteins contain a number of sites phosphorylated by different protein kinases. Protein kinase SDK1 (which is a product of caspase cleavage of δ isoform of protein kinase C) [54], protein kinase A (PKA) activated by sphingosine (but not by cAMP) [55], certain isoforms of protein kinase C [5], protein kinase B (PKB/Akt) [56], as well as protein kinase JNK [57], Bcr (the product of oncogene Breakpoint cluster region protein) [58], and α isoform of casein kinase I (CKI) [59] phosphorylate Ser58, Ser63, Ser184, and Thr232 of 14-3-3. Phosphorylation seems to affect the structure and properties of 14-3-3; however, despite many efforts a clear-cut answer to the question how phosphorylation affects 14-3-3 functioning remains elusive.Let us analyze the effect of phosphorylation of different sites on the structure and properties of 14-3-3. It is well accepted that the extent of Ser63 phosphorylation is rather low and therefore seems to be not very important [60]. However, Ser58, Ser184, and Thr232 are effectively phosphorylated in vivo [8, 54, 59] and therefore analysis of phosphorylation of these sites is of great interest. It is worthwhile mentioning that these sites are not completely conservative and therefore not all isoforms of 14-3-3 contain Ser or Thr in the corresponding positions. Moreover, due to the difference in the primary structure the same enzyme (SDK1) phosphorylates Ser58 of 14-3-3ζ and is unable to phosphorylate the corresponding residues of the ε and γ isoforms of 14-3-3 [54].
There is no doubt that Ser58 of 14-3-3 is phosphorylated by a number of different protein kinases both in vitro and in vivo. However, there is no agreement on the question how Ser58 phosphorylation affects the structure and properties of 14-3-3. According to Powell et al. [56], protein kinase B (PKB/Akt) phosphorylates Ser58 of 14-3-3ζ, and this modification does not affect the stability of the 14-3-3 dimer. This conclusion contradicts later published data. For instance, Gu et al. and Powell et al. found that PKA- or MAPKAP2 kinase-catalyzed phosphorylation of Ser58 of 14-3-3ζ or the S58E/D mutation mimicking phosphorylation lead to dimer dissociation and accumulation of 14-3-3 monomers [18, 36]. Therefore, it was postulated that phosphorylation of Ser58 located at the intersubunit contact (Fig. 3) plays an important role in the regulation of the oligomeric state of 14-3-3. Investigation of the S58E point mutant mimicking phosphorylation revealed that the replacement of Ser by Glu leads to dimer dissociation, decreases the thermostability, and increases the proteolytic susceptibility of 14-3-3ζ [34]. These changes are especially pronounced at low protein concentration and might indicate that Ser58 phosphorylation provokes dissociation of the 14-3-3 dimer. Phosphorylation of Ser58 not only induces dissociation of the 14-3-3 dimer, but it also decreases the interaction of 14-3-3 with the transcriptional factor p53 [18]. The S58D mutation mimicking phosphorylation also strongly impairs the interaction of 14-3-3ζ with Raf-1 [36] and cyclin D1 [61]. It is probable that the decrease in protein–protein interaction is due to the fact that only dimers of 14-3-3 form tight complexes with the protein targets. Further investigations will be needed for elucidation of the effect of Ser58 phosphorylation on the regulation of the activity of 14-3-3.
Investigation of the physicochemical properties of 14-3-3ζ mutant S184E revealed that this mutation mimicking phosphorylation increased the thermostability and slightly increased the Stokes radius without affecting the dimeric structure and susceptibility to proteolysis [34]. It seems probable that Ser184 phosphorylation is not accompanied by large changes in the protein structure. Nevertheless, induced by DNA damage and catalyzed by c-Jun NH2-terminal kinase (JNK) phosphorylation of Ser184 of 14-3-3ζ leads to dissociation of proapoptotic protein Bax from its complex with 14-3-3. Liberated Bax migrates to mitochondria, increases permeability of the mitochondrial membranes, and induces apoptosis [57]. Moreover, JNK-induced phosphorylation of 14-3-3ζ results in dissociation of Bad, c-Abl, and FOXO3a from their complexes with 14-3-3, and this also increases the probability of apoptosis. In this case both Bad and Bax migrate to mitochondria, whereas transcriptional factor FOXO3a and tyrosine kinase c-Abl moves to the cell nucleus. In response to DNA damage these proteins initiate transcription of other proapoptotic proteins finally leading to apoptosis [62]. On the other side, phosphorylation of Ser184 increases the affinity of 14-3-3 to other protein targets. For instance, phosphorylation of Ser184 increases stability of the complex formed by 14-3-3 and protein kinase C, affecting the activity of this enzyme [63].
Analysis of the physicochemical properties of the T232E mutant of 14-3-3ζ has shown that this mutation mimicking phosphorylation induces only a small increase in Stokes radius and sedimentation coefficient and is not accompanied by significant changes in the secondary, tertiary, or quaternary structure, thermostability, or susceptibility of the 14-3-3 to proteolysis [34]. Nevertheless, phosphorylation of Thr232 located in the C-terminal part of 14-3-3 decreases its interaction with different protein substrates. It is probable that this effect is due to the fact that the C-terminal ends can overhang the internal 14-3-3 channel and in this way interact with the sites responsible for the binding of protein substrates (see below) [15, 64]. It is supposed that such an auto-inhibitory effect can be modified by phosphorylation of Thr232 leading to reorientation of the C-terminal end of the 14-3-3 [65]. Circadian rhythms lead to the cyclic phosphorylation of Thr232. Phosphorylation at this site decreases the activating effect of 14-3-3 on arylalkylamine N-acetyltransferase (AANAT) and in this way inhibits conversion of serotonin to melatonin [65]. Phosphorylation of Thr232 impedes normal binding of 14-3-3 with cRaf [59]. The changes in 14-3-3–cRaf interaction might play an important role in the regulation of the Ras/Raf signal pathway.
Summing up, we conclude that phosphorylation affects the physicochemical properties of 14-3-3 and can be involved in the regulation of the interaction of 14-3-3 with different protein targets. However, it is worth mentioning that the interaction of 14-3-3 with protein substrates is predominantly regulated by phosphorylation of the protein targets. Specific phosphorylation sites of protein targets are recognized by 14-3-3 and form the site of tight interaction of the protein substrates with 14-3-3.
Mechanisms of Interaction of 14-3-3 with Protein Substrates. Sequences Recognized by 14-3-3
Numerous investigations have shown that phosphorylation of Ser and/or Thr usually increases the interaction of target proteins with 14-3-3 significantly. Proteomic investigations revealed more than 300 potential protein partners of 14-3-3 [3]. The interaction of 14-3-3 with the target proteins is based on recognition of specific consensus sequences in the ligand molecules. 14-3-3 tightly interacts with different proteins having at least one of two consensus motifs. These motifs contain phosphoserine (pS) (or phosphorylated threonine, pT) and are designated as motif I (RSXpS/pTXP) and motif II (RXY/FXpS/pTXP). Phosphorylated serine can be replaced by phosphorylated threonine in these motifs [66]. In addition, there is the third motif (motif III) that is also recognized by 14-3-3. This motif has the structure pS/pTX1-2-COOH, where X is any amino acid residue (except Pro) and is located on the very C-terminal end of the protein targets [67]. It is supposed that if the site of 14-3-3 binding is located in the middle of the target protein molecule then Pro (or Gly) should be located in position +2 relative to phosphorylated Ser or Thr (compare motifs I and II). The presence of Pro (or Gly) in this position provides for a sharp turn or increased flexibility of the polypeptide chain that is necessary for the proper positioning of the target in 14-3-3 channel. In the case of motif III the site of 14-3-3 binding is located on the very C-terminal end of the target protein and therefore the presence of Pro (or Gly) in position +2 is not necessary. This fact significantly increases the repertoire of proteins interacting with 14-3-3 [67].It is worth mentioning that in addition to the central phosphorylated Ser, motif I contains a second Ser located in position –2. This residue can also be phosphorylated. In the case of p53 and CDC25C, phosphorylation of this site decreases their interaction with 14-3-3 [68].
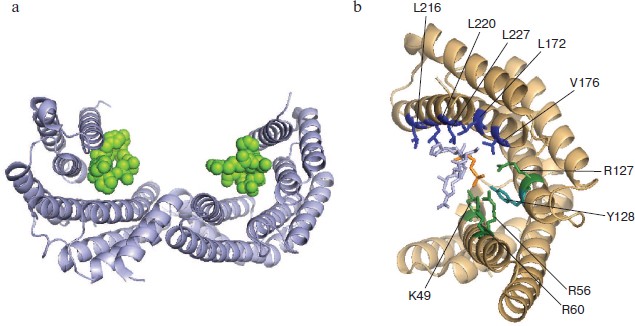
Model phosphopeptides and point mutations have provided important information on amino acid residues of 14-3-3 involved in target binding (Fig. 5; see color insert) [44, 69]. Each L-shaped 14-3-3 monomer contains an amphipathic groove (Fig. 5a) that plays an important role in target binding. The main role in the binding of target protein phosphopeptides belongs to the so-called pocket consisting of positively charged residues K49, R56, R60, R127, and Y128. These residues coordinate phosphoserine -pS- of the peptide interacting with 14-3-3 (Fig. 5b) [15, 32]. Zhang et al. demonstrated the importance of these residues in the interaction of 14-3-3 with Raf-1 and ExoS [69]. Positioning of the substrate molecule and stabilization of the complex formed is achieved with participation of hydrophobic residues of α-helices 5, 7, and 9 located on the inner wall of 14-3-3 dimer (Fig. 5b) [15]. Mutation of Leu172, Val176, and Leu220 belonging to α-helix 9 impairs the interaction of 14-3-3 with Raf-1 [70]. Mutations of Leu216 and Leu227 also decrease the interaction of 14-3-3 with Raf-1, but the effect of this mutation is not so pronounced [70]. Amino acid residues covering the inner surface of the 14-3-3 dimer are very conservative [15, 19]. This probably indicates a common principle of recognition of the protein ligands by all members of the 14-3-3 family [5].
As mentioned earlier, 14-3-3 forms stable dimers containing two potential sites of target binding. This means that 14-3-3 can simultaneously bind two protein targets or cooperatively interact with two sites of the same protein target. Yaffe et al. demonstrated that the presence of two sites containing phosphoserine and recognized by 14-3-3 leads to 30-fold increase in stability of complexes formed by target phosphopeptide and 14-3-3 [66]. Similar results were obtained by Obsil et al. [71]. These authors have shown that the fragment of transcriptional factor FOXO411-213 phosphorylated at Thr28 and Ser193 binds dimer of 14-3-3 with high affinity (Kd about 30 nM). If the same fragment is phosphorylated at only one site, the dissociation constant of the complex formed by this fragment and 14-3-3 is increased to 290-650 nM [71]. Simultaneous phosphorylation of two residues (Ser346 and Ser368) of the ε isoform of protein kinase C leads to significant increase in its binding to 14-3-3 compared with the binding of protein kinase C phosphorylated at either one of these sites [72].Fig. 5. Complex formation of 14-3-3 and phosphopeptide. The PyMOL program was used to show X-ray crystallographic data obtained with the complex of 14-3-3ζ and phosphopeptide (PDB ID 1QJB). a) Ribbon model of the complex formed by dimer of 14-3-3 (gray helices) with two bound phosphopeptides (green). b) Structure of amphipathic groove of 14-3-3 monomer (beige helices) involved in ligand binding. Residues forming hydrophobic surface are indicated in blue and positively charged residues are in green. The numbering corresponds to the ζ isoform of 14-3-3. The phosphorylated residue of the ligand is indicated in orange, and the rest of the phosphopeptide is gray.
