REVIEW: Multiparametric Determination of Genes and Their Point Mutations for Identification of Beta-Lactamases
M. Yu. Rubtsova1*, M. M. Ulyashova1, T. T. Bachmann2, R. D. Schmid2, and A. M. Egorov1
1Chemical Faculty, Lomonosov Moscow State University, 119991 Moscow, Russia; E-mail: mrubtsova@gmail.com2Institute of Technical Biochemistry, University of Stuttgart, Stuttgart, Germany
* To whom correspondence should be addressed.
Received February 1, 2010; Revision received April 19, 2010
More than half of all currently used antibiotics belong to the beta-lactam group, but their clinical effectiveness is severely limited by antibiotic resistance of microorganisms that are the causative agents of infectious diseases. Several mechanisms for the resistance of Enterobacteriaceae have been established, but the main one is the enzymatic hydrolysis of the antibiotic by specific enzymes called beta-lactamases. Beta-lactamases represent a large group of genetically and functionally different enzymes of which extended-spectrum beta-lactamases (ESBLs) pose the greatest threat. Due to the plasmid localization of the encoded genes, the distribution of these enzymes among the pathogens increases every year. Among ESBLs the most widespread and clinically relevant are class A ESBLs of TEM, SHV, and CTX-M types. TEM and SHV type ESBLs are derived from penicillinases TEM-1, TEM-2, and SHV-1 and are characterized by several single amino acid substitutions. The extended spectrum of substrate specificity for CTX-M beta-lactamases is also associated with the emergence of single mutations in the coding genes. The present review describes various molecular-biological methods used to identify determinants of antibiotic resistance. Particular attention is given to the method of hybridization analysis on microarrays, which allows simultaneous multiparametric determination of many genes and point mutations in them. A separate chapter deals with the use of hybridization analysis on microarrays for genotyping of the major clinically significant ESBLs. Specificity of mutation detection by means of hybridization analysis with different detection techniques is compared.
KEY WORDS: antibiotic resistance, beta-lactamases, gene determination, single nucleotide polymorphism, hybridization analysis, microarraysDOI: 10.1134/S0006297910130080
Abbreviations: bp, base pair; dNTPs, deoxyribonucleoside triphosphates; ESBL, extended spectrum β-lactamase; HRP, horseradish peroxidase; PBP, penicillin binding proteins; PCR, polymerase chain reaction; RFLP, restriction fragment length polymorphism; RT-PCR, real time PCR; SNP, single nucleotide polymorphism; SSCP, single strand conformation polymorphism; Tm, melting temperature.
Microorganisms of the Enterobacteriaceae family are among the
most widespread pathogens of infectious diseases including hospital
infections. Beta-lactam antibiotics, which at the present time make up
more than half of antibiotics used in the world, are applied as
antibacterial drugs for treatment of these diseases. However, more and
more frequent cases of clinical inefficiency of drug therapy by this
group of antibiotics appear due to development of drug resistance among
the microorganisms. This biological phenomenon is called
“antibiotic resistance”. The problem of bacterial
resistance to antimicrobial preparations emerged practically
simultaneously with the beginning of use of antibiotics in the 1940s.
Already a year after the beginning of the use of penicillin, an enzyme
destroying this antibiotic, penicillinase, was detected in
Staphylococcus aureus. Data concerning microorganisms resistant
to certain antibiotic groups appeared in the 1970s, while already at
the end of that century microbial strains resistant to all known
antibiotics were described. Thus, the study of problems associated with
resistance to antibiotics as well as searching for ways of addressing
these problems became prominent for the world.
The main mechanism of emergence of resistance of the Enterobacteriaceae family to beta-lactam antibiotics is the appearance in their genes of chance mutations able to change the spectrum of bacterial enzyme activities. Bacterial enzymes able to cleave beta-lactam antibiotics are known as beta-lactamases. Beta-lactamases comprise a superfamily of genetically and functionally different enzymes joined by the ability to destroy the beta-lactam ring, which results in the loss by the antibiotic of its antimicrobial activity. Gram-negative bacteria of the Enterobacteriaceae family that are resistant to the third generation of cephalosporins are now the main problem in clinical practice. They produce the so-called extended spectrum beta-lactamases (ESBL) [1]. The first ESBLs, derivatives of the broad spectrum beta-lactamases, were discovered in the mid 1980s. Now over 300 such enzymes are described, and the number is constantly increasing.
ESBLs have been detected in all members of the Enterobacteriaceae family as well as in Pseudomonas aeruginosa and Acinetobacter baumannii. In most cases the ESBL genes are localized on plasmids, and this is the reason for extremely rapid spreading of resistant pathogens all over the world [2, 3]. This makes difficult correct diagnosis and choice of an adequate method of treatment, which increases the number of cases of inefficient therapy, lengthening and increasing the cost of therapy [4, 5]. Thus, the need for efficient laboratory diagnosis of ESBL production by Gram-negative microorganisms is obvious. The difficulty of detection of beta-lactamase in routine practice should be considered as an important feature of these enzymes. Standard clinical methods of determination of the nature of pathogens causing infectious diseases and revealing their resistance to antibiotics are based on phenotypic characteristics of microbial pathogens. At best, these methods of ESBL detection make it possible to determine that the enzyme is present, but they cannot give information about any particular enzyme. Besides, it is difficult to use these methods in analysis of simultaneous multiple resistance to several groups of antibiotics, whereas the number of such cases is increasing.
