REVIEW: Telomerase: Structure, Functions, and Activity Regulation
M. I. Zvereva*, D. M. Shcherbakova, and O. A. Dontsova
Faculty of Chemistry, Lomonosov Moscow State University, 119999 Moscow, Russia; E-mail: zvereva@genebee.msu.ru* To whom correspondence should be addressed.
Received January 25, 2010; Revision received May 1, 2010
Telomerase is the enzyme responsible for maintenance of the length of telomeres by addition of guanine-rich repetitive sequences. Telomerase activity is exhibited in gametes and stem and tumor cells. In human somatic cells proliferation potential is strictly limited and senescence follows approximately 50-70 cell divisions. In most tumor cells, on the contrary, replication potential is unlimited. The key role in this process of the system of the telomere length maintenance with involvement of telomerase is still poorly studied. No doubt, DNA polymerase is not capable to completely copy DNA at the very ends of chromosomes; therefore, approximately 50 nucleotides are lost during each cell cycle, which results in gradual telomere length shortening. Critically short telomeres cause senescence, following crisis, and cell death. However, in tumor cells the system of telomere length maintenance is activated. Besides catalytic telomere elongation, independent telomerase functions can be also involved in cell cycle regulation. Inhibition of the telomerase catalytic function and resulting cessation of telomere length maintenance will help in restriction of tumor cell replication potential. On the other hand, formation of temporarily active enzyme via its intracellular activation or due to stimulation of expression of telomerase components will result in telomerase activation and telomere elongation that can be used for correction of degenerative changes. Data on telomerase structure and function are summarized in this review, and they are compared for evolutionarily remote organisms. Problems of telomerase activity measurement and modulation by enzyme inhibitors or activators are considered as well.
KEY WORDS: telomerase, senescence, TERT, telomerase RNA, telomerase reaction cycle, telomerase activity, telomerase regulatorsDOI: 10.1134/S0006297910130055
Abbreviations: CTE, C-terminal TERT domain (C-terminal extension); IFD, TERT domain (insertion in fingers domain); Pif1p, helicase of Saccharomyces cerevisiae; TER, telomerase RNA; TERRA, RNA transcribed from telomeric DNA (telomeric repeat-containing RNA); TERT, telomerase reverse transcriptase; TLC1 RNA, telomerase RNA of Saccharomyces cerevisiae; TWJ, Y-like structural element of RNA (three way junction).
Telomeres are DNA–protein structures that are localized at the
ends of eukaryotic chromosomes. They protect the linear ends of
eukaryotic chromosomes against degradation and fusion, thus maintaining
genome stability. The cell replication apparatus is not able to provide
for complete replication of chromosome ends; also, telomeres are
subject to the action of nucleases and other destructive factors. As a
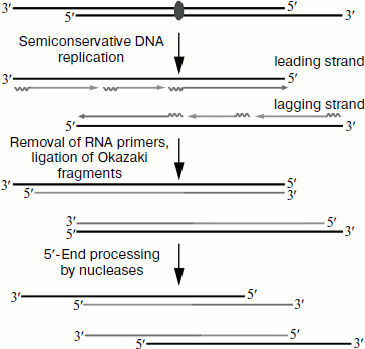
result, telomeres shorten during each cell division (Fig. 1). In most organisms the main mechanism of telomere
length maintenance is completion of DNA telomere repeats by telomerase
[1]. This enzyme elongates the chromosome
3′-end, whereas the complementary strand is completed by DNA
polymerases.
Telomerase is a ribonucleoprotein complex [2]. The core enzyme includes telomerase reverse transcriptase and telomerase RNA containing a template site for DNA elongation. The telomerase complex also contains a number of auxiliary components that provide for functioning of telomerase in vivo. Some of these components are necessary for telomerase attachment to the telomere at a certain cell cycle phase [3], while others are required for regulation of telomerase activity [4]. Some proteins are necessary for maturation of the telomerase complex and degradation of its components [5]. The amount of telomerase in the different types of cells undergoes fine regulation [6, 7]. This is important because telomere shortening in human cells and finally senescence will result in restriction of cell division potential [8]. There are data showing that activation of telomerase is associated with the development of cancer [9], and that it is active in cells exhibiting potential for unlimited division. It is known that telomerase is active in 85% of cancer tumors, while in the other 15% of cases different mechanisms of telomere length maintenance based on recombination are active [10]. It should be noted that telomerase activity is not found in usual somatic tissues. As a result, already beginning from the first years of investigation of telomerase the enzyme was considered as a universal target that could be used in the development of anticancer therapy. So the search for an efficient inhibitor of telomerase activity became relevant to therapies for malignant proliferative diseases.Fig. 1. Telomere shortening due to under-replication and processing in each cell division.
During recent decades the appearance of new and somewhat successful anti-telomerase strategies has been observed. The presence of active telomerase in gametes and stem cells is a stumbling-block on this way. However, it should be noted that telomeres in tumor cells are significantly shorter than in gametes and stem cells. Telomere shortening causes death of tumor cells when telomerase activity is inhibited much earlier than that of normal stem cells. This suggests that there is a “therapeutic window” for safe usage of telomerase inhibitors, and these might be selective and universal antitumor drugs. Human cancer cells can be much more sensitive than normal cells to damaging effects of telomerase inhibitors, and telomeres of these cells are more rapidly reduced to critical length. Short telomeres stimulate cell senescence. Such directed induction of tumor cell senescence became an attractive therapeutic strategy for tumor patients upon which not only active spreading of tumor cells is blocked, but it results in tumor cell death [11]. Apoptosis eliminates tumor cells immediately and irreversibly. Therefore, chemotherapy directed toward cell senescence in malignant tumors is increasingly developed. An increase in sensitivity of telomerase activity testing technologies and improvement of methods for sample preparation has made it possible to extend the possibilities of telomerase activity determination. Telomerase activity has been studied in all types of tissues [12]. Besides increase in telomerase activity observed in tumor cells, it is supposed that the enzyme can also function in “normal cells” with very precisely adjusted regulation [13]. Although telomere length reduction and following initiation of senescence is the usual fate of somatic cells, it is possible that the lack of certain telomerase components results in early phenotypic senescence with loss of function at the cellular and system levels [14].
The main criterion of telomerase efficiency is the number of telomeric repeats at the ends of telomeres. Telomere length reduction is a symptom of many diseases and can be both the result of primary telomerase dysfunction (like those caused by mutations in the main telomerase components, hTERT, hTR, or by disturbance in telomere-organizing systems) and the result of premature telomere loss induced by different factors. Inborn dyskeratosis is of the first type. It was the first identified human genetic disease caused by disturbance in the system of telomere length maintenance [15]. This disease is characterized by skin hyperpigmentation, epithelium keratinization, nail dystrophy, and progressive aplastic anemia. In most cases autosomal diseases are due to mutations in the H/ACA region of human telomerase RNA [16], while X-chromosome-linked cases emerge due to mutations in protein dyskerin leading to disturbance in telomerase complex assembly. The recently discovered mutation in the telomerase reverse transcriptase domain is associated with the dominated form of disease, which emphasizes the importance of telomerase functioning during the development of this disease [17].
Mutations in telomere-binding proteins result in chromosome instability and premature senescence syndrome. The presence of short telomeres is also an important symptom of different genetic diseases.
There are cases when telomere shortening is the secondary effect of the disease. Acquired immune deficiency syndrome is among these [18]. Recently diseases of the cardiovascular system have also been associated with telomere length-dependent senescence [19], and telomere length reduction in coronary endothelial cells has been found in patients with heart ischemia [20] while telomere shortening was also detected in patients with keloid diseases [21]. These data make understandable the increasing interest of researchers and pharmacologists in this enzyme [22]. It would be ideal to learn how to regulate telomerase in certain tissues and at different times to compensate in this way telomere shortening.
Not so much shortening in itself but rather the impossibility to maintain definite structure of the telomere DNA–protein complex at the expense of shortening results in cell division cessation at a certain stage (senescence phenotype). This is characteristic of somatic tissue cells of mammals and other multicellular organisms exhibiting a definite number of divisions, the Hayflick limit [23]. Telomere length correlates with the cell proliferative potential. The existence of the enzyme preventing telomere shortening was predicted long before its discovery by the Russian scientist A. M. Olovnikov [24]. He suggested naming this enzyme telomerase.
It is impossible to cover in a review everything presently known about different aspects of telomerase and telomere functioning. By the present time over 10 thousand papers with key words “telomere” and “telomerase” are published. Since it is impossible to consider all of them, we shall analyze in more detail the telomerase complex composition and structure concentrating our attention on comparison of numerous data for evolutionarily remote organisms such as yeasts and humans. We shall briefly discuss structural features of telomeres, because they are the main substrate of telomerase, and methods for estimation of telomerase activity. The available data on telomerase structure and function are analyzed in the light of the action of known enzyme inhibiting or activating pharmacological agents, which is necessary for understanding principles of creation of artificial telomerase regulators such as its inhibitors and activators.
TELOMERASE STRUCTURE AND FUNCTION
As mentioned above, telomerase is a particular reverse transcriptase working in a complex with special telomerase RNA. Telomerase substrates in its reaction are deoxynucleotide 5′-triphosphates and the telomere 3′ terminus (in tests in vitro it is DNA-oligonucleotide containing the sequence corresponding to telomeric repeats of chromosomes). The particular property distinguishing telomerase from different RNA-dependent DNA polymerases is the use of a fixed region of special telomerase RNA as template for telomere elongation. Telomerase RNA interacts with telomere not only at this template region, but additionally in the so-called “anchor site”. Telomerase is able to add several telomeric repeats during a single act of attachment to oligonucleotide substrate [25].
The cycle of in vitro telomerase reactions (Fig. 2) includes the following stages: primer binding, elongation, translocation, and dissociation. In the case of consecutive transition of the enzyme from condition (1) through conditions (2) and (3) to condition (4), telomerase adds one telomeric repeat to the primer. Transition (4)-(2) corresponds to translocation II, i.e. to addition of several telomeric repeats without separation from the primer (transition (4)-(1) in Fig. 2).
This figure shows translocation processes I and II. The ability for translocation is connected with enzyme processivity. Two types of telomerase processivity are distinguished [26]. Processivity I is the telomerase capability for RNA–DNA duplex translocation in the active center after each nucleotide addition at the stage of elongation. Processivity II is telomerase capability for translocation relative the bound DNA primer after addition of one telomeric repeat, after which the primer again becomes capable of elongation (transition (4)-(2) in Fig. 2). The two types of processivity differ from each other in principle. If in the first case there is simultaneous translocation of RNA–DNA duplex relative the enzyme active center, then in the other 3′ terminus of DNA should change its position relative to the RNA template.Fig. 2. Telomerase reaction cycle. TERT, catalytic subunit (gray circle points to anchor site); TER, telomerase RNA with template site (gray rectangle). Figures in the frame designate telomerase position relative to the primer at different stages: 1) enzyme is not bound to primer; 2) primer annealing; 3) elongation stage; 4) completion of a single telomeric repeat synthesis. Dotted arrows point to possible processes of primer dissociation during enzyme functioning.
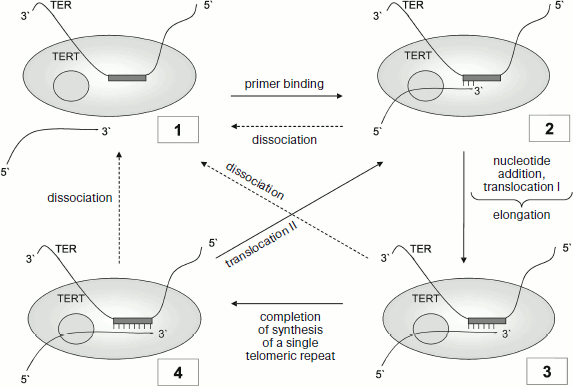
The efficiency of translocation stages I and II in in vitro reaction is different for telomerases from different organisms and can vary depending on isolation conditions of the fraction exhibiting telomerase activity [27-29].
Yeast telomerase in vitro makes pauses and stops primer elongation after addition the next nucleotide. Thus, it is possible to speak about inefficient translocation I and absence of translocation II. The observed heterogeneity of telomeres in S. cerevisiae, added by telomerase, is associated with type I processivity [30].
Human and protozoan telomerases in vitro exhibit type II processivity. They are able to add hundreds of nucleotides to telomeric substrate via multiple completions of telomeric repeats along their RNA template [31, 32]. Telomerases of a number of different organisms such as mouse [33] and various yeasts [34, 35] do not exhibit type II processivity in vitro. It was shown for S. cerevisiae telomerase that after telomeric primer elongation the enzyme remains bound to its own substrate [36]. However, the same enzyme in vivo is able to add to the telomere more than 100 nucleotides during a single cell cycle [37]. This contradiction can be explained by the fact that in vivo the enzyme exhibits type II processivity or by non-processive elongation resulting in multiple dissociation of the enzyme and new association with the telomere again for new elongation.
It was shown that yeast telomerase is able to elongate for more than one repeat primers containing non-telomeric sequence in the region remote by 4-6 nucleotides from the 3′ end. In this case the enzyme does not add several telomeric repeats similarly to the type II human and protozoan telomerases, but slips further along the template [38].
However, there is a different opinion concerning difference in processivity between telomerases of yeast and other organisms. It is supposed that the difference in processivity is not qualitative but rather quantitative [39].