The target protein might contain one or several sites of 14-3-3 binding. It is supposed that the presence of only one of the above-mentioned consensus sites provides for recruitment of 14-3-3 (the so-called “gatekeeper phosphorylation site”) and formation of a rather weak complex where 14-3-3 is not able significantly change the structure and properties of its protein partner. If the target protein contains a second 14-3-3 binding site, then the stability of the complex between 14-3-3 and the target protein is drastically increased and 14-3-3 significantly affects the structure and properties of the ligand [73].
The majority of proteins interacting with 14-3-3 have the above-mentioned consensus motifs containing phosphoserine or phosphothreonine (or sequences similar to these motifs). However, there are a number of proteins lacking the above-mentioned motifs and even not undergoing phosphorylation and nevertheless interacting with 14-3-3. For example we can mention toxic exoenzyme S (ExoS) (ADP-ribosyltransferase) of Pseudomonas aeruginosa having 14-3-3 binding sequence 424DALDL428, which differs from the earlier-described consensus motifs [74]. Another example is phosphatase CDC25B [75] and serum and glucocorticoid-activated protein kinase SGK1 [48] whose interaction with 14-3-3 is independent of phosphorylation.
It seems probable that 14-3-3 contains several sites participating in target binding. For instance, 14-3-3 contains an additional site of target binding located on α-helix 7 (residues 163-187) that seems to be involved in the biding of Raf-kinase [76]. The binding of phosphorylated tryptophan hydroxylase is provided predominantly through contacts formed by residues 171-213 of the η isoform of 14-3-3 [77], i.e. through sites located far from the traditional sites of phosphopeptide binding. Finally, interaction of 14-3-3ζ with platelet receptor of von Willebrand factor involves residues 202-231 belonging to the α9-helix of 14-3-3, i.e. with a site different from that responsible for the binding of consensus phosphopeptides [76].
The presented data show that the structure of 14-3-3 contains several sites involved in target protein binding. The structure of these sites can be very different, and this can be due to the multisite nature of the protein–protein interaction and to difference in the structure of protein targets.
PARTICIPATION OF 14-3-3 IN REGULATION OF CYTOSKELETON AND CELL
MIGRATION
Although 14-3-3 interacts with more than 300 proteins targets [3], we will restrict this review by analyzing the interaction of 14-3-3 with different proteins forming cytoskeleton or involved in attachment of cytoskeleton to the membrane or to the extracellular matrix. It is important to emphasize that the data of the literature are very extensive and contradictory. This is partially due to the difference and incompatibility of the methods used for elucidation of interaction of 14-3-3 with target proteins. For instance, proteomic methods provide information on the possibility of interaction of 14-3-3 with given proteins. However, the question remains whether this possibility is realized in the living cell and what happens if this interaction is indeed realized. On the other hand, biochemical and immunochemical methods are often used for in vitro experiments. These methods provide detailed information on protein–protein interaction and on the effect of these interactions on the properties of the proteins. However, these methods cannot guarantee that the analyzed processes indeed occurs in the cell and play any significant role in its vital activity.
Interaction of 14-3-3 with Proteins of Actin Filament
The proteome data indicate that 14-3-3 interacts with actin [78, 79]. Fluorescent and confocal microscopy indicate that in astrocytes 14-3-3γ is located close to actin filaments, and ischemia and apoptosis are accompanied by changes in the binding of 14-3-3 to F-actin [80]. These morphological data are often interpreted as straightforward evidence of direct interaction of 14-3-3 with actin. We suppose that this conclusion is premature. Colocalization of 14-3-3 and F-actin can be due not only to direct interaction between these two proteins, but to the interaction of 14-3-3 with certain proteins located on actin filament. In good agreement with this viewpoint, the data of Birkenfeld at al. [81] indicate that in the cell 14-3-3 and actin are located close to each other, but all attempts to reveal direct interaction of recombinant 14-3-3 and actin by means of co-sedimentation were unsuccessful. It was postulated that 14-3-3 predominantly interacts not with F-actin itself, but with certain actin-binding proteins.
Interaction of 14-3-3 with Proteins Involved in Regulation of Polymerization and Depolymerization of Actin
Interaction of 14-3-3 with proteins of the actin-depolymerizing-factor (ADF) group and particularly with cofilin has been investigated in detail. Cofilin, a small protein with molecular mass of about 21 kDa, binds to F-actin, affects the structure of actin monomer inside fibrillar actin, and in this way destabilizes the actin filament [82]. This means that cofilin is involved both in fragmentation of long actin filament and accumulation of short filaments that can be used as nuclei for formation new actin filaments. Cofilin is phosphorylated at Ser3 by LIM-kinase I and II (LIMK I, LIMK II) as well as by testicular protein kinase (TESK) [82, 83]. The efficiency of cofilin phosphorylation is rather high, about 30% of intracellular cofilin being phosphorylated [82].Phosphorylated cofilin does not bind and is unable to fragment F-actin. 14-3-3 interacts with phosphorylated cofilin and prevents its dephosphorylation by protein phosphatases [81, 84]. Thus, 14-3-3 conserves cofilin in the phosphorylated state and in this way stabilizes actin filaments, protecting them from cofilin-induced fragmentation. This effect can be important for reorganization of actin filaments in non-muscle cells and might play an important role in cytoskeleton-dependent clusterization of acetylcholine receptors during synapse formation [85]. However, the question whether 14-3-3 participates in reorganization of cytoskeleton in smooth and striated muscle remains unanswered. The concentration of F-actin in muscle cells is very high, and therefore it is difficult to predict the effect of cofilin (only part of which is in the phosphorylated state) on assembly and disassembly of the contractile apparatus and cytoskeleton.
The participation of cofilin in reorganization of cytoskeleton becomes even more complicated if the role of protein phosphatases is taken into account. Protein phosphatases of the slingshot family and the protein phosphatase chronophin are involved in dephosphorylation of cofilin [83]. Slingshot phosphatases bind to F-actin and therefore effectively dephosphorylate cofilin, causing fragmentation of actin filaments [86]. They are phosphorylated by p21-activated protein kinase (PAK4) [86] and by protein kinase D1 [87]. Phosphorylated slingshot interacts with 14-3-3, and this interaction hampers the binding of slingshot to actin filaments [86-88] (see Fig. 6). Removal of slingshot from an actin filament or formation of a tight complex with 14-3-3 inhibits the protein phosphatase activity of slingshot [86, 89].
The p21-activated protein kinase PAK4 as well as MAPKAP-kinase 2 phosphorylate and activate LIM-kinase [86, 90]. Phosphorylated LIM-kinase interacts with 14-3-3, and this interaction affects the intracellular distribution of LIM-kinase [81]. It is worth mentioning that TES-kinase is also phosphorylated (probably by MAP-kinases) [91]. Phosphorylation also improves the interaction of TES-kinase with 14-3-3 [91] and induces redistribution of TES-kinase inside the cell [92]. Finally, protein phosphatases (and among them slingshot) dephosphorylate and in this way inhibit enzymatic activity of protein kinases (for example, LIM-kinase) participating in phosphorylation of cofilin (Fig. 6).Fig. 6. Scheme of possible involvement of 14-3-3 in cofilin-dependent regulation of actin cytoskeleton. Cofilin (in the center of the scheme) in the dephosphorylated state promotes depolymerization of actin (F-actin to G-actin transition). Cofilin can be phosphorylated by LIM- and TES protein kinases (LIMK and TESK), and phosphorylated cofilin is inactive in F-actin depolymerization. Phosphorylated cofilin (cofilin-P) interacts with 14-3-3, and this prevents cofilin dephosphorylation catalyzed by slingshot protein phosphatase. Protein kinases phosphorylating cofilin (LIM- and TES-kinases) are phosphorylated by other protein kinases (PAK4, MAPKAP2), and after phosphorylation they also interact with 14-3-3. Slingshot protein phosphatase also undergoes phosphorylation, which leads to 14-3-3 binding and inhibition. See text for details.
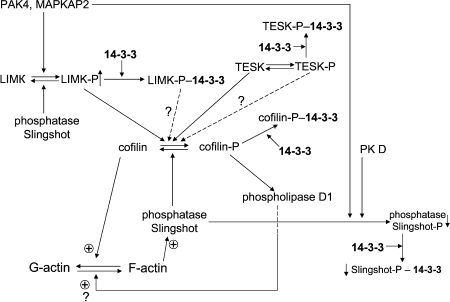
Recently published data indicate that phosphorylated cofilin (that was supposed to be inactive) is able to activate phospholipase D1. This leads to accumulation of phosphatidic acid, which through badly characterized intermediates somehow affects assembly and disassembly of actin cytoskeleton [93] (see Fig. 6). The role of 14-3-3 in the effect of phosphorylated cofilin on phospholipase D1 remains uninvestigated. Summing up, we conclude that 14-3-3 affects localization and activity of enzymes participating in phosphorylation and dephosphorylation of cofilin and interacts with phosphorylated cofilin. Taking into account that phosphorylation affects cofilin-dependent processes of F-actin polymerization and depolymerization, we might conclude that 14-3-3 indirectly (through cofilin) modulate assembly and disassembly of actin cytoskeleton.
One can suppose that the protein targets that form tight complexes with 14-3-3 and are present in high concentration inside cell will (under certain conditions) become able to displace phosphorylated cofilin from its complexes with 14-3-3 and in this way will also indirectly affect actin cytoskeleton. It was supposed that the small heat shock protein with apparent molecular mass 20 kDa (Hsp20, HspB6) might be involved in these processes. The data of Dreiza et al. [94, 95] indicate that a short N-terminal peptide of Hsp20 containing a site phosphorylated by cyclic nucleotide-dependent protein kinases tightly interacts with 14-3-3. Phosphorylated N-terminal peptide of Hsp20 displaces cofilin from its complexes with 14-3-3, and the liberated cofilin is rapidly dephosphorylated by slingshot, thus leading to reorganization of actin cytoskeleton. In subsequent investigation it was shown that not only N-terminal peptide, but intact phosphorylated Hsp20 interacts with 14-3-3 [96]. These data make attractive the hypothesis that hormone-induced phosphorylation of Hsp20 will be accompanied by displacement of cofilin from its complexes with 14-3-3, and this will lead to reorganization of actin cytoskeleton and smooth muscle relaxation [95]. However, taking into account the complexity of the cofilin–14-3-3 system (see Fig. 6) [97], this interpretation seems to be oversimplified, and additional efforts should be undertaken to confirm or to disprove this hypothesis.