The geographical diffusion of β-lactamases and emergence of multiple resistances make necessary simultaneous determination of several genes and their mutations, i.e. multiparametric analysis. Molecular-biological methods for gene analysis are useful for solution of this kind of problem. Investigations in chemistry and biochemistry of nucleic acids during last decades not only resulted in revolutionary advances in biology, but they exerted a pronounced effect on development of new methods in biotechnology and gene engineering, including technologies that can be applied for multiparametric analysis of gene structure and detection of point mutations at the molecular level. Recently several reviews have been published that deal with the resistance of microorganisms to beta-lactam antibiotics and the spreading and properties of beta-lactamase family enzymes [6-9]. However, there are no reviews concerning molecular-biological methods for detection of beta-lactamases enabling simultaneous detection of several antibiotic resistance determinants. The goal of this review was to generalize all available data in this field. Separate sections deal with methods for multiparametric hybridization analysis for detection of several antibiotic resistance determinants and ESBL identification.
ANTIBIOTIC RESISTANCE CAUSED BY BETA-LACTAMASES
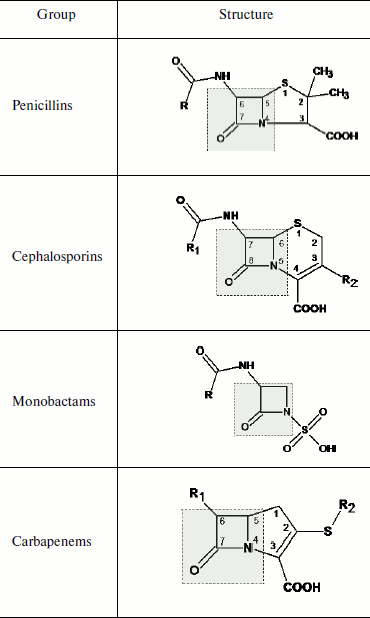
The term “antibiotic resistance” characterizes resistance of bacterial pathogens of infectious diseases to antibiotics and is a particular case of a more common event of “antimicrobial resistance”, resistance of pathogens of different nature infectious (bacteria, viruses, protozoa) to drug preparations. One of the most widely used classes of antibiotics is beta-lactam antibiotics (beta-lactams). Beta-lactams are polar hydrophilic compounds penetrating into bacterial cells through porin channels of the outer membrane [10]. Beta-lactams are divided into several groups based on their chemical structures having a beta-lactam ring as the component in common (Table 1). Cephalosporins are most often used for treatment of infections caused by Gram-positive and Gram-negative bacteria. Antibiotics of the penicillin group are most often used for treatment of infections caused by Gram-positive microbial pathogens. The use of carbapenems as wide spectrum antibiotics has recently increased.
Table 1. Structure of the main groups of
beta-lactam antibiotics (the beta-lactam ring is designated by the
dotted line)
The action mechanism of beta-lactam includes binding to penicillin-binding proteins (PBP) – trans- and carboxypeptidases involved in formation of peptidoglycan chains of the inner bacterial membrane. The interaction of PBP with beta-lactam antibiotics results in disturbance of peptidoglycan synthesis, cessation of cell division, and cell death. The binding of the antibiotic to PBP is based on the affinity of the beta-lactam structure to that of the PBP active site. Owing to this, the presence of a beta-lactam ring is necessary for the antibacterial activity of the antibiotic. Upon the interaction of the beta-lactam with PBP, an acyl-enzyme complex is formed and the C–N bond in the four-membered cycle of the beta-lactam ring is broken.
Point mutations emerged during evolution of some PBP, which resulted in appearance of beta-lactamases able to hydrolyze the lactam ring of antibiotics. Comparative analysis of all PBP and beta-lactamase sequences supports a hypothesis concerning the existence of a common precursor of these proteins [11].
The following mechanisms of emergence of bacterial resistance to beta-lactam antibiotics have been revealed: (i) synthesis of beta-lactamases destroying these antibiotics [11-13]; (ii) lowering the bacterial outer membrane permeability due to the loss of or decrease in porin expression [14]; (iii) change in PBP structure [15]; (iv) active antibiotic extrusion from the microbial cell (efflux system) [16, 17]. Beta-lactamase synthesis is considered as the main mechanism providing resistance of clinically important strains of Gram-negative bacteria to beta-lactam antibiotics [6, 18, 19]. Genetic mutations resulting in replacement of just a few amino acids in a protein sequence alter the enzyme structure resulting in significant broadening of the antibiotic spectrum subjected to hydrolysis [20, 21]. Mutations can emerge rather quickly; there are cases when microorganisms became resistant to antibiotics during the course of treatment.
Resistance can be inborn or acquired. The real natural resistance is characterized by the absence from microorganisms of a target for revealing antibiotic activity. Some microorganisms are able to produce chromosome-encoded beta-lactamases, for example, Klebsiella pneumoniae produces beta-lactamase SHV-1 and Enterobacter cloacae, Enterobacter aerogenes, Citrobacter freundii, Serrata spp., and Pseudomonas aeruginosa produce C class beta-lactamases. The ability of individual bacterial strains to retain viability at antibiotic concentrations inhibiting the bulk of the microbial population is considered as acquired or secondary resistance. Situations are possible when most of the population exhibits the acquired resistance. Formation of acquired resistance is always due to acquiring of new genetic information or to alteration of the native gene expression level. Most often secondary resistance determinants are acquired together with mobile genetic elements. The resistance of Enterobacteriaceae family members to beta-lactam antibiotics is acquired resistance. The ESBL genes are transferred by different mobile genetic elements like plasmids, transposons, IS-elements, class 1 integrons, and the ISCR1 element containing integrons [22, 23]. The variety of mechanisms of genetic transfer contributes to rapid spreading of these genes. From the point of view of diagnosis, just detection of emergence and transfer of acquired resistance comprises the main problem because it is impossible to forecast its presence in the infectious pathogen.