Chang et al. [40] created a special system for in vivo investigation of the yeast telomerase processivity and of the mechanism of efficient elongation of the shortest telomeres. In the case of simultaneous expression of the wild-type telomerase RNA and RNA with mutated template region the telomere elongation was followed in a single round of the cell cycle. Telomeric repeats inserted with involvement of two types of telomerase RNA were distinguishable, and their frequency makes it possible to draw a conclusion concerning the character of telomerase functioning. It was found that on the average telomerase is not processive. However, processive elongation was detected on the shortest telomeres, which appeared to be dependent on a mammalian AMP ortholog, Tel1p kinase.
It should be added to processes shown in Fig. 2 that enzyme-associated nuclease activity was found in yeast [41, 42], protozoan [27], and human [43, 44] telomerases.
Main Components of Telomerase Complex
Telomerase RNA (TER) contains template region and different functionally important secondary structure elements involved in template region restriction, protein subunit binding, and partially carrying out catalytic and other functions [2, 45]. Telomerase reverse transcriptase (TERT) contains a catalytically important domain resembling that of reverse transcriptases, as well as only telomerase-specific domains necessary for TER and DNA substrate binding and for functional activity of telomerase [2, 46]. TER and TERT form the core enzyme. These components are enough to provide functional activity of telomerase in vitro. In vivo functioning requires auxiliary proteins, some of which are included in the holoenzyme. Despite high interest in telomerase and importance of its study in applied aspect, structural data on TERT, TER, and other telomerase proteins have become available relatively recently due to complication of telomerase investigation (very low intracellular enzyme content, difficulties in isolation of its components in soluble form and in sufficient amount, etc.). In this section dealing with TER and TERT the main attention is given to recently obtained results.
Telomerase reverse transcriptase (TERT). Domain structure. The amino acid sequence of telomerase catalytic subunits is similar to that of reverse transcriptases. Conservatism of amino acid residues responsible for catalysis, nucleotide binding, and ribo- and deoxynucleotide recognition is found in viral reverse transcriptases [25].
The reverse transcriptase domain TERT differs from corresponding domains of reverse transcriptases by the IFD site localized between A and B′ motifs conservative for reverse transcriptases (Fig. 3). This site is important for the functioning of yeast telomerase in vivo. Mutations in it result in lowering of the enzyme activity in vitro [47].
The search for functionally important regions in protein primary structure and the detection in them of amino acid residues whose replacement disturbs any telomerase property in most works were based on homology between TERT of different organisms [48-50]. Approaches not using such homology (e.g. single gene evolution [51]) gave similar results.Fig. 3. TERT domain structure.
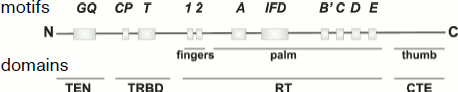
Functional protein domains and sites within them have various names. On the whole, four functional domains can be distinguished within the TERT structure: (i) N-terminal domain containing moderately conservative GQ block (hypomutable domain I according to the classification in [51]) that is now called the TEN domain [52]; (ii) RNA-binding domain (TRBD domain) with conservative motives CP, QFP, and T (hypomutable domains II, III, and IV); (iii) reverse transcriptase domain (RT domain) containing seven conservative domains and an IFD site; (iv) lowly conserved C-terminal domain (CTE domain).
It is a rather complicated problem to obtain soluble TERT in amounts sufficient for crystallization, which makes difficult structural investigations of this protein. Therefore, there are still only few data on the protein structure. In general there are no data on structural organization of the whole complex. Pronounced progress has been recently achieved in TERT studies because there appeared structures of separate TERT domains [52, 53] and of the whole TERT molecule, without the TEN domain [54], obtained by X-ray analysis. Thorough biochemical investigations combined with directed mutagenesis, based on new structural data, have significantly extended concepts concerning the mechanism of TERT functioning [39, 55]. It became possible to crystallize the catalytic subunit of red flour beetle Tribolium castaneum whose genome has been recently sequenced [56]. TERT of T. castaneum does not contain a TEN domain. The other domains (TRBD, RT, CTE) form a ring. Motifs involved in substrate binding and catalysis are localized within the ring. From 7 to 8 base pairs of RNA–DNA duplex can be localized within the ring. The whole structure has much in common with structures of retrovirus reverse transcriptases, viral RNA polymerases, and B family DNA polymerases [54].
Structural differences between various TERT mainly concern N- and C-terminal sites. These data seem especially interesting because no significant TERT sequence conservatism is observed in terminal regions in various organisms.
Let us consider known structural aspects and functions of TERT domains.
Reverse transcriptase domain (RT). In accordance with affiliation to reverse transcriptases, TERT contain seven reverse transcriptase-specific conservative motifs in this domain. The distinctive feature of telomerases is a large insertion between motifs A and B, the IFD. When following the analogy of telomerase being in the “right hand” form, these two motifs will be localized in the “palm” and “fingers” domains. The IFD site got its name because it appeared to be an insertion in the “fingers” domain (Insertion in Fingers Domain [56]). The affiliation of TERT to reverse transcriptases has been confirmed in numerous works [32, 57, 58]. Conservative amino acid residues responsible for catalysis were found. These are three aspartic acid residues localized in motifs A and C [58].
The RT domain in T. castaneum TERT consists of two subdomains (“palm” and “fingers”) constructed of β-sheets and α-helices, which is characteristic of retroviral reverse transcriptases, virus RNA polymerases, and B family DNA polymerases. The polymerase “thumb” domain corresponds in this structure to the CTE domain.
Comparison of T. castaneum TERT structure with that of HIV reverse transcriptase was indicative of their high similarity with the exception of the presence in TERT of the IFD motif. This motif consists of two antiparallel α-helices playing an important role in structural arrangement of the two other helices, possibly being involved in direct contact with RNA–DNA duplex.
The probable position of a nucleotide-binding region was detected by comparison of TERT and HIV reverse transcriptase structures and by identification of conservative residues. The region is localized at the junction of the “palm” and “fingers” subdomains.
TERT of different organisms were analyzed using mutagenesis. Interesting mutations causing telomere elongation [59] were found in the RT domain Est2p (yeast TERT domain) in addition to mutations disturbing enzyme function and complex assembly [47-51]. Mutations in motif E increase processivity concerning addition of nucleotides [59]. There are mutations in subdomain “fingers” (motifs 1 and 2) after which telomerase is not inhibited in vivo by helicase Pif1p [60]. In general, it is known that helicase Pif1p activity removes telomerase from the telomere. This has been shown in vitro upon introduction of Pif1p into the reaction of primer elongation by telomerase [61]. In this case telomerase processivity decreases. A similar effect of mammalian helicase (PIF1) on in vitro telomerase activity was also shown for human telomerase [62]. In the yeast strain pif1Δ, telomeres in vivo are longer than those in the wild-type strain. This elongation is telomerase-dependent [61]. Overexpression of Pif1p, on the contrary, stimulates telomere shortening [63]. Such interesting Pif1p function as inhibition of telomere addition by telomerase to double-stranded breaks was found as well [64]. Mutations in Est2p, disturbing Pif1p action in vivo, are indicative of interaction of these proteins, perhaps indirectly [60].
TEN domain. Interaction of TERT with DNA substrate. Already before crystallization of the TEN domain of the Tetrahymena thermophila telomerase TERT there existed proofs showing that the TEN domain in yeast and human TERT represents a separate structural domain [49, 65]. The N-terminal domains necessary for telomerase functioning in vitro and in vivo do not exhibit similarity with any regions in other proteins; therefore, it is impossible to submit a possible structure of this region on the basis of homology proceeding only from amino acid sequence.
Recently the TEN domain of T. thermophila TERT has been crystallized [52]. It appeared to have a new structural domain. Conservative amino acid residues have been identified and their functional importance was shown. Most of these residues are grouped in the groove on the domain surface. These residues are necessary for telomerase catalytic activity, and some of them are involved in specific interaction with single-stranded region of DNA substrate for elongation by telomerase. Positively charged residues at the domain C terminus are involved in unspecific interaction with RNA. Features of the whole TEN domain are interaction with single-stranded RNA as well as its ability to bind RNA, which is necessary for functioning of telomerase.
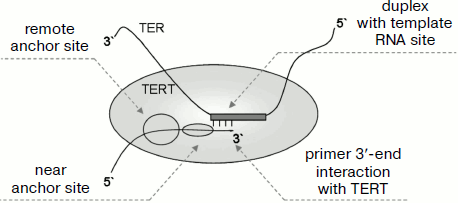
Now we shall consider in more detail what is known about the interaction of TERT with DNA substrate. Telomerase interacts with substrate not only in the place of its 3′-end annealing in the TER template region, but also with its 5′ end in the so-called “anchor site”. It is also supposed that the 3′ end of the primer directly interacts with TERT. In human [66], yeast [67], and protozoan [27] telomerases the remote anchor site (~16-21 nucleotide residues from the primer 3′ end localized in the catalytic site) and the near anchor site localized closer to the primer 3′ end (~4-14 nucleotide residues from 3′ end). Quite a number of data show that the anchor site is localized in the TEN domain. This is confirmed using chemical cross-linking [52, 68] as well as by mutagenesis [52, 66].
Yeast telomerase exhibits increased efficiency in elongation of short primers (~9 nucleotides) and primers with “mutations” (non-telomeric sequence) in the region of near the anchor site [67]. This is due to the fact that upon dissociation from primer telomerase is released from the stable complex with it and again becomes active and capable of a new primer elongation. This effect practically disappears at decreased primer concentration.
Telomerase of T. thermophila exhibits increased ability to elongate short primer [69]. Apparently, like telomerase of S. cerevisiae, it easily dissociates from short substrates after elongation, and therefore it is able to elongate more primer molecules in unit time when it is in excess [68]. Similar primer length effects on its elongation efficiency were also found for human telomerase [66].
The contribution of the near and remote anchor sites to the interaction of processive human and protozoan telomerases and of non-processive yeast telomerase with primer is different. It is found that the remote anchor site of yeast telomerase only slightly influences primer binding. Thus, yeast telomerase [67], unlike the human enzyme [70], practically does not elongate primers with enough extended (8-15 nucleotides) non-telomere sequence at the 3′ end and telomere sequence at the 5′ end (15 nucleotides). There are not enough data on the functional role and localization of the remote anchor site. Perhaps it is formed not at the expense of TERT but at the expense of other components of the complex. Endogenous and in vitro reconstructed T. thermophila telomerase behave differently. It appeared that type II processivity in endogenous enzyme increases upon primer elongation, but this is not characteristic of the reconstructed enzyme. It is possible that the remote anchor site, restraining primer from dissociation and stimulating its processive elongation, is involved in endogenous T. thermophila telomerase functioning [71]. Interaction in the remote anchor site (20-22 nucleotides from the primer 3′ end) of telomerase of the protozoan species Euplotes aediculatus was directly confirmed using chemical cross-links [72].
There are facts showing that the primer 3′ end directly interacts with TERT. It is known that the primer binding by human telomerase depends on its 3′ end position on the RNA template [73]. Euplotes aediculatus telomerase is able to elongate chimeric primers with non-telomeric 3′ end. Chimeric primers cannot form duplex with RNA template. Elongation of these primers starts from the beginning of the template region [74]. This may happen at the expense of the interaction of the primer 3′ end with TERT and its positioning relative the RNA template. Yeast telomerase inefficiently binds and elongates primers in which the last two nucleotides at the 3′ ends are non-telomeric (non-complementary to the template) [36]. In this case interaction of primer 3′ end with TERT may be disturbed.
It has been recently shown directly by fluorescent analysis of single molecules that DNA substrate in the absence of TER forms a stable complex with human TERT [75].
Removal of the TEN domain from TERT makes telomerase practically inactive [52, 76]. Some conservative residues in the TEN domain play a structural role. Most of them are grouped at opposite sides of the domain surface. At one side they form a deep groove, while the other side consists of α-helices whose conservative residues are mainly acidic or hydrophobic.
For T. thermophila telomerase, interaction of DNA primer with the TEN domain was found using the chemical cross-linking technique [52, 55]. This unstable interaction is not fixed by such methods as binding on filters and electromobility shift assay [55]. It was shown that TERT free of the TEN domain interacts with DNA primer, and the efficiency of this interaction corresponds to one third of the efficiency of the interaction of the full-sized TERT [55]. This shows that the TEN domain is not the only place for binding DNA primer. Amino acid residues responsible for binding are localized close to each other in domain structure. They are grouped in the neighborhood of a groove at one side of the domain surface. Interesting data have been obtained showing that the function of the TEN domain is not limited only to binding DNA primer. A mutation was found that has no effect on primer binding but influences the enzyme activity [55]. Contact of the DNA primer with Trp187 localized at the periphery of the TEN domain was found. This contact was detected at any one of three primer 3′-end positions on the template (at the beginning of template region, in the middle, or closer to its 5′ end). Taking into account that the primer is not able to stretch upon elongation and the enzyme active center remains in the same place, this result can be explained only with the assumption that due to mobility of the TEN domain, the position of the latter relative to the active site changes [55]. These data are especially interesting because there is no full-sized TERT structure including the TEN domain. Taking into account the supposed length and geometry of RNA–DNA duplex in the active site, it can be concluded that the distance between Trp187 and Asp residues in the active center changes from 17 to 27 Å depending on the primer 3′ end position on the template region [55]. Data on the structure of this domain suggest that the mobility of the TEN domain is achieved due to flexibility of its C terminus [52]. Owing to such mobility, RNA–DNA duplex can undergo translocation in the active center upon primer elongation. All supposed variants of primer interaction with telomerase are shown in Fig. 4.