There are some data indicating that the phosphorylated small heat shock protein Hsp27 (HspB1) somehow induces polymerization of actin and formation of stress-fibers [98, 99]. We suppose that this viewpoint is rather questionable (see for example [100]), and in addition under in vitro conditions Hsp25 (avian homolog of Hsp27) does not affect actin polymerization [101]. Nevertheless, if phosphorylated Hsp27 somehow affects actin polymerization, then it is worthwhile to analyze Hsp27 phosphorylation and to elucidate the probable effect of 14-3-3 on this process. It is known that Hsp27 is phosphorylated by a number of protein kinases [102], and MAPKAP2 and probably MAPKAP5 play an important role in phosphorylation of this protein [99]. Recently published data indicate that 14-3-3ε interacts with MAPKAP5 and by inhibiting its activity prevents phosphorylation of Hsp27, thus blocking assembly of actin filaments [103]. We think that these data need further confirmation and deserve further detailed verification. However, if these data are correct, then one might conclude that 14-3-3 can indirectly regulate assembly of actin filaments by affecting activity of protein kinases phosphorylating proteins regulating actin polymerization.
Participation of 14-3-3 in Regulation of Protein Motors Interacting with Actin Filaments
The contractile activity of smooth muscle and of non-muscle cells is regulated via phosphorylation of myosin regulatory light chains [104, 105]. In the smooth muscle and non-muscle cell the productive interaction of myosin and actin is possible only after phosphorylation of myosin light chain by myosin light chain kinase (MLCK), and relaxation occurs only after dephosphorylation of myosin light chains by myosin light chain phosphatase (MLCP). Thus, the contractile activity of smooth muscle and non-muscle cells is under coordinated control of two antagonistic enzymes, namely kinase and phosphatase of myosin light chains.Myosin light chain phosphatase consists of three components. A subunit with molecular mass of 105-120 kDa provides for the binding of phosphatase to myosin filaments, a subunit with molecular mass 36-37 kDa possesses protein phosphatase activity, and finally a subunit with molecular mass 20-21 kDa plays an incompletely understood regulatory role [105, 106]. The largest subunit of MLCP is phosphorylated by a number of protein kinases (among them Rho-kinase), and this phosphorylation inhibits phosphatase activity [105]. Phosphorylation of Ser472 of the largest subunit of myosin light chain phosphatase catalyzed by Rho-kinase provokes interaction with 14-3-3β, thus leading to dissociation of phosphatase from myosin filament and inhibition of its activity [107]. Thus, 14-3-3 removes phosphatase from myosin filaments and inhibits its catalytic activity. Both these processes increase the level of myosin light chain phosphorylation and provoke contraction or retard (or prevent) relaxation of smooth muscles.
As mentioned earlier, 14-3-3 interacts with and regulates the activity of many protein kinases [78, 108]. It has been long known that 14-3-3 affects the activity of protein kinase C [19, 72, 109]. One isoform of protein kinase C is involved in phosphorylation of heavy chains of Dictyostelium discoideum myosin, and in this way regulates formation of ordered myosin filaments. 14-3-3 forms a tight complex with this isoform of protein kinase C and thus indirectly regulates the contractile activity of Dictyostelium [110].
In summary, we conclude that 14-3-3 probably does not directly interact with myosin, but by controlling activity of protein kinases and protein phosphatases 14-3-3 can indirectly affect assembly of myosin filaments and the enzymatic activity of myosin.
14-3-3 and Proteins of Cell–Extracellular Matrix Interaction and Cell–Cell Adhesion
Different elements of cytoskeleton participate in fixation of the cell to the extracellular matrix and in intercellular junctions. Heterodimeric integral membrane proteins, integrins, play an important role in the interaction of the cell with the extracellular matrix and intercellular adhesion molecules, and in this way control cell movement. Integrins have a rather large extracellular domain, a short transmembrane sequence, and a small C-terminal cytosolic domain. Eighteen isoforms of α-chains and eight isoforms of β-chains of integrins are described in the literature. These isoforms are combined with formation of 24 different heterodimers providing for interaction of cells with different types of extracellular matrix [111, 112]. Under normal conditions α- and β-chains of integrin form tight heterodimeric complexes. Upon phosphorylation and interaction with protein partners, the interaction of α- and β-chains of integrin is changed. This leads to formation of a complex of focal contacts containing many different protein kinases and adapter proteins as well as proteins providing attachment of actin filaments and clusterization of activated heterodimeric integrins [112].At present 42 potential protein partners interacting with the short (70-90 residues) C-terminal cytosolic domain of the β-chain of integrin are described in the literature [111]. It is obvious that these proteins are not able to interact simultaneously with the short C-terminal domain of integrin β-chain, and therefore these proteins compete with each other for binding to the same sites on the β-chain. The C-terminal cytosolic domain of integrin β-chain contains two sites with the primary structure NXXY. These sites are separated by a short sequence enriched in Ser and Thr that undergo phosphorylation by a number of different protein kinases [111]. Most of the protein partners interact with one of the two Tyr-containing motifs or with the sequence enriched with Ser or Thr. For instance, talin interacts with the first Tyr-containing motif and provides for attachment of actin filaments to integrin. Phosphorylation of Tyr located in the first motif decreases the interaction of talin with the β-chain of integrin. Therefore, the same site of the β-chain of integrin binds another protein, tensin, which not only attaches actin filaments to integrin but also attracts to the focal contact PDK1 and Akt protein kinases [111]. Something similar occurs with the sequence enriched in Ser and Thr and connecting two Tyr-containing sites. In the non-phosphorylated state this Ser/Thr rich sequence of integrin β-chain interacts through hydrophobic contacts with the 21st immunoglobulin-like domain of filamin. This interaction leads to attachment of actin filaments via filamin to integrin and inhibits cell migration [113]. After phosphorylation of the Ser/Thr sequence by protein kinases the interaction of this sequence with filamin is diminished, and in contrast its interaction with different 14-3-3 isoforms is dramatically increased [111]. By an incompletely understood pathway this leads to activation of small G-proteins Rac1 and cdc42, reorganization of cytoskeleton, and increased cellular migration [111]. Phosphorylation of the Ser/Thr rich sequence of integrin β-chain does not directly affect its interaction with talin. However, the interaction with 14-3-3 induced by integrin phosphorylation leads to replacement of talin from its complex with integrin [114], thus indirectly affecting cytoskeleton and cell migration.
Protein–protein interactions of the cytoplasmic domain of integrin α-subunits have been analyzed in less detail. Nevertheless, it has been shown that the α4 subunit of integrin is phosphorylated at Ser978, and this provokes its interaction with 14-3-3 [115]. Moreover, binding of 14-3-3 promotes binding of the universal adapter protein paxilin and formation of triple complex consisting of integrin, 14-3-3, and paxilin. Formation of this complex might affect the orientation of the α- and β-chains of integrin and their interaction with partner proteins and the regulation of small G-proteins (Rac1, cdc42), which are involved in regulation of actin polymerization and lamellipodia formation [115].
As already mentioned, focal contacts contain many proteins and among them adapter proteins and protein kinases. p130Cas (Cas) is one such adapter protein that interacts with Pro-rich sequences of different proteins located in the focal adhesion. Cas can be phosphorylated at Tyr residues and is therefore able to interact with proteins containing the so-called SH2 domain responsible for recognition and binding of phosphotyrosine. All these properties promote interaction of Cas with focal adhesion kinase (FAK) as well as with tyrosine protein phosphatases [116]. Activation of integrin receptor is accompanied by phosphorylation of Cas at serine residues and promotes interaction of Cas with 14-3-3 [116]. It is supposed that 14-3-3 somehow affects transduction of the signal from integrin to cytoskeleton via reversible binding of phosphorylated Cas, which participates in redistribution of protein kinases and protein phosphatases and in this way induces reorganization of cytoskeleton [112].
Cell–cell adhesion is provided by integral membrane proteins such as different kinds of cadherins, desmogleins and desmocollins. Extracellular domains of these proteins interact with each other and “glue” neighboring cells. The cytoplasmic domain of these proteins is involved in the binding of different elements of cytoskeleton (actin filaments and intermediate filaments) to the intercellular contacts or to the sites of interaction of the cell with extracellular matrix [117]. Proteins of the armadillo family provide contacts between actin filaments and cadherins, desmogleins and desmocollins. The armadillo family consists of different catenins, plakoglobins, and plakophilins. All these proteins contain in their primary structure conservative Arm repeats and different in size and structure variable N- and C-terminal parts [117]. Some members of the armadillo family, namely β- and δ-catenins, after phosphorylation interact with different isoforms of 14-3-3 [118, 119].
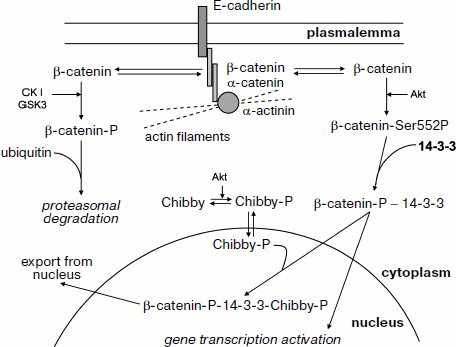
Let us analyze in more detail the interaction of β-catenin with 14-3-3. β-Catenin interacts with the cytoplasmic domain of E-cadherin and provides for its binding to α-catenin, which is directly involved in attachment of actin filaments to the sites of cell adhesion (Fig. 7) [120]. Some β-catenin is tightly bound at adherens junctions and desmosomes, whereas another part of β-catenin remains free and is distributed between cytosol and nucleus. Under normal conditions the portion of β-catenin remaining free is rather small since it undergoes phosphorylation catalyzed by casein kinase I and glycogen synthase kinase. Phosphorylation catalyzed by these protein kinases leads to interaction of β-catenin with certain partner proteins, ubiquitination, and subsequent proteasomal degradation of β-catenin [119, 120]. Hormone-dependent activation of protein kinase Akt (protein kinase B) leads to phosphorylation of β-catenin at Ser552 and its translocation from extracellular junctions to cytosol [120]. β-Catenin phosphorylated at this site interacts with 14-3-3, and this promotes its translocation to the nucleus where β-catenin forms complexes with certain transcriptional factors and activates certain genes (Fig. 7) [119-121]. Overall, these events can lead to uncontrolled proliferation and neoplastic transformation.
The presented data mean that 14-3-3 prevents premature proteolysis of β-catenin and promotes its translocation to the nucleus. The picture becomes even more complicated by the presence of a special protein, Chibby [121]. Chibby interacts with β-catenin, blocking its transcriptional activity. After phosphorylation by protein kinase Akt, Chibby forms a ternary complex consisting of phosphorylated Chibby, 14-3-3, and phosphorylated β-catenin. Formation of this complex promotes nuclear export of β-catenin and decreases its transcriptional activity (Fig. 7) [119, 121]. Thus, 14-3-3 affects β-catenin distribution in the cell and, depending on the isoform, concentration and the presence of phosphorylated Chibby regulates its transcriptional activity and participation in cytoskeleton formation.Fig. 7. Participation of β-catenin in intercellular junctions and regulation of gene transcription. Interacting with E-cadherin and α-catenin, β-catenin is involved in attachment of actin filaments to the region of intercellular junctions. β-Catenin is located both in the region of intercellular junctions (i.e. close to the cell membrane) and in the free state in cytosol. Phosphorylation of catenin by casein kinase I (CKI) and glycogen synthase kinase 3 (GSK3) leads to β-catenin ubiquitination with subsequent proteasomal degradation. Phosphorylation by protein kinase B (Akt) at Ser552 promotes interaction of β-catenin with 14-3-3, translocation of this complex to the nucleus, and activation of certain genes. Chibby protein phosphorylated by protein kinase B (Akt) is able to form a ternary complex with phosphorylated β-catenin and 14-3-3. Formation of this ternary complex promotes nuclear export of β-catenin and in this way prevents activation of gene transcription. See details in text.