Classification of Beta-Lactamases
Over 500 different beta-lactamases have been described, and this number is rapidly increasing each year. This enzyme family itself can be called a superfamily because it joins several huge groups or subfamilies differing in enzymatic properties. They are united on the basis of their ability to hydrolyze beta-lactam antibiotics, whereas differences include enzyme origin, amino acid sequences, spectrum of substrate specificity, catalytic parameters, and sensitivity to inhibitors. Numerous attempts have been made to classify these enzymes. The first methods of classification were based on comparison of their functional activity, namely on the ability of the enzyme to cleave different classes of beta-lactam compounds (penicillins, cephalosporins, monobactams, carbapenems), sensitivity to inhibitors, and differences in biochemical parameters [24-26]. As a result, a scheme of beta-lactamase classification by several functional groups was designed [27]. A different beta-lactamase classification, molecular classification based on a collection of common structural features, was proposed as well [28]. Four molecular classes are distinguished according to the level of homology, the presence of conservative regions in the enzyme structure, and structure of the active center. Each classification has its merits and shortcomings, but none is exhaustive; therefore, they are often used together. Data on beta-lactamase classification using division into molecular classes and into functional groups are given in Table 2.
Table 2. Beta-lactamase classification
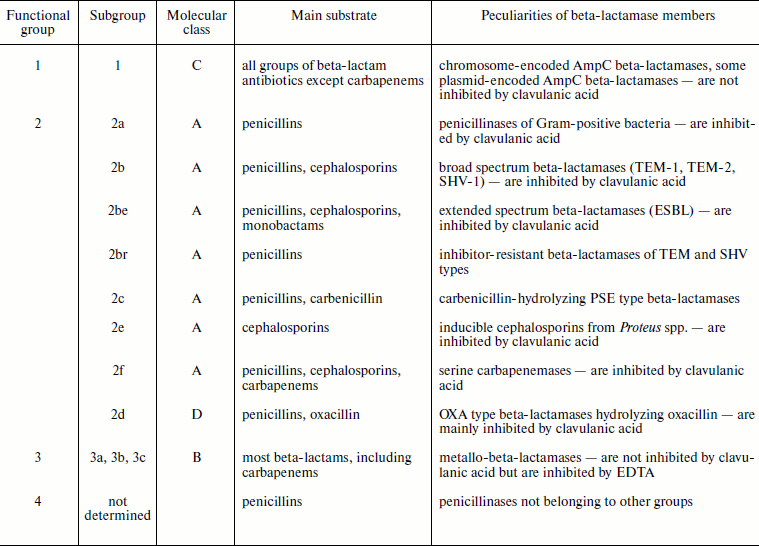
The first functional group includes enzymes of C molecular class (AmpC type beta-lactamases) that can be encoded both by chromosomes and plasmids [30]. A specific feature of this group is their higher activity towards cephalosporins compared to penicillins, and these enzymes also exhibit low sensitivity to inhibitors.
The second functional group, which is most comprehensive and diverse, includes beta-lactamases of A and D classes. Genes of enzymes of this group are incorporated into plasmids, and therefore efficiency of their transfer among different strains and, as a result, their spreading rates are very high. Group 2a includes beta-lactamases of Gram-positive bacteria Staphylococcus spp. and Bacillus spp. These enzymes are the most efficient against penicillins (with the exception of oxacillin and its analogs). The best-known members of group 2b are beta-lactamases TEM-1, TEM-2, and SHV-1. These enzymes also most efficiently hydrolyze penicillins (except ureidopenicillins). Subgroup 2be includes so-called extended spectrum beta-lactamases (ESBL) that are mutants of TEM-1 and SHV-1 enzymes and are able to cleave efficiently both penicillins and cephalosporins of generations I-IV. The same group includes numerous beta-lactamases of CTX-M type efficiently hydrolyzing cefotaxime. A separate group 2br includes TEM and SHV type inhibitor-resistant mutants. Group 2c includes PSE beta-lactamases of Gram-negative bacteria able to hydrolyze carbenicillin. Group 2d includes OXA type beta-lactamases that are members of a separate molecular class D. Some of these enzymes have the ESBL phenotype. They appear mainly in P. aeruginosa and A. baumannii strains. It is assumed that mutation in position 167 is the key one. Genes of group 2e cefalosporinases are localized on chromosomes. They can be under control of both inducible (P. vulgaris, C. diversus) and constitutive (Bacteroides spp., Stenotrophomonas maltophilia) promoters. Group 2f is formed by carbapenemases, i.e. by beta-lactamases able to hydrolyze carbapenems.
The third functional group includes zinc-containing beta-lactamases. Owing to this they are called metallo-beta-lactamases [31, 32]. According to molecular classification, they belong to class B. Enzymes of this group efficiently cleave all types of beta-lactam antibiotics including carbapenems. They are resistant to inhibitors of clavulanic acid, sulbactam, and tazobactam, but in culture they can be inhibited by chelating agents like EDTA.
All beta-lactamases can be divided by mechanism of action into serine (containing serine in the enzyme active center) and metalloenzymes. Serine enzymes include beta-lactamases of molecular classes A, C, and D, whereas class B beta-lactamases are metalloenzymes.
Most attention has been attracted recently to the problem of plasmid-encoded ESBL [3, 33]. The term ESBL was initially used to designate TEM and SHV type beta-lactamase mutants of 2be functional subgroup able to hydrolyze oxyiminocephalosporins. Later the meaning of this term was significantly extended. The following enzymes were included into the ESBL group: enzymes with similar profile of substrate specificity but structurally different from CTX-M and VEB type enzymes like TEM and SHV type mutants with border activity towards cephalosporins such as TEM-12; beta-lactamases with different resistance level, not included into functional subgroup 2be, such as OXA type beta-lactamases and AmpC beta-lactamase mutants with increased enzymic activity towards cefepime [34].