Although the Trp187 residue of T. thermophila TERT directly interacts with DNA primer, it is not necessary for catalytic activity [55]. Deterioration of the interaction with primer is found upon mutation of different amino acid residues located at a distance from Trp187 [55], which influences the telomerase activity. It is supposed that a wide variety of amino acid residues are involved in DNA primer binding. They are localized on the surface of the TEN domain in the groove region, which is confirmed by mutagenesis and analysis of the domain structure [52].Fig. 4. Interaction of primer with telomerase.
The TEN domain interacts nonspecifically with RNA [77, 78]. It is supposed [52] that the nonspecific interaction of the TEN domain transforms to specific interaction within the whole enzyme structure.
Based on proposed structures, homology, and calculations and taking into account electrostatic interactions, a structure for the yeast TEN domain in which the position of conservative residues with confirmed involvement in the yeast telomerase anchor site formation was designed [39].
A number of mutations in TERT intended to influence the ability of telomerase to interact with DNA in the anchor site were designed on the basis of the T. thermophila TEN domain structure [52] and then investigated [79]. One mutation (L14A) resulted in the loss of type II enzyme processivity but had no effect on the interaction of the enzyme with primer and addition of nucleotides to the end of the template region. Replacement of the residue next to L14A resulted in 50% decrease in processivity. Recently a model has been proposed that explains the role in processivity of the anchor site and, in particular, of found residues [79]. The processivity is provided due to the interaction of the TEN domain with the primer 5′ end and catalytic domain during elongation. This interaction is disturbed at the moment of translocation, and then it emerges again. It is supposed that L14A is involved in this process.
Mutations resulting in telomere elongation compared to the wild-type strain were also found in the yeast TEN domain Est2p [80]. These mutations do not influence the telomerase enzymatic properties in vitro. The only participant of the process of telomere elongation by telomerase, Tel1p, is necessary for more pronounced telomere elongation. It is supposed that the N-terminal domain Est2p somehow interacts with Tel1p upon telomere elongation by telomerase [80]. Mutations resulting in changes in the association of the telomeric protein Rap1p with double-stranded telomeric DNA were also found among mutations in the TEN domain Est2p [81]. The mechanism of this is not yet clear.
TRBD domain. First of all, TERT differs from other reverse transcriptases by its ability to use internal RNA template upon addition of telomeric repeats. In human telomerase TEN domain (RID1 by a different classification) interacts with pseudoknot TER, while the TRBD domain (RID2 according to another classification) interacts with P6.1 hairpin of the CR4-CR5 domain. The latter interaction is critical for enzyme assembly [82].
The structure of the isolated TRBD domain of T. thermophila has been determined [53]. The RNA binding TRBD domain contains mainly helical motifs. This structure is unique. Motif QFP is not involved in RNA binding but has a structural function, while conservative motifs CP and T directly participate in RNA binding. Conservative amino acid residues are arranged so two pockets were formed at the surface. One pocket is narrow, restricted by hydrophobic residues, and specific for single-stranded RNA binding (T pocket), while the other is wider and can bind RNA duplex (T-CP pocket) [53]. It is supposed that the TRBD domain interacts with helix I in T. thermophila TER structure as a double-stranded RNA and with 5′-boundary element of T. thermophila TER as a single-stranded RNA.
The detailed pattern of amino acid residue positions explains effects of the above-described mutations in the TRBD domain of TERT [53]. Both RNA-binding pockets cover the perimeter of the whole domain surface.
It is known that in addition to the TRBD domain, motif CP2 at the boundary of the TEN and TRBD domains is involved in binding TER, also called the 5′-boundary element [83, 84]. It is not clear how the TRBD domain and CP2 motif binding is regulated and what the functional importance of the interaction of the 5′-boundary element with CP2 motif is.
It is difficult to draw conclusions based of the T. thermophila TRBD domain structure concerning the interactions in yeast and human telomerases necessary for enzyme assembly and activity. The point is that RNA elements interacting with TERT differ in different organisms, although different TERT exhibit homology between each other. We shall go back to this problem in a section dealing with TER.
The interesting nucleolar protein PinX1p, competing with TLC1 RNA (TER of yeast S. cerevisiae) for interaction with Est2p (TERT of yeast S. cerevisiae), was found among proteins interacting with telomerase complex components [85]. A human homolog of yeast PinX1p was also found, protein PinX1 [86, 87]. This protein is a negative regulator of telomerase and interacts with hTERT. TERT may interact with PinX1 via the same domain as with TER, i.e. the TRBD domain. The role of the interaction of TERT with PinX1 is still unknown. It is supposed that in this way TERT not bound to TER is “preserved” in an inactive state [85].
CTE domain. In the T. castaneum TERT structure the CTE domain is accurately positioned relative the other domains and represents the so-called “thumb” according to generally accepted classification of polymerase domains. The domain structure is not similar to structures of corresponding reverse transcriptase domains. It was shown that the CTE domain of TERT is a new structural domain [54]. Within the structure the CTE domain is structurally close to the TRBD domain. Such organization of TERT domains results in formation of a central “hole” of sufficient width for accommodation of double-stranded 7-8 bp long nucleic acids, which well agrees with experimental data on the length of duplex in the telomerase active center [88]. Gillis et al. [54] modeled the RNA–DNA duplex position in the predicted TERT structure. According to this model, one helix of the CTE domain interacts with the small groove of RNA–DNA duplex. The accuracy of this model is supported by earlier experimental facts based on mutagenesis [89, 90].
It was supposed previously that the CTE domain is not important for yeast telomerase functioning in vivo [51]. However, later data are indicative of the importance of this domain for Estp2 protein stability and efficiency of DNA substrate elongation [59, 89]. In human telomerase mutations in the CTE domain disturb telomerase functioning similar to some mutations in the TEN domain [91, 92].
Nuclease activity of telomerase. Telomerases exhibit nuclease activity towards oligonucleotide substrates. Endonuclease activity was detected in yeast telomerase fraction that had been purified by several stages using different kinds of affinity chromatography [41]. In vitro reconstructed human [43, 44] and protozoan [93] telomerases also exhibited nuclease activity. Nuclease activity of yeast telomerase depends on nucleotide concentration [41]. However, the telomerase domain responsible for nuclease activity has not been identified.
Telomerase probably introduces a break at the border of paired and unpaired bases of primer and of the TER template region [41, 43]. Human telomerase is even capable of complete cleavage of telomeric oligonucleotide depending on its position on the template upon annealing [43]. It should be noted that if nonhydrolyzable internucleotide bonds are introduced into preferable cleavage sites, then the enzyme is able to cleave DNA substrate in different places [41, 44]. Chimeric primers can also be destroyed relatively far from the 3′ end, at the border of telomeric and non-telomeric regions [41, 44]. The remaining telomeric ends are then elongated by the enzyme. As mentioned above, yeast telomerase, unlike the human enzyme, is not able to elongate primers with sufficiently extended 3′-terminal non-telomeric sequence. Such primers are also poor substrates for nuclease activity. It may be that in human telomerase the remote anchor site is involved in the interaction with such primers [44].
Transferase activity. It was shown that in the presence of Mn2+ yeast and human telomerase can work as terminal transferase, i.e. it is able to join nucleotides independently of template [94]. Even in such unusual role, telomerase prefers GT-rich substrates that are telomere-like at the 5′ ends. The terminal-transferase activity explains a number of physiological processes. Thus, it is known that overexpression of mammalian telomerase reverse transcriptase provokes premature cell senescence and cancer, which cannot be explained by the effect of the protein on telomere length. Among possible explanations for this is the supposition that TERT overexpression reveals usually concealed terminal-transferase protein activity and, as a result, it has a destructive effect on cell physiology. It is not known whether the intracellular concentration of Mn2+ is high enough for such telomerase transformation in normal or pathological conditions. On the other hand, it is possible that there are still unidentified small molecules that could produce the same result [94].
Protective functions of telomerase. An interesting phenomenon was found in a number of works. Mutants of core components of yeast and human telomerase influenced cell growth and phenotype independently of their effect on telomere length [38, 95]. The Nobel Prize winner of 2009 Elizabeth Blackburn suggested the following explanation of this phenomenon: in addition to elongation of telomere ends, telomerase exhibits functions protecting telomeres [96]. By now rather many works have appeared showing that not only telomere shortening results in senescence, but rather disturbance of their structure and protection. The disturbance of telomere structure is accompanied by appearance of G-rich protruding ends, the emergence of which is indicative of disturbance of the telomere protecting function and degradation of the C-rich strand [97, 98]. The phenotype of G-rich protruding ends is observed upon disturbance of protective telomeric proteins (Cdc13p, Ku70/Ku80) [98-100].
It was shown for the yeast Candida albicans that removal of EST2 genes (yeasts C. albicans are diploid) results both in telomere shortening and increase in amount of G-rich protruding telomere ends [101]. Unlike this, deletions of C. albicans genes EST1 and EST3, homologous to genes of S. cerevisiae, result in telomere shortening but not in disturbance of their structure and appearance of G-rich protruding ends [101].
Protective functions of C. albicans telomerase were also confirmed in experiments showing that catalytically inactive TERT is able to inhibit accumulation of G-rich protruding ends, and it appeared that not only TERT but TER as well is involved in the C. albicans telomerase protective function [102]. The mechanism of this process is still not clear.
The fact that, unlike for C. albicans, removal of the TER- and TERT-encoding genes from S. cerevisiae does not disturb telomere structure can be explained either by very rapid transition of these yeasts to senescence or by existence in them of some alternative mechanisms of telomere structure protection [102].
Telomerase activity independent of catalytic activity is also observed in mouse tissues after removal of TER. In this case stimulation of cell proliferation by TERT was noted [103]. This was recently explained. It appears that TERT is involved in transcription of genes of the Wnt-β-catenin signal pathway that stimulates proliferation of embryonic and stem cells [104]. In fact, this function of TERT consists in coordination of the telomere maintenance apparatus in dividing cells using telomerase with expression of genes necessary for proliferation.
Telomerase RNA (TER). Secondary structure and its individual components. In different organisms (protozoa, yeasts, vertebrates) TER significantly differ by size and sequence. The processed mature TER of yeasts S. cerevisiae (TLC1 RNA) consists of 1167 nucleotide residues (n.r.) while unprocessed TER consists of approximately 1300 n.r. [105, 106]. TER of protozoa and mammals are noticeably shorter, about 150 [107] and 450 n.r. [108], respectively. Despite differences in the RNA size and sequence of nucleotide residues, total TER function makes researchers to look for structural elements in common. Phylogenetic analysis made possible to identify TER secondary structures for all above-mentioned types of organisms: protozoa [107] and vertebrates [108], as well as yeasts S. cerevisiae [105, 106, 109, 110].
In addition to template region, TER contains secondary structure elements necessary for catalytic functions, type I and II processivity, as well as elements necessary for maturation, telomerase stability, and TER localization.
All structures contain the so-called central domain including pseudoknot, template, and 5′-boundary element (in protozoa TBE, template boundary element). Protozoan TER also contains template recognition element (TRE), the element of recognition by catalytic TERT subunit of the beginning of the template region. All TER also contain a site that binds proteins responsible for TER maturation and stability. These elements differ because these proteins and TER maturation pathways differ as well.
The TER of yeasts differ from protozoan and human TER by larger size, and it is also found that about 50% of the structure is not associated with its in vivo activity. Importantly, the element present in protozoan and human TER, the so-called trans-activating domain, is not found in TER of yeasts. The large size and the high degree of TLC1 RNA evolutionary variability for a long time made it difficult to determine its spatial structure. Decoding of nucleotide sequences of genomes of yeasts closest to S. cerevisiae opened the way to works on determination of the TLC1 RNA secondary structure on the basis of phylogenetic analysis [105, 106, 109]. It appeared that in the case of so pronounced evolutionary variability of the whole sequence, less than half of TLC1 RNA nucleotides (about 500 n.r. of 1200-1300 n.r.) are important for telomerase functioning in vivo [111].
From the above we conclude that despite the difference in the sequence of TER from various organisms, there are common structural elements necessary for the function, and their presence was confirmed by the analysis of TER from various evolutionarily distant organisms like protozoan Tetrahymena [107], yeast Saccharomyces [105, 106], mammalian, and human [108].
TER structural elements responsible for catalysis and TERT binding. All TER contain elements interacting with TERT. First we shall consider elements arranged in the central domain. Most of them are common for TER of different organisms.
TER not only serves as template for catalysis, but also take part in it. In mammalian and protozoan telomerases TER contribute to telomerase processivity [29, 112]. It was shown that nucleotide residues in the TLC1 RNA pseudoknot also make their contribution by direct involvement in catalysis.
Template site. Interaction of TER with DNA primer. Usually the length of the TER template site is approximately equal to the length of one and a half copies of the telomeric repeat, but it can be longer or shorter [26]. The TER template region itself and its limits are defined by the TER secondary structure. At the 3′ end of this region there is the primer annealing site, while at the 5′ end there is a boundary element separating the template region from the rest of the molecule. The TRE element is also described in the T. thermophila TER [113], which serves for TER template site recognition by the TERT catalytic subunit.
