REVIEW: Cross-Linked Nucleic Acids: Isolation, Structure, and Biological Role
V. A. Efimov# and S. V. Fedyunin*
Shemyakin and Ovchinnikov Institute of Bioorganic Chemistry, Russian Academy of Sciences, ul. Miklukho-Maklaya 16/10, 117997 Moscow, Russia; E-mail: eva@mx.ibch.ru; chlorin@yandex.ru# Deceased.
* To whom correspondence should be addressed.
Received March 31, 2010; Revision received June 2, 2010
This review includes literature data on main types of reagents inducing covalent cross-links of nucleic acids. Reactivity of cross-linking agents, preferable sites for their binding, and methods for determination of location of cross-links in duplex are discussed. Biological responses of cells to cross-linking in nucleic acids, i.e. replication and transcription blocking, onset of repair processes, and apoptotic cell death are considered, as well as application of cross-linking reagents as medicinal drugs for solving molecular-biological problems.
KEY WORDS: nucleic acids, cross-link, cross-linking agentDOI: 10.1134/S0006297910130079
Abbreviations: in designations of oligodeoxyribonucleotides (like GNC) prefix “d” and bond type (5′→3′) are omitted; N, any nucleotide; NA, nucleic acid; PNA, peptide–nucleic acids.
Cell functioning first of all requires normal transcription and
replication of genomic DNA. The main stage in these processes is
untwisting of DNA double helix. In the case of covalent cross-linking
between DNA strands this process becomes impossible, which causes
blocking of replication and transcription and cessation of cell
division [1-3]. However, there
is an intracellular complex of repair enzymes for elimination of
different damages in nucleic acids (NA) that is activated in response
to cross-linking reagents. The mechanism for removal of cross-linked
DNA regions are different for prokaryotes and eukaryotes and are mainly
based on repair by homologous recombination and synthesis on damaged
template, and in this case the above-mentioned processes are usually
possible with assistance of excision repair enzymes [3-8]. Besides repair systems,
there are also intracellular “tracking” systems that at any
time estimate the functional state of DNA, the existence of damage, and
the possibility of their timely elimination. In this case the
cross-link repair requires considerably more time and energy expenses
compared to other damages, and therefore in the case of a large number
of cross-links their timely removal is impossible and finally cell
death caused by apoptosis is observed [9-13]. It is found that even a single difficult to
repair cross-link is able to kill a bacterial or yeast cell, whereas 40
cross-links may be enough for death of a mammalian cell [3].
Cross-links between NA strands can emerge in response to both exogenous chemical reagents and to endogenous compounds. Despite different chemical nature, all of them function mainly as alkylating (or arylating) reagents. Cross-linking reagents are now used as antitumor preparations and as tools for investigation of features of biological behavior of cross-linked NA duplexes [14].
The main types of cross-linking reagents and their use for production of cross-linked NA duplexes, as well as cell biological reactions to appearance in them of cross-linked NA, are briefly considered in this review.
CROSS-LINKING AGENTS
Low Molecular Weight Compounds with Electrophilic Groups
The history of chemical reagents capable of NA cross-linking began during the First World War in connection with elaboration and trials of chemical weapons. It was found that the poison gas sulfurous mustard was able to inhibit cell division, which heralded its subsequent use as an antitumor preparation (see review [15]). Further intensive investigations in this field resulted in design of less toxic yperite analogs, nitrous yperites. The first use of these substances in clinical practice was in 1946 [16], after which numerous compounds of this type were prepared and studied [15, 17].The best studied class of cross-linking agents is the nitrous yperites, the typical member of which is embiquine (1) (Fig. 1). It was shown that the GNS sequence is the specific site of yperite joining [18]. In this case the guanine residues at N7 position in the DNA large groove of both strands of the duplex are alkylated. The proposed mechanism of this process includes intermediate formation of a positively charged cyclic compound (2).
Due to high reactivity towards different biological substrates, nitrous yperites exhibit rather low selectivity. Numerous works were published concerning the improvement of efficiency and selectivity of action of this class of compounds. First of all, this concerns the design of hypoxy-selective derivatives, therapy by which is based on the fact that cells positioned in the depth of most solid tumors are oxygen-deficient due to poor blood supply and contain a large content of reductases. These compounds in themselves are nontoxic, but upon reduction they undergo activation, and the lifetime of the reduced derivative in conditions of solid tumors significantly increases. Yperite (3) and conjugates of embiquine quaternary salt with various nitroheterocyclic compounds such as derivative (4) can be classified among these compounds. Nitrous yperite (3) is activated at the expense of N-oxide reduction to amine [19], and in the second case release of embiquine is the result of reducing fragmentation [20].
Parker et al. demonstrated a principally new approach to construction of hypoxy-selective yperite derivatives [21]. In the case of reduction of Cu2+ to Cu+ in compound (5) (Fig. 1) the less stable complex is formed with following release of active preparation. This process is possible only in cells with increase content of reductases, and unlike compound (2) the formed 1,4,7,10-tetraazacycladecane yperite already did not undergo oxidation to inactive precursor.Fig. 1. Nitrous yperites (1-6), epoxide-containing compounds (7, 8), s-triazine derivatives (9, 10), and phenyl compounds (11-14) capable of cross-link formation in DNA.
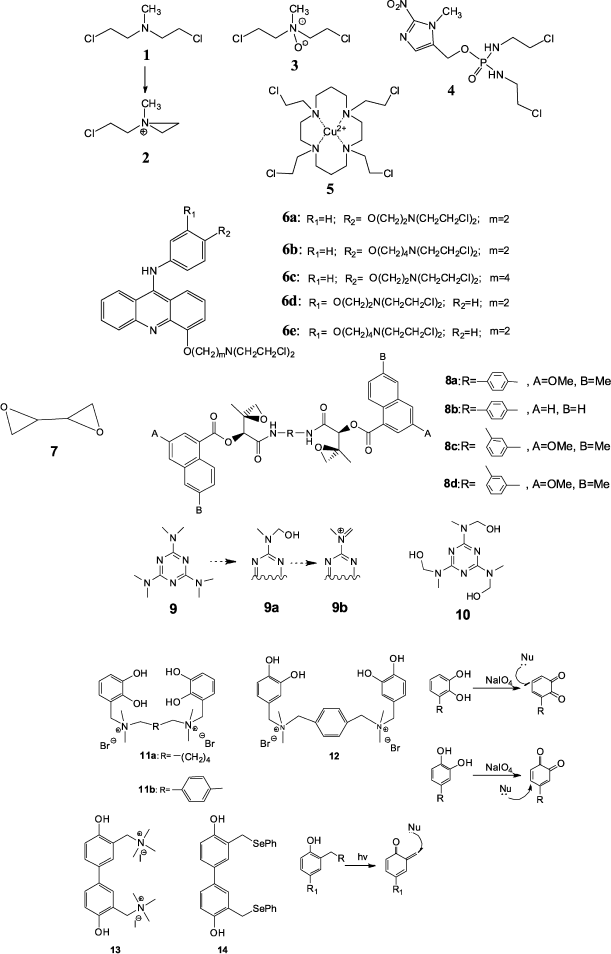
A different strategy for increasing yperite selectivity is their conjugation with different aromatic compounds able to interact with DNA via intercalation. An example of such agents is conjugates of nitrous yperites with 9-aminoacridine exhibiting the high DNA-binding constant [22, 23]. Recently new derivatives of 9-anilinoacridine (6a-6e) carrying nitrous yperite residues in aniline and acridine rings have been prepared (Fig. 1) [24]. They exhibit pronounced cytotoxicity against CCRF-CEM cells of human lymphoblast leukemia.
Derivatives of epoxides, ethylenimines, and phosphamides exhibit similar effects on cancer cells [25]. Diepoxybutane (DEB) (7) is the simplest among numerous epoxide-containing alkylating agents (Fig. 1). The mechanism of cross-linking by diepoxybutane, similar to the mechanism of action of nitrous yperites, was proposed by Millard and White [26]. It was shown that the GC fragment is the preferable point for cross-linking. In this case the most efficient of DEB stereoisomer is the S,S-isomer [27]. DEB also exhibits lower efficiency in cross-linking at sequences GGCC, TGCA, TCGA, CCGG, and TATA. Higher molecular weight dimeric analogs of the natural antitumor antibiotics azinomycins can also be attributed to epoxide-containing compounds. These compounds exhibit higher cross-linking and antitumor activities compared to their monomeric precursors. It was shown that compounds (8a-8d) (Fig. 1), having rigid aromatic spacers [28], exhibit higher toxicity compared to earlier obtained derivatives with different length aminoalkyl linkers [29].
Some s-triazine derivatives are able to cross-link two DNA strands. Among them hexamethylmelamine (9) and trimelamol (10) (Fig. 1) exhibit pronounced antitumor activity [30].
Hexamethylmelamine (9) is an example of an agent activated by oxidation. It appeared that its oxidation by cytochrome P-450 with formation of the methylol fragment (9a) takes place in the body. It is supposed that it is able to undergo transformation to the active imino ion (9b) responsible for alkylating activity. Then a similar fragment is generated in another part of the molecule, and finally a cross-link is formed in DNA [31]. Another triazine derivative trimelamol (10) exceeds hexamethylmelamine by solubility in water [32]. However, the mechanism of cross-link formation and preferable binding sites for these compounds is not yet precisely known [33].
In recent years a number of cross-linking agents that contain phenol fragments have been prepared (11-13) [34-36]. The proposed alkylation mechanism includes formation of quinone type intermediary compounds that are good Michael acceptors and interact with nucleophilic groups of nucleic bases at the β-carbon atom relative to the keto-group. Formation of reactive intermediates is induced by oxidation with sodium periodate in the case of derivatives (11) and (12) or by UV light in the case of compound (13) (Fig. 1). The advantage of these compounds is good solubility in water and high cross-linking activity. Upon bis-phenol (13) treatment of human tumor cells, the cells undergo apoptosis, which can be used later for elaboration of apoptosis-induced antitumor preparations [34].
Compounds Based on Transition Metals
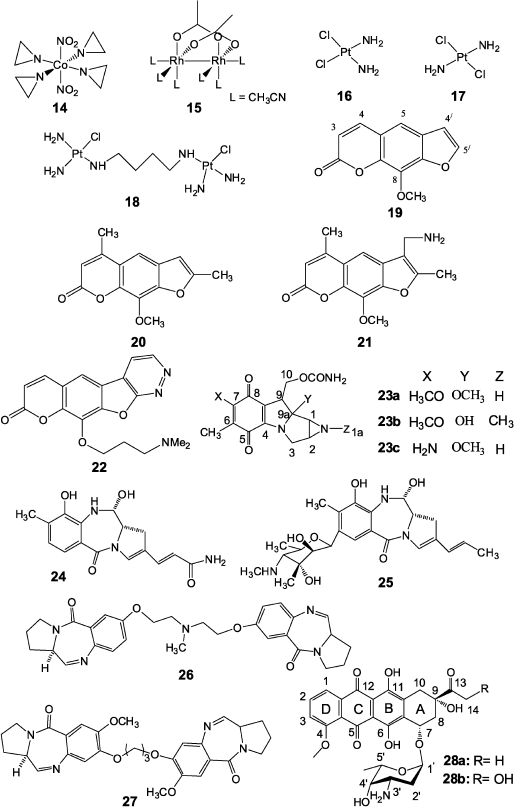
Numerous works have been published on production and application in cancer chemotherapy of transition metal compounds, e.g. such agents as complexes of cobalt(II) (14) [37] and rhodium(II) (15) [38]. Formally, the above-mentioned copper compound (5) can be attributed to the same group. Such platinum compounds as cis-diaminodichloroplatinum (16), trans-diaminodichloroplatinum (17), and type (18) dimeric compounds (Fig. 2) are among the most widely used in antitumor therapy.Among all products of cis-isomer (16) interaction with double-stranded DNA, single-stranded links (65%) at GG sequences are prevalent, while only a few cross-links are present in GC regions [39]. In this case the link greatly distorts DNA structure and both cytosine residues in GC sequence are completely eliminated from the duplex [40]. NMR data show that the cross-link is localized in the major groove of the duplex and stimulates local transformations of dextrorotatory DNA B-form to the levorotatory Z-form. Detailed description of methods for joining the platinum agent to DNA and emerging cell reactions are given in a review by Jung and Lippard [41].
