REVIEW: Biochemical Polymorphism of the Growth Hormone System Proteins and Its Manifestations in Human Prostate Cells
S. S. Shishkin*, K. V. Lisitskaya, and I. N. Krakhmaleva
Bach Institute of Biochemistry, Russian Academy of Sciences, Leninsky pr. 33, 119071 Moscow, Russia; E-mail: shishkin@inbi.ras.ru* To whom correspondence should be addressed.
Received January 12, 2010; Revision received March 11, 2010
The basic mechanisms are considered that are responsible for producing biochemical polymorphism of human proteins realized at three basic levels: the structures of genome and genes; the transcription and maturation of transcripts; the postsynthetic formation of functionally active protein products of gene expression. The data on biochemical polymorphism of growth hormone (GH) and some other proteins that are directly or indirectly necessary for its functioning and support this polymorphism by polylocus, polyallelism, alternative splicing, and various postsynthetic modifications are analyzed. The role of polymorphic proteins of the GH system is discussed in formation of a variety of oligomeric molecular structures of this system (multicomponent transport complexes, receptors, and endocellular protein ensembles involved in the regulation of gene expression). It is emphasized that such structural polymorphism significantly influences the biological effects in various parts of the GH system during physiological processes and in tumors, in particular in prostate cancer.
KEY WORDS: biochemical polymorphism, growth hormone system, prostate cancerDOI: 10.1134/S0006297910130043
Abbreviations: GH, growth hormone; GHR, growth hormone receptor; IGFs, insulin-like growth factors; IGFBPs, insulin-like growth factor binding proteins; IRSs, insulin receptor protein substrates; MAP, mitogen-activated protein kinases; PC, prostate cancer; SNPs, single nucleotide polymorphisms; STAT, a protein family of signal transducers and activators of transcription.
Biochemical polymorphism of proteins has been under study for about 50
years, and these studies were initially associated with the discovery
of isoenzymes and their active study in the limits of biochemical
genetics [1, 2]. During the
last two last decades of the previous century the pronounced
biochemical polymorphism was impressively shown not only for enzymes,
but also of proteins with other functions, in particular, with hormonal
ones [2-4]. In 1991 G. Baumann
[4], who has been attentively studying growth
hormone (GH) and some proteins supporting its functioning, has even
proposed a special term for polymorphic hormonal proteins –
isohormones.
At the beginning of this century, which is considered by many authors as the entrance of biology in a special postgenomic stage of development [5-7], studies on polymorphism acquired a new quality. This was due to the successful finishing of the international “Human Genome” project [8, 9] and appearance of a spectrum of new branches of science, so-called “-omics” (genomics, transcriptomics, proteomics, metabolomics, etc.) [7, 10, 11], as well as of bioinformatics. Constantly growing databases on biopolymers of humans and many other organisms have been created, e.g. in the National Center of Biotechnological Information (NCBI, www.ncbi.nlm.nih.gov) and the Swiss Institute of Bioinformatics (Swiss-Prot, www.expasy.org). This was the start for an unprecedented enlargement of studies on biochemical polymorphism of human protein hormones and also of other proteins performing hormonal functions. This can be well illustrated by results of searching in the PubMed database of NCBI works using combinations of key words “polymorphism human hormone”, which reveal more than 1700 works published during only the last three years. Some authors started reevaluating the significance of protein polymorphism for normal development of organisms and in pathology, including prostate cancer (PC) [10, 12-14].
Growth hormone (GH) and some proteins supporting its functioning are known to play a crucial role in the control of cell proliferation and on its disorders involved in carcinogenesis [4, 13, 14]. These proteins and their genes are now a subject of a special attention in connection with a pronounced tendency for increase in the incidence of PC [13, 14]. Having in mind the importance of the human GH system for solution of various medical problems, we think it is urgent to consider and generalize the basic results of studies on polymorphism of proteins of this system.
MOLECULAR MECHANISMS OF PROTEIN POLYMORPHISM
Studies on biochemical polymorphism of proteins resulted in the creation of some general ideas about molecular mechanisms of this phenomenon and revealed its importance for the development of healthy individuals and also for development of various pathologies including carcinogenesis. There is no doubt that in many cases the biochemical polymorphism of human proteins is caused genetically [1-3, 15]. Even early studies on isoenzymes have shown that they are products of expression of different but closely related genes. Then similar related genes were shown to exist for hemoglobins and many other nonenzymatic proteins. Accumulation of experimental data made it evident that the gene multiplicity or polylocus is one of the commonest mechanisms determining the biochemical polymorphism of proteins [2-4]. During the work on the “Human Genome” project and upon its finishing it became clear that due to gene duplication in humans, thousands of groups of closely related genes were produced encoding proteins with significant resemblance in structure (e.g. similar or identical domains) [8, 9]. Such a resemblance is thought to indicate common evolutionary origin; such genes are combined into specific gene families, and their products into the corresponding protein families [9, 16-18].
The development of DNA technologies has resulted in a flow of studies on different manifestations of DNA polymorphism in genes and, in particular, of single nucleotide polymorphisms (SNPs). In the human genome the number of known SNPs is already higher than 10 million [19, 20]. Certain (so-called nonsynonymous) SNPs in the gene exons and some other types of DNA polymorphism are also shown to be a direct genetic cause of the biochemical polymorphism of proteins (polyallelism). Most generally, the genetic variability (variability of DNA sequences) can manifest itself in both production of qualitatively different isoforms and in changes in their quantitative ratios up to complete absence of individual isoforms [2, 21, 22]. Such variability can lead to pathology or to predisposition for certain diseases [19, 22]. However, there are some cases of DNA polymorphism when a complete inhibition of an individual protein is not accompanied by development of a disease (e.g. the absence of α-actinin 3 caused by the appearance of a premature stop-codon [23]).
Studies on another group of mechanisms of biochemical polymorphism of proteins were associated with the so-called biological mini-revolution of the 1980s, which was marked by the discovery of the exon–intron structure of genes in eukaryotes. During this period and later, on starting genomic projects the transcript splicing, alternative splicing, and also some other noncanonical mechanisms of genetic information realization were revealed [2, 3, 24]. In the 1980s the expression of individual genes was shown to be associated with generation of not one but of a number of proteins with similar functions but with significantly different structure [25, 26]. Alternative splicing was acknowledged as one of the most important mechanisms functioning on the level of transcripts and responsible for generation of certain sets of proteins as a result of expression of individual genes, i.e. protein polymorphism.
Some authors have tried to assess the significance of alternative splicing in genomic information in humans. Thus, Mironov et al. reported in 1999 that ~35% of identified genes can be expressed with alternative splicing [27]. In 2001 some data appeared suggesting alternative splicing of 42% of human genes [28]. In 2005 Lee and Wang [29] described the probability of alternative splicing even in 80% of all genes. Concurrently, it was established that many alternative transcripts were rapidly destroyed and were not used as templates for protein synthesis [30]. And finally, comparative studies on genomes of five eukaryotes published in 2009 indicated that at least 15% of human genes were expressed with alternative splicing [31].
Protein polymorphism is also significantly related to alternative promoters, which have been detected in 58% of so-called transcriptional units encoding proteins [32] (thus, alternative promoters were found in the gene of GH [33]). This molecular mechanism is responsible for formation of several different transcripts on the expression of unique genes and thus significantly contributes not only to normal development but also to some pathologies. Thus, each of the genes encoding proteins p63 and p73 has two promoters, and these proteins are known as important components of apoptosis and carcinogenesis and as members of the family of p53 protein, which is a major suppressor of tumor growth [34].
So-called mRNA editing is another mechanism acting at the level of transcripts [2, 35, 36]. Many data accumulated by the end of previous century have shown the distribution of mRNA editing in various protozoans, yeasts, worms, insects, and in mitochondria of higher eukaryotes [36]. In particular, in humans this phenomenon is studied in detail in for expression of the gene of apolipoprotein B100 (apo-B100). The major product of this gene is a rather large protein – apo-B100 with molecular weight of 549 kDa. Another immunologically related protein termed apo-B48 with molecular weight of 264 kDa was found in blood lipoproteins. The shortened chain of apo-B48 is synthesized because of a single nucleotide substitution in codon 2153, which converts a glutamine codon into the termination signal [37]. The production of the untimely stop codon is a result of deamination of the cytidine base in position 6666 of apoB mRNA and appearance of uridine in this position [38, 39]. An enzyme catalyzing this reaction and termed site-specific cytidine deaminase has also been detected. This enzyme is active in apoB mRNA editing being within a specific multienzyme complex of C/U editosome [39].
The mRNA editing occurs also with involvement of site-specific adenosine deaminases converting certain adenine residues to inosine (A-to-I) [40]. During the “postgenomic period” some authors analyzing the human transcriptome concluded that the majority of the produced RNAs had sites destined for editing the corresponding transcripts by site-specific adenosine deaminases [41]. Note that some data suggest that RNA editing can promote invasiveness and other signs of malignant growth in some tumor cells, in particular of PC [42, 43]. Thus, Martinez et al. found increased levels of the A-to-I editing enzymes in the cells of cultured PC lines [43]. Moreover, these authors also detected editing sites in the transcript of the gene of androgen receptors and revealed the presence of RNA with the corresponding nucleotide substitutions.
Overall, results of transcriptome studies suggested the existence of some mechanisms functioning on the level of RNA and providing for pronounced polymorphism of proteins. This conclusion was convincingly supported by Carnini et al. [44]. They found that in the human genome only 8365 of 32,129 so-called open reading frames for coding proteins (i.e. 26%) were responsible for formation of two or more related proteins.
The terminal (postsynthetic) stage of the genetic information processing contributes significantly to the biochemical polymorphism of proteins. This stage includes some processes in the synthesized polypeptide chains that result in production of functionally active proteins [1, 2, 25]. Many mechanisms have been found that are involved in changing the structure of newly synthesized chains at this stage. These mechanisms associated either with destruction of peptide bonds or with formation of new covalent bonds are determined as posttranslational or postsynthetic modifications. Moreover, at the postsynthetic stage some mechanisms are known which do not involve changes in covalent bonds but are necessary for formation of functional activity of the newly synthesized proteins. Thus, it has been known for a long time that for some enzymes generation of homo- or heteropolymers is an important or even necessary stage for functioning (and also for adequate regulation of functional activity) [1, 2, 45, 46]. Similar data have also been obtained for nonenzymatic proteins [47]. In particular, mechanisms leading to dimerization (and other forms of oligomerization) are revealed in some components of the human GH system and are intensively studied [4]. The probability of formation of structural–functional polymorphism of some proteins caused by differences during the folding stage is also under discussion [48, 49].
Mechanisms of generation of human protein isoforms due to different posttranslational modifications have been studied in thousands of experimental works and considered in hundreds of reviews. First of all, among them several modifications of the N-terminal amino acid sequences that have been identified in many human proteins should be emphasized [50]. Such modifications include removal of N-terminal methionine by specific aminopeptidases, N-α-acetylation, binding of some other fatty acids (myristic, palmitic, etc.), etc. Acylation by fatty acids also occurs on other amino acid residues. The combined use of proteomics and bioinformatics was favorable for creation of special programs for detecting (predicting) the abovementioned modifications in various proteins with the information included in the genomic databases [51].