The template region plays an important role in catalysis. Some mutations in it can result in significant change in enzyme activity in vivo and in vitro. Trinucleotide substitutions in different places of the TLC1 RNA template region decrease the enzyme activity, sometimes up to its complete loss. Mutation causing “slipping” along the template during primer elongation is also found [38]. Point mutations in the template site of T. thermophila TER result in inaccurate insertion of nucleotides and early primer dissociation.
It should be noted that in some cases mutations in the TER template region have no effect on enzymatic activity. Thus, upon replacement of the whole template region of T. thermophila TER the telomerase activity is retained but its in vitro processivity is lost [114]. It is supposed that nucleotide-specific TER–TERT interactions in the template region are not obligatory for the ability of T. thermophila telomerase to elongate primer within the limits of a single telomeric repeat. However, if DNA template is used instead of RNA template, then non-processive telomerase can be obtained with very low efficiency of primer elongation [115].
On replacing the template region in TLC1 RNA by the template region of human TER, homogeneous repeats corresponding to the new template are found in telomeres. This suggests that the base sequence in the TLC1 RNA template is responsible for heterogeneity of telomeric repeats in yeast chromosomes; different variants of primer annealing are possible on this sequence upon primer binding for elongation [30].
The role of base sequence in the telomeric primer annealing site in enzyme processivity seems interesting [32, 111]. Unlike non-processive mouse telomerase, the human enzyme in which this region is longer (five nucleotides) than in mouse telomerase (two nucleotides) exhibits type II processivity in vitro. This difference is explained by efficiency of primer annealing upon translocation: the sequence in the primer annealing site should coincide with the 5′-terminal sequence of the template [29]. However, processivity is defined not only by the primer annealing site. For example, this site in yeast telomerase is extended enough (five nucleotides), but the enzyme does not exhibit type II processivity in vitro [29].
It seemed quite likely that the length of the duplex region between template and primer increases as nucleotides join the primer, but this supposition was disproved by a number of works. In E. aediculatus and human telomerases [73] there is no correlation between the number of base pairs in the duplex that can be potentially formed by primer with template site and with efficiency of its binding by the enzyme. It was shown by chemical testing of the TLC1 RNA template site that in yeast telomerase the number of paired bases between substrate and RNA template site remains constant and equal to seven upon primer elongation [88]. In this case, as elongation proceeds new pairs are formed from the primer 3′-end, while the chain untwists from the 5′-end.
It was shown for the human enzyme that the interaction of the primer with telomerase depends on the position of the primer on the template upon annealing [73]. It is supposed that this is due to the interaction of the primer 3′ end with TERT.
5′-Boundary element. This element in the TER structure retards primer elongation beyond a certain region on the template site. It is a helix restricting the single-stranded region (of yeast [109, 116] and human [132] telomerase) or specific sequence (protozoan telomerase [84]).
Are the mechanisms of action of 5′-boundary TER elements in different organisms similar? In protozoa this element is a specific, efficiently TERT-binding nucleotide sequence, while in yeasts and mammals it is a stem and hairpin in RNA secondary structure [116, 117]. The yeast 5′-boundary element binds TERT and is directly adjacent to the template site. There may be different mechanisms of action of 5′-boundary element in protozoa, yeasts, and mammals. This structure restricts mobility of the TER template site within the protozoan telomerase active center due to RNA–protein interactions. Such template site mobility is necessary during primer elongation because template position relative to active center should change upon addition of each nucleotide. In human telomerase, mobility of the TER template site is restricted due to RNA–RNA interactions in secondary structure. It is supposed that in yeast the telomerase 5′-boundary element restricts not only RNA mobility, but also the accessibility of the single-stranded region because the hairpin stem in 5′-boundary element in TER is directly adjacent to the template.
The TER of the yeast Schizosaccharomyces pombe, TER1, was recently found [118, 119]. This RNA 1213 n.r. in length independently interacts with SpEst1 and SpEst2 (orthologs Est1p and Est2p of S. cerevisiae). The 5′-boundary element of this RNA was analyzed. Like for S. cerevisiae telomerase, not nucleotide sequence of this site was important but rather formation by it of double-stranded RNA helix, which mechanically prevents further primer elongation. However, it appeared that a part of paired region within the helix includes a template region, i.e. 5′-boundary element comprising double-stranded RNA, which has to be partly untwisted upon elongation. Heterogeneity of telomeric repeats of some yeasts is associated with this peculiarity.
Pseudoknot in TER structure. Interaction of TER with TERT. A pseudoknot in the secondary structure of TER of different organisms is localized identically relative to the template site [45]. It was found that not only secondary TER structure is important for telomerase functioning, but rather nucleotide sequence of highly conservative sites within the pseudoknot and single-stranded sites among them [45, 109, 120].
Recently several works appeared reporting the tertiary structure of some protozoan and human TER elements.
In the central domain of human TER helices P2b and P3 as well as loops J2b/3 and J2a/3 form a pseudoknot. The tertiary structure of the pseudoknot in the TER central domain and its correlation with experimental data obtained on the basis of mutagenesis were first established directly in 2005 for human telomerase [121]. Structural and mutation analyses indicated the presence of a triple helix in this region. Telomerase activity strictly correlates with the stability of the triple helix. In this case, if some data are in favor of the dynamic pseudoknot structure [122], others point to its static nature [120].
Recently formation of triple helix in the central domain has also been shown for TLC1 RNA of S. cerevisiae [110]. Distortion of this structure resulted in decrease in in vitro telomerase activity and telomere shortening in vivo. In this case binding to Est2p did not change. The hypothesis was put forward and confirmed experimentally that triple helix is not important for Est2p binding, but it is involved in catalysis due to the template-primer helix orientation using 2′-OH groups. Similar participation in catalysis of pseudoknot in human telomerase RNA was shown. The role of triple helix is not restricted to catalysis; its much more important function is as a structure drawing together the template site duplex and primer with the active center of TERT.
In telomerase RNA of yeasts S. cerevisiae and K. lactis a pseudoknot interacts with the catalytic protein subunit. In mammalian TER not only the pseudoknot but another conservative structure in a region remote from the pseudoknot, hairpin P6.1 of highly conservative domain CR4-CR5, are necessary for TERT binding and in vivo and in vitro telomerase functioning. No similar structure was found in TLC1 RNA. Domain CR4-CR5 together with hairpin P6.1 are classified as so-called “trans-activating domains”.
Localization of helices P6a and P6b as well as of loop J6 between them was detected in the tertiary structure of the CR4-CR5 domain [123]. The most important for telomerase functioning hairpin P6.1 contains at the loop end three nucleotides whose base residues are exposed to the solution. In vivo chemical testing of their structure has shown that they are inaccessible for modification. This suggests that they are involved in interactions with protein or RNA.
In mammalian [120, 124] and protozoan TER a pseudoknot in secondary structure is necessary for processivity upon primer elongation, and in the case of protozoan telomerase not only pseudoknot, but also hairpin IV are important. It is interesting that in human TER the replacement of the pseudoknot by the analogous structure from T. thermophila telomerase results in formation of in vitro non-processive enzyme with low activity. It is supposed that such replacement disturbs the interaction of remote hairpin P6.1 with the pseudoknot structure and possibly with TERT.
NMR spectroscopy of short protozoan TER analogs revealed the structure of hairpins II and IV [125, 126]. In hairpin II the very base of the hairpin is necessary for telomerase functioning and restriction of synthesis along the template for TERT binding, while the hairpin end is not important for activity. Hairpin IV is necessary for interaction with TERT and auxiliary protein p65 and for enzyme processivity [112, 127, 128]. Hairpin IV is considered as a TERT trans-activating domain; due to unpaired GA looping-out, it forms a strongly bent structure [125, 126].
Features of TER functioning in different organisms. It has been found for protozoan telomerase of T. thermophila that the TEN domain of TERT interacts with hairpin IV and element TRE, while the TERT domain TRBD interacts with 5′-boundary element TBE [126]. Complex assembly and hairpin IV bending in the region of GA looping out are stimulated by protein p65. These data draw researchers closer to understanding the mechanism of telomerase action, but by now only a model of assembled telomerase can be proposed. Pseudoknot TER of T. thermophila, conservative among different TERs, does not play an important role in binding of TER to TERT.
Although some analogy between protozoan and mammalian telomerase concerning the existence of remote TER elements necessary for interaction with TERT can be followed, common character of their function is not very obvious. Unlike protozoa, in mammals and humans the pseudoknot of TER is involved in interaction with TERT, and in this case it interacts with the TEN domain of TERT, while remote element CR4-CR5 interacts with the TRBD domain [78]. The opposite situation is observed in protozoan telomerase: the TEN domain interacts with remote hairpin IV and the TRBD domain interacts with 5′-boundary element TBE of the central TER domain.
The problem of the principal difference between mammalian and yeast TERs is still not solved. Many authors try to find analogies and to determine a general mechanism of telomerase functioning in different organisms [109, 129].
During investigation of K. lactis TER structure it was proposed to consider a new element in telomerase RNA structures, namely the point of junction of three helices, TWJ (Three Way Junction), in the terminal RNA arm [129]. It was shown that this element is important for telomerase activity, mutations in it resulting in telomere shortening. Some mutations in this site, remote from the template site, result in loss of in vitro telomerase activity. Overexpression partially inhibits in vivo the effect of mutation in TWJ. Brown et al. [129] compared this structural element with CR4-CR5 of mammalian TER. As already stated above, telomerase RNA of yeasts and higher eukaryotes differ not only by length but by the presence in mammalian TER of the TERT firm binding site (CR4-CR5 with P6.1 hairpin) localized separately from the central domain that also binds TERT. An analogy may be possible, but no direct interaction between the TWJ element of the terminal arm with TERT was found.
The proposed analogies of elements in yeast and mammalian telomerase RNA structure are disproved by the following fact. As mentioned above, Cech et al. [130] obtained in their laboratory RNA called miniT (shortened TLC1 RNA (500 n.r.)) that functioned in vivo, but telomeres were shortened. This RNA together with Est2p was used for the first time for yeast telomerase in vitro reconstruction [130]. An even shorter microT can also be used for in vitro reconstruction of functional telomerase, and in this case it is actually represented by only the central domain of TLC1 RNA. It was shown for mammalian enzyme that the “minimal” TER necessary for in vivo reconstruction of active telomerase must include, in addition to the central domain, the CR4-CR5 domains [120]. These data show that yeast telomerase RNA can be in general free of CR4-CR5 domain analog. Then how can one explain the fact that each telomerase is active if their TER noticeably differ from each other, and, on the contrary, TERT are sufficiently homologous? Since theoretically in telomerase reconstructed in vitro no components except TER and TERT should be necessary for enzyme activity, it is reasonable instead of looking for general motifs in RNA to search for differences in proteins. It is also possible that components of rabbit reticulocyte lysate (RRL), in which reconstruction is carried out, are involved in activity of in vitro reconstructed telomerases. Besides, still unidentified components should be considered in analysis of mechanisms of telomerase activity. Thus recently new components have been found which are involved in enzyme assembly and associated with it from lysate [131]. These are ATPases pontin and reptin.
TER secondary structure elements necessary for its maturation and stability as well as for in vivo assembly with TERT. TER biogenesis. As stated above, TER secondary structure elements interacting with proteins necessary for maturation, stability, and assembly with TERT differ in different organisms. Pathways of TER maturation and stabilization are also different.
The initial protozoan TER transcript is synthesized by RNA polymerase III and is not processed in this case. In the protozoan T. thermophila TER interacts with p65 protein included in telomerase [128, 132, 133]. This protein contains N-terminal La-motif (RNA binding motif), RRM, and C-terminal domains. All of them interact with stem I/stem IV elements in TER. Protein p65 stimulates the interaction of TER and TERT, i.e. it stimulates telomerase complex assembly as well as stabilization of TER. In another protozoan E. aediculatus TER also binds protein p43 containing La-motive. It was shown that upon in vitro complex reconstruction protein p65 significantly stimulates formation of active telomerase [5, 128]. Protein p65 interacts with conservative GA looping out from hairpin IV, thus promoting its bending.
In mammals initial transcript is synthesized by RNA polymerase II, then TER is capped at the 5′ end, modified, and processed at the 3′ end [134, 135]. TER processing and stability depend on H/ACA motif localized at the 3′ end of the molecule. Motif H/ACA is also included in small nucleolar RNA involved in posttranscriptional modification of noncoding RNA. Four proteins necessary for RNA accumulation and stability—dyskerin, NHP2, NOP10, and GAR1—interact with this motif. All these proteins are included in telomerase complex [15, 136-138]. Disturbance of human TER maturation is associated with such genetic disease as inborn dyskeratosis. And TER also contains the CAB motif responsible for TER localization in Kayala bodies [135].
TER of S. cerevisiae (TLC1 RNA) resemble in many parameters small nuclear RNA (snRNA) involved in splicing. Initial transcript is synthesized by RNA polymerase II, is polyadenylated, TMG cap is added to the 5′ end, and then it is processed to the mature molecule with removal of the poly(A) end [139, 140].
Polyadenylated precursor of TLC1 RNA comprises about 5-10% of the total TLC1 RNA [140, 141]. TLC1 RNA contains uridine-rich consensus motif RAU4-6GR (R is a purine base) of yeast snRNA, which interacts with Sm proteins. The same motif was found in noncoding small nuclear RNA of yeasts (snRNA). Mutation in the Sm-protein binding site in TER or decrease in amount of one of them (for example, Sm D1) results in sharp decrease in amounts of TLC1 RNA and telomerase. It was shown that Sm proteins are included in the holoenzyme [139].