Unlike its cis-isomer, the trans-isomer (17) mainly forms products of single attachment to DNA and as a result it exhibits very low antitumor activity. However, it has been shown in recent works [42] that its toxicity can be increased to the level of compound (16) in the case of simultaneous UV radiation in the interval of 320-380 nm.
To increase the efficiency of cross-linking agents based on platinum compounds, different research groups prepared dimeric compounds (18) exhibiting high efficiency, including action against tumors resistant to therapy by cis-diaminodichloroplatinum [43]. It is noted in this case that such compounds are characterized by higher cross-linking activity compared to monomeric analogs [44].
Psoralens
Psoralens are natural furocoumarins and members of the class of photoactivated cross-linking agents. The best-studied psoralens are 8-methoxypsoralen (19), 4,5′,8-trimethylpsoralen (20), and 4′-aminomethyl-4,5′,8-trimethylpsoralen (21) (Fig. 2) [8, 45]. Initiation of cross-linking by psoralens in DNA duplexes includes the stage of intercalation of planar tricyclic compound between planes of two base pairs, TA and AT, and photon absorption with reaction of photocyclo-attachment of the furan cycle 4′,5′ double bond or 3,4 double bond of coumarin to the thymidine 5,6 double bond. In this case the cross-link is formed only when the attachment initially involves a furan fragment. Otherwise, the coumarin ring is destroyed and the resulting product is unstable and no cross-link is formed. It was shown using NMR spectroscopy that the cross-link causes significant strand deformation and local untwisting by 25° in the point of psoralen joining, but it has no significant effect on the duplex as a whole, and the latter retains B-form already at the distance of three base pairs from the cross-link [46]. It should be noted that the DNA cross-links by psoralens are rather chemically stable and they can be easily reproduced on duplexes formed by synthetic oligonucleotides. Therefore, the mechanism of cross-link repair in DNA is often studied using cross-links formed by these compounds.
Via et al. [47] reported the preparation of a new pyridazine psoralen derivative (22) exhibiting significant toxicity but together with high chemotherapeutic activity compared to 8-methoxypsoralen. They believe that results can contribute to further successful elaboration of therapeutic psoralen derivatives with tetracyclic structure.Fig. 2. Compounds based on transition metals (14-18), psoralen derivatives (19-22), and antitumor antibiotics (23-28) exhibiting cross-linking activity.
Antibiotics
Natural antitumor antibiotics occupy a special place among cross-linking preparations. In 1956 in Japan mitomycins A and B [48] and two years later mitomycin C [49] were isolated from Streptomyces caespitosus culture fluid. These compounds are mitosan (23) derivatives and differ in the nature of substituents in positions 1a, 7, and 9a of the molecule [50] (Fig. 2). Of these antibiotics only mitomycin C (23c) began to be widely used in cancer chemotherapy from 1974. Numerous investigations showed that activation of mitomycin C with subsequent DNA cross-link formation is the result of a multistage process [1, 51], while the structure of mitomycin C adduct with DNA was identified in 1987 [52]. It appeared that antibiotic (23c) joins at the site of the CG dimer in the minor DNA groove at the N2 atoms of guanine residues with involvement of C1 and C10 positions of the antibiotic molecule. Data on molecular modeling, NMR spectroscopy, and mobility in PAGE of DNA duplexes cross-linked by mitomycin C show that this antibiotic, unlike bifunctional alkylating agents of nitrous yperite type, practically do not introduce serious deformations into the structure of DNA B-form, causing only slight widening of the minor groove in the cross-link site [53, 54].Activation of mitomycin C can involve different reductases, including those containing a dithiole active center [55]. At the same time, experiments on mitomycin C reduction by thiols were not successful [56]. However, Manuel et al. have recently shown that reduction by thiols is possible at pH close to the pKa of corresponding thiol [57]. Studying this reaction mechanism will make it possible in the future to understand in detail the scheme of mitomycin C activation by reductases having a dithiole active center.
More efficient and selective mitomycin C derivatives have been synthesized, among which dimeric derivatives should be noted in which two mitomycin residues are bound by linkers of differing chemical nature and site of joining to the antibiotic molecule. Some of these dimers significantly increased regioselectivity of these preparations and the amount of cross-links formation compared to the monomeric antibiotic [58, 59].
Pyrrole-benzodiazepine dimers, synthetic derivatives of natural anthramycin (24) and sibiromycin (25) precursors [60], but unlike them able to form cross-links, are also antibiotics able to form cross-links in DNA. The type (26, 27) dimers (Fig. 2) containing an aliphatic cross-link three or five carbon atoms in length exhibit high cross-linking activity, and they are specific for DNA sequences that are much more frequent in coding genome regions comparing to non-coding ones (for example, AGATTCT, GGATCC) [61].
Among other antibiotics capable of covalent bonding with DNA, antitumor antibiotics of the anthracycline series should be noted that were discovered in 1950 in culture liquid of Streptomyces purpurascens [62], and later they were also discovered in many different streptomycete strains. Several thousands of anthracycline analogs and derivatives have now been synthesized, and some of them are used in practical oncology. First of all, this concerns daunorubicin (daunomycin) and doxorubicin (adriamycin) [60]. Daunorubicin (28a) is a glycoside consisting of aglycone (4-metoxy-6,7,9,11-tetraoxy-9-aceto-7,8,9,10-tetrahydrotetracenquinone) and amino sugar (3-amino-2,3,6-trideoxy-L-lixose) (daunosamine) (Fig. 2). Doxorubicin (28b) differs from daunorubicin by an additional hydroxyl group at the C14 atom of the molecular skeleton. Common for anthracycline series antibiotics is the presence of a strong anthraquinone type chromophore exhibiting characteristic fluorescence as well as the presence in the aglycon C9 position of a hydroxyl group playing an important role in the biological activity of these compounds. The mechanism of cytotoxicity of anthracycline series antibiotics is not completely elucidated [61, 62]. It is known that these antibiotics intercalate in DNA between cytosine and guanine bases [63]. Planar tetracyclic aglycone of daunorubicin is intercalated between base pairs, the aglycone “D” ring is protruded into the major groove of DNA, while the 3′ amino sugar joined by a glycoside bond is localized in the minor groove in the immediate vicinity (~3.09 Å) of the guanine N2 atom. This is a prerequisite for formation of a covalent bond between the daunosamine 3′ amino group and the N2 atom of guanine upon action of a small bifunctional electrophilic reagent such as formaldehyde, as shown by authors of works [64, 65] who found methylene cross-link between two adjacent amino groups that covalently binds the antibiotic to DNA. According to data reported in [66], very different compounds can serve as a source of formaldehyde, such as Tris-HCl in various buffers, spermine (usually associated with DNA in cells), and finally doxorubicin itself (due to interaction with hydrogen peroxide formed in the organism during Fenton-type redox processes). Data of works [67, 68], using mass spectrometry for analysis of formaldehyde content in cells treated and not treated by antibiotics, favor this mechanism. The level of formaldehyde in treated cells was higher and correlated with the concentration of the preparation. Thus, it turned out that from a formal point of view antibiotics doxorubicin and daunorubicin form a covalent bond only with one DNA strand, whereas aglycone intercalates into the other DNA strand and they form with each other two hydrogen bonds. This type of interaction was called virtual (or quasi) cross-link [66]. These results lead to rational design of new analogs and derivatives of anthracycline antibiotics exhibiting improved antitumor properties. In particular, a series of dimeric derivatives joined at the 3′ amino groups using various linkers was synthesized. This caused a 104-fold increase in the affinities of the antibiotic to DNA and thus, in perspective, allowing significant reduction of doses of drugs based on these antibiotics [70, 71]. Dimeric conjugates of anthracycline antibiotics with formaldehyde—doxoform, daunoform, and epidoxoform—are extremely active against tumor cells resistant to the original antibiotics [72-74].
Endogenous Cross-Linking Agents
Cross-links in cellular DNA can emerge not only in response to exogenous cross-linking agents, but also as a result of interaction of DNA with endogenous agents. In the case of the action of α,β-unsaturated aldehydes (enals), in particular of acrolein, croton aldehyde, and 4-hydroxynonenal, by-products of oxidative degradation of unsaturated lipids, initially 1,N2-γ-hydroxypyrano-dG adducts are formed, which produce cross-links upon interaction with amino group of guanine of neighboring strand in neighboring C·G and G·C base pairs in the 5′-CpG-3′ sequence [2].Nitric oxide synthesized in human body and carrying out a number of important functions is also able to form cross-links. Upon its interaction with water nitrous acid is formed that diazotizes N2 guanine residues. The resulting diazo-salts are replaced by guanine amino groups of the complementary strand, which results in appearance of a covalent bond in 5′-CpG-3′ sites of the strands [75].
DETECTION OF CROSS-LINK STRUCTURE. PREPARATION OF DUPLEXES WITH
DESIRED CROSS-LINK POSITION
Many compounds capable of causing cross-link formation in DNA have been synthesized. Determination of mechanism of action and preferable binding site of such agents is impossible without methods for detection of the position of the cross-link in the NA molecule.
Electrophoresis under denaturing conditions is used for detecting cross-links in nucleic acid molecules and approximate estimation of the number of cross-linked duplexes. In this case cross-linked duplexes are easily detected by the presence of bands with lower electrophoretic mobility compared to corresponding single-stranded DNA fragments [76]. A method including exonuclease cleavage of the duplex and isolation of the minimal possible length nucleic acid fragment containing the cross-link is used for exact determination of structure of a cross-linked NA fragment [26]. Preliminary synthesis of an oligonucleotide containing a predetermined sequence and cross-link site is used as well. The control duplex free of this site is obtained in parallel. Then this duplex is treated with cross-linking agent, and the presence or absence of the cross-link is detected by gel electrophoresis. After obtaining information on the cross-link position in double-stranded NA and preferable binding site of a certain agent, more precise information concerning structural features of the cross-linked site is obtained using X-ray analysis [77], molecular modeling, NMR [78-80], and mass spectrometry [81].
Cross-linked double-stranded NA can be used as models for studying cellular processes associated with DNA and RNA functioning as well as for design of affinity reagents and “traps” for proteins binding to double-stranded DNA [82]. For example, if the mechanism of enzyme action supposes double helix untwisting, then the presence of a covalent bond between strands prevents such untwisting. So cross-linked duplexes can be powerful inhibitors of proteins whose functioning requires untwisting of double helix [82]. It should be also noted that high affinity just to double-stranded DNA containing covalent bonds is characteristic of the repair system proteins responsible for removal of these bonds; therefore, cross-linked duplexes are actively used for isolation of this type of proteins and detailed investigation of features of their functioning [42]. Methods for preparing covalently linked NA duplexes using different reagents are considered in detail in reviews [14, 83].
There are now three main strategies of directed introduction of a cross-link into a particular sequence of the duplex. First, it is oligonucleotide postsynthetic modification. Already synthesized oligonucleotide duplex is treated with a cross-linking agent having known site-specificity. Then the target cross-linked duplex is purified of byproducts including cross-links in one strand, products of the mono-attachment of the agent, and the initial oligonucleotide. This method is used to obtain cross-links with such agents as nitrous yperites. Another method includes single-stranded oligonucleotide hybridization with complementary sequence containing in a definite place monoderivative of the cross-linking agent. Modification is introduced either postsynthetically or during elongation of the oligonucleotide strand. Then cross-link formation is initiated after hybridization. Thus, in the case of psoralen the cross-linking is stimulated by UV-radiation, while in the case of mitomycin C the cross-link is formed in response to incubation of the duplex in oxygen-free medium in the presence of a reducing agent.