Among protein modifications different types of limited and site-specific proteolysis are particularly widespread due to the presence in the human genome of genes encoding ~600 corresponding enzymes [52]. The functional multiversity of these modifications was shown in different physiological and pathological processes, i.e. in apoptosis and carcinogenesis [52, 53].
Special regions detected in the primary structure of some protein products of translation removed later by posttranslational site-specific proteolysis presented another example of non-Mendelian protein variability and determined a new front for studies on mechanisms of protein polymorphism [54]. Note that upon the proteolytic excision of the removed region the remaining fragments were cross-linked to one another. By analogy with transcript splicing this mechanism was termed protein splicing, the removed regions inteins (from protein introns), and the cross-linked fragments exteins (from protein exons) [54, 55].
Thus, biochemical polymorphism of proteins is manifested, first, by existence (in different individuals or in the same individual) of related proteins with different structure but with similar molecular functions (the structural or quality polymorphism). Second, this polymorphism is manifested by stable changes in different individuals in both the quantities of certain proteins (up to their full absence) and in their functional activity (up to its full absence). The quality polymorphism can also be due to different causes including genetic changes, e.g. SNPs in the corresponding genes. Third, many proteins function as oligomers with a specific quaternary structure and also as components of different supramolecular complexes; therefore, biochemical polymorphism can be caused by generation in different individuals of the same species of structurally different protein oligomers or other complexes to perform the same molecular function. Note that the efficiency of such polymorphic protein structures can be either the same or can vary, up to the full loss of the functional activity.
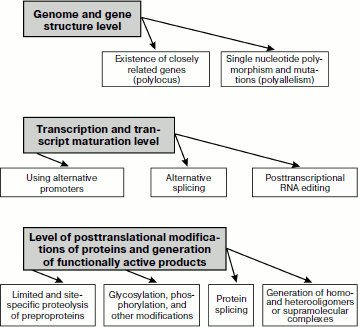
Overall, generation of biochemical polymorphism of proteins is provided for by essentially different mechanisms that manifest themselves on three basic levels (Fig. 1): of genome and gene structures; of transcription and transcript maturation; and at the level of postsynthetic formation of functionally active protein products of gene expression. These complex processes result in creation of functionally connected but structurally different protein ensembles functioning in specialized tissues and organs of multicellular organisms.
Fig. 1. Basic mechanisms responsible for biochemical polymorphism of proteins.
GENERAL CHARACTERISTICS OF HUMAN GROWTH HORMONE SYSTEM PROTEINS AND MANIFESTATIONS OF THEIR BIOCHEMICAL POLYMORPHISM
Growth hormone and a number of other proteins (directly or indirectly supporting its functioning) are responsible for various molecular and cellular effects finally resulting in the organism’s growth and development [22, 56, 57]. These proteins form a kind of axis or a system, which triggers and controls the collection of metabolic processes resulting in growth and associated with cell differentiation [56-58]. The GH system influences both stem cells [59] and cells with different types of differentiation [60], in particular cells of prostate tissue [61]. Disorders in functioning of the GH system can lead to severe hereditary diseases [22, 56] or be involved in pathogenesis of many diseases, including malignant tumors [58, 61, 62].
The GH system functions through successive molecular processes with involvement of tens of other proteins and peptides. Components of this system participate in triggering of GH secretion, its transport in the bloodstream, transmission of the hormonal signal in target cells (intracellular signaling), and, finally, in purposeful changes of gene expression in target cells [22, 56, 63, 64] (Fig. 2; see color insert).
Figure 2 shows that the GH system is subdivided into two branches, the basic branch and lateral branch, and also three special regulatory links determined by the actions of: i) somatoliberin (hypothalamic releasing factor of GH, or somatocrinin (GHRH)); ii) somatostatin (SST, SRIF); and iii) ghrelin (GHRL). Note that each of these regulatory links consists of a chain of molecular events influencing GH secretion [64, 65].Fig. 2. Scheme of functioning of the GH/IGF system (after [56] and [64]). Legend: GH, growth hormone; GHBP, GH-binding protein; GHR, GH receptor; IGF-1, insulin-like growth factor 1; IGFBP, IGF-binding proteins; GHRH-R, somatoliberin receptor; GHS-R, ghrelin receptor; SST-R, somatostatin receptors; ALS, acid-labile subunit; IGF1-R, IGF-1 receptor; s-g1-5, signaling mechanisms triggered by various activated receptors (1-5).
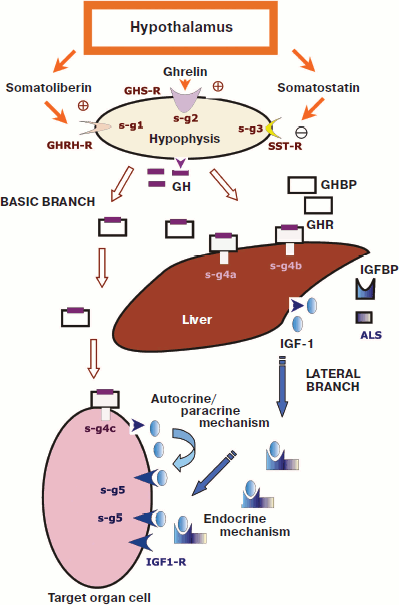
The lateral branch is a result of GH action on many target cells, which respond by synthesis and secretion of a hormone-like protein that was named insulin-like growth factor 1 (IGF-1) [22, 56, 67]. Some authors emphasize this situation and indicate the involvement of IGF-1 in the system by calling it the GH/IGF axis or system. However, IGF-1 can be also produced independently of GH and can play its own role in the regulation of cell proliferation [56, 67, 68]. In particular, there are reports about patients with a shortage of GH not allowing its determination in the blood serum even upon various stimulations, whereas a sufficient level of IGF-1 could be recorded, along with some other components of the GH/IGF system [69].
Basic Branch Proteins
In the GH/IGF system GH is considered to be the central actor, which produces highly differentiated somatotrophic cells of the hypophysis. Synthesis of GH is encoded by the gene GH (or GH1, which is unique in the haploid set). This gene is located on chromosome 17 and is a member of a cluster consisting of five related genes located on a relatively short region of the nucleotide sequence [70]. Among the genes of this cluster the gene GHV is the most similar in structure to the GH1 gene [4, 22], and the GHV gene is sometimes called GH2 (e.g. record 139,240 in OMIM NCBI). In the last century the gene GHV and the other three genes of this cluster (CSL, CSA, and CSB) were thought to be expressed virtually only in placenta cells [70]. However, recent data indicate that at least some of these four genes can be expressed in cancer cells [71]. Thus, the polylocus can contribute to the biochemical polymorphism of GH.The role of polyallelism is revealed by the finding in gene GH1 of 130 SNPs, 18 of which are located in the encoding regions, and among the latter 11 SNPs are nonsynonymous (SNP NCBI). The incidence of SNPs in the GH1 gene in different populations is under study in various countries. In particular, a group of Spanish authors [72] has recently studied in detail 25 such SNPs (a rare allele of which was characterized by incidence higher than 1%) in a representative sample of healthy subjects of both genders with normal height (n = 307). But in subjects with some genotypes a significant decrease in height was recorded along with an increase recorded in others.
The GH1 gene expression is associated with alternative splicing, which produces two-to-four transcripts different in length [13, 73, 74]. There are also reports about another sharply shortened transcript (an isoform or variant 5) lacking most of the GH sequence (regions corresponding to exons 2, 3, and 4) ([75], NP_072056 NCBI).
Alternative splicing and some other mechanisms (including various postsynthetic modifications) result in multiplicity of GH1 protein products, which were detected even in the 1980s and are still under intensive investigation [4, 13, 22, 75, 76]. According to data of enzyme immunoassay, among the GH isoforms detected in the bloodstream about 70-75% is a 22-kDa protein, and this isoform is considered to be the major one; 5-10% is constituted by a 20-kDa protein that is a product of translation of an alternative transcript of the GH1 gene [4, 64, 76]. The other material is distributed mainly between dimeric and oligomeric fractions, which contain corresponding aggregations generated due to both covalent bonds and weak (noncovalent) interactions. Small quantities of isoforms with individual deamidated amino acid residues (Q137→E, 152N→D) were found, as well as glycosylated isoforms [77, 78] and protein products of other alternative transcripts. Note that even the protein product of a very short transcript (an isoform or variant 5) was detected. This 5-kDa product consists of 43 a.a. and displays insulin-like functional activity [74]. Amino acid sequences of protein products produced on translation of five transcripts of the GH1 gene are presented in Swiss-Prot record P01241.
The major isoform of GH consisting of 191 a.a. is produced from a larger precursor protein (217 a.a.), i.e. is a result of limited proteolysis. Formation of two disulfide bonds and phosphorylation of two serine residues are also considered to be important postsynthetic modifications of the major GH isoform (Swiss-Prot P01241).
Qualitative variability of blood contents of different GH isoforms is very important for manifestations of biological activity and plays a significant role during aging and in different pathologies including malignant tumors [79, 80]. The half-life of the major GH isoform in blood is about 13 min [81], and on such a rapid elimination of the hormone the regulation of GH secretion is fundamentally important for functioning of the GH/IGF system.
Somatoliberin is one of stimulators of GH secretion (Fig. 2). It consists of 44 a.a. [65, 82] and is encoded as a prepropeptide by the GHRH gene, which contains 73 SNPs (SNP NCBI). The expression of the GHRH gene is associated with an alternative splicing and results in two isoforms of the prepropeptide. However, the subsequent processing that includes the removal of the N- and C-terminal sequences results in generation of the same functionally active factor (Swiss-Prot P01286). Somatoliberin is mainly produced in the hypothalamus, although the GHRH gene expression is also found in cells of the small intestine, immune system, placenta, and in some tumors [65, 83, 84]. The ectopic production of somatoliberin can increase its level in blood and promote the development of acromegalia [84].
Biological effects of somatoliberin (enhancement of anabolic processes, synthesis of GH, increase in GH secretion, etc.) are due to the interaction of this peptide with the specific membrane receptor GHRH-R encoded by the GHRHR gene ([85], Swiss-Prot Q02643). The GHRHR gene contains 242 SNPs, 10 of which are inside exons, and six nonsynonymous ones (SNP NCBI); and at least seven mutations in this gene are known leading to various GH deficiencies (139191 OMIM NCBI). The expression of the GHRHR gene is associated with an alternative splicing resulting in appearance of at least two transcripts in somatotrophic cells of the hypophysis, which are main targets for somatoliberin ([86], Gene NCBI). The protein product of the GHRHR gene is at first synthesized as a precursor of 423 a.a., from which due to postsynthetic modifications the N-terminal signaling peptide of 22 a.a. is removed and N-glycosylation occurs. In the structure of the mature GHRH-R seven transmembrane domains and a region for interaction with G-proteins are described (Swiss-Prot Q02643). It is also supposed that a shortened GHRH-R isoform (isoform b) can be synthesized during translation of the alternative splicing-variant of the transcript encoding a polypeptide chain of 337 a.a. ([86], NP_001009824 NCBI).