Some cells have highly specialized cytoskeleton and a special set of proteins responsible for attachment of cytoskeleton to integral membrane proteins. However, the main principles of protein–protein interactions remain the same. For instance, podocytes (specialized kidney cells) have complex actin cytoskeleton, damaging or improper functioning of which leads to proteinuria (the loss of proteins with urine). Podocytes contain special integral membrane proteins such as P-cadherin, nephrin, podocins, etc. and special adapter proteins such as earlier mentioned catenins and synaptopodin. Synaptopodin directly or through α-actinin attaches actin filaments to the integral membrane proteins [122, 123]. Synaptopodin is phosphorylated by cAMP-dependent protein kinase and Ca2+-calmodulin-dependent protein kinase and after phosphorylation forms a tight complex with 14-3-3. This complex provides correct attachment of actin filaments to the integral membrane proteins and correct functioning of podocytes. Dephosphorylation by calcineurin increases susceptibility of synaptopodin to proteolysis, leading to disorganization of cytoskeleton and proteinuria [123].
The molecules of cell adhesion CD44 and Na+/H+-exchanger (NHE) also participate in formation of intercellular junctions. These cell adhesion proteins also interact with intracellular cytoskeleton, and this interaction is provided with participation of a special group of proteins belonging to the so-called FERM family. This abbreviation was formed by utilization of the first letter of the names of four proteins belonging to this family, namely, the protein of 4.1 band (four, F), ezrin (E), radixin (R), and moesin (M). The proteins of this family contain conservative FERM-domain and special sites for spectrin and actin binding [124]. Recently certain other proteins were added to this family, and among them Merlin and DAL-1 (differentially expressed in adenocarcinoma of the lung), both of which belong to the 4.1B group of FERM proteins [125, 126]. Both of these proteins participate in regulation of proliferation and neoplastic transformation. It is worth mentioning that the above-mentioned talin, providing attachment of actin filaments to integrin, also contains a FERM-domain [127]. The proteins of the FERM family are distributed between cytosol and intercellular junctions, where they attach actin filaments to integral membrane proteins. Translocation between cytosol and membrane is regulated by phosphorylation of specific sites of FERM proteins and by changes in the level of phosphatidylinositols in the cell membrane [127, 128]. DAL-1 and the proteins of the 4.1 group interact with different isoforms of 14-3-3 [124, 125]. Special sites located in the FERM-domain participate in this interaction, which is not dependent on FERM-protein phosphorylation. To our knowledge the consequences of this interaction remain incompletely understood. However, one can suppose that binding of 14-3-3 might affect intracellular distribution of FERM proteins and their interaction with other protein partners. All these events can induce changes in intercellular interactions and cellular motility.
Interaction of 14-3-3 with Small G-Proteins and Protein Kinases Participating in Cytoskeleton Regulation
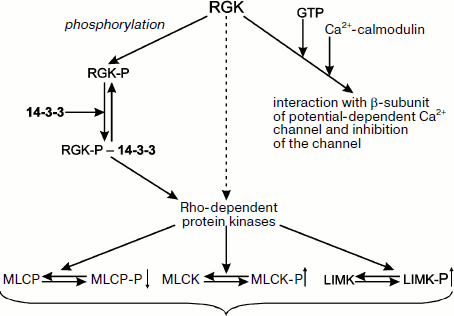
Small G-proteins play an important role in transduction of hormonal signal from receptors inside of the cell. Among small G-proteins belonging to the Ras family, there is a small group designated as RGK, following the names of four proteins belonging to this group, namely Rem (also called Rem1 or Ces), Rem2, Rad, and Gem/Kir [129, 130]. The proteins of the RGK group are different from the other members of the Ras family. The RGK proteins do not contain fatty acid residues (important for attachment of small G-proteins to the membrane) and have low GTPase activity and unique N- and C-terminal sites [129, 130]. The RGK proteins play diverse intracellular roles and interact with a wide number of different protein partners [131]. For instance, RGK proteins interact with the β-subunit of potential-dependent calcium channels and inhibit functioning of these channels. The sites of GTP binding and interaction with β-subunit of calcium channel overlap, and therefore the depth of inhibition depends on the presence of guanine nucleotide in the active site of RGK as well as on the binding of calcium-saturated calmodulin with the site located in the C-terminal part of the RGK molecule (Fig. 8) [129, 130]. RGK proteins can be phosphorylated at a number of sites. Two phosphorylated Ser residues located in the very N- and C-terminal parts of RGK are recognized by 14-3-3 [131]. Thus, phosphorylation makes possible tight bidentate binding of RGK proteins with 14-3-3 [132]. Interaction with 14-3-3 results in redistribution of RGK proteins between nucleus and cytosol, increased stability of RGK, and conformational changes in the GTP-binding site of RGK [129, 130]. RGK proteins (both in the free state and in the complex with 14-3-3) interact and affect Rho-dependent protein kinases [129]. The β-isoform of Rho-kinase phosphorylates and activates myosin light chain kinase and LIM-kinase involved in cofilin phosphorylation. In addition, Rho-kinase phosphorylates and inhibits activity of myosin light chain phosphatase (Fig. 8) [129]. It is probable that the α-isoform of Rho-kinase also participates in regulation of these enzymes. If the scheme presented in Fig. 8 is correct, then RGK proteins seem to be involved in formation of stress fibers and focal contacts. Taking into account that 14-3-3 affects stability and intracellular distribution of RGK, one can suppose that 14-3-3 indirectly affects formation of cytoskeleton and cell motility.
Earlier we analyzed the interaction of 14-3-3 with integrins and the probable role of 14-3-3 in transduction of the signal from integrins to adapter proteins such as filamin, talin, and tensin. The protein composition of focal contacts is very complicated, and in addition to adapter proteins it includes a number of small G-proteins and protein kinases. Detailed mechanisms of interaction of all these proteins remain poorly understood. Nevertheless, we will try to discuss some hypotheses formulated in the literature.Fig. 8. Scheme of possible involvement of 14-3-3 in RGK protein functioning. RGK proteins interact with calcium-calmodulin and bind GTP. This can lead to changes in intracellular distribution of these proteins and to inhibition of potential-operated calcium channel. Phosphorylation of RGK proteins provides their interaction and tight complex formation with 14-3-3. RGK proteins affect the catalytic activity of Rho-kinases phosphorylating and activating myosin light chain kinase (MLCK), phosphorylating and inhibiting catalytic activity of myosin light chain phosphatases (MLCP) and phosphorylating and activating LIM-kinase responsible for cofilin phosphorylation. Regulating activity of Rho-kinases, the RGK–14-3-3 complex affects formation of stress fibers and focal contacts as well as cell shape and migration.
Insulin and many growth factors binding to their receptors activate phosphatidylinositol 3-kinase, which is responsible for phosphatidylinositol-3,4,5-trisphosphate (PIP3) synthesis. Accumulation of PIP3 results in translocation to the membrane of Akt protein kinase, which phosphorylates a large number of different protein substrates, among them integrin subunits, guanine nucleotide exchange factor (GEF), small G-protein Rac1, and protein kinase Pak [133, 134]. The sites phosphorylated by Akt and having the primary structure RXRXX(S/T) [135] are often recognized by different isoforms of 14-3-3. These events lead to accumulation in the region of focal contacts of a number of phosphorylated proteins, some of which interact with 14-3-3. The focal contacts contain the small G-protein Rac1 [136], which is in the activated state due to the presence of activating guanine nucleotide exchange factor (GEF). Activated Rac1 stimulates protein phosphatases that dephosphorylate cofilin, and in this way promote disassembly of actin filaments. At the same time, Rac1 in the complex with 14-3-3β stimulates p21-activated protein kinase (PAK) that also leads to reorganization of cytoskeleton [133]. Recently described Kank protein is also located in focal contacts [134]. This protein inhibits actin polymerization and undergoes phosphorylation catalyzed by Akt. Phosphorylated Kank interacts with 14-3-3 and in this way competes with other protein targets of 14-3-3 in the region of focal contacts. All these interactions again can lead to reorganization of actin cytoskeleton [134].
In summary, we conclude that 14-3-3 interacts with many G-proteins and protein kinases participating in reorganization of cytoskeleton and therefore can indirectly affect intercellular junctions, attachment of cells to extracellular matrix, and cell migration.
Interaction of 14-3-3 with Proteins of Microtubules
Microtubules are formed by ordered polymerization of tubulin dimers, and the data of the literature indicate that 14-3-3 can interact with both α- and β-tubulins [78]. However, the details of this interaction and its physiological significance remain incompletely understood.Microtubule assembly is regulated by a large number of different proteins designated as MAP (microtubule-associated proteins). One of the proteins involved in regulation of microtubule association is the so-called τ-protein. This protein belongs to a group of proteins with intrinsically unordered structure [137-139]. Six isoforms of τ-protein are predominantly expressed in the nervous system. These isoforms are the result of alternative splicing and differ in their length from 352 to 441 amino acid residues. All isoforms of τ-protein contain acidic N-terminus, a Pro-rich sequence located in the middle of the protein, and three or four sites with similar structure located in the C-terminal part of molecule [140]. It is supposed that these three or four C-terminal repeats are involved in the interaction of τ-protein with tubulin. The Pro-rich sequence seems to be located along microtubules, and the acidic N-terminal tail hangs free [141] affecting translocation of motor proteins along microtubules [142] and/or interaction between neighboring microtubules [143]. Affecting the rate of microtubule assembly [144], the interaction between microtubules, and translocation of kinesin and dynein along microtubules [142], τ-protein plays an important role in the normal functioning of cytoskeleton.
There are more than 19 sites of phosphorylation in τ-protein. These sites can be phosphorylated by a number of different protein kinases (cAMP-dependent (PKA), neuronal cdc2-like protein kinase (NCLK), mitogen-activated protein kinase (MAPK), glycogen synthase kinase 3 (GSK3), protein kinase C (PKC), phosphorylase kinase, calmodulin-dependent protein kinase (CaCMK), casein kinases of the first and second type (CK I and II), kinases of MARK/Par-1 family, etc.) [145-148]. The sites of phosphorylation are located along the whole length of the polypeptide chain of τ-protein [146, 149]. Therefore phosphorylation might affect many properties of τ-protein. For instance, phosphorylation affects the interaction of τ-protein with microtubules [150] and the susceptibility of τ-protein to proteolysis [151, 152]. Especially important is the effect of phosphorylation on the ability of τ-protein to form different in form and size aggregates, the so-called paired helical filaments (PHFs) and neurofibrillary tangles (NFTs) [153, 154].