The next method can be formally classified as the two above-mentioned methods. It includes introduction during solid-phase synthesis of complementary strands of a modified link containing an active electrophilic group [84] or its precursor [85]. Then the strands are hybridized along with treatment of the duplex with a bifunctional nucleophilic reagent such as a diaminoalkane.
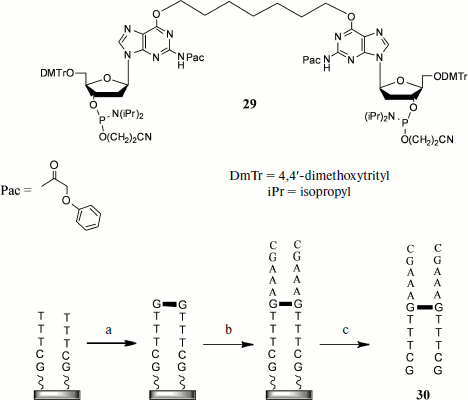
Another group of methods involves the introduction of cross-links using the solid-phase method of oligonucleotide synthesis. The method including preparation of an oligonucleotide with the alkyl cross-link at oxygen atoms of guanine residues of opposite strands can serve as an example [86] (Fig. 3). The synthesis proceeds as follows. The first stage includes simultaneous elongation of two oligonucleotide strands in 3′→5′ direction using the nucleoside 3′-O-phosphite-amides. Then at a certain step a dimer (29) consisting of two protected deoxyguanosine residues joined to each other at heterocycle O6 atoms is introduced into condensation, and in this case the standard isobutyroyl protective group of the heterocyclic base is replaced by the phenoxyacetyl (Pac) group that makes it possible to unblock under milder conditions. Thus, at this stage the cross-link is introduced at a certain oligonucleotide site. Then elongation of both strands continues to a chosen length, after which the duplex is unblocked and purified by HPLC.
During recent years cyclo-attachment of terminal alkynes to azides, catalyzed by copper(I) salts [3+2] and called the “click-reaction”, has become widely used [87]. This reaction is characterized by mild conditions, high yields, the possibility of using a broad pH interval, and inertness towards most functional groups present in biological substrates. Owing to this, the “click-reaction” is now used in numerous fields of molecular biology, chemistry, and biochemistry, in particular for design of NA conjugates with proteins [88], for their immobilization on solid carriers [89], for introduction into NA of different labels [90], etc.Fig. 3. Synthesis on solid base of cross-linked oligonucleotide duplexes including cross-link introduction by simultaneous attachment of dimeric oligonucleotide to both growing chains: a) joining of bis-phosphite-amide (29); b) addition of 3′-O-phosphite-amides until reaching desired oligonucleotide length; c) release from carrier and unblocking.
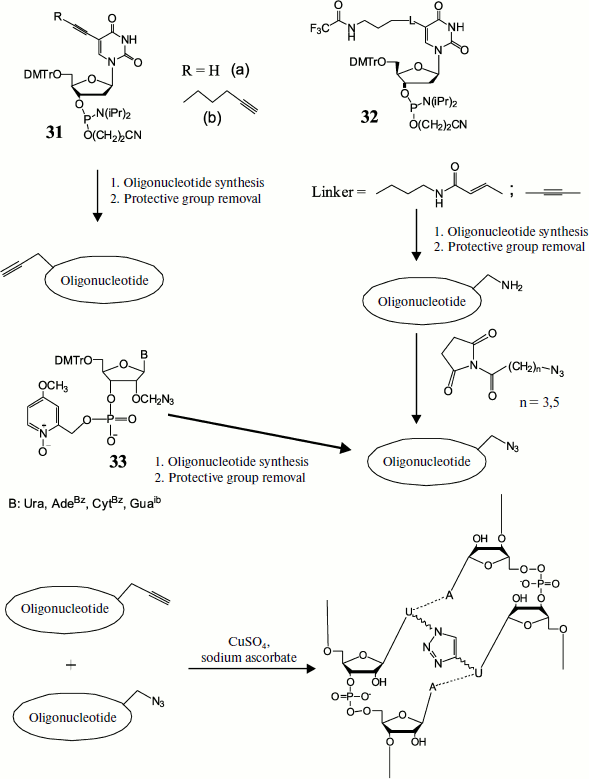
The possibility of applying this method for preparing DNA duplexes containing different length cross-links between two modified uracil residues was demonstrated by Zocalka et al. [91]. They prepared phosphite amide monomers, some of which contained alkynyl groups (31) and others contained blocked amino groups (32) (Fig. 4). Azide groups were introduced by postsynthetic treatment of amino-containing oligonucleotides by N-hydroxysuccinimide esters of 4-azidobutanic and 6-azidohexanic acids. We have proposed a different method for introduction of an azide group into oligonucleotides using the type (33) monomer and phosphotriester route of oligonucleotide synthesis [92]. In work [91] nine oligonucleotides with identical base sequence were obtained, some of which contained a link with alkynyl group, while others contained complementary azide groups in different positions. Upon hybridization the azide group in one strand and alkynyl in the other were found in different duplexes at different distances. Two oligonucleotides free of modifications were used as reference in measuring melting temperature. Formation of cross-linked duplexes was shown by denaturing gel electrophoresis. The rate of the cross-link formation reaction was dependent on both linker length and position of the octadienyl group in the sequence, but on average cross-link formation was rather rapid. In this case no cross-link formation was observed in the absence of copper catalyst. Thermal denaturation showed that all cross-linked double-stranded oligonucleotides had melting temperature exceeding that of unmodified duplexes, on average by 30°C.
Fig. 4. Preparation of cross-linked duplex using the click-reaction. Preliminarily obtained monomers, some of which contain alkyne (31) and others protected amine (32) or already ready azide (33), are introduced into oligonucleotide chain at a certain stage of synthesis. After removal of protective groups the amino group-containing oligomers are modified by ω-azido acid residues. Then azide and alkynyl oligonucleotides are hybridized in pairs and cross-linked using the [3+2] dipolar cycloaddition reaction.
REACTIONS OF BIOLOGICAL CELLS TO FORMATION OF CROSS-LINKS IN DNA
Preservation of the native state of DNA is necessary for normal cell activity. Therefore, there are special systems continuously monitoring the condition of DNA and removing damage that constantly occurs due to chemical agents, UV radiation, and other factors [93]. Such damage includes products of the interaction of DNA with cross-linking agents. The appearance of cross-links in DNA stimulates a number of cell reactions that may differ depending on the cell cycle phase, number of cross-links, existence of genetic drawbacks, etc. [3].
It is known that transcription and replication require untwisting of the DNA molecule. In the presence of a covalent bond between strands untwisting becomes impossible, and this blocks these processes. In the case of replication there emerges the so-called stalled replication fork. This launches repair mechanisms that in initial stages involve a site-specific endonuclease splitting off double strand of daughter DNA containing leader strand. However, this event is observed only in eukaryotic cells [6]. Such DNA damages are not classical breaks of double helix like those emerging in response to reactive oxygen species or UV radiation, but rather they are “single-stranded breaks (nicks)” [94]. It has been shown recently that during initial nuclease treatment classical double-stranded breaks are possible as well [95].
In addition to the above-mentioned events, cell division ceases. It is known that the regular sequence of changes in the cell cycle stages occurs upon interaction of cyclin-dependent kinases with cyclins. Cells in G0 phase are able to enter the cell cycle in response to growth hormones launching intracellular signal cascade, finally leading to transcription of cyclin and cyclin-dependent kinase genes. Cyclin-dependent kinases are activated only upon interaction with appropriate cyclins.
Each cell cycle stage except G0 is characterized by the presence of a corresponding cyclin, while the cyclin/cyclin-dependent kinase complex launches a cascade of phosphorylation reactions regulating the change in the cell cycle phases. By termination of a certain phase the corresponding cyclin undergoes rapid destruction by proteinases. There are check points for monitoring of completion of each cell cycle phase. Many factors, including DNA damage, can influence the passing of the check point by the cell. There are at least three check points of the cycle in which the presence of DNA damage is checked: a point in G1 (G1/S) when the intact DNA state is checked before entering S phase; a check point in S phase where correctness of DNA replication is determined, and a check point in G2 (G2/M) where damages are checked that were omitted during previous check points or were obtained at following stages of the cell cycle. In G2 phase the completeness of DNA replication is detected and cells with incomplete DNA replication do not enter mitosis. DNA damage, in particular, replication fork blocking due to the presence of a cross-link, is detected by the hMOF, TIP60, and MRE11 proteins carrying out signal transduction to transducing PIKK (phosphatidylinositol 3-kinase-dependent kinases): ATM (ataxia-telangiectasia mutated), ATR (ataxia telangiectasia and Rad3 related), and DNA-PK (DNA-dependent protein kinase). It is supposed that the three types of protein kinases are interchangeable but simultaneously exhibit different specificity. Thus, ATM is first of all responsible for formation of reaction to double-stranded breaks in DNA, whereas ATR is activated in response to numerous different DNA damages, including replication fork blocking [96]. Activated kinases, in turn phosphorylate p53 protein (one of key components in launching apoptotic cascade [97]) and effector molecules like kinases Chk1 and Chk2. They phosphorylate a number of proteins including complex cyclin/cyclin-dependent protein kinase, Cdc5, 14, 25, 45, etc., which results in cell cycle arrest. Besides, kinases Chk1 and Chk2 activate the cell repair complex. After activation the signal mechanism together with the cell cycle arrest molecular machine are in activated form until the damage is repaired and the system of specific phosphatases restores intracellular molecular order [98]. Numerous investigations have shown that cross-linking agents mainly cause cell cycle arrest in G2 phase [99].
If opportune elimination of DNA damage is impossible, then the apoptosis process is launched via a series of signal and mediator proteins and protein systems. It should be noted that although it is still not known exactly how and when a cell distinguishes between launching a repair process or apoptotic systems, nevertheless numerous data reveal key participants of both processes [100].
Cross-Link Repair in DNA
Repair of damaged DNA is now one of most intensely studied fields of biochemistry and molecular biology, and there is even a specialty journal dealing with these problems (DNA Repair). Depending on their complication, different DNA damages are removed with involvement of numerous proteins and protein complexes. Thus, O6-methylated guanines are easily and quickly removed by O6-methylguanine-DNA-methyltransferase [101], whereas repair of double-stranded breaks and cross-links requires participation of multiple proteins and protein complexes, both directly involved in damage removal and those carrying out signal and/or regulatory functions, because one and the same protein can simultaneously fulfill several functions, which makes it significantly more difficult to study such processes. It is also necessary to note that complex DNA damage is eliminated by a succession of simpler methods and proceeds differently in prokaryotic and eukaryotic cells [5, 8]. The detailed analysis of all methods of elimination of DNA damage is beyond the scope of this review, and below we shall consider only the main steps in cross-link repair.
Nucleotide excision repair. Nucleotide excision repair (NER) recognizes and corrects severe DNA damage that blocks replication and transcription [102]. Main proteins involved in NER in E. coli are Uvr(A, B, C) and Pol I. UvrA and UvrB form complex (UvrA)2UvrB that recognizes the damaged site, and UvrB joins DNA, whereas UvrA dissociates and then UvrC joins the DNA–UvrB complex. Then phosphodiester bonds surrounding the damaged site are cleaved, and after that (UvrB)(UvrC) complex dissociates and the damaged site is replaced by DNA polymerase Pol I.