TLC1 RNA undergoes 3′-terminal processing [105, 106]. Several forms of this RNA differing in the length of the 5′-terminal part have been found. The possibility of modification of base C651 in vivo was shown. Details of TLC1 RNA biogenesis are not clear yet, but there are data showing that during biogenesis this RNA can migrate between the cell nucleus and the cytoplasm [142, 143].
TER1 of Schizosaccharomyces pombe has much in common with TLC1 RNA of S. cerevisiae [118, 119]. Recently the process of biogenesis in S. pombe was decoded, and it appears that complete splicing is not necessary for TER1 maturation, but only its first stage without following exon ligation [144]. Such incomplete splicing was described for the first time. A similar process may also be responsible for maturation of different telomerase RNA.
Additional Proteins of Telomerase Complex
Additional proteins have been identified in telomerase complex of different organisms that are necessary for its functioning. For example, telomerase complex of S. cerevisiae consists not only of catalytic subunit (Est2p) and the RNA molecule (TLC1) serving as template for reverse transcription, but contains additional subunits Est1p and Est3p [145]. They are not required for in vitro telomerase activity but are necessary for its activity in vivo [146].
Potential RNA-recognizing motif RRM is present in the primary structure of Est1p [147]. This protein along with Est2p establishes contact with telomerase RNA near the template site, which points to its involvement in formation of the enzyme active center. Est1p is able to form a stable TLC1-containing complex even in the absence of Est2p or Est3p. It was also shown that Est1p interacts with the 3′-terminal region of telomeric DNA [148]. However, in vivo Est1p is able to bind telomeric DNA only in complex with Cdc13p. Thus, Est1p is a mediator connecting telomeric complex with Cdc13p [149]. Est3p is a constant component of telomerase complex, and moreover the association of Est3p with the enzyme requires intact condition of the catalytic center. The interaction of Est3p with Cdc13p significantly increases the accessibility of telomerase to the telomere [150].
Besides TERT and TER, additional telomerase components were also found in human telomerase. The human genome contains three Est1 orthologs, two of which (Est1A, Est1B) encode proteins involved in telomerase regulation [150]. No orthologs of Est3 have been identified.
TELOMERE-BINDING PROTEINS
Proteins specifically binding telomeric sequence and factors interacting with these proteins form a dynamic ribonucleoprotein structure. This structure has a protective role; it participates in telomere length regulation and is responsible for gene silence at telomeric and telomere-side sites. Also, telomere structures serve as targets for inhibitors that prevent telomerase binding to telomere [151, 152].
Yeast telomere DNA consists of a 250-350 bp long double-stranded region with C1-3A/TG1-3 sequence and a short single-stranded protruding end with TG1-3 sequence. Yeast telomeric repeats ((TG)0-6TGGGTGTG(G))n [30]) are heterogeneous, unlike the homogeneous repeats of mammalian telomeres (TTAGGG)n [153].
In yeasts the telomere double-stranded region directly binds protein Rap1p [154], and the single-stranded region binds Cdc13p [155]. Rap1p interacts with complex of Sir (silent information regulators) proteins that are responsible for heterochromatin formation in the subtelomeric region. Protein Rap1p also interacts with telomere proteins Rif1p and Rif2p. These proteins are associated with the telomere during the whole cell cycle and are negative regulators of telomerase [156].
Protein Cdc13p interacts with the 3′ protruding single-stranded end. This protein regulates telomerase access to the telomere. It is known to be able to interact with two different protein complexes oppositely influencing the ability of telomerase to elongate telomeres. As stated above, Cdc13p is necessary for attachment of telomerase to the telomere. This function involves interaction with the telomerase component Est1p [61]. On the other hand, Cdc13p interacts with protein complex Stn1p–Ten1p inhibiting telomere elongation by telomerase. Similarity between Rpa2p and Stn1p structures and between biochemical characteristics of the whole complex Cdc13p–Stn1p–Ten1p with RPA protein complex (Rpa1p–Rpa2p–Rpa3p, respectively) was found [157]. The heterotrimer Cdc13p–Stn1p–Ten1p interacts with complex of DNA polymerase α necessary for synthesis of complementary C strand in telomeric DNA [158].
Heterodimer of Ku-proteins binds at the border of single-stranded and double-stranded telomere regions. Ku-proteins are involved in different ways in maintenance of telomere length. They protect the ends of telomeres. Due to interaction with Ku-proteins of specific stem-loop region of TLC1, the catalytic subunit of telomerase appears to be bound to telomeres in the G1 phase of the cell cycle [159].
In mammals telomeric DNA is more closely packed in nucleosomes compared to other eukaryotes, and some nucleosomes carry heterochromatin markers [160]. Proteins TRF1 and TRF2 interact with double-stranded telomeric DNA, and protein POT1 and its partner TPP1 interact with the single-stranded region. Protein TRF2 binds RAP1. Proteins binding single-stranded and double-stranded DNA interact with each other via TIN2 protein interacting with TRF1 and TRF2 as well as with TPP1 protein. This overall structure on mammalian telomeres was named shelterin because it carries out protective functions [161].
Telomeres of dividing S. pombe cells are more similar in arrangement to telomeres of higher eukaryotes compared to telomeres of S. cerevisiae [162]. Rap1p of S. pombe does not directly bind double-stranded telomeric DNA but interacts with telomere-binding protein Taz1p, whereas protein Pot1p is bound to single-stranded region. The 3′-protruding end on telomeres of ciliate protozoa binds proteins TEBPα and TEBPβ. These proteins are orthologs of proteins POT1 and TPP1, respectively [162].
The 3′ protruding G-rich strand can form on complex structures of telomeres, the G-quadruplexes [163, 164]. These are G-quartets in stacking conformation (packed in parallel), i.e. planar structures of four guanines forming Hoogsteen pairs and localized in the same DNA strand. Formation of such structures on telomeres can be a problem for DNA replication and telomere elongation by telomerase. Telomeric DNA of vertebrates can form in addition to G-quadruplexes a complex loop-like structure. T-Loop is formed when 3′-single-stranded end penetrates the double-stranded region where the displaced second strand forms an internal D-loop [165, 166].
Unlike yeast protein Cdc13p, mammalian protein POT1, depending on its position relative to the 3′ end, when binding to DNA not only does not prevent but even stimulates telomere elongation by telomerase. If POT1 is not localized at the very end of the DNA, then it destroys G-quadruplexes and allows telomerase to elongate telomeres [167].
Despite differences in character of binding of protein complexes to telomere ends, an interesting similarity was found between telomere complexes of different organisms. Mammalian TPP1 contains an OB-fold domain resembling protozoan protein domain TEBPβ, while POT1 contains another OB-fold domain resembling protozoan protein domain TEBPα working in tandem with TEBPβ. So, there seems to be similarity of protozoan TEBPα–TEBPβ complexes and mammalian POT1–TPP1 complexes. The recently discovered similarity between OB-fold domain of S. cerevisiae protein Est3p and an analogous domain in TPP1 protein [168] seems even more interesting. Extending this analogy, one can propose Est1p for the role of POT1. No other analogies have been yet found between Est3p and TPP1 for attraction of telomerase to telomere, but this fact requires attention and makes reasonable further search of such analogies.
An interesting fact was recently found. In S. cerevisiae [169], human [170], and mouse [171] cells polymerase II transcribes telomeric repeats and RNA products called TERRA (Telomere Repeat-Containing RNA or telomeric RNA) are formed. TERRA is transcribed from C-rich strand and is associated with telomeric chromatin. Increased amount of TERRA in human cells correlates with telomere loss, whereas in yeasts this inhibits telomerase. The mechanism of TERRA functioning and its association with telomeres and role in oncogenesis are still not clear (see more detail about TERRA in [172]).
In yeasts telomeres are assembled in clusters and associated with nuclear membrane. The significance of such localization has not been finally elucidated, but it may be associated with telomere replication and essential for correct assembly of this intricate DNA–protein complex after replication [172].
TESTING OF TELOMERASE ACTIVITY
Knowledge of structural and functional features of telomerase opens the way for the search for telomerase inhibitors and activators. First of all the question arises concerning ways of measuring changes in telomerase activity. This was not a simple problem in the past, but recently new methods based on physical principles of detection have simplified the testing of telomerase activity [173]. In the first step any measurement of telomerase activity is based on detection of products of its activity upon specific oligonucleotide elongation using deoxyribonucleoside triphosphates as monomers [174]. The oligonucleotide has a sequence of one and the half telomere repeat, which allows the oligonucleotide to serve as a good substrate for elongation by telomerase. To exclude the activity of DNA-dependent DNA polymerases a control reaction with pre-processing RNase for splitting the RNA is used, since after the removal of the RNA-template the activity of telomerase cannot be detected. A specific feature of telomerase is the low copy number per cell [175] and hence low activity even in tumor cells where telomerase is activated by different pathway [176]. At first sight, it seems simpler to measure effects of inhibitors on activity of isolated enzyme. However, such direct test for kinetic studies is not generally available because it is suitable only for cell extract with an increased amount of telomerase by overexpression of telomerase components in the cell or by concentration with antibody to a telomerase component [177]. Direct assay involves the use of α32P-labeled deoxyribonucleoside triphosphates as monomer [174]. Total radioactivity of label attached to specific oligonucleotide is proportional to telomerase activity. The reaction mixture can be analyzed by electrophoresis, which gives additional information about enzyme processivity. Due to the inconvenience of using α32P-labeled nucleotides with high specific activity and by low sensitivity, the method is not widely used. There are several other methods for telomerase activity determination, and there are commercially available kits for measurement of its enzymatic activity. These methods are indirect and based on signal enhancement of specific primer extension by telomerase using polymerase chain reaction (PCR) or other methods. PCR was first used by Kim and Wu for enhancement of signal from products of telomerase activity [178]. The method was called the TRAP-test. Using information about low telomerase specificity to the substrate, they replaced oligonucleotides with natural telomere sequence by substrate primer (TS) with partially telomeric sequence. This made it possible to enhance the signal in the PCR pitch from telomerase-dependent elongation of oligonucleotide substrate without the pseudo-positive signal, e.g. like that due to formation of PCR products caused by primer self-annealing or internal repeat annealing. This method is not quantitative, but nevertheless owing to its sensitivity and reproducibility it has become most widespread [179]. Then, at the stage of signal amplification real time PCR was used, which made it possible to achieve quantitative estimation and design protocols (for review see [179]). Using the PCR-based methods of telomerase activity measurement for investigation of telomerase inhibitors, one should bear in mind that telomerase is a specialized DNA polymerase. Thus, there is a chance that the studied substance of supposed telomerase inhibitor can also inhibit the process of primer elongation catalyzed by DNA-dependent DNA polymerase in PCR, and therefore additional controls are necessary.
Three different modern sensing platforms for the analysis of telomerase activity in human cells have been described [173]. The first method involves the incorporation of hemin into the G-quadruplex structure generated by the extended specific telomeric oligonucleotide associated with CdSe-ZnS quantum dots and the resulting electron transfer quenching by the hemin units. The second method involves the electronic detection of the extended telomerase substrate on a field effect transistor device. The third method includes surface plasmon resonance detection for the amplification of the signal by the binding of Au nanoparticle labels to the telomeres. The sensitivities of methods are from hundreds to tens of cells per microliter. It makes these novel methods able to compete with previous direct assay of telomerase activity and opens new possibilities for screening substances as telomerase activity regulators.
ACTIVATION OF TELOMERASE
The mechanisms of telomerase activation in cancer progression are diverse, and they have been analyzed in review [176]. We will focus here on low molecular weight activators that may in the future be a basis for anti-aging therapeutic interventions involving transient somatic activation of endogenous telomerase to restore telomere length [180].
The main restriction in use of low molecular weight compounds directly interacting with telomerase and enhancing its activity is the necessity for retention of residual telomerase activity in tissue where a potentially therapeutic effect of such compounds can be revealed. A possible target for such compounds is stem cells of regenerating tissues characterized by constant moderate telomerase activity. Another target can be lymphocytes also having their own low level of telomerase activity. This process has been recently considered in detail [181]. Molecules increasing telomerase activity could restore proliferative ability of blood cells along with some other functions.
Among the most promising telomerase activators are low molecular weight compounds similar to a major saponin extracted from the root of Astragalus membranaceus – cycloastragenol (referred before as TAT2) and its derivatives. For TAT153 derivative now developed by the Geron company there is positive data on its oral availability in an animal model of idiopathic pulmonary fibrosis (http://www.geron.com).
TA-65, a telomerase activator originally isolated from the Chinese plant Astragalus, was tested in a health maintenance program and exhibited at low nanomolar levels moderate activation of telomerase in human keratinocytes, fibroblasts, and immune cells. Analysis of biomarkers of aging suggests improvements in the cardiovascular system, metabolism, and bone mineral density [182].
Mitochondrial dysfunction increasing the content of reactive oxygen species (ROS) was considered as the main determinant of telomere-dependent aging at the single cell level [183]. Antioxidants delay the beginning of vascular aging in a telomere-dependent pathway. In vitro ROS decrease the level of nuclear protein hTERT and telomerase activity in endothelial cells. This is accompanied by early emergence of senescence phenotype, while incubation with antioxidant N-acetylcysteine blocks the nuclear export of hTERT to the cytosol [184]. Tocopherol, a known antioxidant, also inhibits telomere length reduction and retains the level of telomerase activity in cells of brain capillary vessels [185].