In earlier publications it was supposed that certain isoforms of 14-3-3 play a role of special adapter that is important for formation of triple complex consisting of τ-protein, 14-3-3, and different protein kinases. Formation of this complex promotes effective phosphorylation of τ-protein by cAMP-dependent protein kinase [146], glycogen synthase kinase 3 [155, 156], and serum and glucocorticoid-inducible protein kinase 1 [48]. In a later publication formation of τ-protein/14-3-3/protein kinase triple complex was not confirmed [157], but it was postulated that 14-3-3 somehow affects phosphorylation of τ-protein.
For a long time it was postulated that phosphorylation is not necessary for productive interaction of τ-protein with 14-3-3 and that the sites of 14-3-3 binding are located close to tubulin-binding sites (the region of conservative repeats) of τ-protein, thus leading to competition of 14-3-3 and tubulin for binding to τ-protein [146]. Recently published data [158, 159] indicate that phosphorylation of Ser214 of the largest isoform of τ-protein (or phosphorylation of homologous Ser156 of the shortest isoform of τ-protein) leads to significant increase in interaction between 14-3-3 and τ-protein. The primary structure at this site of phosphorylation located in the Pro-rich region correlates well with the structure of consensus sequences recognized by 14-3-3 in other protein targets [158, 159], and therefore involvement of this site in 14-3-3 binding is not unexpected. However, mutated τ-protein with replacement of Ser156 by alanine and phosphorylated by cAMP-dependent protein kinase interacted with 14-3-3 stronger than unphosphorylated wild type protein [160]. These data were interpreted as an indication that phosphorylation of not only Ser156, but of certain other sites is also important for interaction of τ-protein with 14-3-3. By means of site directed mutagenesis, it was shown that at least two additional sites (Ser235 and Ser267 of the shortest isoform of τ-protein) phosphorylated by cAMP-dependent protein kinase and located in the region of conservative repeats, involved in the interaction of τ-protein with tubulin, play an important role in formation of tight complexes between τ-protein and 14-3-3. Thus, several sites of τ-protein participate in the interaction with 14-3-3, and the stability of the complex formed depends on the phosphorylation of τ-protein at several sites (located both in the Pro-rich region and in conservative repeats important for tubulin binding) [160].
Under certain conditions τ-protein can undergo aggregation, forming aggregates of different form and size. Neurological diseases that are accompanied by accumulation of aggregates of τ-protein are called tauopathies. This group of diseases combines Alzheimer’s disease, Pick’s disease, frontotemporal dementia, and certain other neurological disorders [154, 161, 162]. Detailed explanation of mechanisms underlying τ-protein aggregation is still lacking, but it is postulated that hyperphosphorylation, oxidation of SH-groups, glycosylation, and limited proteolysis can increase the probability of τ-protein aggregation. Polyanions and fatty acids can also promote formation of τ-protein aggregates [154, 163]. Recently published data indicate that neurofibrillary tangles formed by aggregated τ-protein contain 14-3-3 [164, 165]. Therefore, the question arises what the effect of 14-3-3 on τ-protein aggregation is. Addition of 14-3-3 promotes aggregation of intact τ-protein and its fragments [166]. It was postulated that competing with tubulin, 14-3-3 induces dissociation of τ-protein from microtubules and in this way provokes τ-protein aggregation leading to formation of paired helical filaments and neurofibrillary tangles. At the same time, formation of 14-3-3–τ-protein complexes might promote phosphorylation of τ-protein by cAMP-dependent protein kinase, and this will somehow prevent formation of insoluble aggregates of τ-protein [166]. Investigations of Japanese authors [159, 167] support this suggestion. 14-3-3 promotes aggregation of unphosphorylated τ-protein and at the same time prevents aggregation of τ-protein phosphorylated by cAMP-dependent protein kinase and protein kinase B [159, 167]. Unfortunately, the molecular mechanisms of 14-3-3 action on τ-protein aggregation remain enigmatic. However, even now we can state that 14-3-3 affects incorporation of τ-protein into microtubules and in this way affects microtubule assembly and translocation of motor proteins along microtubules. 14-3-3 can affect aggregation of τ-protein, leading to development of different neurological forms of tauopathies. Taking into account that τ-protein is not only a component of microtubules but interacts and affects the structure of actin microfilaments [168], we can conclude that 14-3-3 might be involved in reorganization of neuronal cytoskeleton and in this way might affect development of different neurodegenerative diseases.
Microtubules form a system of “rails” used for translocation of motor proteins carrying different cargoes from the nucleus to periphery or in the opposite direction. It has been shown that one kinesin, namely KIF1C, present in the form of monomer or in the form of heterodimer and participating in transportation of cargoes from Golgi to endoplasmic reticulum, is able to interact with 14-3-3 [169]. The site of interaction is located in the C-terminal part of KIF1C, and the strength of interaction depends on phosphorylation of Ser1092 of KIF1C, which is phosphorylated by casein kinase II [169]. The proteins interacting with the C-terminal part of kinesin might affect the functioning of the motor domain, which is usually located in the N-terminal part of kinesin. Moreover, the proteins bound to the C-terminal portion of kinesin are located close to cargoes that are carried by kinesin, and therefore they can be important for choosing proper organelles (or vesicles) that will be bound and transported by this motor protein. Binding of 14-3-3 can promote formation of dimers of KIF1C [169] and in this way affect the properties of kinesin. Further investigations have shown that 14-3-3 is also able to interact with kinesin light chain 2, being a component of a heterotetrameric form of conventional kinesin KIF5B [170]. The light chains are also located in the C-terminal part of this kinesin isoform and participate in attachment of cargoes transported by kinesin. 14-3-3 binding is dependent on phosphorylation of Ser575 located in the peptide with primary structure RARSS575LNFLN, which is significantly different from the structure of the sites usually recognized by 14-3-3 (see above). The presented data mean that 14-3-3 interacts with different isoforms of kinesin and the sites of 14-3-3 binding are usually located close to the site of cargo binding. This agrees with the viewpoint that 14-3-3 is used as a universal adapter protein or is a member of complicated protein complexes involved in cargo attachment and transportation in the cell [171, 172]. To our knowledge the detailed role of 14-3-3 in cargo attachment to kinesin remains poorly investigated.
In summary, we conclude that 14-3-3 can indirectly affect assembly and disassembly of microtubules, translocation of motor proteins along microtubules, and probably interaction of microtubules and microfilaments as well as aggregation of certain microtubule-associated proteins.
14-3-3 and Proteins of Intermediate Filaments
Three types of filaments, namely microfilaments (formed by actin and having diameter of about 8 nm), microtubules (formed by tubulin and having diameter of about 25 nm), and so-called intermediate filaments having diameter of about 10-12 nm, are the main components of cytoskeleton [173, 174]. There are six classes of proteins forming intermediate filaments: two classes of cytokeratins (so-called acidic and basic keratins); proteins of muscle, mesenchymal, and glial intermediate filaments (desmin, vimentin); proteins of neurofilaments; lamins forming nuclear envelope; and proteins of the so-called orphan intermediate filaments of crystalline lens [173, 174].The proteins of intermediate filaments have similar structure. They contain a highly conservative central part tending to form coiled-coil and different in size N- and C-terminal parts [175]. Dimers of proteins of intermediate filaments (stabilized by coiled-coil structures) are assembled into mature intermediate filaments where contacts between neighboring dimers are formed by the interaction of variable N- and C-terminal parts of the molecule. Although the intermediate filaments are very stable and mechanically strong, these elements of cytoskeleton undergo a constant and dynamic process of subunit exchange [174]. Subunit exchange and assembly and disassembly of intermediate filaments are dependent on phosphorylation of sites located in the N- and C-terminal variable parts of intermediate filament proteins [176]. As a rule phosphorylation destabilizes protein–protein interaction and promotes disassembly of intermediate filaments.
In S/G2/M phases of the cell cycle certain cytokeratins (cytokeratins K8/K18) undergo hyperphosphorylation (it is probable that protein kinase cdc2 plays an important role in this process) [177]. Phosphorylated cytokeratins K8/K18 interacts with 14-3-3, and this prevents incorporation of cytokeratins into intermediate filaments. Interaction with 14-3-3 becomes possible only after phosphorylation of Ser33 of cytokeratin K18 [178]. The primary structure at phosphorylated Ser33, RPVSSAAS33VY, is significantly different from that in the consensus sequences recognized by 14-3-3 in the other protein substrates. It is supposed that interaction with 14-3-3 might affect both intracellular distribution of cytokeratins and their ability to assemble into intermediate filaments [177-179]. Treating of cells with the inhibitor of protein phosphatases calyculin A is accompanied by increase in the portion of phosphorylated vimentin. Phosphorylation of the sites located in the N-terminal part (residues 1-96) leads to interaction of vimentin with 14-3-3 [180]. The concentration of vimentin in the cell is rather high, and the complex formed with 14-3-3 is very tight. Therefore phosphorylated vimentin effectively competes with the other protein targets of 14-3-3, influences distribution of 14-3-3 in the cell, and affects different processes controlled by 14-3-3 [180].
Glial fibrillary acidic protein (GFAP) together with vimentin forms intermediate filaments and plays an important role in formation of cytoskeleton of astrocytes. Glial acidic proteins both being free in cytosol and being incorporated into intermediate filaments interacts with 14-3-3. This interaction is dependent on phosphorylation of Ser8 of GFAP [181]. The primary structure at Ser8, RRITS8AR, is different from the consensus sequences usually recognized by 14-3-3 and from the sequence recognized by 14-3-3 in cytokeratins. As in the case with cytokeratins, interaction with 14-3-3 hampers incorporation of glial acidic protein into intermediate filaments and in this way significantly affects integrity of intermediate filaments and dynamics of their assembly and disassembly [181]. It is worthwhile to mention that no less than 1% of cell proteins are represented by different proteins of intermediate filaments [180]. Therefore, phosphorylation of intermediate filament proteins can lead to the binding of large quantities of 14-3-3, and this will indirectly affect catalytic activity of protein phosphatases (cdc25 protein phosphatase) and protein kinases (Raf, Akt) regulated by 14-3-3 [173, 174]. Thus, interaction of intermediate filament proteins with 14-3-3 regulates assembly and disassembly of important elements of cytoskeleton as well as multiple processes depending on phosphorylation and dephosphorylation of different intracellular proteins.
CONCLUSION
In summary, we conclude that the proteins of the 14-3-3 family are universal adapters able to interact with many different cellular proteins. Activity of 14-3-3 can be regulated in different ways. First, 14-3-3 activity can be regulated by changing of the rate of synthesis and proteolytic degradation. Second, the cell can synthesize a number of different 14-3-3 isoforms that differ in their ability to form homo- or heterodimeric complexes and to interact with protein targets. Third, proteins of the 14-3-3 family undergo different types of posttranslational modification (such as phosphorylation and acetylation), significantly affecting their structure and properties. Fourth, both homo- and heterodimers of 14-3-3 contain at least two sites involved in the binding of phosphorylated ligands having special primary structure. This makes preferential the binding of protein targets having two (or more than two) sites of phosphorylation. In addition the presence of two sites of phosphopeptide binding makes possible the binding of two protein targets, each of which containing a single site of phosphorylation. 14-3-3 is able to bind not only protein targets having phosphorylated Ser/Thr residues located in specific consensus motifs, but also many other proteins phosphorylated at unusual sites or even unphosphorylated proteins. We suppose that further investigation of the mechanism of recognition will be an important subject of future investigations.