The binding of somatoliberin with GHRH-R activates the target cells of adenylate cyclase and increases in the level of cAMP [85, 87, 88]. An important role in this mechanism is ascribed to the C-terminal part of the GHRH-R molecule and its interaction with G-proteins. For regulation of the cAMP-mediated signaling, phosphodiesterases are very important: they hydrolyze this secondary messenger and can act as factors inhibiting this signaling. A number of phosphodiesterase isoforms (combined into 11 families) is found, and their appearance is shown to be due to polylocus, alternative splicing, alternative promoters, and other mechanisms determining the biochemical polymorphism of proteins [89]. Data on polymorphism of phosphodiesterases in prostate cells and of the signaling-associated hormones of the GH/IGF system will be considered in the next section.
Another natural peptide activator of GH secretion was found in 1999 by Kojima et al. [90] during studies of the action mechanism of small synthetic molecules called growth-hormone secretagogues (GHSs) capable of stimulating GH release from somatotrophic cells and interacting with a special membrane receptor called GHS-R (discriminated from GHRHR). During their search for a natural ligand for GHS-R, these authors isolated from rat stomach and identified as a natural hormone a regulator of GH secretion, a polypeptide consisting of 28 a.a. Considering its ability for growth hormone release, they called this peptide ghrelin [90, 91].
Human ghrelin is initially synthesized inside a preproprotein, which is encoded by the GHRL gene (605353 OMIM NCBI). Subsequent site-specific and limited proteolysis generates from this protein of two products, ghrelin and obestatin, and functioning of the latter seems to be not associated with the GH/IGF system [92]. The GHRL gene has been shown to include 74 SNPs, and only one of which (a nonsynonymous one) occurs inside the exon sequence. The GHRL gene is expressed with alternative promoters and an alternative splicing occurs. A number of transcripts are produced, and some of them seem to be not translated (after GENE NCBI).
The ability to synthesize ghrelin is shown not only in stomach cells but also in cells of different parts of the brain and of some other organs, and in many tumor cells including PC [91, 93-95]. To acquire its functional activity, ghrelin has to undergo postsynthetic modifications [91, 95]. In particular, such modifications are acylation of the Ser3 residue by octanoic acid, which occurs in the stomach tissues. Several ghrelin isoforms are found in blood, and this hormone has been shown to influence cell proliferation [91, 94, 95].
During its functioning, ghrelin interacts with a special receptor (GHS-R), and this (under the influence of G-proteins) results in stimulation of phospholipase C and increase in the content of intracellular Ca2+ [91]. The receptor of ghrelin is encoded by the GHSR gene (located on chromosome region 3q26.31). On its expression due to alternative splicing, at least two transcripts are produced. One of them (isoform 1a) is responsible for synthesis of a polypeptide chain consisting of 366 a.a. and functioning as a receptor (GENE NCBI; Swiss-Prot Q92847). The extracellular N-terminal region of the receptor is glycosylated. GHS-R is detected not only in somatotrophic cells of the hypophysis but also in the cells of the hypothalamus, hippocampus, and some other brain sections, as well as in cells of stomach, small intestine, kidneys, placenta, and some tumors [91, 94, 95].
It is known from the 1970s that in hypothalamus the polypeptide hormone somatostatin (growth hormone release-inhibiting factor, SST, SRIF) is synthesized, which inhibits GH secretion and thus is an antagonist of somatoliberin and ghrelin [96]. The somatostatin gene SST encodes a polypeptide chain consisting of 116 a.a. that is a preproprotein subjected to postsynthetic modifications resulting in alternative production from its C-terminal region of two functionally active peptides consisting either of 28 or 14 a.a. (somatostatin-28 or somatostatin-14, respectively) ([97], Swiss-Prot P61278). Active isoforms of somatostatin circulate in the bloodstream [96, 97]. Somatostatin not only inhibits the secretion of GH but also performs some other functions in both the nervous system and peripheral tissues, in particular it influences cell proliferation normally and in cancer [96, 98].
Somatostatin receptors are combined into a special protein family of six members. These membrane proteins detectable in differently differentiated cells [96, 98, 99] are encoded by five genes SSTR1-SSTR5 with different exon–intron structure and located on different chromosomes (GENE NCBI). The best studied among them is the SSTR2 gene, which contains 130 SNPs, only four of them found in exons (three SNPs are nonsynonymous) (SNP NCBI). The SSTR2 gene is expressed with an alternative splicing and generation of two isoforms, SSTR2A and SSTR2B (respectively 369 and 356 a.a.), which undergo postsynthetic modifications including glycosylation of the N-terminal extracellular domain and acylation by palmitic acid of the C-terminal cytoplasmic domain ([99], Swiss-Prot P30874). The somatostatin receptors function on interacting with G-proteins, and this leads to generation of cAMP and also to triggering of other types of signaling [96, 98]. Respectively, the biochemical polymorphism of all signaling processes additionally contributes to the variety of biological effects of somatostatin, and consequently, of the whole GH/IGF system.
In the action of GH by the basic branch some protein products of the same gene of the growth hormone receptor (the GHR gene) are especially important. This gene and its products are now rather well studied [22, 56, 100, 101].
First, the GHR gene has been shown to direct synthesis of a transmembrane protein, and just this full-size product (the mature protein consists of 620 a.a. and its precursor contains 638 a.a.) forms a functionally active dimer and acts as a GH receptor (GHR) [22, 100].
Second, on GHR expression due to alternative splicing, 26 nucleotides can be removed in exon 9, which results in the appearance of a stop codon in position 280. As a result, a short protein GHR (GHRtr or GHR1-279) is produced that lacks 97.5% of the intracellular domain [101]. Two more products of the alternative splicing of the GHR gene are also known, one of which appears on excision of exon 3 (GHRd3) and the other (GHR1-277) appears due to use of the alternative acceptor site of exon 9 [102]. The physiological significance of these products remains under discussion.
Third, the limited proteolysis upon removal of the signaling peptide leads in human to detachment of a large fragment of the sequence (the extracellular domain), which enters into the bloodstream and functions as a special transporter and stabilizer of GH. This product is called GH-binding protein (GHBP) [22, 56]. Some data suggest that GHRtr is most effectively processed in GHBP [103].
Moreover, postsynthetic modifications are associated with a pronounced glycosylation of GHR (by five amino acid residues in the N-terminal extracellular region) and production of three intramolecular disulfide bonds (Swiss-Prot P10912).
The presence of GHR (with differently pronounced synthesis and different contents of individual isoforms) is recorded in most human tissues; therefore, different tissues of internal organs, including the prostate, are targets of GH [22, 56].
As a mediator of signal transmission from GH to cells, GHR first of all activates tyrosine protein kinase JAK2 [22, 56]. This enzyme of the protein kinase family JAK (Janus kinases) has two very similar domains, one of which possesses catalytic activity and the other is pseudocatalytic. Just because of this feature these enzymes were given the name of the two-faced god Janus [104]. In JAK2, 942 SNPs were found including 23 inside exons in which 12 SNPs are nonsynonymous, i.e. cause amino acid substitutions providing for biochemical polymorphism of a protein product due to polyallelism (SNP NCBI). Up to now there are no data on alternative splicing at the JAK2 gene expression. However, the protein product JAK2 consisting of 1132 a.a. is phosphorylated postsynthetically, and moreover is capable of autophosphorylation (Swiss-Prot O60674).
Phosphorylation of JAK2 is accompanied by acquisition by this protein of enzymatic activity that allows it to initiate a number of signaling mechanisms. First, the signaling mechanisms triggered by JAK2 include phosphorylation of proteins of the STAT family and cascade processes that arise later [104, 105]. The family of human STAT proteins is represented by seven proteins with highly homologous primary structures that play an important role in the regulation of cell proliferation [106]. Phosphorylated STAT proteins (in particular 1, 3, 5a, and 5b) form dimers capable of penetrating into the cell nucleus and interacting with specific sequences in DNA that leads to activation of transcription of certain genes. This results in enhancement of different anabolic processes in the cell. The mechanism involving proteins STAT5a/b is thought to be the most important for postnatal growth [105].
Second, in addition to STAT phosphorylation, the GH signal transmitted into the cell through GHR is involves phosphorylation of insulin receptor substrate proteins (IRSs) [56, 107]. These proteins, in particular IRS-1 and IRS-2, can associate with phosphatidylinositol-3′-phosphate kinase, which triggers a chain of events initiated not only by GH but also by IGF-1 (see below) and by some other regulatory factors [56, 108].
Third, signaling mechanisms triggered through GHR and JAK2 also activate mitogen-activated kinases (MAP kinases), which in turn provide for some anabolic effects [56, 107, 109]. Four types of cascade reactions initiated through MAP kinases are characterized, and some of them are involved in pathogenesis of tumors and other common diseases [109-111]. Other important effects of GHR on the target cell are also known, in particular the enhancement of Ca2+ entrance into it from the intercellular space through a special voltage-dependent L-type transmembrane channel with a subsequent cascade of Ca2+-activated events and also activation of phosphoprotein kinase C (PKC) [56, 112].
These signaling processes induce of a spectrum of genes [56, 111] including the abovementioned gene encoding IGF-1 and some genes associated with carcinogenesis (in particular c-fos, serine proteinase inhibitor 2.1 (Spi2.1), etc.).
Thus, through the basic branch a very broad spectrum of GH effects on variously differentiated cells is realized. The biochemical polymorphism of proteins involved in this branch (caused by polylocus, polyallelism, alternative splicing, different postsynthetic modifications, etc.) significantly contributes to the final biological effects creating a variety of physiological and pathological processes.
Lateral Branch Proteins
Synthesis of IGF-1 induced by GH was found in the cells of liver, muscles, and some other tissues including prostate [13, 22, 56, 62, 63]. According to modern concepts, IGF-1 functions in the human organism not only as a growth factor but also as an endocrine agent secreted into the bloodstream and involved in normal and pathological processes of various target cells [13, 56-58]. Thus, Laron [113] proposed considering IGF-1 as a growth-regulating hormone. The level of serum IGF-1 has been shown to correlate with growth and weight characteristics in humans and some other mammals. Functioning of IGF-1 is provided by various proteins, which are combined by some authors into a special IGF-1 axis or a lateral branch in the GH/IGF system [13, 56-58].The IGF1 gene has been studied for about 25 years. It was found to contain 890 SNPs, and only five of them are inside exons including three nonsynonymous ones, and only one SNP leads to an amino acid substitution in the final protein product (67Ala→Thr in the sequence of 70 a.a.) (SNP NCBI, Swiss-Prot P01343-1). The IGF1 gene has been shown to contain three types of mutations, which can either cause severe inborn developmental defects or be manifested rather slightly (147440 OMIM NCBI). The works in this field are continuing, and in 2009 a new type of mutations was described [114] that sharply affects the ability of IGF-1 to bind with its receptor IGF1-R (see below).