In yeast cells NER function involves proteins of four complexes, NEF1-4. Complex NEF2 including proteins Rad4 and Rad23, like Rad14 protein of NEF1 complex are involved in damage recognition, while endonucleases Rad1-Rad10 (NEF1) and Rad2 (NEF3) make cuts near the damaged site. Then DNA is synthesized by DNA polymerases Pol2 and Pol δ and ligated by DNA ligase at the final stage. Complex NEF4 consisting of proteins Rad7 and Rad16 mediates excision repair in untranscribed DNA regions [103]. Helicases Rad3 and Rad25, performing local untwisting of double-stranded DNA, are involved in formation of cuts by endonucleases.
In human cells proteins RPA, XPA, and XPC recognize damage, and then helicase TFIIH is attached and locally untwists DNA in the damaged site. Then proteins XPG and XPF/ERCC1 cut from 3′ and 5′ ends of the damaged site, respectively. As a result, a 24-32-membered oligonucleotide is removed and then, like in previous cases, the formed gap is filled by RFC/PCNA proteins and Pol δ/ε polymerases with following ligation by DNA ligase I [5].
Repair based on homologous recombination. The main function of this repair mechanism is elimination of double-stranded breaks in DNA, which are often formed following exposure to cross-linking agents [104]. Repair by homologous recombination is used by the cell mainly in S and G2 cell cycle phases when sister chromatids are accessible, whereas in phases G1 and M another repair mechanism operates, nonhomologous end joining [104-106].
In E. coli cells a damaged site recognized by RecABC complex that prepares the substrate for RecA protein pairing homologous DNA molecules. RecABC has nuclease and helicase functions, it untwists DNA and degrades strands after which RecA joins the treated DNA from the 3′ end. Then in complex with SSB (single strand binding protein) it searches for homology in undamaged DNA, which is followed by joining of the molecules, and DNA polymerase I restores the DNA sequence. The resulting structure is transformed to two new strands by RuvC protein together with RuvAB.
In yeast cells the DNA strand initially treated by an unknown nuclease, after which the nuclease–helicase complex Rad50/MRE11/XRS2 prepares the resulting DNA-substrate. Then proteins Rad51 (yeast homolog of bacterial protein RecA) and Rad52 together Rad54 and Rad55,57 promote DNA untwisting and strand hybridization between donor and damaged DNA. Cleavage of formed recombinant structures proceeds in different ways with involvement of nuclease mus81-mms4 and/or a protein analogous to RuvABC complex called resolvase A.
In mammalian cells the above-mentioned functions are fulfilled, like in yeast cells, by protein Rad51, while nuclease–helicase complex Rad50/MRE11/XRS2 includes NBS1 protein instead of XRS2. Besides, proteins XRCC2 and XRCC3 take part in the process, which repair breaks and cross-links and interact with rad51 protein as well. It should be emphasized that mechanisms of repair by homologous recombination in eukaryotic cells are still not fully studied, and their intensive study continues [8].
Synthesis on damaged template. Main function of this process is elimination of damage blocking replication. Unlike previous types of DNA damage repair, significant errors are possible in this type of repair. In its simplest form it includes three stages: 1) DNA polymerase carrying replication is blocked when it encounters damage on one strand, in this case the enzyme is removed from the replication fork; 2) instead of the usual polymerase, a special DNA polymerase capable of synthesis on damaged template is introduced. It synthesizes a short fragment passing through the damaged site of the parental strand; 3) then the special DNA polymerase is released and synthesis is resumed by the usual DNA polymerase.
In mammalian, bacterial, and yeast cells functions of special polymerase are carried out by different proteins. Thus, in mammalian cells proteins of the Y family are the best studied of this type of polymerases, but these enzymes are not involved in cross-link repair. In E. coli DNA polymerase II fulfills functions of synthesis on damaged template, while in yeasts Rev3 and Rev7 form the heterodimer polymerase ζ, a member of the DNA polymerase B family [8].
Cross-link repair in E. coli. Experiments have shown that cross-link repair is a complex and multistage process. Duplexes with cross-links formed by 8-methylpsoralen, mitomycin C, or nitrous yperite were used in the main works studying mechanisms of cross-linked DNA repair in bacterial, yeast, and mammalian cells. Double-stranded DNA cross-linked by other reagents is used rather rarely.
Unlike in eukaryotic cells, the process of cross-link repair in E. coli cells is rather well studied [5]. It can include the process of nucleotide excision repair and either homologous recombination or synthesis on damaged template. It has been shown that in the case of psoralen cross-link endonuclease complex Uvr(A)2BC (also designated as UvrABC, see detail about enzyme complex functioning [107]) first makes two cuts in one DNA strand from the furan side of the cross-link, which results in a short oligonucleotide covalently joined to the other DNA chain via the psoralen bridge (Fig. 5a). With the 5′-exonuclease activity of DNA polymerase I (Pol I) not able to synthesize on this template, a small region near the above-mentioned oligonucleotide is removed, then protein RecA is oligomerized on the resulting single-stranded site by joining in the 5′→3′ direction and forming of a helical protein thread. The site containing the cross-link is placed into this thread. Homologous pairing of two DNA molecules forms DNA heteroduplex with a three-stranded site. Thus, exchange by strands, made easier by RecA, begins from the single-stranded site and continues towards the oligomeric thread formation. Then Pol I synthesizes a new strand on the basis of donor DNA strand in the 3′→5′ direction, after which Uvr(A)2BC cuts the damaged strand and the cross-link is removed in the form of a covalently bound oligonucleotide duplex, while DNA polymerase finishes the process by completion of the gap formed in one of the recipient strands.
Biochemical and genetic experiments on the example of the yperite cross-link repair have also shown the existence of an alternative pathway of cross-link elimination occurring in the absence of accessible homologous DNA or in E. coli cells deficient in synthesis of RecA protein [108]. This mechanism is shown in Fig. 5b. At the first stage protein complex Uvr(A)2BC hydrolyzes phosphodiester bonds from both ends of the cross-link site, and the resulting gap is then filled by DNA polymerase β. Then Uvr(A)2BC introduces breaks in the second strand, the cross-linked oligonucleotide duplex is removed, and the gap is filled by DNA polymerase I.Fig. 5. Methods of cross-link repair in E. coli including stages of homologous recombination (a) and synthesis on damaged template (b).
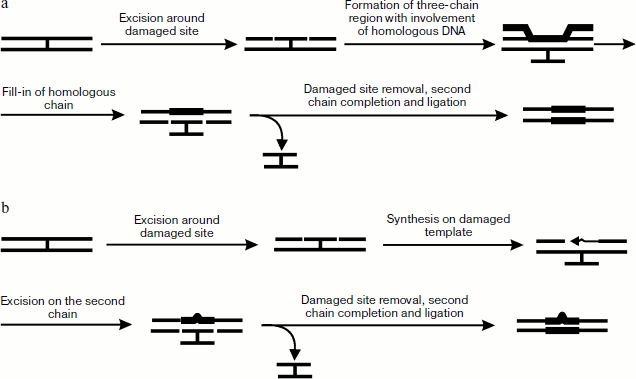
In addition to the described main proteins of the bacterial repair apparatus, different proteins like RecB, C, D, F, G, O, and R can also be involved in cross-link elimination. Thus RecFOR facilitates RecA joining to DNA and accelerates homologous pairing of two DNA molecules, while RecABC is necessary when double-stranded breaks are formed during cross-linked DNA repair (which, in turn, can be the result of nuclease action or most often this happens upon blocking of the replication fork movement in the presence of a cross-link). Proteins RecG or RuvABC are responsible for separation of recombinant intermediate structures [5].
Cross-link repair in eukaryotic cells. In eukaryotic cells the repair process is much more complex and includes numerous proteins. This makes much more difficult investigation of the mechanisms involved. The situation is even more complicated because some proteins participating in these processes, in addition to their main role, fulfill several other functions at different stages of the process. Nevertheless, although exact mechanisms of cross-link repair are still not resolved, in recent years much experimental and theoretical material concerning these processes and their participants has accumulated. Data from the literature on repair processes in yeast and mammalian cells are generalized in reviews [5, 6, 8] along with description of all known protein participants and their functions.
In cells of the best studied strain of budding yeasts, Saccharomyces cerevisiae, functions of endonucleases of the excision stage of the repair process are carried out mainly by products of genes RAD1-4, RAD10, RAD14, and RAD25, and in this case proteins Rad1 and Rad10 together comprise endonuclease complex cutting the DNA strand from the 5′ end, whereas Rad2 cuts the DNA strand from the 3′ end. Helicases Rad3 and Rad25 locally untwist the double helix in the site of action of the nuclease.
Like in bacterial cells, the main repair step in yeast cells is either homologous recombination or synthesis on damaged template. In the case of repair by homologous recombination key participants are products of genes RAD51, RAD52, and RAD54 [5] as well as regulatory elements (see in detail [109]).
Another process, synthesis on damaged template, is under control of products of genes RAD6 and RAD18, which fulfill their regulatory function via modification of proliferating cell nuclear antigen (PCNA) (lysine ubiquitination in position 164) involved both in replication and in repair of damaged DNA [110]. It is an auxiliary factor of DNA polymerase δ (the main DNA polymerase of eukaryotic cells) and by interaction with it, significantly increasing its activity. When necessary, PCNA is modified by Rad6–Rad18 complex and DNA polymerases ζ and/or η (Pol ζ and/or Pol η) capable of synthesis on damaged template begin to act [8].
Numerous data also show that in addition to the above-mentioned genes, PSO genes (psoralen-sensitive) are also involved in cross-link repair. This group now includes 10 genes known as PSO1-10. The existence of these genes was first found about two decades ago when a pso1 mutant strain of S. cerevisiae was found exhibiting increased sensitivity to psoralen [111]. Later it was shown that the PSO1 gene is allelic to REV3 (encodes catalytic subunit of polymerase ζ) and thus is involved in processes of post-replicative repair. The PSO2 gene product takes part in cross-link repair by nonhomologous end joining. This repair mechanism is also used by the cell upon cross-link elimination but to a lesser extent than in homologous recombination (Fig. 6a). Upon blocking the movement of the polymerase complex in the presence of a cross-link, the enzyme topoisomerase I, whose main function is prevention of accumulation of positive supertwists upon replication, generates cruciform DNA structures on both sides from the site with the cross-link, especially in the presence of palindrome sequences. Then enzyme complex Mre11p–Rad50p–Xrs2p hydrolyzes phosphodiester bonds in single-stranded regions of the cruciform structures of two strands, which results in hairpin formation. Then these structures are cleaved with involvement of the PSO2 gene product, cross-linked double-stranded region is removed, and then treatment and following ligation of the double-stranded break ends proceed with the participation of DNA ligase IV and Xrcc4.
It should be also noted that only eight members of the PSO gene group products are directly involved in cell responses to DNA damage, whereas Pso6 and Pso7 are not involved in processes associated with metabolism of nucleic acids (see in detail [112]).Fig. 6. Cross-link repair in mammalian cells. a) Mechanism of cross-linked DNA repair including nonhomologous end joining; b) main mechanism of cross-link repair.