The binding of target proteins leads to a number of different consequences. The target proteins bound to 14-3-3 become protected from proteolysis and/or dephosphorylation, change their intracellular distribution, and finally two different protein targets can be drawn together. The last feature is especially important if one target protein is an enzyme (for instance, protein kinase) and the second one is its substrate. As already mentioned, 14-3-3 interacts with a number of different targets; therefore, changes in the concentration of one protein target can affect the interaction of 14-3-3 with the other protein targets. This is especially important in the case of proteins present at a rather high concentration in the cell. All this means that 14-3-3 is a highly effective regulator of many intracellular processes and makes further investigation of 14-3-3 very promising.
In a rather short review it is impossible to describe the structure, properties, and functions of 14-3-3 in detail. The interested reader can find more details in the earlier published excellent reviews dealing with the history of discovery and investigation of 14-3-3 [5], mechanism of functioning and regulation of 14-3-3 activity [182, 183], specific functions of 14-3-3 in the nervous system [23], the role of 14-3-3 in apoptosis and survival [184], 14-3-3 as a possible oncogene [185], and the role of 14-3-3 in oncogenesis [186].
The authors are deeply indebted to N. V. Solov’eva for careful scientific editing.
This investigation was supported by the Russian Foundation for Basic Research (grant No. 10-04-00026).
REFERENCES
1.Boston, P. F., Jackson, P., Kynoch, P. A., and
Thompson, R. J. (1982) J. Neurochem., 38, 1466-1474.
2.Muslin, A. J., Tanner, J. W., Allen, P. M., and
Shaw, A. S. (1996) Cell, 84, 889-897.
3.Mackintosh, C. (2004) Biochem. J.,
381, 329-342.
4.Moore, B. W., Perez, V. J., and Gehring, M. (1968)
J. Neurochem., 15, 265-272.
5.Aitken, A. (2006) Sem. Canc. Biol.,
16, 162-172.
6.Ichimura, T., Isobe, T., Okuyama, T., Yamauchi, T.,
and Fujisawa, H. (1987) FEBS Lett., 219, 79-82.
7.Ichimura, T., Isobe, T., Okuyama, T., Takahashi,
N., Araki, K., Kuwano, R., and Takahashi, Y. (1988) Proc. Natl.
Acad. Sci. USA, 85, 7084-7088.
8.Aitken, A., Howell, S., Jones, D., Madrazo, J., and
Patel, Y. (1995) J. Biol. Chem., 270, 5706-5709.
9.Toker, A., Sellers, L. A., Amess, B., Patel, Y.,
Harris, A., and Aitken, A. (1992) Eur. J. Biochem., 206,
453-461.
10.Aitken, A., Ellis, C. A., Harris, A., Sellers, L.
A., and Toker, A. (1990) Nature, 344, 594.
11.Martens, G. J., Piosik, P. A., and Danen, E. H.
(1992) Biochem. Biophys. Res. Commun., 184,
1456-1459.
12.Hirsch, S., Aitken, A., Bertsch, U., and Soll, J.
(1992) FEBS Lett., 296, 222-224.
13.Van Heusden, G. P., Wenzel, T. J., Lagendijk, E.
L., de Steensma, H. Y., and van den Berg, J. A. (1992) FEBS
Lett., 302, 145-150.
14.Xiao, B., Smerdon, S. J., Jones, D. H., Dodson,
G. G., Soneji, Y., Aitken, A., and Gamblin, S. J. (1995) Nature,
376, 188-191.
15.Liu, D., Bienkowska, J., Petosa, C., Collier, R.
J., Fu, H., and Liddington, R. (1995) Nature, 376,
191-194.
16.Jones, D. H., Ley, S., and Aitken, A. (1995)
FEBS Lett., 368, 55-58.
17.Shen, Y., Godlewski, J., Bronisz, A., Zhu, J.,
Comb, M., Avruch, J., and Tzivion, G. (2003) Mol. Biol. Cell,
14, 4721-4733.
18.Gu, Y.-M., Jin, Y.-H., Choi, J.-K., Baek, K.-H.,
Yeo, C.-Y., and Lee, K.-Y. (2006) FEBS Lett., 580,
305-310.
19.Aitken, A. (2002) Plant Mol. Biol.,
50, 993-1010.
20.Yano, M., Nakamuta, S., Wu, X., Okumura, Y., and
Kido, H. (2006) Mol. Biol. Cell, 17, 4769-4779.
21.Kockel, L., Vorbruggen, G., Jackle, H., Mlodzik,
M., and Bohmann, D. (1997) Genes Dev., 11, 1140-1147.
22.Tommerup, N., and Leffers, H. (1996)
Genomics, 33, 149-150.
23.Berg, D., Holzmann, C., and Riess, O. (2003)
Nat. Neurosci., 4, 752-762.
24.Shankardas, J., Senchyna, M., and Dimitrijevich,
S. (2008) Mol. Vis., 14, 2604-2615.
25.Pennington, C., Chohan, G., Mackenzie, J.,
Andrews, M., Will, R., Knight, R., and Green, A. (2009) Neurosci.
Lett., 455, 56-59.
26.Xiao, T., Ying, W., Li, L., Hu, Z., Ma, Y., Jiao,
L., Ma, J., Cai, Y., Lin, D., Guo, S., Han, N., Di, X., Li, M., Zhang,
D., Su, K., Yuan, J., Zheng, H., Gao, M., He, J., Shi, S., Li, W., Xu,
N., Zhang, H., Liu, Y., Zhang, K., Gao, Y., Qian, X., and Cheng, S.
(2005) Mol. Cell Proteom., 4, 1480-1486.
27.Green, A. J., Thompson, E. J., Stewart, G. E.,
Zeidler, M., McKenzie, J. M., MacLeod, M. A., Ironside, J. W., Will, R.
G., and Knight, R. S. (2001) J. Neurol. Neurosurg. Psychiatry,
70, 744-748.
28.Qi, W., Liu, X., Chen, W., Li, Q., and Martinez,
J. D. (2007) Mol. Carcinog., 46, 847-856.
29.Jang, J. S., Cho, H. Y., Lee, Y. J., Ha, W. S.,
and Kim, H. W. (2004) Oncol. Res., 14, 491-499.
30.Nakanishi, K., Hashizume, S., Kato, M., Honjoh,
T., Setoguchi, Y., and Yasumoto, K. (1997) Hum. Antibodies,
8, 189-194.
31.Benzinger, A., Popowicz, G., Joy, J., Majumdar,
S., Holak, T., and Hermeking, H. (2005) Cell Res., 15,
219-227.
32.Gardino, A., Smerdon, S., and Yaffe, M. (2006)
Sem. Cancer Biol., 16, 173-182.
33.Robinson, K., Jones, D., Patel, Y., Martin, H.,
Madrazo, J., Martin, S., Howell, S., Elmore, M., Finnen, M. J., and
Aitken, A. (1994) Biochem. J., 299, 853-861.
34.Sluchanko, N., Chernik, I., Seit-Nebi, A.,
Pivovarova, A., Levitsky, D., and Gusev, N. (2008) Arch. Biochem.
Biophys., 477, 305-312.
35.Verdoodt, B., Benzinger, A., Popowicz, G. M.,
Holak, T. A., and Hermeking, H. (2006) Cell Cycle, 5,
2920-2926.
36.Powell, D., Rane, M., Joughin, B., Kalmukova, R.,
Hong, J.-H., Tidor, B., Dean, W., Pierce, W., Klein, J., Yaffe, M., and
McLeish, K. (2003) Mol. Cell. Biol., 23, 5376-5387.
37.Woodcock, J. M., Murphy, J., Stomski, F. C.,
Berndt, M. C., and Lopez, A. F. (2003) J. Biol. Chem.,
278, 36323-36327.
38.Wilker, E., Grant, R., Artim, S., and Yaffe, M.
(2005) J. Biol. Chem., 280, 18891-18898.
39.Liang, X., Butterworth, M., Peters, K., Walker,
W., and Frizzell, R. (2008) J. Biol. Chem., 283,
27418-27425.
40.Alvarez, D., Callejo, M., Shoucri, R., Boyer, L.,
Price, G., Zannis, and Hadjopoulos, M. (2003) Biochemistry,
42, 7205-7215.
41.Ostrerova, N., Petrucelli, L., Farrer, M., Mehta,
N., Choi, P., Hardy, J., and Wolozin, B. (1999) J. Neurosci.,
19, 5782-5791.
42.Kim, T. D., Choi, E., Rhim, H., Paik, S. R., and
Yang, C. H. (2004) Biochem. Biophys. Res. Commun., 324,
1352-1359.
43.Athwal, G., and Huber, S. (2002) Plant J.,
29, 119-129.
44.Rittinger, K., Budman, J., Xu, J., Volinia, S.,
Cantley, L. C., Smerdon, S. J., Gamblin, S. J., and Yaffe, M. B. (1999)
Mol. Cell, 4, 153-166.
45.Kumagai, A., and Dunphy, W. G. (1999) Genes
Dev., 13, 1067-1072.
46.Brunet, A., Kanai, F., Stehn, J., Xu, J.,
Sarbassova, D., Frangioni, J., Dalal, S., DeCaprio, J., Greenberg, M.,
and Yaffe, M. (2002) J. Cell Biol., 156, 817-828.
47.Muslin, A. J., and Xing, H. (2000) Cell
Signal., 12, 703-709.
48.Chun, J., Kwon, T., Lee, E. J., Kim, C. H., Han,
Y. S., Hong, S. K., Hyun, S., and Kang, S. S. (2004) Mol. Cells,
18, 360-368.
49.Das, A. K., Cohen, P. W., and Barford, D. (1998)
EMBO J., 17, 1192-1199.
50.Fu, H., Subramanian, R. R., and Masters, S. C.
(2000) Annu. Rev. Pharmacol. Toxicol., 40, 617-647.
51.Nomura, M., Shimizu, S., Sugiyama, T., Narita,
M., Ito, T., Matsuda, H., and Tsujimoto, Y. (2003) J. Biol.
Chem., 278, 2058-2065.
52.Won, J., Kim, D. Y., La, M., Kim, D., Meadows, G.
G., and Joe, C. O. (2003) J. Biol. Chem., 278,
19347-19351.
53.Choudhary, C., Kumar, C., Gnad, F., Nielsen, M.
L., Rehman, M., Walther, T. C., Olsen, J. V., and Mann, M. (2009)
Science, 325, 834-840.
54.Megidish, T., Cooper, J., Zhang, L., Fu, H., and
Hakomori, S. (1998) J. Biol. Chem., 273, 21834-21845.
55.Ma, Y., Pitson, S., Hercus, T., Murphy, J.,
Lopez, A., and Woodcock, J. (2005) J. Biol. Chem., 280,
26011-26017.
56.Powell, D., Rane, M., Chen, Q., Singh, S., and
McLeish, K. (2002) J. Biol. Chem., 277, 21639-21642.