The comparison of the IGF1 nucleotide sequences with the genes of two related proteins, the insulin-like growth factor 2 (IGF-2) and insulin, and also data on common features in molecular mechanisms of functioning of these proteins [115] and biological effects including the influence on glucose metabolism [57] resulted in the conclusion of their common evolutionary origin [56, 115].
In 1989 Rotwein et al. [116] described the IGF1 gene structure containing five exons. However, later an alternative promoter and one more exon were found [117, 118]. Figure 3 presents a general concept about the IGF1 gene structure and its expression using alternative promoters and the alternative splicing resulting in production of some transcript isoforms ([117, 118], GENE NCBI).
The reading of information from isoforms IGF-1-a and IGF-1-b results in production of two protein precursors (preproproteins), which contain 153 and 195 a.a. and undergo posttranslational processing (which includes proteolysis and production of three disulfide bonds) resulting in formation and releasing into the bloodstream of the same final product consisting of 70 a.a. (Swiss-Prot P01343).Fig. 3. General scheme of the IGF1 gene structure and products of its expression using alternative promoters and alternative splicing ([117, 118], GENE NCBI).
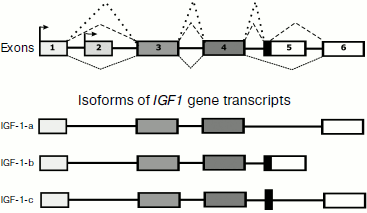
The mRNA of IGF-1-c gives another final product that consists of 110 a.a. (CAR81472 Protein NCBI) and contains in its sequence a special C-terminal region partially encoded by an alternatively spliced fragment of the intron [118]. This protein product was called MGF (mechano-growth factor) because its synthesis was observed to pronouncedly increase in muscles upon intensive exercise [118]. The physiological significance of MGF seems not to be limited to muscle tissues, thus, some data suggest its protective effect in ischemia of brain cells [119].
The basic IGF-1 isoform (70 a.a.) functions by both endocrine and autocrine/paracrine mechanisms (Fig. 2) [56, 113]. The major part of IGF-1 is secreted into the bloodstream and circulates mainly in complexes with specific insulin-like growth factor-binding proteins (IGFBPs) [120, 121]. In humans six IGFBPs are found, and these proteins that are essentially similar in structure and functions form a special family [120, 122]. All members of this family have in the primary structure two conservative domains (N- and C-terminal ones) with cysteine- and proline-enriched consensus sequences and a relatively variable median area and are also able to bind IGF-1 with high affinity. On synthesis of each IGFBP a precursor protein is initially produced, then signaling peptides are eliminated and the final product enters into the bloodstream. Four of six IGFBPs undergo postsynthetic glycosylation and three IGFBPs undergo phosphorylation [120, 123].
The major part of circulating IGF-1 (~75-80%) is found in the complex with IGFBP-3 and with the so-called acid-labile subunit (ALS) [122, 123]. The corresponding gene (IGFALS) contains two exons; 109 SNPs and some mutations leading to hereditary diseases are found in its sequence. ALS is a leucine-enriched glycoprotein with molecular weight of ~85 kDa. The IGF-1 complex with IGFBP-3 and ALS has molecular weight of 150 kDa and a relatively long lifetime for proteins of the GH/IGF system in the bloodstream – about 12-16 h. The concentration of this complex depends on GH and decreases with deficiency of endogenous GH. Nearly all remaining IGF-1 is thought to form complexes with other proteins, and only ~1% of IGF-1 circulates in the plasma in the free state [124].
Nine more proteins are known in humans that are somewhat similar to IGFBP in structure and capable of binding IGF-1 but with significantly lower affinity [120]. These proteins are called IGFBP-related proteins (IGFBP-rPs). The diversity of IGFBP and IGFBP-rPs combined into the same superfamily demonstrates the significance of protein polymorphism for the GH/IGF system.
IGFBPs play an important role in physiological processes associated with cell proliferation and are involved in pathogenesis of many diseases including tumorigenesis [119, 125]. Further studies of polymorphism of proteins transporting IGF-1 are promising for more detailed knowledge about the regulation of various anabolic processes in the human body.
Many manifestations of biochemical polymorphism are found in membrane receptors capable of recognizing IGF-1 and transmitting to target cells the signal from a given growth factor. The function of IGF-1 receptor is mainly performed by the protein product of the IGF1R gene [56, 113, 115, 123]. This gene belongs to a family of genes encoding membrane receptors possessing a cytoplasmic domain with tyrosine kinase activity (Swiss-Prot P08069). The IGF1R gene is already known to contain more than 3400 SNPs of which 86 are in the area of exons, and 25 of them are nonsynonymous (SNP NCBI). The latter lead to amino acid substitutions and thus are responsible for a pronounced contribution of polyallelism to biochemical polymorphism of the IGF-1 receptor.
Expression of the IGF1R gene is recorded in cells with very different types of differentiation: from muscle and liver cells to those of placenta (147370 OMIM NCBI). The expression of the IGF1R gene is also shown in prostate cells and in different tumors, and many authors indicate an immediate involvement of the IGF-1 receptor in carcinogenesis and in some other pathologies [126, 127].
The expression of the IGF1R gene produces a protein precursor of the receptor, which consists of 1367 a.a. and undergoes a number of postsynthetic modifications (Swiss-Prot P08069). First, through limited and site-specific proteolysis the protein precursor loses the 30-a.a. signaling peptide and is cut into two polypeptide chains that are later converted into α- and β-subunits of the mature receptor (706 and 627 a.a.). Second, a number of disulfide bonds are produced which, in particular, provide for the joining of α- and β-subunits and formation of a functionally active receptor as a tetrameric complex. Third, the repeated glycosylation of extracellular regions of both α- and β-subunits has been shown. And finally, four tyrosine residues are subjected to autocatalytic phosphorylation during functioning in the intracellular region of the β-subunit of the mature IGF-1 receptor; one serine and one threonine residues are also phosphorylated. During the last ten years several models have been proposed of domain and three-dimensional structure of the IGF-1 receptor [126, 128, 129].
It should also be noted that the biochemical polymorphism of the IGF-1 receptor is significantly contributed to by its ability to produce heterotetramers with structure similar to that of insulin receptors [129, 130]. These heterotetramers are different from homotetramers in the ability to bind IGF-1. Some data indicate that they can bind insulin and the related IGF-2 even more effectively [129, 130]. The presence of heterotetramers (in different quantities relatively to homotetramers) is found in the cells of different organs and tissues [129, 130].
Signaling processes triggered by the activated receptor IGF-1 have been studied for more than a decade. Numerous studies revealed some proteins involved in these processes and characterized by a pronounced biochemical polymorphism [56, 131]. The main substrates for IGF-1R are the abovementioned IRS proteins [56, 112]. At least four members of this family of proteins are involved in activation of the signal from IGF-1 and thus provide a contribution of the polylocus to the biochemical polymorphism of this process. The best-studied participant is IRS-1 [131], its gene located in q36 of chromosome 2 containing only two exons and 372 SNPs (38 SNPs and 14 of them are nonsynonymous) (GENE SNP NCBI). Moreover, mutations in the gene of IRS-1 are also known that cause amino acid substitutions associated with type II diabetes and/or with a risk of early atherosclerotic cardiovascular diseases ([132], 147545 OMIM NCBI).
IRS-1 is a polypeptide chain consisting of 1242 a.a. that contains a number of domains and special motifs with high contents of serine, glycine, and proline residues (Swiss-Prot P35568). The postsynthetic phosphorylation of IRS-1 accompanied by binding with specific adaptor proteins (e.g. GRB2) can be considered as one of the most important stages in the signaling induced by IGF-1 via IGF-1R. This results, in particular, in a chain of reactions leading to generation of inositol-3,4,5-triphosphate, which is known as a secondary messenger activating in turn specific serine/threonine kinases, or RAC serine/threonine-protein kinases denoted in the English literature by the abbreviation AKT after an oncogene [112].
AKTs phosphorylate some substrates including proteins involved in protein synthesis and in gene transcription, and this increases cell proliferation and viability [112, 133]. Moreover, the activity of AKTs enhances glucose transport into cells and inhibits intracellular proteinases [133, 134]. In mammals AKTs are represented by three isoforms determined, respectively, by three different genes. AKT1, or serine/threonine kinase (RAC-alpha serine/threonine-protein kinase), is intensively studied in connection with problems of cancer [133] because at least one amino acid substitution (17E→K) in it is believed to be associated with the appearance of differently located malignant tumors [135, 136].
In addition to IRS proteins, activated IGF-1R also phosphorylates another group of proteins known as transforming proteins of the SHC family (with molecular weight of 46, 52, and 66 kDa). The structure of these proteins is characterized by the presence in the C-terminal region of the SH2-domain and of an area enriched with glycine and proline [56, 137]. These three proteins are considered as isoforms because they are produced due to alternative splicing on expression of the same gene SHC1. Their presence is recorded in cells with various types of differentiation in the cytoplasm and mitochondrial matrix ([137], Swiss-Prot P29353). Upon phosphorylation induced by IGF-1R, the SHC family proteins trigger a cascade of reactions activating cell proliferation. The involvement of these proteins in malignant transformation of cells is also shown.
As compared to the abovementioned data on the IGF-1 signaling mechanisms involving phosphorylation of various proteins, the detection of activation by this factor of calcium-dependent serine/threonine protein phosphatase 3, or calcineurin, was rather unexpected [112, 138, 139]. This enzyme is a heterodimer containing a catalytic subunit (A) with molecular weigh of ~60 kDa and a regulatory subunit (B or Ca2+-binding protein) with molecular weight of 19 kDa. The human genome contains three genes encoding subunit A (PPP3CA, PPP3CB, PPP3CC – 114105, 114106, 114107 OMIM NCBI, respectively), which include several hundreds of SNPs. Polylocus and polyallelism as well as alternative splicing on expression of these genes are responsible for the pronounced biochemical polymorphism of calcineurin. Subunit B also contributes to this phenomenon despite its possessing only one gene but more than 500 SNPs and alternative splicing on expression (601302 OMIM NCBI).
Calcineurin hydrolyzes phosphoester bonds in molecules of transcription factors that are known as nuclear factors of activated T cells (NF-AT). Then NF-ATs diffuse into the cell nucleus, interact with certain sites in DNA, and trigger expression of some genes. Calcineurin-induced signaling reactions can be related with development of hypertrophies [138] and with pathogenesis of some inflammatory diseases. However, calcineurin inhibitors are also shown to stimulate proliferation of some tumor cells [139].
Thus, numerous studies of proteins of the lateral branch of the human GH/IGF system revealed their biochemical polymorphism (caused by different molecular mechanisms), which directly influences functioning of cell structures and cell proliferation.
Manifestations of biochemical polymorphism in the ten most important participants of the human GH/IGF system are generalized in the table.