Dissemination of Class A Extended Spectrum Beta-Lactamases
Dissemination of beta-lactamases, including ESBL, is actively studied in many countries. The number of publications concerning ESBL abundance in separate clinics, regions, and countries constantly increases. Data obtained in Europe [35-37], North [38] and South America [39], Canada [40], Asian region countries [41, 42], and in the Near East [43] are indicative of increased resistance to beta-lactam antibiotics among pathogens of nosocomial and outpatient infections, and of significance especially ESBL abundance, which is a serious problem for public health. Comparative analysis of these data indicates that the ESBL fraction among E. coli and Kl. pneumoniae pathogens in countries of East and South Europe, South America, and Asia has already reached 30% and more; in countries of North Europe, North America, and Canada it is on average significantly lower and does not exceed 10-15%. Although TEM and SHV type beta-lactamases were mainly detected in Europe in the 1990s, the situation changed sharply in the beginning of the XXI century and at the present time the CTX-M type is prevalent in Europe, Asia, and South America [44-49]. The increase in the CTX-M type beta-lactamase fraction is exponential, and it is already called pandemic [50]. It is supposed that such high rate of spreading is due both to emergence of new mutants and efficient gene transfer within plasmids and mobile genetic elements. In the USA the situation is not changed, the SHV and TEM type beta-lactamases are the most widespread, and the CTX-M type enzymes were detected only in patients infected outside the country [35, 51].
Antibiotic resistance, including that to beta-lactam antibiotics, is also investigated in Russia, and results have been published in a number of works [52-55]. It is noted that spreading of beta-lactam resistance among pathogens of the Enterobacteriaceae family reaches on average 50-60% for nosocomial infections, and it has already become a national problem. Increase in the number of strains simultaneously containing genes of two, three, and even four beta-lactamase types is found. The number of such strains already reaches 30%. The ratio of ESBL types revealed in Russia correlates with data on different ESBL type spreading in Eastern Europe: the CTX-M beta-lactamases are significantly prevalent (about 80%), and beta-lactamases of SHV type are rather common. Enzymes of the CTX-M-1 subcluster are the most often found CTX-M type genes. In recent years significant increase in the CTX-M-9 subcluster beta-lactamase frequency has been recorded [52, 56].
Variety of the Main Types of Class A Extended Spectrum Beta-Lactamases
On the basis of available data on molecular mechanisms of bacterial resistance to beta-lactam antibiotics and abundance of different beta-lactamase molecular classes and functional groups, special attention of researchers is given to investigation of three main ESBL types: beta-lactamases of TEM, SHV, and CTX-M types. All contain serine in the enzyme active center and have molecular mass about 29 kDa. The main structural features of these enzymes will be briefly considered below.
TEM type beta-lactamases. The first characterized enzyme of this group was TEM-1 penicillinase detected in E. coli cells isolated from blood of an infected patient [57]. Later TEM-1-containing plasmids were found in strains of different members of the Enterobacteriaceae family, in strains of Pseudomonas aeruginosa, Haemophilus influenzae, Neisseria gonorrhoeae, etc. Several years later the TEM-2 enzyme was isolated, which differs by a single amino acid mutation Gln39Lys with the same profile of substrate specificity [58]. However, already the next detected enzyme TEM-3 was characterized by a second mutation Glu104Lys in addition to the already described mutation in position 39. This enzyme was able to hydrolyze cephalosporins, and thus it had a wider spectrum of substrates [58]. Now over 170 members of this type of enzymes have been described and the beta-lactamase database (http://www.lahey.org/studies) is steadily growing. The primary structure of TEM-1 includes 288 amino acid residues. All described derivatives of TEM type beta-lactamases are variants of TEM-1 beta-lactamase and differ from original enzyme by single amino acid substitutions (from one to seven). Mutations are found in 60 positions, but the frequency of mutations in each position is very different. Ninety TEM type enzymes are ESBL. Figure 1 shows the distribution of known mutations in amino acid sequence of TEM-1 beta-lactamase, which is based on information available in the GenBank international database. Most frequent are mutations in positions 21, 39, 69, 104, 164, 182, 238, 240, 244, 265, and 275. Among them mutations in positions 104, 164, 238, and 240 are keys for extension of substrate specificity. Introduction of substitutions Glu104Lys, Arg164Ser(His), and Glu240Lys into TEM-1 results in change in total charge of the protein globule. Mutation Gly238Ser results in appearance of enzymes able to destroy with equal efficiency cefotaxime and ceftazidime, while enzymes with mutation Arg164Ser are more active towards ceftazidime and less efficient against cefotaxime [6]. There are mutations resulting in appearance of inhibitor-resistant enzymes (IRT, inhibition resistance phenotype, subgroup 2br) [60]. The following positions of mutations define resistance to inhibitors: 69, 244, 275, and 276 [61]. Combination of the first and second type mutations makes it possible to obtain TEM variants exhibiting both types of resistance, ESBL + IRT. Six pairs of beta-lactamases (TEM-1 and TEM-98, TEM-3 and TEM-14, TEM-10 and TEM-23, TEM-30 and TEM-99, TEM-34 and TEM-97, TEM-63 and TEM-64) are structurally identical. The use of random and directed mutagenesis [62] as well as of insertional mutagenesis [63] made it possible to obtain artificially synthesized mutants with extended activity spectrum and to even forecast the emergence of new subtypes of TEM beta-lactamases before their clinical isolation.