Extracts of natural objects are also used for telomerase activation. Thus, extract of Ginkgo biloba delayed the beginning of aging due to stimulation of telomerase formation via transduction of signals of the PI3k/Akt pathway Moreover, pretreatment with PI3K inhibitor significantly attenuated the Ginkgo biloba extract-induced telomerase activity [186].
During the last ten years no new low molecular weight telomerase activators were reported. Even for known substances the molecular mechanism of action remains unknown. Elaboration of methods for therapy of degenerative diseases should include searching for ways to regenerate tissue that can be stimulated by telomerase-activating substances [187]. So, there is an urgent need for more investigations of telomerase that will promote creation of new activators that can be used in therapy of degenerative diseases in the near future.
INHIBITION OF TELOMERASE
Inhibition telomerase in by regulation in the cell is summarized in review [176]. The main focus here is relatively low molecular weight components that may serve as a basis for anticancer action.
All presently available inhibitors of telomerase activity can be divided into three groups based on their action and chemical properties. They are nucleoside and nucleotide (nucleos(t)ide) analogs working as substrate inhibitors, various low molecular weight compounds with different mechanisms of action, and oligonucleotide-based inhibitors blocking producing of active telomerase complex. Let us consider all three groups.
Nucleos(t)ide analogs are well known inhibitors for DNA-polymerases. Inhibition of DNA polymerization is due to the impossibility of incorporation of the next nucleotide residue because of competitive binding of these substances in the enzyme active site. Since the catalytic subunit of telomerase is RNA-depended DNA-polymerase or a reverse transcriptase and there are antivirus preparations such as those for therapy of HIV-infections blocking reverse transcriptase, the idea to check inhibitory activity of such preparations towards telomerase became apparent. The most widely used inhibitor of HIV reverse transcriptase is azidothymidine (AZT). AZT was the first nucleos(t)ide analog tested for telomerase inhibition. Positive effect was observed if leukemia T cells were treated with AZT, which was indicative of expediency of this nucleoside as an anti-telomerase agent [188]. Such treatment resulted in loss of telomerase activity and in telomere length reduction. Since pharmacological properties of AZT were already well studied, it was possible to complete investigation in this field during treatment of patients with T cell leukemia/lymphoma. Studies of nucleoside analogs as telomerase inhibitors continued in numerous works (a table of comparison is given in review [22]). The most active inhibitors of this class are, according to analysis of patents, acrylated nucleosides and guanine derivatives [189].
Another class of telomerase inhibitors consists of a group of low molecular weight compounds with structures different from nucleos(t)ides. This group includes substances influencing mainly hTERT [190]. Some of them are used in HIV therapy. There is a lot of information about function and inhibition of retroviral reverse transcriptases [191], a logical continuation of these investigations was testing of telomerase inhibition by components targeting retroviral reverse transcriptases: rubromycin and similar compounds [192]. Rubromycins and purpuromycin appeared to be powerful inhibitors (50% inhibitory concentrations are as low as 3 μM). Additional efforts to develop more effective antiviral agents target other specific enzymes in the virus replication cycle. It was reported that some quinolines form a family of integrase inhibitors [193]. The antiviral activity of quinolines became the basis for testing anti-telomerase activity of these compounds [194]. The mechanism of action of this class of compounds is now well understood, because it was shown that benzo(h)quinoline derivatives work as G-quadruplex binding agents [195]. Good examples of quinolones—floxacin and levofloxacin—moderately inhibited telomerase activity [196]. The rhodocyan dye analog MKT077, which accumulates in tumor cells and in mitochondria, was used as a basic structure for design of a more powerful telomerase inhibitor named FJ5002 (IC50 = 2 μM) [197]. The long-term cultivation of human leukemia cell line at high FJ5002 concentration was accompanied by progressive loss of telomeres, stimulation of aging, and cell crisis.
A current hypothesis concerning the mechanism of action of these compounds suggests stabilization of special quadruplex structures at telomere ends, blocking telomerase binding to its substrate [198, 199]. These inhibitors block telomerase either due to disturbance of protein–nucleic acid interactions supporting telomere structure, or due to blocking the substrate (telomere) binding to enzyme (telomerase). Unfortunately, quadruplexes can be formed not only on telomeres but also in different G-rich genome regions, and therefore it is often impossible to predict the effect of G-quadruplex DNA structure blocking by selective ligands. Because of this, the synthesis of such inhibitors is not now considered as a promising direction. However, many new inhibitors have been identified on the basis of quadruplex stabilization [200]. BIBR1532, a simple synthetic compound, a naphthalene derivative of benzoic acid (IC50 is 93 nM), is a promising non-nucleoside telomerase inhibitor, which stabilizes quadruplex structures [201]. Treatment of cancer cells with this preparation devoid of high cytotoxicity results in progressive telomere reduction. After long-term treatment with this substance cell proliferation arrest and emergence of aging symptoms, including morphological, mitotic, and chromosomal disturbances, were observed. The BIBR1532 preparation was active under natural conditions; its introduction into nude mice lowered tumorigenic potential of preliminarily inoculated tumor cells.
In 2005 it was shown that the natural lactone helenalin is a telomerase inhibitor [202]. The mode of action of this cytostatic agent is not clear. Perhaps helenalin affects telomerase activity through its interaction with nuclear factor κB and regulation of hTERT level. Curiously, polyunsaturated fatty acids inhibit telomerase via direct interaction with the catalytic subunit, and at the same time they switch off expression of the hTERT gene [203].
A unique approach to telomerase inhibition was recently proposed—the use of low molecular weight substances involved in recognition of RNA/DNA heteroduplexes formed upon the interaction of telomerase RNA with chromosome ends [204].
It was shown [205] that the main catechin of green tea (Epigallocatechin gallate) not only directly inhibits telomerase in a concentration-dependent manner, but also induces apoptosis in cells of tumor of head and neck via inhibiting the telomerase activity.
The most interesting potential inhibitor was found upon screening of 16,000 organic compounds [206]. It is an isothiozolin derivative (50% inhibition is achieved at 1 μM concentration) that is a noncompetitive inhibitor relative to substrate and deoxynucleotide triphosphates. It possesses remarkable selectivity. It simultaneously has no effect on DNA polymerase and HIV reverse transcriptase. Glutathione and dithiothreitol enhance its inhibitory activity. This fact suggests telomerase inhibition by affecting cysteine residues in the catalytic subunit.
The third class of inhibitors is oligonucleotides. The main components for targeting telomerase by oligonucleotides are hTR and messenger RNA for hTERT. The template region hTR serves as the target for antisense oligonucleotides. Complete accessibility of this region was demonstrated during investigation of its secondary structure [122, 207]. New technologies, namely the use of siRNA, ribozymes, and aptamers open great possibilities for design of inhibitors based on oligonucleotides. There are two main problems for usage of oligonucleotides as therapeutic substances: (i) problems of their delivery and (ii) their low stability in cell. These problems were solved in part by creation of chemically modified oligonucleotides retaining the bulk of biological activity. The detailed description of chemical modifications of oligonucleotides is beyond the framework of this review (see reviews [208, 209]).
Despite the large amount of information on telomerase inhibitors, transition from telomerase inhibiting compounds obtained by researchers to successful introduction of preparations into clinical practice proceeds slowly.
For the telomerase inhibitor Imetelstat (oligonucleotide GRN163L complementary to the template part of telomerase RNA) clinical trials were supported by Geron Corporation (California). In 2010 they began phase II trials for four different malignancies. Its safety was proved and optimal doses were selected.
Thus, the relatively few clinical trials that have been performed have given encouraging results. The development of the field of anticancer therapy on the basis of telomerase inhibition becomes a promising direction. Significant progress was achieved only in therapy using oligonucleotides. Other low molecular weight inhibitors are waiting their time.
An alternative possibility is the use in the nearest future of hTERT regulatory functions [210]. The hTERT or telomerase RNA regulatory properties, independent of polymerizing activity, are still not completely studied, but already interesting results have been obtained that can be used to develop anticancer therapeutic agents. Already used telomerase inhibitors may affect different telomerase functions (in addition to maintenance of telomere length). Understanding of these functions will allow elaboration of more efficient inhibitors.
Two different effects were described for in vitro cultivated tumor cells in the presence of different kinds of telomerase inhibitors. Sometimes certain substances exhibited the long-term effect reducing the telomere length, whereas in other cases telomerase inhibition resulted in rapid loss of telomere length. There is an explanation for these at first sight contradictory observations. If an inhibitor plays the role of a substance affecting telomerase as DNA polymerase and does not concern any other non-canonical enzyme function [211] or telomere structure, then the long-term treatment should cause telomere reduction and stimulation of aging or apoptosis. However, if the cell already has short telomeres or if the inhibitor also influences alternative telomerase functions or telomere structure, then rapid response is observed. Inhibition of hTERT telomerase activity using siRNA directed against hTERT mRNA is of the second type of inhibition resulting in rapid inhibition of cancer cell growth [212].
Telomere length correlates with the cell proliferative potential [23]. A hypothesis by Olovnikov [24] suggests that maintenance of telomere length is responsible for the proliferative potential. However, telomere length is connected with telomere structure. This parameter is influenced not only by active telomerase, but also by amount and ratio of the telomere structural proteins and by all regulatory proteins as well [213]. Modulation of binding properties of these proteins can change access of telomerase to telomeres and influence their structure and cell proliferative potential. In particular, proteins directly interacting with components of telomerase core enzyme also influence the cell proliferative potential. It is found that dyskerin is an essential component of active telomerase. Data on its interaction with telomerase and with telomerases activity are summarized in the recently published review by Mason et al. [214]. It is possible that searches for inhibitors directly interacting with dyskerin and preventing its binding to telomerase will begin in the near future. In other words, the complex processes of telomerase assembly, regulation, and modification of telomere structure open many ways for designing telomerase activity modulators.
CONCLUSION
Telomerase has recently become the basis for development of different therapies. This review considers features of telomerase structure and functioning for evolutionarily rather remote organisms like human and yeast as well as some inhibitors and activators of the enzyme that might be used for therapy in the future.
Recently different ways of blocking the telomerase-catalyzed process of telomere length maintenance were found on the basis of fundamental investigations of the mechanism of telomerase activity. The first is direct inhibition either of the catalytic subunit (hTERT) or the RNA component of telomerase complex (hTERC). The second is blocking telomere binding to telomerase.
The search of telomerase activators is a relatively new field. Development of modern directions in medicine associated with application of stem cells in therapy may soon make such activators a center of enhanced attention. So far only a few substances exhibiting such properties have been identified.
Further search and creation of new compounds modulating telomerase activity can be successful only with fuller understanding of the general mechanism of enzyme function and the extension of concepts concerning the telomerase structural components, which is impossible without generalization of already accumulated data and new investigations in this field.
This work was supported by Russian Federation Ministry of Education and Science (grant P800) and the Russian Foundation for Basic Research (grants 09-04-12071-ofi_m and 08-04-01220).
REFERENCES
1.Greider, C. W., and Blackburn, E. H. (1987)
Cell, 51, 887-898.
2.Shcherbakova, D. M., Zvereva, M. E., Shpanchenko,
O. V., and Dontsova, O. A. (2006) Mol. Biol. (Moscow),
40, 1-15.
3.Osterhage, J. L., Talley, J. M., and Friedman, K.
L. (2006) Nat. Struct. Mol. Biol., 13, 720-728.
4.Hsu, M., Yu, E. Y., Singh, S. M., and Lue, N. F.
(2007) Eukaryot. Cell, 6, 1330-1338.
5.Collins, K. (2006) Nat. Rev. Mol. Cell.
Biol., 7, 484-494.
6.Mozdy, A. D., Podell, E. R., and Cech, T. R. (2008)
Mol. Cell. Biol., 28, 4152-4161.
7.Cristofari, G., and Lingner, J. (2006) Embo
J., 25, 565-574.
8.Denchi, E. L. (2009) DNA Repair (Amst.),
8, 1118-1126.
9.Janknecht, R. (2004) FEBS Lett., 564,
9-13.
10.Bollmann, F. M. (2007) Cancer Treat. Rev.,
33, 704-709.
11.Schmitt, C. A. (2007) Biochim. Biophys.
Acta, 1775, 5-20.
12.Masutomi, K., et al. (2003) Cell,
114, 241-253.
13.Kaszubowska, L. (2008) J. Physiol.
Pharmacol., 59, Suppl. 9, 169-186.
14.Flores, I., Benetti, R., and Blasco, M. A. (2006)
Curr. Opin. Cell. Biol., 18, 254-260.
15.Mitchell, J. R., Wood, E., and Collins, K. (1999)
Nature, 402, 551-555.
16.Mitchell, J. R., Cheng, J., and Collins, K.
(1999) Mol. Cell. Biol., 19, 567-576.
17.Armanios, M., et al. (2005) Proc. Natl. Acad.
Sci. USA, 102, 15960-15964.
18.Chen, G., Tai, A. K., Lin, M., Chang, F.,
Terhorst, C., and Huber, B. T. (2007) Eur. J. Immunol.,
37, 663-674.
19.Minamino, T., and Komuro, I. (2007) Circ.