Manifestations of biochemical polymorphism in ten components of the
growth hormone (GH)/insulin-like growth factor 1 (IGF-1) system in
humans after NCBI and Swiss-Prot databases (www.ncbi.nlm.nih.gov/ and www.expasy.org/)
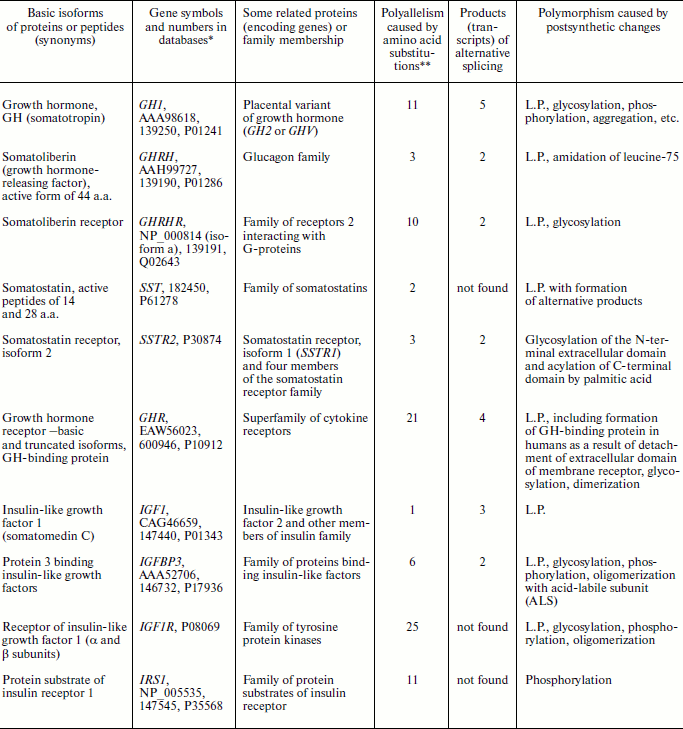
Note: L.P., limited and/or site-specific proteolysis.
*Numbers are in the following order: NCBI Protein, OMIM, and
Swiss-Prot.
**After databases of SNP NCBI and Swiss-Prot.
ISOFORMS OF GROWTH HORMONE SYSTEM PROTEINS IN PROSTATE CELLS DURING
GROWTH, DEVELOPMENT, AND CARCINOGENESIS
The prostate, which plays an important role in reproduction, has a complex histological structure. It includes cells with different types of differentiation and also stem cells, and the development of prostate during ontogenesis and is functioning are controlled by many endocrine factors [22, 56, 140]. Certainly, androgenic hormones and their receptors are given special attention in both the normal state of the prostate and in cases of its tumors [140, 141]. However, as mentioned, some authors recorded in the prostate cells synthesis of many proteins of the GH/IGF system. For the first time it was recorded in 1987 by Prieto and Carmena [142] who found that epithelial cells of the rat prostate could effectively bind GH, and later these cells intensively captured leucine. Based on these and some other findings, the authors concluded that the GH receptor should be present on membranes of prostate epithelial cells. It was also concluded that GH could regulate anabolic processes in the prostate cells through its receptor. These conclusions were confirmed by many works including those performed on human prostate cells. The paper by Ballesteros et al. [143] is one of the most comprehensive works concerning this question. They not only revealed in the prostate three transcripts of the GHR gene (known isoforms of mRNA) but compared their contents in the prostate with the contents in other tissues and in some cultures of human cells (liver, muscles, adipose tissue, kidneys, fibroblasts, lymphocytes, etc.). Along with the major full-size transcript of the GHR gene (GHRf1), the production of isoforms GHR1-279 and GHR1-277 was shown in the prostate. These isoforms seemed to be products of alternative splicing and to play a special role in the GH/IFG system, as noted above. Moreover, the ratio of isoforms of these transcripts rather specifically varied in the studied tissues [143] and seemed to be associated with cell differentiation.
Expression of the GHR gene was found by different groups of authors in various cells of malignant tumors of the prostate in humans and animals [145]. By now GHR gene expression is clearly shown in both biopsy of malignant and benign tumors of human prostate and in the cells of all studied strains of prostate carcinoma [146, 147]. Thus, in 2004 Weiss-Messer et al. [146] studied mRNA of GHR in the cells of benign prostate hyperplasia (BPH) and prostate adenocarcinoma and also in cell cultures LNCaP, PC3, and DU145. This mRNA was detected in both the tissue samples and the cultured cells, and its level in the carcinoma tissues was 80% higher than in the BPH cells. In 2009 GH, estradiol, and triiodothyronine were reported to stimulate production of different isoforms of the GHR gene transcripts in LNCaP cells [147]. Note that GH stimulated the production of both GHRf1 and the short isoform GHR1-279, whereas estradiol and triiodothyronine mainly stimulated synthesis of the short isoform. The evaluation of GHR gene expression and the observed polymorphism of the products allowed the authors to conclude that they would be promising as targets for drugs directed to decrease and/or prevent the development of human prostate tumors.
Unexpected data were obtained by investigations of transcripts of some genes of the GH/IGF system in cell lines PC3, DU145, LNCaP, and ALVA41 [148]. Using real-time PCR, Chopin et al. demonstrated the presence in these cells not only of isoforms of the GHR gene transcripts but also of GH mRNA. The sequencing revealed that this mRNA was represented by expression products of the GH1 gene (pituitary isoform) and of the GH2 gene (or GHV, placental isoform). Note that generation of the corresponding protein products (GH and GHR) was also detected using immunochemistry. Therefore, it was suggested that at least in cancer cells of the prostate the GH/IGF system could function by endocrine and also by autocrine/paracrine mechanisms [148].
This hypothesis was confirmed in another work of the same group of Australian researchers who succeeded in detecting mRNA for GHRH and GHRH-R in several cell lines of human prostate cancer (DU145, LNCaP, PC3) [149]. Antagonists of GHRH inhibited the growth of prostate cancer cells, possibly due to inhibition of GHRH-R and of the GH/IGF system functioning by the autocrine/paracrine mechanism. In 2005 the main conclusions of the Australian researchers were confirmed by American authors [150] who found in the prostate cancer cells not only the major transcript of the GHRHR gene but also its isoform produced by alternative splicing. Because GHRHR gene expression was also recorded in lung cancer cells, Havt et al. [150] concluded that GHRH and GHRH-R should play an important role in pathophysiology of human malignant tumors.
The presence of autocrine/paracrine mechanism in the GH/IGF system in prostate cancer is also evidenced by data of the cited above Australian researchers about the presence in four lines of prostate cancer of transcripts encoding ghrelin and two isoforms of the ghrelin receptor (1a, 1b) [151]. The presence of ghrelin and the receptor 1a isoform was confirmed immunochemically in all four cell lines. In the cultured cells of PC3, ghrelin caused a 33% increase in proliferation. On analysis of cDNA of a library obtained from normal cells of human prostate, the authors concurrently found transcripts only of GHS-R1a and did not detect transcripts of ghrelin and isoform 1b of the receptor. Thus, it is possible that the autocrine/paracrine mechanism of the GH/IGF system in prostate cancer is rather a specific consequence of changes in gene expression during carcinogenesis.
Similar results but detailing some aspects of the GHRL and GHSR gene expression in the cultured prostate cancer cells and also in specimens of malignant and benign prostate tumors were published by Cassoni et al. [152]. In all tissue specimens from prostate cancer and in half of specimens with BPH they found transcripts of the GHRL gene, although no protein product was detected by immunochemical analysis. Then in PC3 cells the presence of ghrelin transcript and its protein product were confirmed, but no corresponding transcripts or ghrelin itself were found in DU145 and LNCaP cells. Studies on ghrelin receptors resulted in particular in finding of mRNA of the GHS-R1b isoform in 50% of specimens with BPH, but no transcripts of the GHSR gene were found in the prostate cancer tissue specimens. However, in DU145 cells transcripts of both isoforms of the ghrelin receptor (1a, 1b) were found, whereas the search for transcripts of the GHSR gene in PC3 and LNCaP cells was unsuccessful. Nevertheless, the specific binding of labeled ghrelin was recorded in both specimens of malignant and benign tumors of prostate and on membranes of cultured DU145 and PC3 cells. Naturally, the authors supposed that in prostate cancer ghrelin receptor isoforms could appear that would be different from the known GHS-R1a and 1b.
In 2008 the abovementioned works and some other publications were analyzed in a review by Lanfranco et al. [94]. They concluded that ghrelin and its receptors capable of providing the autocrine/paracrine mechanism of the GH/IGF system should play an important role in prostate cancer (as well as in other forms of cancer). Moreover, in 2009 an increased content of ghrelin was found in blood serum of patients with prostate cancer, and therefore this hormone was suggested to be involved in the tumor cell proliferation [153].
Surprisingly, synthesis and secretion of somatostatin, which is the third regulatory hormone that can influence triggering of the GH/IGF system, were also found in cells of human prostate cancer. In particular, in cells of the PC3 and LNCaP lines the SST gene transcript and somatostatin secretion into the culture medium were recorded [154]. And the somatostatin secretion level and proliferative activity were inversely proportional. Somatostatin receptors in the prostate cancer cells were reported in the end of the last century [155], and later isoforms of these proteins were actively studied by many authors. All five known isoforms of somatostatin receptors were recently detected by immunochemical methods in prostate cancer cells, but their amounts were significantly different in the cell subpopulations [156]. Reports about the presence of somatostatin and its receptors in prostate cancer cells can be considered as additional evidence for existence in these cells of both endocrine and autocrine/paracrine mechanisms for the GH/IGF system and also as a confirmation of importance of biochemical polymorphism of proteins providing for this mechanism.
It was mentioned above that the signaling of positive inducers of GH (GHRH and ghrelin) and also the inhibiting effect of somatostatin include a stage of cAMP production. Phosphodiesterases are involved in the control of the level of this secondary messenger, and more than ten isoforms of phosphodiesterases are present in human prostate cells [89]. Thus, the biochemical polymorphism of these enzymes also contributes to functioning of the GH/IGF system.
Some proteins of the lateral branch of the GH/IGF system were also found in different cells of the prostate. Thus, in 1993 Pietrzkowski et al. [157] reported about the ability of cell lines PC3, DU145, and LNCaP to secret IGF-1 and also to synthesize the IGF-1 receptor. Soon, Kimura et al. [158] confirmed that these cell lines had the IGF-1 receptor, but there were only trace amounts of mRNA of IGF-1. Nevertheless, they revealed the generation of IGF-2 (an IGF-1 related factor) and the IGF-2 receptor. In the same work synthesis of some members of the IGFBP family was also shown. It was finally concluded that isoforms of different components of the lateral branch of the GH/IGF system could play an important role during prostate carcinogenesis. Later different cells of prostate were shown to produce IGF-1 [159, 160]. Therefore, both endocrine and autocrine/paracrine mechanisms of IGF-1 (and/or IGF-2) were suggested to exist during tumorigenesis in prostate [160, 161].
A possible endocrine influence of IGF-1 (and/or IGF-2) on progression of prostate cancer has been under discussion for many years [162]. An important argument in favor of such a mechanism is the increased level of IGF-1 in blood of patients with prostate cancer, but contradicting data are also known. In the review of 2009, Rowlands et al. [121] analyzed more than two thousand reports on the problem during the period of 1966-2007 and came to a conclusion about pronounced correlation of prostate cancer with IGF-1 and its poor association with IGFBP3. The authors also noted that the importance of other members of the IGFBP family (-1 and -2) and of IGF-2 was shown in relatively few studies of the risk of prostate cancer.