SHV type beta-lactamases. Enzymes of this type emerged second after TEM type beta lactamases. The peculiarity of this type of beta-lactamases produced by Klebsiella pneumoniae is the presence of SHV-1 beta-lactamase encoded by chromosomes [1]. In this case the SHV-1 enzyme, encoded by the gene localized on plasmids, was widespread. The SHV-2 mutant differing by a single mutation Gly238Ser was the first described ESBL [64].Fig. 1. Positions and types of amino acid substitutions in TEM type beta-lactamases described before January 2010.

The database http://www.lahey.org/studies/ contains information about 126 mutants of SHV type beta-lactamases. They are derivatives of SHV-1 enzyme and differ from the latter not only by the presence of point mutations, but by deletions (in SHV-9 and SHV-10) or inserts (in SHV-16) as well. Figure 2 shows the distribution of mutations recently detected in amino acid sequence of SHV-1 beta-lactamase using information from the GenBank database. Mutations are found in 79 positions; a deletion in position 54 in SHV-9 and an insert 163DRWET167 in SHV-16 are also described. Depending on the substitution type, like in the case of TEM enzymes, mutant SHV differ by the profile of substrate specificity towards cephalosporins. Most frequent are mutations in positions 35, 238, and 240 and they are considered as the key ones for changes in the substrate specificity profile. Beta-lactamase SHV-10 with IRT resistance phenotype is described. It differs from ESBL SHV-9 by a single amino acid substitution, Ser130Gly, but it is not an ESBL itself.
CTX-M type beta-lactamases. Beta-lactamases of CTX-M type received their name because they more efficiently hydrolyze cefotaxime compared to ceftazidime. The first enzyme of this type was isolated in 1989 from an E. coli strain [65]. Later it was called CTX-M-1. Genes of this enzyme group have plasmid localization. Over 90 enzymes of this type are known. Among the A class ESBL, the CTX-M group of beta-lactamases is the most heterogeneous concerning amino acid sequence and, respectively, the encoding gene structures. The CTX-M type beta-lactamases are now divided into five subclusters [66]. Each subcluster consists of the main enzyme (CTX-M-1, CTX-M-2, CTX-M-8, CTX-M-9, and CTX-M-25) and its mutants differing by one to several mutations. Distribution of CTX-M type enzymes by subclusters and mutations described for each subcluster is shown in Fig. 3. The homology between CTX-M type beta-lactamases and other types of A class enzymes is marginally pronounced (below 40%) [67]. Much higher homology (over 70%) is observed for the chromosome-encoded enzymes from Klebsiella oxytoca, Citrobacter diversus, Proteus vulgaris, and Serratia fonticola [68]. This suggests that the plasmid-encoded enzymes of CTX-M type originated from enzymes whose genes are incorporated in chromosomes. The difference between CTX-M type beta lactamases on one side and the TEM and SHV type enzymes on the other side concerning their substrate specificity is that all of CTX-M type lactamases hydrolyze cephalosporins, i.e. they are ESBL. Differences between CTX-M type enzymes concern changes in catalytic activity towards different cephalosporins such as cefotaxime, ceftazidime, and cefepime. It was shown that there are also key mutations for enzymes of this type, such as those in positions 167 and 240, leading to changes in the substrate specificity profile. It is noted that the CTX-M type beta-lactamases are often revealed in infections in outpatients [19].Fig. 2. Positions and types of amino acid substitutions in SHV type beta-lactamases described before January 2010.

Fig. 3. Division of CTX-M type beta-lactamases into subclusters and positions of amino acid substitutions in them described before January 2010.

Problems of Laboratory Diagnosis of ESBL
The difficulty of detection of ESBL in clinical practice should be considered as their important feature. Standard microbiological methods of determination of infectious pathogen nature and resistance to antibiotics are based on phenotypic characteristic of microbial pathogens. They are based on estimation of the antibiotic minimal inhibitory concentration (MIC) necessary for inhibition of cell growth in culture as tested by different methods using panels of antibiotics and their combinations [69, 70]. According to the antibiotic MIC level, the National Committee for Clinical Laboratory Standards (NCCLS) (USA) established criteria of sensitivity of Gram-negative microorganisms to the third generation cephalosporins only for E. coli and Klebsiella spp. However, ESBL production is described in practically all members of the Enterobacteriaceae family and in a number of other Gram-negative microorganisms. These methods require rather much time, and data concerning antibiotic sensitivity of certain strains can also be interpreted ambiguously. Neither traditional microbiological method based on estimation of microbial phenotype provides for detection of ESBL in all strains. The “inoculum effect” or a sharp increase in the antibiotic MIC upon increase in the pathogen inoculation dose was found, i.e. the determined level of sensitivity can depend on the cell concentration in culture. Resistance determination is significantly complicated in the presence of several microbial resistance determinants, and the number of such cases constantly increases. For example, production by E. coli strains of AmpC beta-lactamases (C class) concealed the presence of ESBL, and they were not detected when E-test (5 samples of 7) or a Phoenix automatic analyzer (3 samples of 7) was used [71]. Pseudo-positive results showing the presence of ESBL in negative samples were also obtained in the same work, and most such kind of results (9 false results for 19 samples) was obtained using diffusion on discs. At best, traditional microbiological methods of ESBL detection make it possible to estimate the fact of the presence of an enzyme, but they cannot give information concerning the presence of a certain enzyme. Therefore, it becomes obvious that the use of phenotypic methods for testing sensitivity of bacterial pathogens is not enough for proper understanding of the nature and properties of infectious agent [72, 73]. The necessity of choosing adequate methods of diagnosis for correct determination of resistance type is actively discussed [74]. This information can be obtained using molecular-biological methods of gene analysis.