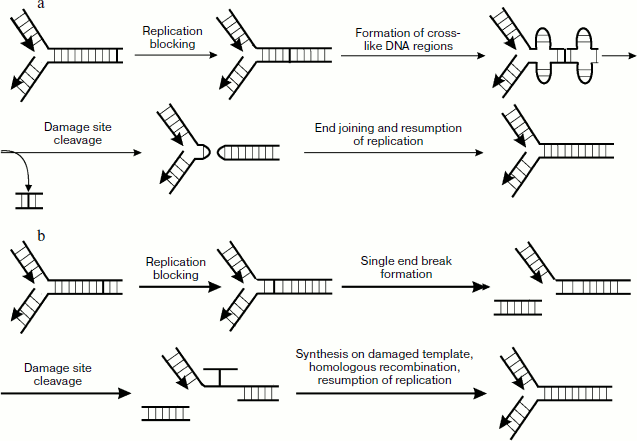
In mammalian including human cells, cross-link elimination in double-stranded DNA follows the same principles but at a far more complex level. A number of genes involved in cross-link repair in mammalian cells such as RAD6, RAD18, RAD54, SNM1, REV3, and REV7 were identified by homologous sequences of these genes in yeast cells. However, many proteins of the yeast repair apparatus (Rad6, Rad23, Rad30, Rad51, and Snm1) have up to several homologs in mammalian cells. It is necessary to emphasize that due to complex packaging of the mammalian genome, increasingly important is regulation of processes connected with recognition of DNA damage and formation of cell responses at the level of chromatin macrostructure (for example, double-stranded breaks cause phosphorylation of histone H2AX, which is widely used as a marker of the mentioned damage [113]). At the same time the role of factors of topoisomerase and helicase functions increases, and some of these factors are multifunctional [114]. All above-mentioned factors make much more difficult the study of mechanisms of cross-link repair in mammalian cells.
The main endonucleases involved in cross-link repair in mammalian cells are XPG and ERCC1-XPF, which hydrolyze phosphodiester bonds from the side closer to the 3′ and 5′ ends, respectively [5]. Gene XPG is a homolog of yeast RAD2, whereas ERCC1 and XPF are homologs of RAD10 and RAD1, respectively. Proteins RAD51, RAD52, and RAD54 are mainly involved at the stage of homologous recombination. Bergstralh and Sekelsky [115] suppose that nuclease ERCC1-XPF is also involved at the stage of homologous recombination, mediating together with other proteins separation of intermediate recombinant structures. Polymerases ζ, η, k [116, 117], Rev and i, as well as products of already mentioned genes RAD6 and RAD18 [8] are key participants of synthesis on damaged template. Participation of polymerase N in synthesis on damaged template in human cells has been reported [118], but its main biological role is still unknown.
Investigations of cell response to cross-linked DNA showed cells mutant in certain genes not involved in the main repair processes exhibit increased sensitivity to cross-linking agents. These mutations cause the so-called Fanconi’s anemia expressed in a syndrome of bone marrow insufficiency. Later, during different experiments on mutant lines over 10 genes were revealed whose products play a pronounced role in recognition of replication fork blocking and formation of the cell response [119]. Products of these genes mainly include proteins with common name FANC and some others that play an important role in cross-link repair. This process occurs mainly in the cell cycle S phase and proceeds as follows. The blocked replication fork is recognized independently by FANCM protein and FANCD2/FANC1 complex. Then a complex consisting of proteins FANCA,B,C,E,F,G,L,M, FAAP24, and FAAP100 dissociates. It was recently shown [120] that this protein complex exists during the whole cell cycle, and its association with different chromatin regions is regulated depending on the presence of damage in DNA and the cell cycle phase [121]. Then the interaction of this complex with FANCM and FANCD2/FANCI ubiquitination are switched on with mediation of UBE2T. After that FANCM with involvement of FANCD2 create substrate for Mus81-Eme1 and ERCC1-XPF endonucleases, the actions of which stimulate cleavage of a daughter strand from the parental strand. In this case a double-strand break is observed, and then a break is formed in one chain of the parental strand close to the cross-link site. This is followed by synthesis on damaged template for sealing the formed gap. FANCD2 stimulates binding to the double-stranded break of homologous recombination proteins completing the repair process (Fig. 5e).
Thus, cell response to cross-linked DNA mainly involves transcription and replication due to the impossibility of further movement of the corresponding enzyme complexes along the strand. Recent studies have shown that the repair mechanism activation is also possible independently of these processes due to local distortion of DNA structure in the cross-link site [122]. In this work biotin-modified radiolabeled oligonucleotide duplexes containing cross-links of different nature were used. Among them were alkyl cross-links N4C-ethyl-N4C in C·C pairing error (designated below as C–C) and in sequences CG and GC (CG, GC). Duplexes containing N3T-ethyl-N3T in T·T pairing error (T–T) and the cross-link formed by 4′-amino-4,5′,8-trimethylpsoralen in TA (TA) sequence were also used. In this case the most pronounced deviations from DNA B form were observed in the case of GC (disturbance of base pairing in two nucleotides from each side of the cross-link site), whereas in CG sequence dimethylene cross-link causes practically no local distortion of double helix as well as in T–T. Cross-link C–C results in mean (compared to CG and GC) deviation from B-form. These cross-link effects on duplex structures are consistent with thermodynamic data. Thus, ethyl cross-link of two cytosines in CG sequence and in C–C pairing error resulted in increase of the duplex melting temperature by 49 and 25°C, respectively, whereas GC cross-link noticeably decreased duplex stability as shown by lowering melting temperatures by 10°C. Thus, cross-links can be arranged in the following order on the basis of the extent of lowering the double helix distortion: GC, C–C, TA, and exhibiting approximately equal effects CG and T–T.
Investigations on HeLa and AA8 cells have shown that repair of these cross-links can be accompanied either by formation of a single excision from the side of each 5′ and 3′ end from the cross-link site or of two excisions from the 5′ end at a short distance from each other. Finally, the main product was a single-stranded oligonucleotide containing the cross-link residue in the form of covalently attached nucleotide. It should be noted that formation of excisions from two sides of the cross-link site did not involve the excision repair enzymes as shown using cell lines deficient in their synthesis. Incubation of cross-linked duplexes with these cells showed that in 1 h in the case of double-stranded oligonucleotide with cross-link in the region of GC sequence, 6% of cross-linked duplex is transformed to the single-stranded product, whereas in the case of CG only 0.7% is transformed. For C–C cross-link and for psoralen cross-link the corresponding values were 2.6 and 1.9%. The data show that the extent of duplex distortion at the cross-link site influences damage recognition by repair enzymes.
In a later work [123] the same authors report about investigation of synthesis on damaged template using the above-mentioned duplexes and cells free of sister chromatids. Besides these duplexes, duplex with N1I-ethyl-N3T cross-link in I·T pairing error (I–T) was used. It appeared that cross-links GC, CG, and C–C were efficiently repaired, while cross-links T–T and I–T blocked repair. According to authors, this event is due to the fact that in the case of N4C-ethyl-N4C fragment ethyl cross-link is beyond the heterocycle zone responsible for hydrogen bond formation, whereas in T–T and I–T the possibility of hydrogen bond formation is blocked. Thus, upon repair of initially formed single-stranded oligonucleotide containing cross-link residue, in the case of N4C-ethyl-N4C fragment the appropriate DNA polymerase can carry out synthesis on damaged template, because there is the possibility of base pairing, whereas residues N3T-ethyl-N3T and N1I-ethyl-N3T do not give such possibility.
The field of investigations connected with the directed effect on participants of cell response to DNA damage for antitumor therapy is now intensely developed (see reviews [7, 124-126]). Investigation of proteins and protein systems involved in covalent cross-link repair and importance of their functions allow elaboration of antitumor preparations aimed at inhibition of these proteins and protein systems and their exclusion from the process. Thus, it is possible either to stop completely or significantly retard cross-link repair in DNA. The use of these preparations in combination with cross-linking agents can significantly increase the efficiency of the latter and thus decrease the dose required for similar therapeutic effect. A simple example of this strategy is the use of sulfanilamide preparations in complex with trimethoprim [127].
Among such target proteins in antitumor chemotherapy there are repair DNA helicases involved in many stages and in different repair processes [114]. Many of them belong to the RecQ helicase family. Mutations of these enzymes can be responsible for rapid aging, genomic instability, and increased risk of carcinogenesis [128]. Directed inhibition of such helicases can significantly increase therapeutic efficiency.
Methods for investigating repair processes. Investigation of mechanisms of repair processes in cells of different organisms usually based on the use of DNA duplexes with cross-links of different nature and different cell lines, mutant at different genes, whose products are supposedly involved in different repair pathways (see reviews [8, 129, 130]).
As mentioned above, even the use of highly specific cross-linking agents does not make possible to exactly localize a covalent cross-link, first, because the reagent specificity sites can be localized in any site of the genome, and second, due to a small fraction of cross-links comparing to other products of interaction with DNA. Owing to this, plasmids [131] and oligonucleotide duplexes [123] containing a covalent bond between strands at specific places are mainly used for studying cell response to cross-linked DNA.
Contemporary fluorescence analysis methods can reveal DNA repair processes in real time. These methods include fluorescence recovery after photobleaching and fluorescence quenching in bleached images [132, 133]. They are based on the use of conjugates of fluorescent proteins with the proteins under study. In this case photobleaching of a fluorophore results in disappearance of fluorescence that is restored some time later. In the fluorescence restoration method, after photobleaching a certain part of the cell is intensely illuminated by light, after which fluorescence intensity is measured before illumination and after fluorescence restoration and their ratio makes it possible to judge the mobility of the studied factor “labeled” by the fluorescent protein. At the same time, this method helps to determine the rate of migration of a molecule into the illuminated region, which with known diffusion constants of molecules of a particular size makes it possible to define whether molecules migrate separately or within different complexes. There is also a method in which a small part of a cell undergoes continuous illumination, and constant monitoring of lowering of the fluorescence level in the region near the illuminated region provides information concerning the transfer of the studied protein over this region. Fluorescence methods of analysis include also fluorescence resonance energy transfer, which reveals interactions between different biomolecules by specific spectral changes [134]. High resolution fluorescence microscopy makes possible to observe different processes directly in living cells and to overcome many restrictions of usual fluorescence microscopy [135].
In addition to the above-mentioned methods, in investigation of repair processes and other cell reactions atomic force microscopy is widely used to observe three-dimensional structure and conformational changes in biomolecules on the nanometer scale in vitro under conditions close to physiological [136]. In particular, this method was used for investigation of conformational changes in Mre11/Rad50/Nbs1 complex involved in recognition of double-stranded breaks upon binding to DNA substrate [137]. X-Ray analysis, NMR spectroscopy, and electron microscopy are also used to obtain information about protein spatial structure [138]. They are widely used for studying different sites responsible for protein–protein and protein–DNA interactions.
While fluorescence recovery after photobleaching and fluorescence quenching in bleached images can be used for observation of the repair process in real time within a cell, other methods of investigation require isolation of repair proteins and intermediate complexes from cell lysate. For this aim a short oligonucleotide containing in a certain site a monoderivative of cross-linking agent is hybridized with single-stranded plasmid. The latter is then completed to the double-stranded one by T7 DNA polymerase and ligated by T4 DNA ligase. This is followed by cleavage by restriction endonuclease at sites located at some distance from both sides of the damaged site. The resulting double-stranded DNA fragment is modified by biotin using DNA polymerase I Klenow fragment and appropriate biotin-containing nucleoside triphosphates, and then it is immobilized on streptavidin-containing magnetic microcarrier. This DNA construct was used as an affinity trap for isolation of repair system proteins [129].