Res., 100, 15-26.
20.Ogami, M., et al. (2004) Arterioscler. Thromb.
Vasc. Biol., 24, 546-550.
21.De Felice, B., Wilson, R. R., and Nacca, M.
(2009) BMC Med. Genet., 10, 110.
22.Tarkanyi, I., and Aradi, J. (2008)
Biochimie, 90, 156-172.
23.Hayflick, L. (1997) Biochemistry (Moscow),
62, 1180-1190.
24.Olovnikov, A. M. (1973) J. Theor. Biol.,
41, 181-190.
25.Kelleher, C., Teixeira, M. T., Forstemann, K.,
and Lingner, J. (2002) Trends Biochem. Sci., 27,
572-579.
26.Lue, N. F. (2004) Bioessays, 26,
955-962.
27.Collins, K. (1999) Annu. Rev. Biochem.,
68, 187-218.
28.Bosoy, D., and Lue, N. F. (2004) Nucleic Acids
Res., 32, 93-101.
29.Chen, J. L., and Greider, C. W. (2003) Embo
J., 22, 304-314.
30.Forstemann, K., and Lingner, J. (2001) Mol.
Cell. Biol., 21, 7277-7286.
31.Morin, G. B. (1989) Cell, 59,
521-529.
32.Greider, C. W. (1991) Mol. Cell. Biol.,
11, 4572-4580.
33.Prowse, K. R., Avilion, A. A., and Greider, C. W.
(1993) Proc. Natl. Acad. Sci. USA, 90, 1493-1497.
34.Cohn, M., and Blackburn, E. H. (1995)
Science, 269, 396-400.
35.Lue, N. F., and Peng, Y. (1997) Nucleic Acids
Res., 25, 4331-4337.
36.Prescott, J., and Blackburn, E. H. (1997)
Genes Dev., 11, 2790-2800.
37.Teixeira, M. T., Arneric, M., Sperisen, P., and
Lingner, J. (2004) Cell, 117, 323-335.
38.Prescott, J., and Blackburn, E. H. (1997)
Genes Dev., 11, 528-540.
39.Lue, N. F., and Li, Z. (2007) Nucleic Acids
Res., 35, 5213-5222.
40.Chang, M., Arneric, M., and Lingner, J. (2007)
Genes Dev., 21, 2485-2494.
41.Niu, H., Xia, J., and Lue, N. F. (2000) Mol.
Cell Biol., 20, 6806-6815.
42.Petrov, A. V., Dokudovskaya, S. S., Sokolov, K.
A., Lavrik, O. I., Favre, A., Dontsova, O. A., and Bogdanov, A. A.
(1998) FEBS Lett., 436, 35-40.
43.Huard, S., and Autexier, C. (2004) Nucleic
Acids Res., 32, 2171-2180.
44.Oulton, R., and Harrington, L. (2004) Mol.
Biol. Cell, 15, 3244-3256.
45.Theimer, C. A., and Feigon, J. (2006) Curr.
Opin. Struct. Biol., 16, 307-318.
46.Autexier, C., and Lue, N. F. (2006) Annu. Rev.
Biochem., 75, 493-517.
47.Lue, N. F., Lin, Y. C., and Mian, I. S. (2003)
Mol. Cell. Biol., 23, 8440-8449.
48.Friedman, K. L., Heit, J. J., Long, D. M., and
Cech, T. R. (2003) Mol. Biol. Cell, 14, 1-13.
49.Xia, J., Peng, Y., Mian, I. S., and Lue, N. F.
(2000) Mol. Cell. Biol., 20, 5196-5207.
50.Bosoy, D., Peng, Y., Mian, I. S., and Lue, N. F.
(2003) J. Biol. Chem., 278, 3882-3890.
51.Friedman, K. L., and Cech, T. R. (1999) Genes
Dev., 13, 2863-2874.
52.Jacobs, S. A., Podell, E. R., and Cech, T. R.
(2006) Nat. Struct. Mol. Biol., 13, 218-225.
53.Rouda, S., and Skordalakes, E. (2007)
Structure, 15, 1403-1412.
54.Gillis, A. J., Schuller, A. P., and Skordalakes,
E. (2008) Nature, 455, 633-637.
55.Romi, E., Baran, N., Gantman, M., Shmoish, M.,
Min, B., Collins, K., and Manor, H. (2007) Proc. Natl. Acad. Sci.
USA, 104, 8791-8796.
56.Richards, S., et al. (2008) Nature,
452, 949-955.
57.Nugent, C. I., and Lundblad, V. (1998) Genes
Dev., 12, 1073-1085.
58.Weinrich, S. L., et al. (1997) Nat.
Genet., 17, 498-502.
59.Peng, Y., Mian, I. S., and Lue, N. F. (2001)
Mol. Cell., 7, 1201-1211.
60.Eugster, A., et al. (2006) Nat. Struct. Mol.
Biol., 13, 734-739.
61.Boule, J. B., Vega, L. R., and Zakian, V. A.
(2005) Nature, 438, 57-61.
62.Zhang, D. H., Zhou, B., Huang, Y., Xu, L. X., and
Zhou, J. Q. (2006) Nucleic Acids Res., 34, 1393-1404.
63.Zhou, J., Monson, E. K., Teng, S. C., Schulz, V.
P., and Zakian, V. A. (2000) Science, 289, 771-774.
64.Mangahas, J. L., Alexander, M. K., Sandell, L.
L., and Zakian, V. A. (2001) Mol. Biol. Cell, 12,
4078-4089.
65.Jacobs, S. A., Podell, E. R., Wuttke, D. S., and
Cech, T. R. (2005) Protein Sci., 14, 2051-2058.
66.Moriarty, T. J., Ward, R. J., Taboski, M. A., and
Autexier, C. (2005) Mol. Biol. Cell, 16, 3152-3161.
67.Lue, N. F., and Peng, Y. (1998) Nucleic Acids
Res., 26, 1487-1494.
68.Lue, N. F. (2005) J. Biol. Chem.,
280, 26586-26591.
69.Hardy, C. D., Schultz, C. S., and Collins, K.
(2001) J. Biol. Chem., 276, 4863-4871.
70.Harrington, L. A., and Greider, C. W. (1991)
Nature, 353, 451-454.
71.Baran, N., Haviv, Y., Paul, B., and Manor, H.
(2002) Nucleic Acids Res., 30, 5570-5578.
72.Hammond, P. W., Lively, T. N., and Cech, T. R.
(1997) Mol. Cell. Biol., 17, 296-308.
73.Wallweber, G., Gryaznov, S., Pongracz, K., and
Pruzan, R. (2003) Biochemistry, 42, 589-600.
74.Melek, M., Greene, E. C., and Shippen, D. E.
(1996) Mol. Cell. Biol., 16, 3437-3445.
75.Alves, D., Li, H., Codrington, R., Orte, A., Ren,
X., Klenerman, D., and Balasubramanian, S. (2008) Nat. Chem.
Biol., 4, 287-289.
76.Lai, C. K., Mitchell, J. R., and Collins, K.
(2001) Mol. Cell. Biol., 21, 990-1000.
77.O’Connor, C. M., Lai, C. K., and Collins,
K. (2005) J. Biol. Chem., 280, 17533-17539.
78.Moriarty, T. J., Marie-Egyptienne, D. T., and
Autexier, C. (2004) Mol. Cell. Biol., 24, 3720-3733.
79.Zaug, A. J., Podell, E. R., and Cech, T. R.
(2008) Nat. Struct. Mol. Biol., 15, 870-872.
80.Ji, H., Platts, M. H., Dharamsi, L. M., and
Friedman, K. L. (2005) Mol. Cell. Biol., 25,
9103-9114.
81.Ji, H., Adkins, C. J., Cartwright, B. R., and
Friedman, K. L. (2008) Mol. Cell. Biol., 28,
2380-2390.
82.Chen, J. L., Opperman, K. K., and Greider, C. W.
(2002) Nucleic Acids Res., 30, 592-597.
83.Miller, M. C., Liu, J. K., and Collins, K. (2000)
Embo J., 19, 4412-4422.
84.Lai, C. K., Miller, M. C., and Collins, K. (2002)
Genes Dev., 16, 415-420.
85.Lin, J., and Blackburn, E. H. (2004) Genes
Dev., 18, 387-396.
86.Zhou, X. Z., and Lu, K. P. (2001) Cell,
107, 347-359.
87.Banik, S. S., and Counter, C. M. (2004) J.
Biol. Chem., 279, 51745-51748.
88.Forstemann, K., and Lingner, J. (2005) EMBO
Rep., 6, 361-366.
89.Hossain, S., Singh, S., and Lue, N. F. (2002)
J. Biol. Chem., 277, 36174-36180.
90.Huard, S., Moriarty, T. J., and Autexier, C.
(2003) Nucleic Acids Res., 31, 4059-4070.
91.Banik, S. S., Guo, C., Smith, A. C., Margolis, S.
S., Richardson, D. A., Tirado, C. A., and Counter, C. M. (2002) Mol.
Cell. Biol., 22, 6234-6246.
92.Armbruster, B. N., Banik, S. S., Guo, C., Smith,
A. C., and Counter, C. M. (2001) Mol. Cell. Biol., 21,
7775-7786.
93.Gandhi, L., and Collins, K. (1998) Genes
Dev., 12, 721-733.
94.Roy, J., Fulton, T. B., and Blackburn, E. H.
(1998) Genes Dev., 12, 3286-3300.
95.Zhu, J., Wang, H., Bishop, J. M., and Blackburn,
E. H. (1999) Proc. Natl. Acad. Sci. USA, 96,
3723-3728.
96.Blackburn, E. H. (2005) FEBS Lett.,
579, 859-862.
97.Maringele, L., and Lydall, D. (2002) Genes
Dev., 16, 1919-1933.
98.Zubko, M. K., Guillard, S., and Lydall, D. (2004)
Genetics, 168, 103-115.
99.Gravel, S., Larrivee, M., Labrecque, P., and
Wellinger, R. J. (1998) Science, 280, 741-744.
100.Garvik, B., Carson, M., and Hartwell, L. (1995)
Mol. Cell. Biol., 15, 6128-6138.
101.Steinberg-Neifach, O., and Lue, N. F. (2006)
Nucleic Acids Res., 34, 2710-2722.
102.Hsu, M., McEachern, M. J., Dandjinou, A. T.,
Tzfati, Y., Orr, E., Blackburn, E. H., and Lue, N. F. (2007) Proc.
Natl. Acad. Sci. USA, 104, 11682-11687.
103.Sarin, K. Y., Cheung, P., Gilison, D., Lee, E.,
Tennen, R. I., Wang, E., Artandi, M. K., Oro, A. E., and Artandi, S. E.
(2005) Nature, 436, 1048-1052.
104.Park, J. I., et al. (2009) Nature,
460, 66-72.
105.Zappulla, D. C., and Cech, T. R. (2004)
Proc. Natl. Acad. Sci. USA, 101, 10024-10029.
106.Dandjinou, A. T., Levesque, N., Larose, S.,
Lucier, J. F., Abou Elela, S., and Wellinger, R. J. (2004) Curr.
Biol., 14, 1148-1158.
107.Romero, D. P., and Blackburn, E. H. (1991)
Cell, 67, 343-353.
108.Chen, J. L., Blasco, M. A., and Greider, C. W.
(2000) Cell, 100, 503-514.
109.Lin, J., Ly, H., Hussain, A., Abraham, M.,
Pearl, S., Tzfati, Y., Parslow, T. G., and Blackburn, E. H. (2004)
Proc. Natl. Acad. Sci. USA, 101, 14713-14718.
110.Qiao, F., and Cech, T. R. (2008) Nat.
Struct. Mol. Biol., 15, 634-640.
111.Livengood, A. J., Zaug, A. J., and Cech, T. R.
(2002) Mol. Cell. Biol., 22, 2366-2374.
112.Lai, C. K., Miller, M. C., and Collins, K.
(2003) Mol. Cell, 11, 1673-1683.
113.Miller, M. C., and Collins, K. (2002) Proc.
Natl. Acad. Sci. USA, 99, 6585-6590.
114.Ware, T. L., Wang, H., and Blackburn, E. H.
(2000) Embo J., 19, 3119-3131.
115.Legassie, J. D., and Jarstfer, M. B. (2005)
Biochemistry, 44, 14191-14201.
116.Seto, A. G., Umansky, K., Tzfati, Y., Zaug, A.
J., Blackburn, E. H., and Cech, T. R. (2003) Rna, 9,
1323-1332.
117.Chen, J. L., and Greider, C. W. (2003) Genes
Dev., 17, 2747-2752.
118.Leonardi, J., Box, J. A., Bunch, J. T., and
Baumann, P. (2008) Nat. Struct. Mol. Biol., 15,
26-33.
119.Webb, C. J., and Zakian, V. A. (2008) Nat.
Struct. Mol. Biol., 15, 34-42.
120.Chen, J. L., and Greider, C. W. (2005) Proc.
Natl. Acad. Sci. USA, 102, 8080-8085; discussion
8077-8089.
121.Theimer, C. A., Blois, C. A., and Feigon, J.
(2005) Mol. Cell, 17, 671-682.
122.Antal, M., Boros, E., Solymosy, F., and Kiss,
T. (2002) Nucleic Acids Res., 30, 912-920.
123.Leeper, T. C., and Varani, G. (2005)
Rna, 11, 394-403.