MOLECULAR-BIOLOGICAL METHODS FOR IDENTIFICATION OF ANTIBIOTIC
RESISTANCE DETERMINANTS
Analysis of the above-described data shows that study of the base sequence of beta-lactamase-encoding genes is necessary for determination of resistance of Gram-negative microorganisms to beta-lactam antibiotics and for identification of gene types and existing mutations. Point mutations in genes (single nucleotide polymorphism (SNP)) are single nucleotide positions in genomic DNA for which in a certain population there are different variants of sequences (alleles) [75]. The detection of SNPs requires either identification of complete base sequence of the gene or structure of its fragments. All presently available molecular-biological methods of base sequence analysis are based on amplification of genes from biological material. Polymerase chain reaction (PCR) is most often used for this. It is necessary to use additional methods for determination of complete or partial structure of DNA synthesized during the reaction. The “gold standard” of gene molecular typing is sequencing or determination of primary structure, i.e. determination of base sequence in the gene strand. Sequencing can be used to identify both known and new gene variants. The main disadvantage of this method is its labor consumption and relatively high cost. The use of sequencing is difficult when it is necessary to detect several genes in the same sample, especially when these genes belong to the same genetic group where distinctions include separate mutations.
Multiplex PCR
For simultaneous amplification of several genes multiplex (multiprimer) PCR or the reaction of co-amplification of several DNA templates in the same reaction medium using several pairs of primers are used. Design of primers and detection of probability of unspecific fragment amplification are very important for increase in specificity of the method. There are now multiplex PCR techniques for simultaneous amplification of gene fragments of three main ESBL types (TEM, SHV, CTX-M) [76]. The method can assign the enzyme gene to a certain beta-lactamase type. More detailed gene differentiation by multiplex PCR is possible at the subcluster level for CTX-M type ESBL, but in this case analysis is carried out only for the same type enzymes (in this case CTX-M) [77, 78]. The multiplex PCR technique becomes increasingly important in combination with different methods providing more detailed structural characteristics of the gene. Thus, the method of multiplex PCR was elaborated for simultaneous amplification of CTX-M type beta-lactamases, but owing to amplification of genes of different subclusters in the form of equal size fragments, further investigation of their structure is necessary for the assignment of gene type [79].
Real-Time Polymerase Chain Reaction
Real-time PCR (RT-PCR) is an actively developing technology. Different types of fluorescent dyes are used as labels. To determine the result of the labeled oligonucleotide hybridization, either donor fluorophore energy transfer to an acceptor with a different emission spectrum or energy transfer to a fluorescence quencher is used.TaqMan technology is based on a method of destructible probes. Oligonucleotide is modified by a fluorophore and fluorescence quencher. In the absence of a target the fluorophore and quencher are drawn together and fluorescence is inhibited. In the presence of a specific template the probe is hybridized to an amplicon, which results in its destruction and emergence of fluorescence due to 5′ exonuclease activity of Taq polymerase (the ability to hydrolyze DNA sequence in the 5′→3′ direction). The use of this approach was described for identification of a subcluster of CTX-M type ESBL genes [80]. The method is characterized by high rate and good productivity compared to standard PCR techniques.
A different variant of RT-PCR is the method of adjacent probes based on use of two oligonucleotide probes hybridized to template DNA in the immediate vicinity of each other. One probe includes a donor fluorophore and the other contains an acceptor fluorophore. Probe hybridization with the template brings the fluorophores together and energy transfer from donor to acceptor (fluorescence resonance energy transfer) occurs. This approach became widely adapted to SNP detection. Thus, its abilities were demonstrated in detection of key mutations in the SHV, TEM, and CTX-M type ESBLs [81-84]. The method is limited by the possibility of simultaneous detection of only a few SNPs due to the limited set of fluorescent labels.
Methods for Analysis of Amplification Products Used for Gene Identification
Restriction fragment length polymorphism (RFLP) method. This method is used for detection of polymorphous DNA regions. To do this, the amplified gene is cleaved by an appropriate restriction endonuclease and changes in restriction sites are determined [85]. For each gene type its own set of restriction products is obtained and analyzed by electrophoresis. The method indicates the presence of variations in base sequence but requires further analysis of fragments or comparison with a separation profile of known gene variants. This approach was applied to identification of SHV type beta-lactamases in Pseudomonas aeruginosa strains by the presence of key mutations in positions 35, 238, and 240 [86]. The method is simple enough in practice and can be used for screening a large number of samples; its limitation is the possibility of detection of only mutations localized in restriction sites as well as complications in revealing numerous mutations.
Method of analysis of conformational distinctions between single-stranded DNA fragments. This method (SSCP, single strand conformation polymorphism) was elaborated for SNP analysis and is based on variations of single-stranded DNA conformation after denaturation. This is demonstrated by changes in electrophoretic mobility. The SSCP shows the presence of changes in nucleotide sequence, but it requires further analysis of fragments or comparison with the separation profile of known gene variants. The method was used for analysis of TEM and SHV type ESBL [87, 88].
Method of time-of-flight mass spectrometry. An alternative to existing methods for detection of polymorphous DNA regions is differential sequencing by time-of-flight mass spectrometry. The ionized DNA molecules are separated from the substrate using MALDI (matrix-assisted laser desorption/ionization) technique, accelerated in an electric field, and sent to a detector via a vacuum chamber. The registered time of motion is inversely proportional to the speed of the molecule, which in turn is in direct proportion to the mass-to-charge ratio of the molecule. The mass-to-charge ratio is unique for each molecule and is defined exclusively by its base sequence; therefore, the sensitivity of the method is very high. The time spent for analysis of each sample by this method is only a few seconds. The possibility of use of this approach for rapid detection of TEM type ESBL by key mutations in positions 104, 164, and 238 was shown [89]. The sensitivity of the method is high, and 50 bacterial cells in a sample are enough for determination. Determination of nucleotide type in mutation position and, respectively, recognition of genes with various substitutions is possible. The method is characterized by high productivity and the possibility of simultaneous analysis of several samples. Among clear advantages of the method is the possibility to detect new mutations that have not been previously described.