Apoptotic Cell Death
Apoptosis is an event of programmed cell death accompanied by a set of typical cytological features (apoptosis markers) and molecular processes that are different in unicellular and multicellular organisms [10]. It differs from another kind of cell death, necrosis, by a number of morphological and biochemical features, in particular, by cell separation from extracellular matrix, condensation of the nucleus, shrinkage of cytoplasmic membrane, and finally, by formation of apoptotic bodies – membrane vesicles with cellular content that are later absorbed by macrophages. In apoptosis a cell seems to be prepared for its future phagocytosis without development of inflammatory reaction, as happens in the case of necrosis. A characteristic feature of apoptosis is DNA fragmentation, not chaotic but rather forming a so-called “apoptotic ladder”. It is a set of different length DNA fragments, the length difference being approximately 200 nucleotides because these fragments are the result of internucleosomal fragmentation by different endonucleases [9]. Development of apoptosis is a very complex and not fully studied process. Because of this, in this review we shall consider only some participants of the apoptotic process; activation of apoptosis and pathways of apoptotic signal transduction are in detail and rather completely considered in reviews [10-13]. In biochemical aspect four main components are distinguished in apoptosis: 1) Cys-Asp proteinases or caspases; 2) so-called death receptors on the cell surface involved in apoptosis activation by certain ligands; 3) mitochondria and incorporated cytochrome c, and 4) special pro- and antiapoptotic proteins [139].Activation of apoptosis can be the result of both external effects and endogenous activation of the apoptotic cascade. Exogenous activation includes, for example, certain ligands (such as FasL) that after joining specific receptors on the cell surface (Fas/CD95) launch a series of processes finally causing activation of proteolytic and nucleolytic enzymes. In this case there can be possible direct activation of the caspase cascade via caspase 8 or signal transduction to mitochondria, which results in further development of the apoptotic process. Endogenous inducers of apoptosis may cause different intracellular damages, in particular, blocked replication fork and double-strand breaks in DNA. In this case the damages are recognized by special proteins that in one variant of exogenous activation further transduce the signal to mitochondria, which play the central role in the further chain of apoptotic events [13]. Different proapoptotic proteins such as AIF (apoptosis-inducing factor) and SMAC (second mitochondria-derived activator of caspases) are released from mitochondria. Besides, cytochrome c is also released into the cytoplasm and after binding to Apaf-1 (apoptotic protease activating factor-1) forms the so-called apoptosome complex that initiates activation of the caspase cascade.
The main proteins involved in the apoptotic signal are caspases (the Cys-Asp proteinase family). In mammals the caspase class is represented by 14 proteins continuously synthesized in the form of proenzymes and activated in apoptosis. Their consecutive activation is the caspase cascade that begins from autocatalytic activation of initiator caspase, which in turn activates effector caspases [140]. The latter cleave actin filaments, inhibit protein synthesis, and activate DNase.
As already mentioned, DNA damage is among the factors responsible for development of apoptosis. Cross-linked DNA is not an exception. It is known from the example of a cross-link formed by cis-platinum that the reason for activation of apoptosis is transcription and replication blocking. In this case there is activation of SAPK (stress-activated protein kinase) protein, also known as JNK (c-Jun N-terminal kinase), and of p38-kinase, which cause AP-1 (activator protein-1) gene expression. The latter stimulates synthesis of FAS-L protein, a FAS receptor ligand. Then their interaction is accompanied by activation of the caspase cascade. It was found that the above-mentioned series of processes begins in 2-4 days after treatment with cis-platinum [141].
In the presence of a cross-link in DNA blocking of the replication fork and formation of double-stranded breaks (single end breaks) also occur. These processes stimulate activation of different signal systems. As mentioned above, ATM and ATR proteins are involved in recognition of these damages. Activation of these proteins is accompanied by their phosphorylation of NBS1 protein that then activates kinases chk1 and chk2. This results in cell cycle arrest. Besides, ATM phosphorylates p53 protein, one of the main participants launching apoptosis caused by DNA damage, and owing to this it is a component of antitumor control of the organism [142]. This protein is simultaneously active in several directions, and it activated gene p21 whose product finally causes cell cycle arrest. It also activates a number of proapoptotic proteins, namely Bax (BCL-2-associated X protein), PUMA (p53 upregulated modulator of apoptosis), and FAS receptor. Protein Bax binds porin in the outer mitochondrial membrane and together they form the channel stimulating cytochrome c release. PUMA, in turn, binds Bcl-2, which prevents the formation of the latter complex with Bax and so even more stimulates cytochrome c release. Altogether, these events activate the caspase cascade.
It should be also noted that besides increase in p53 concentration, activation of repair systems by the same signal proteins ATM/ATR also occurs. It is supposed that the final fate of a cell depends on complex relationships of all the above-mentioned signal and regulatory elements having either pro- or antiapoptotic functions [4].
CROSS-LINKED RNA–RNA AND DNA–RNA DUPLEXES AND THEIR
APPLICATION IN MOLECULAR BIOLOGY
The first works on RNA cross-linking were published already at the end of the 1950s and dealt with effects of formaldehyde, glyoxal, and ketoxal on nucleic acid of tobacco mosaic virus [143, 144]. Later cross-links of RNA strands by formaldehyde were used in detection of tRNA secondary structure by Bayev et al. [145]. Creation of cross-links in RNA molecules was also used in detection of tertiary structure of ribosomal RNA. For obtaining cross-linked rRNA, mainly UV-induced formation of cross-links due to the presence in certain sites of 4-thiouridine residues was used. These residues were introduced into the strand upon ribosomal RNA reconstruction by ligation of fragments already containing 4-thiouridine links, or upon microbiological synthesis of RNA using an E. coli strain deficient in pyrimidine synthesis grown in a medium containing 4-thiouridine. The cross-link sites in RNA were determined both using reverse transcription (in the cross-link site the process is stopped, and then the synthesized oligomer is analyzed) and after enzymic degradation by RNase H within RNA/DNA duplex with following analysis of the resulting fragments by denaturing gel electrophoresis. A conclusion concerning spatial structure of ribosomal RNA was drawn on the basis of cross-link localization and number, internal dynamics of its strands during ribosome functioning, mechanisms of RNA interaction with different macromolecules, etc. [146, 147]. A similar technique is also applicable to investigation of different types of RNA.
The cross-link of DNA–RNA duplexes can help in increasing the efficiency of antisense therapy [148, 149]. In this method a specific antisense oligodeoxyribonucleotide carrying a 4,5′,8-trimethylpsoralen residue at the end or in the middle of the strand undergoes hybridization with target RNA with subsequent UV irradiation (366 nm). The formed covalent cross-link sharply increases thermal stability of heteroduplex, which in turn results in significant increase in the efficiency of antisense therapy. Hairpin-type modified antisense oligonucleotides with psoralen fragment at the 5′ end were synthesized [148]. They consisted of two fragments with a specific base sequence joined by a linker of four thymidine residues. These oligonucleotides were complementary to homopurine sites of mRNA encoding c-Myc protein, which causes rapid growth of B16-F0 melanoma cells. Decrease in the expression level of this protein sharply retards cell growth. The above-mentioned modified oligonucleotides are able to produce triplex structures with target RNA due to involvement in formation of Watson–Crick duplex that then takes part in Hoogsteen complex formation with the second oligonucleotide part (Fig. 7a). Owing to their ability to form similar structures, these oligonucleotides exhibit high specificity, efficiency, and selectivity. Experiments on B16-F0 cells with immunodetection of c-Myc protein showed a very low level of its expression after treatment by such modified antisense oligonucleotides.
A different approach suggests the use of oligonucleotides carrying a psoralen fragment attached to the 2′ hydroxyl of a riboadenosine link via a methylene or methoxymethylene bridge (Fig. 7b). It was found that in the case of oligonucleotide in which 4,5′,8-trimethylpsoralen is joined via a methylene cross-link, the covalent bonding efficiency is about 35%, whereas in oligonucleotide in which psoralen is attached via a methoxymethylene linker it reaches 75% [149].Fig. 7. Oligonucleotides containing modifications and capable of covalent bonding with RNA. a) Self-complementary oligonucleotide containing oligo-T-linker in the middle of the chain and a psoralen residue at the 5′ end, and the scheme of its interaction with target mRNA. b) Oligonucleotide containing in the middle of the chain an adenosine residue modified at the 2′ hydroxyl by psoralen. c) Cross-link formation during thymine link reactions within PNA containing a phenylselenyl group.
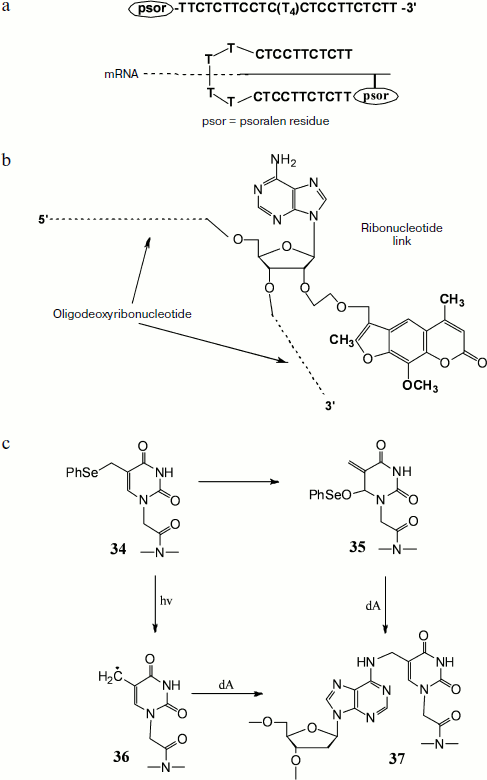
In addition to the described modified oligonucleotides, oligonucleosides whose monomers are joined by methylphosphonate residues are also used for inhibition of protein expression. Upon binding to mRNA, these compounds are able to block ribosome joining upon initiation of protein synthesis or at the stage of elongation. The advantage of these oligomers over usual oligonucleotides is their resistance to intracellular nucleases. An oligonucleoside with methylphosphonate links containing a psoralen residue was also prepared, and its ability to inhibit translation of mRNA in vitro encoding rabbit α and β globins was demonstrated [150]. Later the use of a 12-membered oligomer carrying a psoralen residue at one end and a rhodamine at the other made it possible to show in experiments with mouse L949 cells that this oligonucleotide could be used for estimation of efficiency of antisense interactions in living cells by fluorescence microscopy [151].
Among the mentioned approaches to antisense therapy there is also the use of peptide–nucleic acids (PNA). Due to their neutrality (they do not carry negative charge at moderate or high pH because they are not acids), constants of PNA-ligand binding to complementary NA strands are usually higher than binding constants of natural NA duplexes. Their advantages also include resistance to intracellular nucleases.
Recent studies have shown that an approach including cross-link formation in the target heteroduplex by introduction of different modifications in PNA oligomers can be used to increase the efficiency of binding to target sequence [152, 153]. Thus, it was shown by Kim and Hong [152] that cross-linking is achieved by introduction of a thymine residue (34) containing a phenylselenyl group with subsequent oxidation or illumination. In the case of oxidation Dimroth rearrangement occurs with formation of an active Michael acceptor (35) that alkylates the exocyclic amino-group of adenine. Illumination results in formation of radical (36), whose subsequent reaction with an adenine residue produces covalent bonding (37) (Fig. 7c).
It was shown by Bombard et al. [154] that cross-linking reagents can be also used for investigation of the mechanism of action of ribozymes. In the presence of Mn2+ and polyU, the shortest member, the pentaribonucleotide GAAACp, undergoes hydrolysis of the phosphodiester bond between guanosine and adenosine residues with formation of guanosine-2′,3′-cyclophosphate and AAACp tetranucleotide [155]. Covalent binding of the pentanucleotide N7 atoms by trans-diaminodichloroplatinum completely inhibited the cleavage reaction, whereas mono-adducts at the same atoms had no effect on reaction rate. This resulted in the conclusion that coordination by magnesium ion of N7 atoms of guanosine and adenosine links is not necessary for the cleavage reactions. At the same time blocking adenosine residues in the second or third position of the Pt(NH3)32+ chain greatly decreased the cleavage reaction rate, which was indicative of the need for complex formation between the second adenosine residue and the 2′-OH link of guanosine with Mn2+ upon formation of the activated complex.
In general, it should be noted that there are still relatively few works on investigation of cross-linked RNA–RNA and RNA–DNA duplexes compared to the numerous literature dealing with cross-linked DNA.