In studies of the endocrine mechanism of IGF-1 during progression of prostate tumors many authors focused their attention on isoforms of IGF-1 and proteins responsible for IGF-1 signaling [159, 161, 162]. This resulted in the viewpoint that prostate cancer and other malignancies should be associated with deregulation of the whole IGF-1 axis [162].
A special field in studies on the role of polymorphism of various proteins of the GH/IGF system in prostate cells during growth, development, and carcinogenesis is represented by works directed to determine associations between different SNPs and other types of DNA polymorphism in the corresponding genes. In particular, promising results were recently obtained in investigation of SNPs in the GHR gene and in the genes encoding individual members of the IGFBP family [163, 164]. A significant association was revealed between the risk of prostate cancer and one SNP in the IRS1 gene leading to the amino acid substitution G972R accompanied by changing protein charge [165]. A similar result was recently obtained for another SNP in the IRS1 gene responsible for a similar amino acid substitution (E917R) [166]. Interesting data concerning SNPs in the genes of somatostatin receptors were published in 2009 by Johansson et al. [167]. Although the authors failed in finding correlation of these SNPs with the risk of prostate cancer, some of SNPs in SSTR5 (especially rs4988483) were shown to be associated with an increased level of IGF-1 in the blood of patients, and this seemed to be related with the tumor growth. Among numerous studies on other types of DNA polymorphism, the comprehensive paper by Chen et al. [168] should be mentioned reporting that heterozygotes in the repeat (AGG)7 in the IGF-1R gene were characterized by an increased risk of prostate cancer.
CONCLUSION
Results of numerous investigations show that manifestations of biochemical polymorphism of growth hormone and of other protein and peptide hormones, as well as of transport proteins, receptors, and signaling components are very important for functioning of the GH/IGF system in humans. This polymorphism is formed on three levels: on the level of genome and gene structure, during transcription and maturation of transcripts, and during postsynthetic formation of functionally active protein complexes. As a consequence, polymorphic proteins of the growth hormone system provide for a pronounced inter-individual diversity of the corresponding molecular structures: multicomponent transport complexes, oligomeric membrane receptors, and intracellular protein ensembles involved in regulation of gene expression. In turn, this structural polymorphism has a significant influence on the biological effects in different links of the GH/IGF system using both the endocrine and autocrine/paracrine mechanisms during physiological processes and in tumorigenesis. Special attention on the role of polymorphism of some proteins of the GH/IGF system is given in the development of prostate cancer. In particular, just in this aspect the family of proteins binding IGF-1 and various oligomeric receptors capable of interacting with it were given special consideration [126, 162, 169, 170]. Recently the gene of the IGF-1 receptor was even proposed to be considered as an oncogene [170].
It should also be noted that available data on the polymorphism of components of the human GH/IGF system indicate that postgenomic techniques are very promising for understanding many urgent biomedical problems, in particular, to determine predisposition to prostate cancer.
This work was supported by State contracts Nos. 373n-08 and 375n-08 of the Department of Science and Industrial Politics of the Moscow City Government, 2008-2009.
REFERENCES
1.Woods, R. A. (1983) Biochemical Genetics
[Russian translation], Mir, Moscow.
2.Beaudet, A. L., Scriver, C. R., Sly, W. S., Cooper,
D. N., Mekusick, V. A., and Schmidke, J. (1989) in The Metabolic
Basis of Inherited Disease (Scriver, C. R., Beaudet, A. L., Sly, W.
S., and Walle, D., eds.) 6th Edn., Mc Graw-Hill Inf. Ser. Chem., Vol.
1, pp. 3-163.
3.Lewin, B. (1987) Genes [Russian
translation], Mir, Moscow.
4.Baumann, G. (1991) Endocrine Rev.,
12, 424-449.
5.Woychik, R. P., Klebig, M. L., Justice, M. J.,
Magnuson, T. R., and Avner, E. D. (1998) Mutat. Res.,
400, 3-14.
6.Anderson, N. G., Matheson, A., and Anderson, L.
(2001) Proteomics, 1, 3-12.
7.Shishkin, S. S. (2002) Vestn. Ros. Akad. Med.
Nauk, No. 4, 11-16.
8.International Human Genome Sequencing Consortium
(2001) Nature, 409, 860-921.
9.Venter, C. J., Adams, M. D., Myers, E. W., Li, P.
W., Mural, R. J., Sutton, G. G., Smith, H. O., Yandell, M., Evans, C.
A., Holt, R. A., Gocayne, J. D., Amanatides, P., Ballew, R. M., Huson,
D. H., Wortman, J. R., Zhang, Q., Korida, C. D., Zheng, X. H., Chen,
L., Skupski, M., et al. (2001) Science, 291,
1304-1351.
10.Hernandez-Boussard, T., Woon, M., Klein, T. E.,
and Altman, R. B. (2006) OMICS, 10, 545-554.
11.Ghosh, D., and Poisson, L. M. (2009)
Genomics, 93, 13-16.
12.D’Amico, F., Biancolella, M., Margiotti,
K., Reichardt, J. K., and Novelli, G. (2007) Pharmacogenomics,
8, 645-661.
13.Baumann, G. P. (2009) Growth Horm. IGF
Res., 19, P.333-340.
14.Mononen, N., and Schleutker, J. (2009) J.
Urol., 181, 1541-1549.
15.Marri, R., Grenner, D., Mejes, P., and Roduell,
V. (1993) Human Biochemistry, Vol. 2 [Russian translation], Mir,
Moscow, pp. 170-185.
16.Orengo, C. A., and Thornton, J. M. (2005)
Annu. Rev. Biochem., 74, 867-900.
17.Buljan, M., and Bateman, A. (2009) Biochem.
Soc. Trans., 37, 751-755.
18.Zhang, F., Gu, W., Hurles, M. E., and Lupski, J.
R. (2009) Annu. Rev. Genom. Hum. Genet., 10, 451-481.
19.Primrose, S., and Twyman, R. (2008) Genomics:
Applications in Human Biology [Russian translation], BINOM, Moscow,
pp. 38-84.
20.Ramensky, V. E., and Syunyaev, Sh. R. (2007) in
Molecular Polymorphism in Human (Varfolomeev, S. D., ed.) [in
Russian], Vol. 1, Moscow, pp. 83-107.
21.Zhang, F., Gu, W., Hurles, M. E., and Lupski, J.
R. (2009) Annu. Rev. Genom. Hum. Genet., 10, 451-481.
22.Phillips, J. A. (1995) in The Metabolic Basis
of Inherited Disease (Scriver, C. R., Beaudet, A. L., Sly, W. S.,
and Walle, D., eds.) 7th Edn., Mc Graw-Hill Professions Division, Vol.
II, pp. 3023-3044.
23.Yang, N., Garton, F., and North, K. (2009)
Med. Sport Sci., 54, 88-101.
24.Shishkin, S. S., and Kalinin, V. N. (1992)
Medical Aspects of Biochemical and Molecular Genetics [in
Russian], VINITI, Moscow.
25.Frezal, J., Munnich, A., and Mitchell, G. (1983)
Hum. Genet., 64, 311-314.
26.Breitbart, R. E., Andreadis, A., and
Nadal-Ginard, B. (1987) Ann. Rev. Biochem., 56,
467-495.
27.Mironov, A. A., Fickett, J. W., and Gelfand, M.
S. (1999) Genome Res., 9, 1288-1293.
28.Modrek, B., Resch, A., Grasso, C., and Lee, C.
(2001) Nucleic Acids Res., 29, 2850-2859.
29.Lee, C., and Wang, Q. (2005) Brief
Bioinform., 6, 23-33.
30.Lewis, B. P., Green, R. E., and Brenner, S. E.
(2003) Proc. Natl. Acad. Sci. USA, 100, 189-192.
31.Eilbeck, K., Moore, B., Holt, C., and Yandell, M.
(2009) BMC Bioinformatics, Feb 23;10:67. P.1-15.
32.Gustincich, S., Sandelin, A., Plessy, C.,
Katayama, S., Simone, R., Lazarevic, D., Hayashizaki, Y., and Carninci,
P. (2006) J. Physiol., 575, 321-332.
33.Courtois, S. J., Lafontaine, D. A., and Rousseau,
G. G. (1992) J. Biol. Chem., 267, 19736-19743.
34.Moll, U. M., and Slade, N. (2004) Mol. Cancer
Res., 2, 371-386.
35.Chen, S.-H., Habib, G., Yang, C. Y., Gu, Z. W.,
Lee, B. R., Wang, S. A. Silberman, S. R., Cai, S.-J., Deslypere, J. P.,
Rosseneu, M., Gotto, A. M., Li, W. H., and Chan, L. (1987)
Science, 238, 363-366.
36.Deichman, A. M. (2001) RNA Editing [in
Russian], Rusaki, Moscow.
37.Kane, J. P., and Havel, R. J. (1989) in The
Metabolic Basis of Inherited Disease (Scriver, C. R., Beaudet, A.
L., Sly, W. S., and Walle, D., eds.) 6th Edn., Mc Graw-Hill Inf. Ser.
Chem., Vol. 1, pp. 1139-1164.
38.Hersberger, M., Patarroyo-White, S., Arnold, K.
S., and Innerarity, T. L. (1999) J. Biol. Chem., 274,
34590-34597.
39.Siddiqui, J. F., van Mater, D., Sowden, M. P.,
and Smith, H. C. (1999) Exp. Cell Res., 252, 154-164.
40.Bass, B. L. (2002) Annu. Rev. Biochem.,
71, 817-846.
41.DeCerbo, J., and Carmichael, G. G. (2005)
Genome Biol., 6, 216 (P1-4).
42.Paz, N., Levanon, E. Y., Amariglio, N.,
Heimberger, A. B., Ram, Z., Constantini, S., Barbash, Z. S., Adamsky,
K., Safran, M., Hirschberg, A., Krupsky, M., Ben-Dov, I., Cazacu, S.,
Mikkelsen, T., Brodie, C., Eisenberg, E., and Rechavi, G. (2007)
Genome Res., 17, 1586-1595.
43.Martinez, H. D., Jasavala, R. J., Hinkson, I.,
Fitzgerald, L. D., Trimmer, J. S., Kung, H. J., and Wright, M. E.
(2008) J. Biol. Chem., 283, 29938-29949.
44.Carninci, P., Kasukawa, T., Katayama, S., Gough,
J., Frith, M. C., Maeda, N., et al. (2005) Science,
309, 1559-1563.
45.Chestkov, V. V., Kovalev, L. I., Shishkin, S. S.,
and Annenkov, G. A. (1985) Vopr. Med. Khim., 31,
60-65.
46.Carafoli, E. (1994) FASEB J., 8,
993-1002.
47.Tzu, J., and Marinkovich, M. P. (2008) Int. J.
Biochem. Cell Biol., 40, 199-214.
48.Sangster, T. A., Lindquist, S., and Queitsch, C.
(2004) Bioessays, 26, 348-362.