High performance liquid chromatography method. The method of high performance liquid chromatography (HPLC) under denaturing conditions was elaborated for analysis of the product formed during amplification, and it was used for detection of CTX-M type ESBL [90]. Genes of CTX-M type beta-lactamases were amplified using multiplex PCR [78], and in this case the fragments of genes belonging to different subclusters differed in size. Then amplified fragments were mixed with an equal amount of DNA of control strains producing CTX-M type beta-lactamases that were characterized by gene typing. Mixtures were quickly heated for denaturation and then cooled at a certain rate for formation of homo- and hetero-DNA duplexes. The duplexes were analyzed by HPLC in an acetonitrile gradient. The temperature was optimized to make possible distinguishing 13 CTX-M beta-lactamase gene variants by the shape of chromatographic peaks. To achieve maximal sensitivity and specificity, it was necessary to analyze duplexes for CTX-M-1 subcluster at two temperatures, 64 and 65°C, for subclusters CTX-M-2 and 25/26 at 64°C, and for CTX-M-9 at 67°C. The method is characterized by high productivity (100 samples in 6 h), and its cost is significantly lower than that for sequencing. The authors of this work also suppose that it will be possible to follow the emergence of new mutations by appearance of anomalous chromatographic peaks. A limitation of the method is that it determines only some mutations.
Pyrosequencing method. Pyrosequencing has been proposed for analysis of mutations. The method is based on detection of pyrophosphate cleavage during DNA synthesis. Pyrophosphate is the first substrate in a cascade of enzymatic reactions resulting in emission of light. The intensity of the emitted light is in proportion with the number of incorporated nucleotides. Detection of emitted light intensity in real time makes possible rapid identification of short gene fragments up to 100 bp. Pyrosequencing was used for SNP detection in genes of CTX-M type ESBL [91] and of GES type ESBL [92]. Determination of genetic subcluster for CTX-M type beta-lactamases was possible during analysis of 13 bp long DNA fragment. Analysis of another 16 bp gene region identified separate subtypes of CTX-M beta-lactamases. The method is characterized by the very high rate of analysis; total determination time, including PCR, is 3 h.
Hybridization. Hybridization on insoluble supports proposed by E. Southern [93] is used for specific detection of individual DNA fragments. This method is often used after DNA fragmentation like that using RFLP, after which electrophoretically separated fragments are transferred onto porous membrane by immunoblotting. Hybridization (complementary interaction of polynucleotide strand under study with labeled oligonucleotide) is used for identification. In this case the labeled oligonucleotide is the “probe”. The label activity is determined in DNA duplexes on the support after washing. Hybridization analysis was used for detection of ESBL and AmpC beta-lactamases upon screening Salmonella enterica and Pseudomonas aeruginosa strains [94, 95]. The use of hybridization in amplified DNA detection significantly increases the specificity of gene fragment determination. Depending on the type of the label introduced into the oligonucleotide probe (fluorescent dyes, haptens), different ways of detection of hybridization complex are used (direct measurement of fluorescence, detection using specific antibody conjugates with enzyme). Approaches developed for gene detection by hybridization on solid phase served as a prototype for design of hybridization analysis on microarrays.
Method of hybridization analysis on microarrays. The first report concerning the technology of biological microarrays for investigation of gene expression level appeared in 1995 [96]. During the past 10 years this technology has significantly developed and gains more areas for its application. A DNA-microarray presents a set of spots distributed in a strictly determined order on a glass or polymer surface of a small size (1-10 cm2); each spot contains single-stranded oligonucleotides with a particular base sequence. The number of such spots and thus the number of different base sequences can reach 1·106/cm2, their length varying from 15 to 1000 nucleotides. The method of hybridization analysis on microarrays is based on amplification of analyzed DNA region with simultaneous introduction of label and following hybridization of the analyzed base sequence on the microchip surface with oligonucleotides or DNA sequences immobilized in a definite order [97]. The difference between hybridization analysis on microarrays and classical hybridization is that oligonucleotides called probes are immobilized on the support and the label is introduced into the analyzed DNA. The result of hybridization of the labeled DNA sample with immobilized oligonucleotides (probes) is determined by the label activity on the support using special devices – high resolution scanners. Mainly passive hybridization is used in DNA microarrays, i.e. the interaction of the DNA target with the immobilized oligonucleotides is a random process and depends on several factors such as the length of DNA probe, chemical composition of labeled DNA target, hybridization temperature, composition of hybridization mixture, and the type of introduced label.
Different types of labels for hybridization analysis on microarrays are used. Radioactive label, usually P33 incorporated into the phosphate residue of the nucleotide introduced into the DNA strand during its synthesis, was proposed for labeling of the analyzed DNA [93]. In this case each DNA molecule contains several atoms of radioactive phosphorus, the maximal number of which can be equal to the number of nucleotides. The presence of several labels within the analyzed DNA increases sensitivity of the hybridization assay. The use of isotope technology in microarrays makes it possible to achieve high sensitivity, although there are some limitations. First of all, they are connected with insufficiently high resolution of the film used as the detector as well as with difficulty of work with radioactive materials.