57.Tsuruta, F., Sunayama, J., Mori, Y., Hattori, S.,
Shimizu, S., Tsujimoto, Y., Yoshioka, K., Masuyama, N., and Gotoh, Y.
(2004) EMBO J., 23, 1889-1899.
58.Clokie, S., Cheung, K., Mackie, S., Marquez, R.,
Peden, A., and Aitken, A. (2005) FEBS J., 272,
3767-3776.
59.Dubois, T., Rommel, C., Howell, S., Steinhussen,
U., Soneji, Y., Morrice, N., Moelling, K., and Aitken, A. (1997) J.
Biol. Chem., 272, 28882-28888.
60.Dubois, T., Howell, S., Amess, B., Kerai, P.,
Learmonth, M., Madrazo, J., Chaudhri, M., Rittinger, K., Scarabel, M.,
Soneji, Y., and Aitken, A. (1997) J. Protein Chem., 16,
513-522.
61.Ahmed, K. M., Fan, M., Nantajit, D., Cao, N., and
Li, J. J. (2008) Oncogene, 27, 6738-6748.
62.Sunayama, J., Tsuruta, F., Masuyama, N., and
Gotoh, Y. (2005) J. Cell Biol., 170, 295-304.
63.Aitken, A., Howell, S., Jones, D., Madrazo, J.,
Martin, H., Patel, Y., and Robinson, K. (1995) Mol. Cell.
Biochem., 149/150, 41-49.
64.Silhan, J., Obsilova, V., Vecer, J., Herman, P.,
Sulc, M., Teisinger, J., and Obsil, T. (2004) J. Biol. Chem.,
279, 49113-49119.
65.Obsilova, V., Herman, P., Vecer, J., Sulc, M.,
Teisinger, J., and Obsil, T. (2004) J. Biol. Chem., 279,
4531-4540.
66.Yaffe, M. B., Rittinger, K., Volinia, S., Caron,
P. R., Aitken, A., Leffers, H., Gamblin, S. J., Smerdon, S. J., and
Cantley, L. C. (1997) Cell, 91, 961-971.
67.Coblitz, B., Wu, M., Shikano, S., and Li, M.
(2006) FEBS Lett., 580, 1531-1535.
68.Dougherty, M., and Morrison, D. (2004) J. Cell
Sci., 117, 1875-1884.
69.Zhang, L., Wang, H., Liu, D., Liddington, R., and
Fu, H. (1997) J. Biol. Chem., 272, 13717-13724.
70.Wang, H., Zhang, L., Liddington, R., and Fu, H.
(1998) J. Biol. Chem., 273, 16297-16304.
71.Obsil, T., Ghirlando, R., Anderson, D. E.,
Hickman, A. B., and Dyda, F. (2003) Biochemistry, 42,
15264-15272.
72.Kostelecky, B., Saurin, A., Purkiss, A., Parker,
P., and McDonald, N. (2009) EMBO Rep., 10, 983-989.
73.Yaffe, M. (2002) FEBS Lett., 513,
53-57.
74.Yasmin, L., Jansson, A., Panahandeh, T., Palmer,
R., Francis, M., and Hallberg, B. (2006) FEBS J., 273,
638-646.
75.Mils, V., Baldin, V., Goubin, F., Pinta, I.,
Papin, C., Waye, M., Eychene, A., and Ducommun, B. (2000)
Oncogene, 19, 1257-1265.
76.Gu, M., and Du, X. (1998) J. Biol. Chem.,
273, 33465-33471.
77.Ichimura, T., Uchiyama, J., Kunihiro, O., Ito,
M., Horigome, T., Omata, S., Shinkai, F., Kaji, H., and Isobe, T.
(1995) J. Biol. Chem., 270, 28515-28518.
78.Pozuelo Rubio, M., Geraghty, K. M., Wong, B. H.,
Wood, N. T., Campbell, D. G., Morrice, N., and Mackintosh, C. (2004)
Biochem. J., 379, 395-408.
79.Liang, S., Yu, Y., Yang, P., Gu, S., Xue, Y., and
Chen, X. (2009) J. Chromatogr. B Analyt. Technol. Biomed. Life
Sci., 877, 627-634.
80.Chen, X. Q., and Yu, A. C. (2002) Biochem.
Biophys. Res. Commun., 296, 657-663.
81.Birkenfeld, J., Betz, H., and Roth, D. (2003)
Biochem. J., 369, 45-54.
82.Bamburg, J. R. (1999) Annu. Rev. Cell Dev.
Biol., 15, 185-230.
83.Huang, T. Y., DerMardirossian, C., and Bokoch, G.
M. (2006) Curr. Opin. Cell Biol., 18, 26-31.
84.Gohla, A., and Bokoch, G. M. (2002) Curr.
Biol., 12, 1704-1710.
85.Lee, C. W., Han, J., Bamburg, J. R., Han, L.,
Lynn, R., and Zheng, J. Q. (2009) Nature Neurosci., 12,
848-856.
86.Soosairajah, J., Maiti, S., Wiggan, O., Sarmiere,
P., Moussi, N., Sarcevic, B., Sampath, R., Bamburg, J., and Bernard, O.
(2005) EMBO J., 24, 473-486.
87.Eiseler, T., Doppler, H., Yan, I. K., Kitatani,
K., Mizuno, K., and Storz, P. (2009) Nature Cell Biol.,
11, 545-556.
88.Nagata-Ohashi, K., Ohta, Y., Goto, K., Chiba, S.,
Mori, R., Nishita, M., Ohashi, K., Kousaka, K., Iwamatsu, A., Niwa, R.,
Uemura, T., and Mizuno, K. (2004) J. Cell Biol., 165,
465-471.
89.Kligys, K., Yao, J., Yu, D., and Jones, J. (2009)
Biochem. Biophys. Res. Commun., 383, 450-454.
90.Kobayashi, M., Nishita, M., Mishima, T., Ohashi,
K., and Mizuno, K. (2006) EMBO J., 25, 713-726.
91.Sarmiere, P. D., and Bamburg, J. R. (2004) J.
Neurobiol., 58, 103-117.
92.Toshima, J. Y., Toshima, J., Watanabe, T., and
Mizuno, K. (2001) J. Biol. Chem., 276, 43471-43481.
93.Han, L., Stope, M. B., de Jesus, M. L., Oude
Weernink, P. A., Urban, M., Wieland, T., Rosskopf, D., Mizuno, K.,
Jakobs, K. H., and Schmidt, M. (2007) EMBO J., 26,
4189-41202.
94.Dreiza, C. M., Brophy, C. M., Komalavilas, P.,
Furnish, E. J., Joshi, L., Pallero, M. A., Murphy-Ullrich, J. E., von
Rechenberg, M., Ho, Y. S., Richardson, B., Xu, N., Zhen, Y., Peltier,
J. M., and Panitch, A. (2005) FASEB J., 19, 261-263.
95.Dreiza, C. M., Komalavilas, P., Furnish, E. J.,
Flynn, C. R., Sheller, M. R., Smoke, C. C., Lopes, L. B., and Brophy,
C. M. (2009) Cell Stress Chaperones, 15, 1-11.
96.Chernik, I., Seit-Nebi, A., Marston, S., and
Gusev, N. (2007) Mol. Cell. Biochem., 295, 9-17.
97.Seit-Nebi, A. S., and Gusev, N. B. (2010) Cell
Stress Chaperones, in press.
98.Damarla, M., Hasan, E., Boueiz, A., Le, A., Pae,
H. H., Montouchet, C., Kolb, T., Simms, T., Myers, A., Kayyali, U. S.,
Gaestel, M., Peng, X., Reddy, S. P., Damico, R., and Hassoun, P. M.
(2009) PLoS One, 4, e4600.
99.Kostenko, S., Johannessen, M., and Moens, U.
(2009) Cell Signal., 21, 712-718.
100.Gusev, N. B., Bogatcheva, N. V., and Marston,
S. B. (2002) Biochemistry (Moscow), 67, 511-519.
101.Panasenko, O. O., Seit Nebi, A., Bukach, O. V.,
Marston, S. B., and Gusev, N. B. (2002) Biochim. Biophys. Acta,
1601, 64-74.
102.Kostenko, S., and Moens, U. (2009) Cell.
Mol. Life Sci., 66, 3289-3307.
103.Tak, H., Jang, E., Kim, S. B., Park, J., Suk,
J., Yoon, Y. S., Ahn, J. K., Lee, J.-H., and Joe, C. (2007) Cell
Signal., 19, 2379-2387.
104.Khapchaev, A. Yu., Shirinsky, V. P., and
Vorotnikiv, A. V. (2003) Uspekhi Biol. Khim., 43,
365-420.
105.Kim, H. R., Appel, S., Vetterkind, S.,
Gangopadhyay, S. S., and Morgan, K. G. (2008) J. Cell Mol. Med.,
12, 2165-2180.
106.Erdodi, F., Ito, M., and Hartshorne, D. J.
(eds.) (1996) in Biochemistry of Smooth Muscle Contraction
(Barany, M., ed.) Academic Press, San Diego-New
York-Boston-London-Sydney-Tokyo-Toronto, pp. 131-142.
107.Koga, Y., and Ikebe, M. (2008) Mol. Biol.
Cell, 19, 1062-1071.
108.Jin, J., Smith, D., Stark, C., Wells, C.,
Fawcett, J., Kulkarni, S., Metalnikov, P., O’Donnell, P., Taylor,
P., Taylor, L., Zougman, A., Woodgett, J., Langeberg, L., Scott, J.,
and Pawson, T. (2004) Curr. Biol., 14, 1436-1450.
109.Van der Hoeven, P. C., van der Wal, J. C.,
Ruurs, P., and van Blitterswijk, W. J. (2000) Biochem. J.,
347, 781-785.
110.Matto-Yelin, M., Aitken, A., and Ravid, S.
(1997) Mol. Biol. Cell, 8, 1889-1899.
111.Legate, K. R., and Fassler, R. (2009) J.
Cell. Sci., 122, 187-198.
112.Harburger, D. S., and Calderwood, D. A. (2009)
J. Cell. Sci., 122, 159-163.
113.Campbell, I. D. (2008) Biochem. Soc.
Trans., 36, 263-266.
114.Takala, H., Nurminen, E., Nurmi, S. M.,
Aatonen, M., Strandin, T., Takatalo, M., Kiema, T., Gahmberg, C. G.,
Ylanne, J., and Fagerholm, S. C. (2008) Blood, 112,
1853-1862.
115.Deakin, N. O., Bass, M. D., Warwood, S.,
Schoelermann, J., Mostafavi-Pour, Z., Knight, D., Ballestrem, C., and
Humphries, M. J. (2009) J. Cell Sci., 122, 1654-1664.
116.Garcia-Guzman, M., Dolfi, F., Russello, M., and
Vuori, K. (1999) J. Biol. Chem., 274, 5762-5768.
117.Bass-Zubek, A. E., Godsel, L. M., Delmar, M.,
and Green, K. J. (2009) Curr. Opin. Cell Biol., 21,
708-716.
118.Mackie, S., and Aitken, A. (2005) FEBS
J., 272, 4202-4210.