49.Fung, K. L., and Gottesman, M. M. (2009)
Biochim. Biophys. Acta, 1794, 860-871.
50.Martinez, A., Traverso, J. A., Valot, B., Ferro,
M., Espagne, C., Ephritikhine, G., Zivy, M., Giglione, C., and Meinnel,
T. (2008) Proteomics, 8, 2809-2831.
51.Meinnel, T., and Giglione, C. (2008)
Proteomics, 8, 626-649.
52.Mahrus, S., Trinidad, J. C., Barkan, D. T., Sali,
A., Burlingame, A. L., and Wells, J. A. (2008) Cell, 134,
866-876.
53.Xu, G., Shin, S. B., and Jaffrey, S. R. (2009) Proc. Natl.
Acad. Sci. USA, 106, 19310-19315.
54.Perler, F. B., Davis, E. O., Dean, G. E., Gimble,
F. S., Jack, W. E., Neff, N., Noren, C. J., Thorner, J., and Belfort,
M. (1994) Nucleic Acids Res., 22, 1125-1127.
55.Sun, W., Yang, J., and Liu, X. Q. (2004) J.
Biol. Chem., 279, 35281-35286.
56.Le Roith, D., Bondy, K., Yakar, S., Liu, J.-L.,
and Butler, A. (2001) Endorcine Rev., 22, 53-74.
57.Holt, R. I., Simpson, H. L., and Sonksen, P. H.
(2003) Diabet Med., 20, 3-15.
58.Rodriguez, S., Gaunt, T. R., and Day, I. N.
(2007) Hum. Genet., 122, 1-21.
59.LaMarca, H. L., and Rosen, J. M. (2008)
Endocrinology, 149, 4317-4321.
60.Thorey, I. S., Hinz, B., Hoeflich, A., Kaesler,
S., Bugnon, P., Elmlinger, M., Wanke, R., Wolf, E., and Werner, S.
(2004) J. Biol. Chem., 279, 26674-26684.
61.Wong, Y. C., Wang, X. H., and Ling, M. T. (2003)
Int. Rev. Cytol., 227, 65-130.
62.Creighton, C. J., Casa, A., Lazard, Z., Huang,
S., Tsimelzon, A., Hilsenbeck, S. G., Osborne, C. K., and Lee, A. V.
(2008) J. Clin. Oncol., 26, 4078-4085.
63.Leger, J., Mercat, I., Alberti, C., Chevenne, D.,
Armoogum, P., Tichet, J., and Czernichow, P. (2007) Eur. J.
Endocrinol., 157, 685-692.
64.Holt, R. I., and Sonksen, P. H. (2008) Br. J.
Pharmacol., 154, 542-556.
65.Anderson, L. L., Jeftinija, S., and Scanes, C. G.
(2004) Exp. Biol. Med. (Maywood), 229, 291-302.
67.Velloso, C. P. (2008) Br. J. Pharmacol.,
154, 557-568.
68.Li, G., Del Rincon, J. P., Jahn, L. A., Wu, Y.,
Gaylinn, B., Thorner, M. O., and Liu, Z. (2008) J. Clin. Endocrinol.
Metab., 93, 1379-1385.
69.Hathout, E. H., Baylink, D. J., and Mohan, S.
(1999) Growth Horm. IGF Res., 9, 272-277.
70.Chen, E. Y., Liao, Y. C., Smith, D. H.,
Barrera-Saldana, H. A., Gelinas, R. E., and Seeburg, P. H. (1989)
Genomics, 4, 479-497.
71.Yamaguchi, N., Oyama, T., Ito, E., Satoh, H.,
Azuma, S., Hayashi, M., Shimizu, K., Honma, R., Yanagisawa, Y.,
Nishikawa, A., Kawamura, M., Imai, J., Ohwada, S., Tatsuta, K., Inoue,
J., Semba, K., and Watanabe, S. (2008) Cancer Res., 68,
1881-1888.
72.Esteban, C., Audi, L., Carrascosa, A.,
Fernandez-Cancio, M., Perez-Arroyo, A., Ulied, A., Andaluz, P., Arjona,
R., Albisu, M., Clemente, M., Gussinye, M., and Yeste, D. (2007)
Clin. Endocrinol., 66, 258-268.
73.Estes, P. A., Cooke, N. E., and Liebhaber, S. A.
(1992) J. Biol. Chem., 267, 14902-14908.
74.Ryther, R. C., Flynt, A. S., Harris, B. D.,
Phillips, J. A., 3rd, and Patton, J. G. (2004) Endocrinology,
145, 2988-2996.
75.Such-Sanmartin, G., Bosch, J., Segura, J., Wu,
M., Du, H., Chen, G., Wang, S., Vila-Perello, M., Andreu, D., and
Gutierrez-Gallego, R. (2008) Growth Factors, 26,
152-162.
76.Jansson, C., Boguszewski, C., Rosberg, S.,
Carlsson, L., and Albertsson-Wikland, K. (1997) Clin. Chem.,
43, 950-956.
77.Haro, L. S., Lewis, U. J., Garcia, M.,
Bustamante, J., Martinez, A. O., and Ling, N. C. (1996) Biochem.
Biophys. Res. Commun., 228, 549-556.
78.Bustamante, J. J., Gonzalez, L., Carroll, C. A.,
Weintraub, S. T., Aguilar, R. M., Munoz, J., Martinez, A. O., and Haro,
L. S. (2009) Proteomics, 9, 3474-3488.
79.Lamberts, S. W., van den Beld, A. W., and van der
Lely, A. J. (1997) Science, 278, 419-424.
80.Borst, S. E. (2004) Age Ageing, 33,
548-555.
81.Sohmiya, M., and Kato, Y. (1992) J. Clin.
Endocrinol. Metab., 75, 1487-1490.
82.Hersch, E. C., and Merriam, G. R. (2008) Clin.
Interv. Aging, 3, 121-129.
83.Zeitler, P., and Siriwardana, G. (2002)
Endocrine, 18, 85-90.
84.Thorner, M. O., Perryman, R. L., Cronin, M. J.,
Rogol, A. D., Draznin, M., Johanson, A., Vale, W., Horvath, E., and
Kovacs, K. (1982) J. Clin. Invest., 70, 965-977.
85.Mayo, K. E. (1992) Mol. Endocrinol.,
6, 1734-1744.
86.Hashimoto, K., Koga, M., Motomura, T., Kasayama,
S., Kouhara, H., Ohnishi, T., Arita, N., Hayakawa, T., Sato, B., and
Kishimoto, T. (1995) J. Clin. Endocrinol. Metab., 80,
2933-2939.
87.Zeitler, P., Stevens, P., and Siriwardana, G.
(1998) J. Mol. Endocrinol., 21, 363-371.
88.Mayo, K. E., Miller, T., DeAlmeida, V., Godfrey,
P., Zheng, J., and Cunha, S. R. (2000) Recent Prog. Horm. Res.,
55, 237-266.
89.Wheeler, M. A., Ayyagari, R. R., Wheeler, G. L.,
and Weiss, R. M. (2005) J. Smooth Muscle Res., 41,
1-21.
90.Kojima, M., Hosoda, H., Date, Y., Nakazato, M.,
Matsuo, H., and Kangawa, K. (1999) Nature, 402,
656-660.
91.Kojima, M., and Kangawa, K. (2005) Physiol.
Rev., 85, 495-522.
92.Zhang, J. V., Ren, P. G., Avsian-Kretchmer, O.,
Luo, C. W., Rauch, R., Klein, C., and Hsueh, A. J. (2005)
Science, 310, 996-999.
93.Olszewski, P. K., Schioth, H. B., and Levine, A.
S. (2008) Brain Res. Rev., 58, 160-170.
94.Lanfranco, F., Baldi, M., Cassoni, P., Bosco, M.,
Ghe, C., and Muccioli, G. (2008) Vitam. Horm., 77,
301-324.
95.Yin, X., Li, Y., Xu, G., An, W., and Zhang, W.
(2009) Acta Biochim. Biophys. Sin. (Shanghai), 41,
188-197.
96.Llona, I., and Eugenin, J. (2005) Biol.
Res., 38, 347-352.
97.D’Alessio, D. A., Sieber, C., Beglinger,
C., and Ensinck, J. W. (1989) J. Clin. Invest., 84,
857-862.
98.Ferone, D., Gatto, F., Arvigo, M., Resmini, E.,
Boschetti, M., Teti, C., Esposito, D., and Minuto, F. (2009) J. Mol.
Endocrinol., 42, 361-370.
99.Tulipano, G., and Schulz, S. (2007) Eur. J.
Endocrinol., 156, Suppl. 1, S3-11.
100.Leung, D. W., Spencer, S. A., Cachianes, G.,
Hammonds, R. G., Collins, C., Henzel, W. J., Barnard, R., Waters, M.
J., and Wood, W. I. (1987) Nature, 330, 537-543.
101.Fisker, S., Kristensen, K., Rosenfalck, A. M.,
Pedersen, S. B., Ebdrup, L., Richelsen, B., Hilsted, J., Christiansen,
J. S., and Jorgensen, J. O. (2001) J. Clin. Endocrinol. Metab.,
86, 792-796.
102.Stallings-Mann, M. L., Ludwiczak, R. L.,
Klinger, K. W., and Rottman, F. (1996) Proc. Natl. Acad. Sci.
USA, 93, 12394-12399.
103.Ross, R. J., Esposito, N., Shen, X. Y., von
Laue, S. L., Chew, P. R., Dobson, M., Postel-Vinay, M.-C., and
Finidori, J. (1997) Mol. Endocrinol., 11, 265-273.
104.Scott, M. J., Godshall, C. J., and Cheadle, W.
G. (2002) Clin. Diagn. Lab. Immunol., 9, 1153-1159.
105.Waters, M. J., Hoang, H. N., Fairlie, D. P.,
Pelekanos, R. A., and Brown, R. J. (2006) J. Mol. Endocrinol.,
36, 1-7.
106.Yang, J., and Stark, G. R. (2008) Cell
Res., 18, 443-451.
107.Carter-Su, C., Rui, L., and Stofega, M. R.
(2000) Recent Prog. Horm. Res., 55, 293-311.
108.Pilecka, I., Patrignani, C., Pescini, R.,
Curchod, M. L., Perrin, D., Xue, Y., Yasenchak, J., Clark, A., Magnone,
M. C., Zaratin, P., Valenzuela, D., Rommel, C., and Hooft van
Huijsduijnen, R. (2007) J. Biol. Chem., 282,
35405-35415.
109.Katz, M., Amit, I., and Yarden, Y. (2007)
Biochim. Biophys. Acta, 1773, 1161-1176.
110.Muslin, A. J. (2008) Clin. Sci. (Lond.),
115, 203-218.
111.Cesena, T. I., Cui, T. X., Piwien-Pilipuk, G.,
Kaplani, J., Calinescu, A. A., Huo, J. S., Iniguez-Lluhi, J. A., Kwok,
R., and Schwartz, J. (2007) Mol. Genet. Metab., 90,
126-133.
112.Glass, D. J. (2003) Nature Cell Biol., 5,
87-90.
113.Laron, Z. (2001) Mol. Pathol.,
54, 311-316.