Thus, there are numerous reagents of different nature and specificity capable of cross-linking in NA. Efficient methods have been developed for preparing cross-linked duplexes, which are used in various molecular-biological investigations, in particular, for studying mechanisms of intracellular repair processes, apoptosis, as well as of other intracellular processes. Knowledge obtained during investigation of repair mechanisms and involved protein systems are used in medicine and pharmacology, in particular, they are the basis for elaboration of new forms of antitumor preparations. The search for compounds exhibiting cross-linking activity is actively continuing.
REFERENCES
1.Rajski, S., and Williams, R. (1998) Chem.
Rev., 98, 2723-2796.
2.Stone, M., Cho, Y., Huang, H., Kin, H., Kozekov,
I., Kozekova, A., Wang, H., Minro, I., Lloyd, R., Harris, T., and
Rizzo, C. (2008) Accounts Chem. Res., 41, 793-804.
3.Scharer, O. (2005) ChemBioChem., 6,
27-32.
4.Su, T. (2006) Annu Rev. Genet., 40,
187-208.
5.Dronkert, M., and Kanaar, R. (2001) Mutat.
Res., 486, 217-247.
6.Lehoczky, P., McHugh, J., and Chovanec, M. (2007)
FEMS Microbiol. Rev., 31, 109-133.
7.Lieberman, B. (2008) Curr. Med. Chem.,
15, 360-367.
8.Noll, D., Mason, T., and Miller, P. (2006) Chem.
Rev., 106, 277-301.
9.Lushnikov, E. F., and Abrosimov, A. Yu. (2001)
Cell Death (Apoptosis) [in Russian], Meditsina, Moscow,
pp. 81-109.
10.Blank, M., and Shiloh, Y. (2007) Cell
Cycle, 6, 686-695.
11.Strasser, A., O’Connor, L., and Dixit, V.
(2000) Ann. Rev. Biochem., 69, 217-245.
12.Roos, W., and Kaina, B. (2006) Trends Mol.
Med., 12, 440-450.
13.Gordeeva, A. V., Labas, Yu. A., and Zvyagilskaya,
R. A. (2004) Biochemistry (Moscow), 69, 1055-1066.
14.Antsipovich, S. I., and Oretskaya, T. S. (1998)
Uspekhi Khim., 67, 274-393.
15.Brookers, P. (1990) Mutat. Res.,
233, 3-14.
16.Gilman, A., and Phillips, F. S. (1946)
Science, 103, 409-411.
17.Ward, K. (1935) J. Am. Chem. Soc.,
57, 914-916.
18.Rink, S., Solomon, M., Taylor, M., Rajur, S.,
McLaughlin, L., and Hopkins, P. (1993) J. Am. Chem. Soc.,
115, 2551-2557.
19.White, I., Suzanger, M., Mattocks, A., Bailey,
E., Farmer, P., and Connors, T. (1989) Carcinogenesis,
10, 2113-2118.
20.Duan, J., Jiao, H., Kaizerman, J., Stanton, T.,
Evans, J., Lan, L., Lorente, G., Banica, M., Jung, D., Wang, J., Ma,
H., Li, X., Yang, Z., Hoffman, R., Ammons, W., Hart, C., and Matteucci,
M. (2008) J. Med. Chem., 51, 2412-2420.
21.Parker, L., Lacy, S., Farrugia, L., Evans, C.,
Robins, D., O’Hare, C., Hartley, J., Jaffar, M., and Stratford,
I. (2004) J. Med. Chem., 47, 5683-5689.
22.Kapuriya, N., Kapuriya, K., Dong, H., Zhang, X.,
Chou, T., Chen, Y., Lee, T., Lee, W., Tsai, T., Naliapara, Y., and Su,
T. (2009) Bioorg. Med. Chem., 17, 1264-1275.
23.Baguley, B. (1991) Anti-Cancer Drug
Design, 6, 1-35.
24.Chen, C., Lin, Y., Zhang, X., Chou, T., Tsai, T.,
Kapuriya, N., Kakadiya, R., and Su, T. (2009) Europ. J. Med.
Chem., 44, 3056-3059.
25.Hendry, J., Rose, F., Homer, R., and Roades, A.
(1948) Br. J. Pharmacol., 6, 235-255.
26.Millard, J., and White, M. (1993)
Biochemistry, 32, 2120-2124.
27.Millard, J., Hanly, T., Murphy, K., and
Tretyakova, N. (2006) Chem. Res. Toxicol., 19, 16-19.
28.LePla, R., Landreau, C., Shipman, M., Hartley,
J., and Jones, G. (2005) Bioorg. Med. Chem. Lett., 15,
2861-2864.
29.Finerty, M., Bingham, J., Hartley, J., and
Shipman, M. (2009) Tetrahedron Lett., 50, 3648-3650.
30.Coley, H. (1997) Gen. Pharmac., 28,
177-182.
31.Jackson, C., Crabb, T., Gibson, M., Godfrey, R.,
Saunders, R., and Thurston, D. (1991) J. Pharm. Sci.,
80, 245-251.
32.Jackson, C., Hartley, J., Jenkins, T., Godfrey,
R., Saunders, R., and Thurston, D. (1991) Biochem. Pharmacol.,
42, 2091-2097.
33.Coley, H., Brooks, N., Phillips, D., Hewer, A.,
Jenkins, T., Jarman, M., and Judson, I. (1995) Biochem.
Pharmac., 49, 1203-1212.
34.Song, Y., Wang, P., Wu, J., Zhou, X., Zhang, X.,
Weng, L., Cao, X., and Liang, F. (2006) Bioorg. Med. Chem.
Lett., 16, 1660-1664.
35.Weng, X., Ren, L., Weng, L., Huang, J., Zhu, S.,
Zhou, X., and Weng, L. (2007) Angew. Chem. Int. Ed., 46,
8020-8023.
36.Song, Z., Weng, X., Weng, L., Huang, J., Wang,
X., Bai, M., Zhou, Y., Yang, G., and Zhou, X. (2008) Chemistry,
14, 5751-5754.
37.Ware, D., Wilson, W., Denny, W., and Rickard, C.
(1991) J. Chem. Soc. Chem. Commun., 1171-1173.
38.Dunham, S., Chifotides, H., Mikulski, S., Burr,
A., and Dunbar, K. (2005) Biochemistry, 44, 996-1003.
39.Malinge, J., Perez, C., and Leng, M. (1994)
Nucleic Acids Res., 22, 3834-3839.
40.Huang, H., Zhu, L., Reid, B., Drobny, G., and
Hopkins, P. (1995) Science, 270, 1842-1845.
41.Jung, Y., and Lippard, S. (2007) Chem.
Rev., 107, 1387-1407.
42.Heringova, P., Woods, J., Mackay, F., Kasparkova,
J., Sadler, P., and Brabec, V. (2006) J. Med. Chem., 49,
7792-7798.
43.Chopard, C., Lenoir, C., Rizzato, S., Vidal, M.,
Arpalahti, J., Gabison, L., Albinati, A., Garbay, C., and Kozelka, J.
(2008) Inorg. Chem., 47, 9701-9705.
44.Qu, Y., Scarsdale, N., Tran, M., and Farrell, N.
(2004) J. Inorg. Biochem., 98, 1585-1590.
45.Ciniino, G., Gamper, H., Isaacs, S., and Hearst,
J. (1985) Ann. Rev. Biochem., 54, 1151-1193.
46.Hwang, G., Kim, J., and Choi, B. (1996)
Biochem. Biophys. Res. Commun., 219, 191-197.
47.Via, L., Gonzalez-Gomez, J., Perez-Montoto, L.,
Santana, L., Uriarte, E., Marciani, S., Gia, M., and Gia, O. (2009)
Bioorg. Med. Chem. Lett., 19, 2874-2876.
48.Hata, T., Sano, Y., Sugawara, R., Matsumae, A.,
Kanamori, K., Shima, T., and Hoshi, T. (1956) J. Antibiot. Ser.
A, 9, 141-146.
49.Wakagi, S., Marumo, H., Tomoka, K., Shimizu, G.,
Kata, E., Kamada, H., Kudo, S., and Fugimoto, Y. (1958) Antibiot.
Chemother., 8, 228-238.
50.Webb, J., Cosulich, D., Mowat, J., Patrick, J.,
Broscha, R., Meyer, W., Williams, R., Wolf, C., Fulmor, W., and
Pidacks, C. (1962) J. Am. Chem. Soc., 84, 3185-3187.
51.Danishefsky, S., and Schkeryantz, J. (1995)
Synlett., 475-490.
52.Tomasz, M., Lifman, R., Chowdary, D., Pawlak, J.,
Verdine, G., and Nakanishi, K. (1987) Science, 235,
1204-1208.
53.Rink, S., Lipman, R., Alley, S., Hopkins, P., and
Tomasz, M. (1996) Chem. Res. Toxicol., 9, 382-389.
54.Norman, D., Live, D., Sastry, M., Lipman, R.,
Hingerty, B., Tomasz, M., Broyde, S., and Patel, D. (1990)
Biochemistry, 29, 2861-2875.
55.Adikesavan, A., and Jaiswal, A. (2007) Mol.
Cancer Ther., 6, 2719-2727.
56.He, Q., Maruenda, H., and Tomasz, M. (1994) J.
Am. Chem. Soc., 116, 9349-9350.
57.Manuel, M. (2009) Chem. Res. Toxicol.,
22, 1663-1668.
58.Paz, M., Kumar, S., Glover, M., Waring, M., and
Tomasz, M. (2004) J. Med. Chem., 47, 3308-3319.
59.Na, Y., Li, V., Nakanishi, Y., Bastow, K., and
Kohn, H. (2001) J. Med. Chem., 44, 3453-3462.
60.Boger, D., and Paianki, S. (1992) J. Am. Chem.
Soc., 114, 9318-3927.
61.Hadjivassileva, T., Stapleton, P., Thurston, D.,
and Taylor, P. (2007) Int. J. Antimic. Agents, 29,
672-678.
62.Brockmann, H., and Bauer, K. (1950)
Naturwissen Schaften, 37, 492-497.
63.Wang, A., Ughetto, G., Quigley, G., and Rich, A.
(1987) Biochemistry, 26, 1152-1163.
64.Gao, Y., Liaw, Y., Li, Y., van der Marel, G., van
Boom, J., and Wang, A. (1991) Proc. Natl. Acad. Sci. USA,
88, 4845-4849.
65.Leng, F., Savkur, R., Fokt, I., Przewloka, T.,
Priebe, W., and Chaires, J. (1996) J. Am. Chem. Soc.,
118, 4731-4738.
66.Taatjes, D., Gaudiano, G., Resing, K., and Koch,
T. (1997) J. Med. Chem., 40, 1276-1286.
67.Kato, S., Burke, P., Fenick, D., Taatjes, D.,
Bierbaum, V., and Koch, T. (2000) Chem. Res. Toxicol.,
13, 509-516.
68.Taatjea, D., Gaudiano, G., and Koch, T. (1997)
Chem. Res. Toxicol., 10, 953-961.
69.Zhang, H., Gao, Y., van der Marel, G., van Boom,
J., and Wang, A. (1993) J. Biol. Chem., 268,
10095-10101.
70.Fox, K., Webster, R., Phelps, R., Fokt, I., and
Priebe, W. (2004) Eur. J. Biochem., 271, 3556-3566.
71.Portugal, J., Cashman, D., Trent, J.,
Ferrer-Miralies, N., Przewloka, T., Fokt, I., Priebe, W., and Chaires,
J. (2005) J. Med. Chem., 48, 8209-8219.
72.Fenick, D., Taatjes, D., and Koch, T. (1997)
J. Med. Chem., 40, 2452-2461.
73.Taatjes, D., Fenick, D., and Koch, T. (1998)
J. Med. Chem., 41, 1306-1314.
74.Koch, T., Barthel, B., Kalet, B., Rudnicki, D.,
Post, G., and Burkhart, D. (2008) Top. Curr. Chem., 283,
141-170.