119.Takemaru, K., Fischer, V., and Li, F. Q. (2009)
Cell Cycle, 8, 210-213.
120.Fang, D., Hawke, D., Zheng, Y., Xia, Y.,
Meisenhelder, J., Nika, H., Mills, G. B., Kobayashi, R., Hunter, T.,
and Lu, Z. (2007) J. Biol. Chem., 282, 11221-11229.
121.Li, F. Q., Mofunanya, A., Harris, K., and
Takemaru, K. (2008) J. Cell Biol., 181, 1141-1154.
122.Faul, C., Asanuma, K., Yanagida-Asanuma, E.,
Kim, K., and Mundel, P. (2007) Trends Cell Biol., 17,
428-437.
123.Faul, C., Donnelly, M., Merscher-Gomez, S.,
Chang, Y. H., Franz, S., Delfgaauw, J., Chang, J. M., Choi, H. Y.,
Campbell, K. N., Kim, K., Reiser, J., and Mundel, P. (2008) Nature
Med., 14, 931-938.
124.Calinisan, V., Gravem, D., Chen, R. P.,
Brittin, S., Mohandas, N., Lecomte, M. C., and Gascard, P. (2006)
Front. Biosci., 11, 1646-1666.
125.Yu, T., Robb, V., Singh, V., Gutmann, D., and
Newsham, I. (2002) Biochem. J., 365, 783-789.
126.Sun, C. X., Robb, V. A., and Gutmann, D. H.
(2002) J. Cell Sci., 115, 3991-4000.
127.Bretscher, A., Edwards, K., and Fehon, R. G.
(2002) Nature Rev. Mol. Cell. Biol., 3, 586-599.
128.Polesello, C., and Payre, F. (2004) Trends
Cell Biol., 14, 294-302.
129.Correll, R. N., Pang, C., Niedowicz, D. M.,
Finlin, B. S., and Andres, D. A. (2008) Cell Signal., 20,
292-300.
130.Beguin, P., Mahalakshmi, R. N., Nagashima, K.,
Cher, D. H., Takahashi, A., Yamada, Y., Seino, Y., and Hunziker, W.
(2005) J. Cell Sci., 118, 1923-1934.
131.Ward, Y., Spinelli, B., Quon, M. J., Chen, H.,
Ikeda, S. R., and Kelly, K. (2004) Mol. Cell. Biol., 24,
651-661.
132.Kelly, K. (2005) Trends Cell Biol.,
15, 640-643.
133.Somanath, P., and Byzova, T. (2009) J. Cell.
Physiol., 218, 394-404.
134.Kakinuma, N., Roy, B. C., Zhu, Y., Wang, Y.,
and Kiyama, R. (2008) J. Cell Biol., 181, 537-549.
135.Yaffe, M. B., Leparc, G. G., Lai, J., Obata,
T., Volinia, S., and Cantley, L. C. (2001) Nature Biotechnol.,
19, 348-353.
136.Kligys, K., Claiborne, J., DeBiase, P.,
Hopkinson, S., Wu, Y., Mizuno, K., and Jones, J. (2007) J. Biol.
Chem., 282, 32520-32528.
137.Mylonas, E., Hascher, A., Bernado, P.,
Blackledge, M., Mandelkow, E., and Svergun, D. I. (2008)
Biochemistry, 47, 10345-10353.
138.Mukrasch, M. D., Bibow, S., Korukottu, J.,
Jeganathan, S., Biernat, J., Griesinger, C., Mandelkow, E., and
Zweckstetter, M. (2009) PLoS Biol., 7, e34.
139.Jeganathan, S., von Bergen, M., Mandelkow, E.
M., and Mandelkow, E. (2008) Biochemistry, 47,
10526-10539.
140.Johnson, G. V., and Stoothoff, W. H. (2004)
J. Cell Sci., 117, 5721-5729.
141.Amos, L. A., and Schlieper, D. (2005) Adv.
Protein Chem., 71, 257-298.
142.Dixit, R., Ross, J. L., Goldman, Y. E., and
Holzbaur, E. L. (2008) Science, 319, 1086-1089.
143.Rosenberg, K. J., Ross, J. L., Feinstein, H.
E., Feinstein, S. C., and Israelachvili, J. (2008) Proc. Natl. Acad.
Sci. USA, 105, 7445-7450.
144.Scott, C. W., Blowers, D. P., Barth, P. T., Lo,
M. M., Salama, A. I., and Caputo, C. B. (1991) J. Neurosci.
Res., 30, 154-162.
145.Timm, T., Marx, A., Panneerselvam, S.,
Mandelkow, E., and Mandelkow, E. M. (2008) BMC Neurosci., 9
(Suppl. 2), S9.
146.Hashiguchi, M., Sobue, K., and Paudel, H. K.
(2000) J. Biol. Chem., 275, 25247-25254.
147.Scott, C. W., Spreen, R. C., Herman, J. L.,
Chow, F. P., Davison, M. D., Young, J., and Caputo, C. B. (1993) J.
Biol. Chem., 268, 1166-1173.
148.Landrieu, I., Lacosse, L., Leroy, A.,
Wieruszeski, J. M., Trivelli, X., Sillen, A., Sibille, N., Schwalbe,
H., Saxena, K., Langer, T., and Lippens, G. (2006) J. Am. Chem.
Soc., 128, 3575-3583.
149.Haase, C., Stieler, J. T., Arendt, T., and
Holzer, M. (2004) J. Neurochem., 88, 1509-1520.
150.Schneider, A., Biernat, J., von Bergen, M.,
Mandelkow, E., and Mandelkow, E. M. (1999) Biochemistry,
38, 3549-3558.
151.Litersky, J. M., and Johnson, G. V. (1995)
J. Neurochem., 65, 903-911.
152.Johnson, G. V., and Foley, V. G. (1993) J.
Neurosci. Res., 34, 642-647.
153.Ballatore, C., Lee, V. M., and Trojanowski, J.
Q. (2007) Nature Rev. Neurosci., 8, 663-672.
154.Iqbal, K., Alonso Adel, C., Chen, S., Chohan,
M. O., El-Akkad, E., Gong, C. X., Khatoon, S., Li, B., Liu, F., Rahman,
A., Tanimukai, H., and Grundke-Iqbal, I. (2005) Biochim. Biophys.
Acta, 1739, 198-210.
155.Agarwal-Mawal, A., Qureshi, H. Y., Cafferty, P.
W., Yuan, Z., Han, D., Lin, R., and Paudel, H. K. (2003) J. Biol.
Chem., 278, 12722-12728.
156.Yuan, Z., Agarwal-Mawal, A., and Paudel, H. K.
(2004) J. Biol. Chem., 279, 26105-26114.
157.Matthews, T. A., and Johnson, G. V. (2005)
Neurosci. Lett., 384, 211-216.
158.Sluchanko, N., Seit-Nebi, A., and Gusev, N.
(2009) Biochem. Biophys. Res. Commun., 379, 990-994.
159.Sadik, G., Tanaka, T., Kato, K., Yamamori, H.,
Nessa, B. N., Morihara, T., and Takeda, M. (2009) J. Neurochem.,
108, 33-43.
160.Sluchanko, N., Seit-Nebi, A., and Gusev, N.
(2009) FEBS Lett., 583, 2739-2742.
161.Avila, J. (2000) FEBS Lett., 476,
89-92.
162.Spires-Jones, T. L., Stoothoff, W. H., de
Calignon, A., Jones, P. B., and Hyman, B. T. (2009) Trends
Neurosci., 32, 150-159.
163.Barghorn, S., and Mandelkow, E. (2002)
Biochemistry, 41, 14885-14896.
164.Sugimori, K., Kobayashi, K., Kitamura, T.,
Sudo, S., and Koshino, Y. (2007) Psychiatry Clin. Neurosci.,
61, 159-167.
165.Layfield, R., Fergusson, J., Aitken, A., Lowe,
J., Landon, M., and Mayer, R. J. (1996) Neurosci. Lett.,
209, 57-60.
166.Hernandez, F., Cuadros, R., and Avila, J.
(2004) Neurosci. Lett., 357, 143-146.
167.Sadik, G., Tanaka, T., Kato, K., Yanagi, K.,
Kudo, T., and Takeda, M. (2009) Biochem. Biophys. Res. Commun.,
383, 37-41.
168.Fulga, T. A., Elson-Schwab, I., Khurana, V.,
Steinhilb, M. L., Spires, T. L., Hyman, B. T., and Feany, M. B. (2007)
Nature Cell Biol., 9, 139-148.
169.Dorner, C., Ullrich, A., Haring, H. U., and
Lammers, R. (1999) J. Biol. Chem., 274, 33654-33660.
170.Ichimura, T., Wakamiya-Tsuruta, A., Itagaki,
C., Taoka, M., Hayano, T., Natsume, T., and Isobe, T. (2002)
Biochemistry, 41, 5566-5572.
171.Taya, S., Shinoda, T., Tsuboi, D., Asaki, J.,
Nagai, K., Hikita, T., Kuroda, S., Kuroda, K., Shimizu, M., Hirotsune,
S., Iwamatsu, A., and Kaibuchi, K. (2007) J. Neurosci.,
27, 15-26.
172.Molla-Herman, A., Boularan, C., Ghossoub, R.,
Scott, M. G., Burtey, A., Zarka, M., Saunier, S., Concordet, J. P.,
Marullo, S., and Benmerah, A. (2008) PLoS One, 3,
e3728.
173.Coulombe, P. A., and Wong, P. (2004) Nature
Cell Biol., 6, 699-706.
174.Eriksson, J. E., Dechat, T., Grin, B., Helfand,
B., Mendez, M., Pallari, H. M., and Goldman, R. D. (2009) J. Clin.
Invest., 119, 1763-1771.
175.Herrmann, H., and Aebi, U. (2004) Annu. Rev.
Biochem., 73, 749-789.
176.Hyder, C. L., Pallari, H. M., Kochin, V., and
Eriksson, J. E. (2008) FEBS Lett., 582, 2140-2148.
177.Liao, J., and Omary, M. B. (1996) J. Cell
Biol., 133, 345-357.
178.Ku, N. O., Liao, J., and Omary, M. B. (1998)
EMBO J., 17, 1892-1906.
179.Ku, N. O., Michie, S., Resurreccion, E. Z.,
Broome, R. L., and Omary, M. B. (2002) Proc. Natl. Acad. Sci.
USA, 99, 4373-4378.
180.Tzivion, G., Luo, Z. J., and Avruch, J. (2000)
J. Biol. Chem., 275, 29772-29778.
181.Li, H., Guo, Y., Teng, J., Ding, M., Yu, A. C.,
and Chen, J. (2006) J. Cell Sci., 119, 4452-4461.
182.Yaffe, M. B. (2002) FEBS Lett.,
513, 53-57.
183.Van Heusden, G. P. H. (2005) IUBMB Life,
57, 623-629.
184.Porter, G. W., Khuri, F. R., and Fu, H. (2006)
Sem. Cancer Biol., 16, 193-202.
185.Tzivion, G., Gupta, V. S., Kaplun, L., and
Balan, V. (2006) Sem. Cancer Biol., 16, 203-213.
186.Morrison, D. K. (2009) Trends Cell
Biol., 19, 16-23.