114.Netchine, I., Azzi, S., Houang, M., Seurin, D.,
Perin, L., Ricort, J. M., Daubas, C., Legay, C., Mester, J., Herich,
R., Godeau, F., and le Bouc, Y. (2009) J. Clin. Endocrinol.
Metab., 94, 3913-3921.
115.Barbieri, M., Bonafe, M., Franceschi, C., and
Paolisso, G. (2003) Am. J. Physiol. Endocrinol. Metab.,
285, E1064-1071.
116.Rotwein, P., Pollock, K. M., Didier, D. K., and
Krivis, G. G. (1986) J. Biol. Chem., 261,
4828-4832.
117.Smith, P. J., Spurrell, E. L., Coakley, J.,
Hinds, C. J., Ross, R. J., Krainer, A. R., and Chew, S. L. (2002)
Endocrinology, 143, 146-154.
118.Hameed, M., Orrell, R. W., Cobbold, M.,
Goldspink, G., and Harridge, S. D. (2003) J. Physiol.,
547, 247-254.
119.Dluzniewska, J., Sarnowska, A., Beresewicz, M.,
Johnson, I., Srai, S. K., Ramesh, B., Goldspink, G., Gorecki, D. C.,
and Zablocka, B. (2000) FASEB J., 19, 1896-1898.
120.Hwa, V., Oh, Y., and Rosenfeld, R. G. (1999)
Endocrin. Rev., 20, 761-787.
121.Rowlands, M. A., Gunnell, D., Harris, R.,
Vatten, L. J., Holly, J. M., and Martin, R. M. (2009) Int. J.
Cancer, 124, 2416-2429.
122.Rosen, C. J., and Pollak, M. (1999) Trends
Endocrinol. Metab., 10, 136-141.
123.Firth, S. M., and Baxter, R. C. (2002)
Endocrin. Rev., 23, 824-854.
124.Guler, H.-P., Zapf, J., Schmid, C., and
Froesch, E. R. (1989) Acta Endocrinol. (Copenh.), 121,
753-758.
125.Mu, L., Katsaros, D., Wiley, A., Lu, L., de la
Longrais, I. A., Smith, S., Khubchandani, S., Sochirca, O., Arisio, R.,
and Yu, H. (2009) Breast Cancer Res. Treat., 115,
151-162.
126.Chitnis, M. M., Yuen, J. S., Protheroe, A. S.,
Pollak, M., and Macaulay, V. M. (2008) Clin. Cancer Res.,
14, 6364-6370.
127.Delafontaine, P., Song, Y. H., and Li, Y.
(2004) Arterioscler. Thromb. Vasc. Biol., 24,
435-444.
128.Dupont, J., Dunn, S. E., Barrett, J. C., and
LeRoith, D. (2003) Recent Prog. Horm. Res., 58,
325-342.
129.Hernandez-Sanchez, C., Mansilla, A., de Pablo,
F., and Zardoya, R. (2008) Mol. Biol. Evol., 25,
1043-1053.
130.Nakae, J., Kido, Y., and Accili, D. (2001)
Endocrin. Rev., 22, 818-835.
131.Le Roith, D. (2003) Exp. Diabesity Res.,
4, 205-212.
132.Jansson, P. A., Pellme, F., Hammarstedt, A.,
Sandqvist, M., Brekke, H., Caidahl, K., Forsberg, M., Volkmann, R.,
Carvalho, E., Funahashi, T., Matsuzawa, Y., Wiklund, O., Yang, X.,
Taskinen, M. R., and Smith, U. (2003) FASEB J., 17,
1434-1440.
133.Vivanco, I., and Sawyers, C. L. (2002)
Nature Rev. Cancer, 2, 489-501.
134.Hajduch, E., Litherland, G. J., and Hundal, H.
S. (2001) FEBS Lett., 492, 199-203.
135.Carpten, J. D., Faber, A. L., Horn, C., Donoho,
G. P., Briggs, S. L., Robbins, C. M., Hostetter, G., Boguslawski, S.,
Moses, T. Y., Savage, S., Uhlik, M., Lin, A., Du, J., Qian, Y. W.,
Zeckner, D. J., Tucker-Kellogg, G., Touchman, J., Patel, K., Mousses,
S., Bittner, M., Schevitz, R., Lai, M. H., Blanchard, K. L., and
Thomas, J. E. (2007) Nature, 448, 439-444.
136.Viglietto, G. (2009) Cell Cycle,
8, 2869-2870.
137.Alam, S. M., Rajendran, M., Ouyang, S.,
Veeramani, S., Zhang, L., and Lin, M. F. (2009) Endocr. Relat.
Cancer, 16, 1-16.
138.Molkentin, J. D., Lu, J. R., Antos, C. L.,
Markham, B., Richardson, J., Robbins, J., Grant, S. R., and Olson, E.
N. (1998) Cell, 93, 215-228.
139.Datta, D., Contreras, A. G., Grimm, M.,
Waaga-Gasser, A. M., Briscoe, D. M., and Pal, S. (2008) J. Am. Soc.
Nephrol., 19, 2437-2446.
140.Thomson, A. A. (2008) Differentiation,
76, 587-598.
141.Sun, Y., Niu, J., and Huang, J. (2009) Am.
J. Transl. Res., 1, 148-162.
142.Prieto, J. C., and Carmena, M. J. (1987)
Cell Biochem. Funct., 5, 63-68.
143.Ballesteros, M., Leung, K. C., Ross, R. J.,
Iismaa, T. P., and Ho, K. K. (2000) J. Clin. Endocrinol. Metab.,
85, 2865-2871.
144.Sinowatz, F., Breipohl, W., Waters, M. I.,
Lincoln, D., Lobie, P. E., and Amselgruber, W. (1991) Prostate,
19, 273-278.
145.Reiter, E., Kecha, O., Hennuy, B., Lardinois,
S., Klug, M., Bruyninx, M., Closset, J., and Hennen, G. (1995)
Endocrinology, 136, 3338-3345.
146.Weiss-Messer, E., Merom, O., Adi, A., Karry,
R., Bidosee, M., Ber, R., Kaploun, A., Stein, A., and Barkey, R. J.
(2004) Mol. Cell Endocrinol., 220, 109-123.
147.Bidosee, M., Karry, R., Weiss-Messer, E., and
Barkey, R. J. (2009) Mol. Cell Endocrinol., 309,
82-92.
148.Chopin, L. K., Veveris-Lowe, T. L., Philipps,
A. F., and Herington, A. C. (2002) Growth Horm. IGF Res.,
12, 126-136.
149.Chopin, L. K., and Herington, A. C. (2001)
Prostate, 49, 116-121.
150.Havt, A., Schally, A. V., Halmos, G., Varga, J.
L., Toller, G. L., Horvath, J. E., Szepeshazi, K., Koster, F., Kovitz,
K., Groot, K., Zarandi, M., and Kanashiro, C. A. (2005) Proc. Natl.
Acad. Sci. USA, 102, 17424-17429.
151.Jeffery, P. L., Herington, A. C., and Chopin,
L. K. (2002) J. Endocrinol., 172, R7-11.
152.Cassoni, P., Ghe, C., Marrocco, T., Tarabra,
E., Allia, E., Catapano, F., Deghenghi, R., Ghigo, E., Papotti, M., and
Muccioli, G. (2004) Eur. J. Endocrinol., 150,
173-184.
153.Malendowicz, W., Ziolkowska, A., Szyszka, M.,
and Kwias, Z. (2009) Urol. Int., 83, 471-475.
154.Zapata, P. D., Ropero, R. M., Valencia, A. M.,
Buscail, L., Lopez, J. I., Martín-Orozco, R. M., Prieto, J. C.,
Angulo, J., Susini, C., Lopez-Ruiz, P., and Colas, B. (2002) J.
Clin. Endocrinol. Metab., 87, 915-926.
155.Reubi, J. C., Maurer, R., von Werder, K.,
Torhorst, J., Klijn, J. G., and Lamberts, S. W. (1987) Cancer
Res., 47, 551-558.
156.Montironi, R., Cheng, L., Mazzucchelli, R.,
Morichetti, D., Stramazzotti, D., Santinelli, A., Moroncini, G.,
Galosi, A. B., Muzzonigro, G., Comeri, G., Lovisolo, J.,
Cosciani-Cunico, S., and Bono, A. V. (2008) Cell Oncol.,
30, 473-482.
157.Pietrzkowski, Z., Mulholland, G., Gomella, L.,
Jameson, B. A., Wernicke, D., and Baserga, R. (1993) Cancer
Res., 53, 1102-1106.
158.Kimura, G., Kasuya, J., Giannini, S., Honda,
Y., Mohan, S., Kawachi, M., Akimoto, M., and Fujita-Yamaguchi, Y.
(1996) Int. J. Urol., 3, 39-46.
159.Kawada, M., Inoue, H., Masuda, T., and Ikeda,
D. (2006) Cancer Res., 66, 4419-4425.
160.Garcia, F. U., Urbanska, K., Koltowski, L.,
Reiss, K., and Sell, C. (2007) Clin. Cancer Res., 13,
3140-3146.
161.Powolny, A. A., Wang, S., Carlton, P. S., Hoot,
D. R., and Clinton, S. K. (2008) Mol. Carcinog., 47,
458-465.
162.Jerome, L., Shiry, L., and Leyland-Jones, B.
(2003) Endocr. Relat. Cancer, 10, 561-578.
163.McKay, J. D., Kaaks, R., Johansson, M., Biessy,
C., Wiklund, F., Balter, K., Adami, H. O., Boillot, C.,
Gioia-Patricola, L., Canzian, F., Stattin, P., and Gronberg, H. (2007)
Cancer Epidemiol. Biomarkers Prev., 16, 169-173.
164.Johansson, M., McKay, J. D., Rinaldi, S.,
Wiklund, F., Adami, H. O., Gronberg, H., Kaaks, R., and Stattin, P.
(2009) Prostate, 69, 1281-1291.
165.Neuhausen, S. L., Slattery, M. L., Garner, C.
P., Ding, Y. C., Hoffman, M., and Brothman, A. R. (2005)
Prostate, 64, 168-174.
166.Lisitskaya, K. V., Krakhmaleva, I. N., and
Shishkin, S. S. (2010) Mol. Genet. Mikrobiol. Virusol.,
No. 2, 32-36.
167.Johansson, M., McKay, J. D., Wiklund, F.,
Rinaldi, S., Hallmans, G., Balter, K., Adami, H. O., Gronberg, H.,
Stattin, P., and Kaaks, R. (2009) Cancer Epidemiol. Biomarkers
Prev., 18, 1644-1650.
168.Chen, C., Freeman, R., Voigt, L. F.,
Fitzpatrick, A., Plymate, S. R., and Weiss, N. S. (2006) Cancer
Epidemiol. Biomarkers Prev., 15, 2461-2466.
169.Pavelic, J., Matijevic, T., and Knezevic, J.
(2007) Indian J. Med. Res., 125, 511-522.
170.Werner, H., and Bruchim, I. (2009) Arch.
Physiol. Biochem., 115, 58-71.