75.Caulfield, J., Wishnok, J., and Tannenbaum, S.
(2003) Chem. Res. Toxicol., 16, 571-574.
76.Jackson, C., Hartley, J., Jenkins, T., Godfrey,
R., Saunders, R., and Thurston, D. (1991) Biochem. Pharmacol.,
42, 2091-2097.
77.Edfeldt, N., Harwood, E., Sigurdsson, S.,
Hopkins, P., and Reid, B. (2004) Nucleic Acids Res., 32,
2785-2794.
78.Edfeldt, N., Harwood, E., Sigurdsson, S.,
Hopkins, P., and Reid, B. (2004) Nucleic Acids Res., 32,
2795-2801.
79.Swenson, M., Paranawithana, S., Miller, P., and
Kielkopf, C. (2007) Biochemistry, 46, 4545-4553.
80.Diakos, C., Messerle, B., del Socorro Murdoch,
P., Parkinson, J., Sadler, P., Fenton, R., and Hambley, T. (2009)
Inorg. Chem., 48, 3047-3056.
81.Candy Chen, H., and Chen, Y. (2009) Chem. Res.
Toxicol., 22, 1334-1341.
82.Kozlov, I. A., Ivanovskaya, M. G., Kubareva, E.
A., and Volkov, E. M. (1996) Mol. Biol. (Moscow), 30,
503.
83.Noll, D., Noronha, A., Wilds, C., and Miller, P.
(2004) Front. Biosci., 6, 421-437.
84.Angelov, T., Guainazzi, A., and Scharer, O.
(2009) Org. Lett., 11, 661-664.
85.Dooley, P., Tsarouhtsis, D., Korbel, G., Nechev,
L., Shearer, J., Zegar, I., Harris, C., Stone, M., and Harris, T.
(2001) J. Am. Chem. Soc., 123, 1730-1739.
86.Wilds, J., Booth, J., and Noronha, A. (2006)
Tetrahedron Lett., 47, 9125-9128.
87.Colb, H., Finn, M., and Sharpless, K. (2001)
Angew. Chem., 113, 2056-2075.
88.Humenik, M., Huang, Y. W., Wang, Y. R., and
Sprinzl, M. (2007) ChemBioChem., 8, 1103-1106.
89.Rozkiewicz, D., Gierlich, J., Burley, G.,
Gutsmiedl, K., Garell, T., Ravoo, B., and Reinhoudt, D. (2007)
ChemBioChem., 8, 1997-2002.
90.Seela, F., and Sirivolu, V. (2007) Helv. Chim.
Acta, 90, 535-552.
91.Zocalka, P., El-Sagheer, A., and Brown, T. (2008)
ChemBioChem., 9, 1280-1285.
92.Efimov, V. A., Aralov, A. V., Fedyunin, S. V.,
Klykov, V. N., and Chakhmakhcheva, O. G. (2009) Bioorg. Chem.
(Moscow), 35, 270-273.
93.Gates, K. (2009) Chem. Res. Toxicol.,
22, 1747-1760.
94.Shrivastav, M., de Haro, L., and Nickoloff, J.
(2008) Cell Res., 18, 134-147.
95.Peng, X., Ghosh, A., van Houten, B., and
Greenberg, M. (2010) Biochemistry, 49, 11-19.
96.Skladanowski, A., Bozko, P., and Sabisz, M.
(2009) Chem. Rev., 109, 2951-2973.
97.Millau, J., Bastien, N., and Drouin, R. (2009)
Mutat. Res., 681, 118-133.
98.Likhacheva, A. S., Rogachev, V. A., Nikolin, V.
P., Popova, N. A., Shilov, A. G., Sebeleva, T. E., Strunkin, D. N.,
Chernykh, E. R., Gelfgat, E. L., Bogachev, S. S., and Shurdov, M. A.
(2008) Vestnik VOGiS, 12, 426-473.
99.Konora, J. (1988) Biochem. Pharmacol.,
37, 2303-2309.
100.Bernstein, C., Bernstein, H., Payne, C., and
Gareval, H. (2002) Mutat. Res., 511, 145-178.
101.Kaina, B., Christmann, M., Naumann, S., and
Roos, W. (2007) DNA Repair, 6, 1079-1099.
102.Reardon, J., and Sancar, A. (2005) Progr.
Nucleic Acid Res. Mol. Biol., 79, 183-235.
103.Verhage, R., Zeeman, A., Degroot, N., Gleig,
F., Bang, D., van de Putte, P., and Brouwer, J. (1994) Mol Cell
Biol., 14, 6135-6142.
104.Shrivastav, M., de Haro, L., and Nickoloff, J.
(2008) Cell Res., 18, 134-147.
105.Ohnishi, T., Mori, E., and Takahashi, A. (2009)
Mutat. Res., 669, 8-12.
106.Burma, S., Chen, B., and Chen, D. (2006) DNA
Repair, 5, 1042-1048.
107.Truglio, J., Croteau, D., van Houten, B., and
Kisker, C. (2006) Chem. Rev., 106, 233-252.
108.Berardini, M., Foster, P., and Loechler, E.
(1999) J. Bacteriol., 181, 2878-2882.
109.Kroghand, B., and Symington, L. (2004) Annu.
Rev. Genet., 38, 233-271.
110.Essers, J., Theil, A., Baldeyron, C., van
Cappellen, W., Houtsmuller, A., Kanaar, and Vermeulen, R. (2005)
Mol. Cell. Biol., 9350-9359.
111.Henriques, J., and Moustacchi, E. (1980)
Genetics, 95, 273-288.
112.Brendel, M., Bonatto, D., Strauss, M., Revers, L., Pungartnik, C., Saffi, J., and Henriques, J. (2003)
Mutat. Res., 544, 179-193.
113.Clingen, P., Wu, J., Miller, J., Mistry, N.,
Chin, F., Wynne, P., Prise, K., and Hartley, J. (2008) Biochem.
Pharmacol., 76, 19-27.
114.Rigu, G., and Robert, B. (2007) Curr. Med.
Chem., 14, 503-517.
115.Bergstralh, D., and Sekelsky, J. (2008)
Trends Genet., 24, 70-76.
116.Minko, I., Harbut, M., Kozekov, I., Kozekova,
A., Jakobs, P., Olson, S., Moses, R., Harris, T., Rizzo, C., and Lloyd,
R. (2008) J. Biol. Chem., 283, 17075-17082.
117.Woodgate, R. (1999) Genes Dev.,
13, 2191-2195.
118.Zietlow, L., Smith, L., Bessho, M., and Bessho,
T. (2009) Biochemistry, 48, 11817-11824.
119.De Winter, J., and Joenje, H. (2009) Mutat.
Res., 668, 11-19.
120.Alpi, A., Langevin, F., Mosedale, G.,
Machida, Y., Dutta, A., and Patel, K. (2007) Mol. Cell Biol.,
27, 8421-8430.
121.Kim, J., Kee, Y., Gurtan, A., and Andrea, A.
(2008) Blood, 111, 5215-5222.
122.Smeaton, M., Hlavin, E., Mason, T., Noronha,
A., Wilds, C., and Miller, P. (2008) Biochemistry, 47,
9920-9930.
123.Smeaton, M., Hlavin, E., Noronha, A., and
Murphy, S. (2009) Chem. Res. Toxicol., 22, 1285-1297.
124.Ljungman, M. (2009) Chem. Rev.,
109, 2929-2950.
125.Powell, S., and Bindra, R. (2009) DNA
Repair, 8, 1153-1165.
126.Zhu, Y., Hu, J., Hu, Y., and Liu, W. (2009)
Cancer Treatment Rev., 35, 590-596.
127.Mashkovsky, M. D. (1998) Officinal Drugs
(Manual for Physicians) [in Russian], Pt. 2, Meditsina, Moscow, pp.
288-290.
128.Brosh, R., and Bohr, V. (2007) Nucleic Acids
Res., 35, 7527-7544.
129.Feuerhahn, S., and Egly, J. (2008) Trends
Genet., 24, 467-474.
130.Nagy, Z., and Soutoglou, E. (2009) Trends
Cell Biol., 19, 617-629.
131.Ben-Yehoyada, M., Wang, L., Kozekov, I., Rizzo,
C., Gottesman, M., and Gautier, J. (2009) Molecular Cell,
35, 704-715.
132.Lippincott-Schwartz, J., Snapp, E., and
Kenworthy, A. (2001) Nat. Rev. Mol. Cell
Biol., 2, 444-456.
133.Shav-Tal, Y., Singer, R., and
Darzacq, X. (2004) Nat. Rev. Mol. Cell
Biol., 5, 855-861.
134.Piston, D., and Kremers, G. (2007) Trends
Biochem. Sci., 32, 407-414.
135.Huang, B., Bates, M., and Zhuang, X. (2009)
Annu. Rev. Biochem., 78, 993-1016.
136.Silva, L. (2005) Curr. Protein Pept.
Sci., 6, 387-395.
137.Moreno-Herrero, F., de Jager, M., Dekker, N., Kanaar, R., Wyman, C., and
Dekker, C. (2005) Nature, 437,
440-443.
138.Recuero-Checa, M., Dore, A., Arias-Palomo, E.,
Rivera-Calzada, A., Scheres, S., Maman, J., Pearl, L., and Llorca, O.
(2009) DNA Repair, 8, 1380-1389.
139.Oleinick, N., Morris, R., and Belichenko, I.
(2002) Photochem. Photobiol. Sci., 1, 1-21.
140.Salvesen, G., and Dixit, V. (1999) Proc.
Natl. Acad. Sci. USA, 96, 10964-10967.
141.Brozovic, A., Fritz, G., Christmann, M.,
Zisowsky, J., Jaehde, U., Osmak, M., and Kaina, B. (2004) Int. J.
Cancer, 112, 974-985.
142.Lane, D. (1992) Nature, 358,
15-16.
143.Staehelin, M. (1958) Biochim. Biophys.
Acta, 29, 410-417.
144.Staehelin, M. (1959) Biochim. Biophys.
Acta, 31, 448-454.
145.Axelrod, V., Feldman, Y., Chuguev, I., and
Bayev, A. (1969) Biochim. Biophys. Acta, 186, 33-45.
146.Juzumiene, D., Shapkina, T., Kirillov, S., and
Wollenzien, P. (2001) Methods, 25, 333-343.
147.Nanda, K., and Wollenzien, P. (2004)
Biochemistry, 43, 8923-8934.
148.Stewart, D., Thomas, S., Mayfield, C., and
Miller, D. (2001) Nucleic Acids Res., 29, 4052-4061.
149.Higuchi, M., Sakamoto, T., Kobori, A., and
Murakami, A. (2006) Nucleic Acids Symp. Ser., 50,
301-302.
150.Kean, J., Murakami, A., Blake, K., Cushman, C.,
and Miller, P. (1988) Biochemistry, 27, 9113-9121.
151.Thaden, J., and Miller, P. (1993) Bioconj.
Chem., 4, 386-394.
152.Kim, Y., and Hong, I. (2008) Bioorg. Med.
Chem. Lett., 18, 5054-5057.
153.Gantchev, T., Girouard, S., Dodd, D.,
Wojciechowski, F., Hudson, R., and Hunting, D. (2009)
Biochemistry, 48, 7032-7044.
154.Bombard, S., Kozelka, J., Favre, A., and
Chottard, J. (1998) Eur. J. Biochem., 252, 25-35.
155.Kazakov, S., and Altman, S. (1992) Proc.
Natl. Acad. Sci. USA, 89, 7939-7943.