124.Ly, H., Blackburn, E. H., and Parslow, T. G.
(2003) Mol. Cell. Biol., 23, 6849-6856.
125.Chen, Y., Fender, J., Legassie, J. D.,
Jarstfer, M. B., Bryan, T. M., and Varani, G. (2006) Embo J.,
25, 3156-3166.
126.Richards, R. J., Wu, H., Trantirek, L.,
O’Connor, C. M., Collins, K., and Feigon, J. (2006) Rna,
12, 1475-1485.
127.Mason, D. X., Goneska, E., and Greider, C. W.
(2003) Mol. Cell. Biol., 23, 5606-5613.
128.Prathapam, R., Witkin, K. L., O’Connor,
C. M., and Collins, K. (2005) Nat. Struct. Mol. Biol.,
12, 252-257.
129.Brown, Y., Abraham, M., Pearl, S., Kabaha, M.
M., Elboher, E., and Tzfati, Y. (2007) Nucleic Acids Res.,
35, 6280-6289.
130.Zappulla, D. C., Goodrich, K., and Cech, T. R.
(2005) Nat. Struct. Mol. Biol., 12, 1072-1077.
131.Venteicher, A. S., Meng, Z., Mason, P. J.,
Veenstra, T. D., and Artandi, S. E. (2008) Cell, 132,
945-957.
132.Witkin, K. L., and Collins, K. (2004) Genes
Dev., 18, 1107-1118.
133.O’Connor, C. M., and Collins, K. (2006)
Mol. Cell. Biol., 26, 2029-2036.
134.Feng, J., et al. (1995) Science,
269, 1236-1241.
135.Fu, D., and Collins, K. (2006) Genes
Dev., 20, 531-536.
136.Mitchell, J. R., and Collins, K. (2000) Mol.
Cell, 6, 361-371.
137.Pogacic, V., Dragon, F., and Filipowicz, W.
(2000) Mol. Cell. Biol., 20, 9028-9040.
138.Dragon, F., Pogacic, V., and Filipowicz, W.
(2000) Mol. Cell. Biol., 20, 3037-3048.
139.Seto, A. G., Zaug, A. J., Sobel, S. G., Wolin,
S. L., and Cech, T. R. (1999) Nature, 401, 177-180.
140.Chapon, C., Cech, T. R., and Zaug, A. J. (1997)
Rna, 3, 1337-1351.
141.Mozdy, A. D., and Cech, T. R. (2006)
Rna, 12, 1721-1737.
142.Teixeira, M. T., Forstemann, K., Gasser, S. M.,
and Lingner, J. (2002) EMBO Rep., 3, 652-659.
143.Gallardo, F., Olivier, C., Dandjinou, A. T.,
Wellinger, R. J., and Chartrand, P. (2008) Embo J., 27,
748-757.
144.Box, J. A., Bunch, J. T., Tang, W., and
Baumann, P. (2008) Nature, 456, 910-914.
145.Hughes, T. R., Evans, S. K., Weilbaecher, R.
G., and Lundblad, V. (2000) Curr. Biol., 10, 809-812.
146.Lingner, J., Cech, T. R., Hughes, T. R., and
Lundblad, V. (1997) Proc. Natl. Acad. Sci. USA, 94,
11190-11195.
147.Zhou, J., Hidaka, K., and Futcher, B. (2000)
Mol. Cell. Biol., 20, 1947-1955.
148.Virta-Pearlman, V., Morris, D. K., and
Lundblad, V. (1996) Genes Dev., 10, 3094-3104.
149.Evans, S. K., and Lundblad, V. (1999)
Science, 286, 117-120.
150.Snow, B. E., Erdmann, N., Cruickshank, J.,
Goldman, H., Gill, R. M., Robinson, M. O., and Harrington, L. (2003)
Curr. Biol., 13, 698-704.
151.Neidle, S. (2009) Curr. Opin. Struct.
Biol., 19, 239-250.
152.Wong, H. M., Payet, L., and Huppert, J. L.
(2009) Curr. Opin. Mol. Ther., 11, 146-155.
153.Meyne, J., Ratliff, R. L., and Moyzis, R. K.
(1989) Proc. Natl. Acad. Sci. USA, 86, 7049-7053.
154.Wright, J. H., and Zakian, V. A. (1995)
Nucleic Acids Res., 23, 1454-1460.
155.Bourns, B. D., Alexander, M. K., Smith, A. M.,
and Zakian, V. A. (1998) Mol. Cell. Biol., 18,
5600-5608.
156.Smith, C. D., Smith, D. L., DeRisi, J. L., and
Blackburn, E. H. (2003) Mol. Biol. Cell, 14, 556-570.
157.Gao, H., Cervantes, R. B., Mandell, E. K.,
Otero, J. H., and Lundblad, V. (2007) Nat. Struct. Mol. Biol.,
14, 208-214.
158.Pennock, E., Buckley, K., and Lundblad, V.
(2001) Cell, 104, 387-396.
159.Fisher, T. S., Taggart, A. K., and Zakian, V.
A. (2004) Nat. Struct. Mol. Biol., 11,
1198-1205.
160.Smogorzewska, A., and de Lange, T. (2004)
Annu. Rev. Biochem., 73, 177-208.
161.De Lange, T. (2005) Genes Dev.,
19, 2100-2110.
162.Gilson, E., and Geli, V. (2007) Nat. Rev.
Mol. Cell. Biol., 8, 825-838.
163.Guedin, A., de Cian, A., Gros, J., Lacroix, L.,
and Mergny, J. L. (2008) Biochimie, 90, 686-696.
164.Johnson, J. E., Smith, J. S., Kozak, M. L., and
Johnson, F. B. (2008) Biochimie, 90, 1250-1263.
165.Griffith, J. D., Comeau, L., Rosenfield, S.,
Stansel, R. M., Bianchi, A., Moss, H., and de Lange, T. (1999)
Cell, 97, 503-514.
166.Wei, C., and Price, M. (2003) Cell. Mol.
Life Sci., 60, 2283-2294.
167.Lei, M., Zaug, A. J., Podell, E. R., and Cech,
T. R. (2005) J. Biol. Chem., 280, 20449-20456.
168.Lee, J., Mandell, E. K., Tucey, T. M., Morris,
D. K., and Lundblad, V. (2008) Nat. Struct. Mol. Biol.,
15, 990-997.
169.Luke, B., Panza, A., Redon, S., Iglesias, N.,
Li, Z., and Lingner, J. (2008) Mol. Cell, 32,
465-477.
170.Azzalin, C. M., Reichenbach, P., Khoriauli, L.,
Giulotto, E., and Lingner, J. (2007) Science, 318,
798-801.
171.Schoeftner, S., and Blasco, M. A. (2008)
Nat. Cell. Biol., 10, 228-236.
172.Luke, B., and Lingner, J. (2009) Embo
J., 28, 2503-2510.
173.Sharon, E., Freeman, R., Riskin, M., Gil, N.,
Tzfati, Y., and Willner, I. (2010) Anal. Chem., 82,
8390-8397.
174.Greider, C. W., and Blackburn, E. H. (1985)
Cell, 43, 405-413.
175.Cohen, S. B., Graham, M. E., Lovrecz, G. O.,
Bache, N., Robinson, P. J., and Reddel, R. R. (2007) Science,
315, 1850-1853.
176.Skvortsov, D. A., Rubtsova, M. P., Zvereva, M.
I., Kisseljov, F. L., and Dontsova, O. A. (2009) Acta Naturae,
49-65.
177.Cohen, S. B., and Reddel, R. R. (2008) Nat.
Meth., 5, 355-360.
178.Kim, N. W., and Wu, F. (1997) Nucleic Acids
Res., 25, 2595-2597.
179.Fajkus, J. (2006) Clin. Chim. Acta,
371, 25-31.
180.Sahin, E., and DePinho1, R. A. (2010)
Nature, 464, 520-528.
181.Lin, J., Epel, E., Cheon, J., Kroenke, C.,
Sinclair, E., Bigos, M., Wolkowitz, O., Mellon, S., and Blackburn, E.
(2010) J. Immunol. Meth., 352, 71-80.
182.Harley, C. B., Liu, W., Blasco, M., Vera, E.,
Andrews, W. H., Briggs, L. A., and Raffaele, J. M. (2010)
Rejuvenation Res., ahead of print.
183.Passos, J. F., et al. (2007) PLoS Biol.,
5, e110.
184.Haendeler, J., Hoffmann, J., Diehl, J. F.,
Vasa, M., Spyridopoulos, I., Zeiher, A. M., and Dimmeler, S. (2004)
Circ. Res., 94, 768-775.
185.Tanaka, Y., Moritoh, Y., and Miwa, N. (2007)
J. Cell. Biochem., 102, 689-703.
186.Dong, X. X., Hui, Z. J., Xiang, W. X., Rong, Z.
F., Jian, S., and Zhu, C. J. (2007) J. Cardiovasc. Pharmacol.,
49, 111-115.
187.Flores, I., and Blasco, M. A. (2010) FEBS
Lett., 584, 3826-3830.
188.Datta, A., Bellon, M., Sinha-Datta, U.,
Bazarbachi, A., Lepelletier, Y., Canioni, D., Waldmann, T. A., Hermine,
O., and Nicot, C. (2006) Blood, 108, 1021-1029.
189.Hajek, M., Matulova, N., Votruba, I., Holy, A.,
and Tloust’ova, E. (2005) Biochem. Pharmacol., 70,
894-900.
190.Sluis-Cremer, N., Temiz, N. A., and Bahar, I.
(2004) Curr. HIV Res., 2, 323-332.
191.Herschhorn, A., and Hizi, A. (2010) Cell
Mol. Life Sci., 67, 2717-2747.
192.Ueno, T., Takahashi, H., Oda, M., Mizunuma, M.,
Yokoyama, A., Goto, Y., Mizushina, Y., Sakaguchi, K., and Hayashi, H.
(2000) Biochemistry, 39, 5995-6002.
193.Mouscadet, J. F., and Desmaele, D. (2010)
Molecules, 15, 3048-3078.
194.Yamakuchi, M., Nakata, M., Kawahara, K.,
Kitajima, I., and Maruyama, I. (1997) Cancer Lett., 119,
213-219.
195.Paritala, H., and Firestine, S. M. (2009)
Bioorg. Med. Chem. Lett., 19, 1584-1587.
196.Yamakuchi, M., Nakata, M., Kawahara, K.,
Kitajima, I., and Maruyama, I. (1997) Cancer Lett., 119,
213-219.
197.Naasani, I., Seimiya, H., Yamori, T., and
Tsuruo, T. (1999) Cancer Res., 59, 4004-4011.
198.Lipps, H. J., and Rhodes, D. (2009) Trends
Cell. Biol., 19, 414-422.
199.Balasubramanian, S., and Neidle, S. (2009)
Curr. Opin. Chem. Biol., 13, 345-353.
200.Neidle, S. (2010) FEBS J., 277,
1118-1125.
201.Damm, K., et al. (2001) Embo J.,
20, 6958-6968.
202.Huang, H. S., Chou, C. L., Guo, C. L., Yuan, C.
L., Lu, Y. C., Shieh, F. Y., and Lin, J. J. (2005) Bioorg. Med.
Chem., 13, 1435-1444.
203.Eitsuka, T., Nakagawa, K., Suzuki, T., and
Miyazawa, T. (2005) Biochim. Biophys. Acta, 1737,
1-10.
204.Rangarajan, S., and Friedman, S. H. (2007)
Bioorg. Med. Chem. Lett., 17, 2267-2273.
205.Wang, X., Hao, M. W., Dong, K., Lin, F., Ren,
J. H., and Zhang, H. Z. (2009) Arch. Pharm. Res., 32,
1263-1269.
206.Hayakawa, N., Nozawa, K., Ogawa, A., Kato, N.,
Yoshida, K., Akamatsu, K., Tsuchiya, M., Nagasaka, A., and Yoshida, S.
(1999) Biochemistry, 38, 11501-11507.
207.Chen, J. L., and Greider, C. W. (2004) Proc.
Natl. Acad. Sci. USA, 101, 14683-14684.
208.Dikmen, Z. G., Ozgurtas, T., Gryaznov, S. M.,
and Herbert, B. S. (2009) Biochim. Biophys. Acta,
1792, 240-247.
209.Gryaznov, S. M. (2010) Chem.
Biodivers, 7, 477-493.
210.Maida, Y., Yasukawa, M., Furuuchi, M.,
Lassmann, T., Possemato, R., Okamoto, N., Kasim, V., Hayashizaki, Y.,
Hahn, W. C., and Masutomi, K. (2009) Nature, 461,
230-235.
211.Parkinson, E. K., Fitchett, C., and Cereser, B.
(2008) Cytogenet. Genome Res., 122, 273-280.
212.Gandellini, P., Folini, M., Bandiera, R., de
Cesare, M., Binda, M., Veronese, S., Daidone, M. G., Zunino, F., and
Zaffaroni, N. (2007) Biochem. Pharmacol., 73,
1703-1714.
213.Beliveau, A., Bassett, E., Lo, A. T., Garbe,
J., Rubio, M. A., Bissell, M. J., Campisi, J., and Yaswen, P. (2007)
Proc. Natl. Acad. Sci. USA, 104, 4431-4436.
214.Gu, B., Bessler, M., and Mason, P. J. (2009)
Cell Cycle, 8, 6-10.