The fluorescent label is now the most widespread in hybridization analysis on DNA microarrays. In this case, dyes directly joined to the sequence of interest can be used as generators of the fluorescent signal [98]. The fluorescent label is usually introduced into DNA during PCR as a component of primers or fluorescent dye conjugates with dCTP or dUTP [99]. Methods of hybridization analysis using nanoparticles of gold with following visual [100] or electrochemical detection [101] have been described.
So-called “indirect” labeling with biotin was proposed, and biotin is revealed by a conjugate of streptavidin with fluorescent dye [102] or with horseradish peroxidase [103] or alkaline phosphatase enzymes [104].
Hybridization analysis on microarrays has significant advantages over traditional molecular-biological methods, which include the possibility of multiparametric determination, i.e. determination of numerous parameters (from tens to hundreds and even thousands of genes and mutations in them) in a single sample due to carrying out numerous parallel reactions on the microchip surface under identical conditions.
The microchip technology is a principally new level of laboratory investigations making possible hundreds and thousands of parallel reactions. Examples of the application of this method for identification and typing of different genes responsible for emergence of antibiotic resistance among microorganisms will be considered in separate sections.
METHODS OF HYBRIDIZATION ANALYSIS ON MICROARRAYS FOR
IDENTIFICATION OF SEVERAL ANTIBIOTIC RESISTANCE DETERMINANTS
Microarrays for Gene Identification
Potentials of hybridization analysis on microarrays providing for multiparametric gene determination were demonstrated upon revealing genes responsible for development of antibiotic resistance, including that to beta-lactam antibiotics.A method was proposed for determination of nine beta-lactamase types (TEM, SHV, PSE, OXA, FOX, MEN, SMY, OXY, and AmpC) on a glass microchip with fluorescent detection [105]. Specific probes of about 1000 bp were amplified by PCR using specific primers. The studied DNA was amplified by multiplex PCR with introduction of fluorescent label. Hybridization was carried out at 70°C for 2 h. The possibility of determination of resistance genes using just a single bacterium as a PCR template was shown. Thus, it is supposed that the sensitivity of the method makes the analysis possible without preliminary culturing of clinical samples. The possibility of detection of two type resistance genes (OXY and PSE) in the same sample was shown experimentally.
A method was developed for determination of 23 resistance genes and 25 virulence genes in Salmonella spp. and E. coli strains [106]. Sequences up to 400-800 bp long, obtained by PCR, were immobilized on a chip of membrane support. Digoxygenin, revealed by the anti-digoxygenin antibody conjugate with alkaline phosphatase, was used as the DNA label. The enzyme on the support was detected colorimetrically. Beta-lactamases of TEM, CMY, and PSE types were revealed among different antibiotic resistance determinants.
A method was proposed for typing St. aureus spp., E. coli spp., and Ps. aeruginosa spp. strains with simultaneous identification of resistance determinants of these bacteria to several groups of antibiotics [107]. About 120 specific probes of 200-800 bp were amplified from recombinant plasmids and applied onto a glass microchip. Probes for pathogen type identification were tested along with probes homologous to regions of genes encoding virulence factors and antibiotic resistance determinants in St. aureus strains to oxacillin-methicillin (mecA), gentamycin (aacA-aphD), erythromycin (ermA), and penicillin (blaZ), and in E. coli strains to penicillin (blaTEM-106) and aminoglycosides (aacC2). Blood samples were cultivated on special media without further DNA amplification. Total DNA labeled by several types of fluorescent dyes was used for analysis. Hybridization time was 18 h at 60°C. Although DNA isolated from blood of infected patient contains bacterial and human DNA mixture, high specificity of identification was achieved. There was 100% coincidence between results of identification on the microchip and phenotyping data for St. aureus spp. resistant to penicillin, oxacillin, erythromycin, and gentamycin. Among advantages of the method the following can be mentioned: i) recombinant plasmids were used for synthesis of the probe panel, which makes easy extension of probe amount and composition; ii) the use of total DNA for analysis; iii) the absence of an amplification stage of the analyzed DNA; iv) preliminary pathogen typing is not necessary. From the point of view of ESBL identification, the use of sufficiently long probes is a disadvantage because it makes it impossible to use this approach for detection of point mutations in genes.
Hybridization analysis on microarrays with colorimetric detection was elaborated for identification of 90 antibiotic resistance genes in Gram-positive microorganisms [108]. For identification of 81 genes, two probes specific for each homologous region of the studied genes were chosen, while for nine genes one probe was chosen for each. Altogether, 137 oligonucleotides of 26-33 bp in length were immobilized on a microchip, and specificity of gene detection was studied for 125 probes. Biotin was used as the label. It was introduced into the DNA during PCR and then was detected on the microchip surface by incubation with streptavidin–peroxidase conjugate and following detection of enzyme activity. The required specificity of analysis was achieved upon hybridization for 1 h at 60°C.
In this work advantages of hybridization on microarrays, which exhibits higher specificity than PCR, are discussed. The use of oligonucleotide probes also has advantages over longer PCR products due both to higher specificity and reduced time of analysis. The possibility of automating different stages of the method undoubtedly is among its positive characteristics: a microchip is placed into a special Array Tube (technology of the Clondiag company), which makes significantly simpler the performance of stages of hybridization, washing, and detection. The impossibility of specific detection of point mutations due to rather long oligonucleotide probes can be considered as a drawback of the method.
The same technology was used in a microarray elaborated for 47 genes responsible for development of resistance to five groups of antibiotics (beta-lactams, aminoglycosides, trimethoprim, sulfonamides, tetracycline, chloramphenicol, and quinolones) in Gram-negative bacteria [109]. The microarray was tested using 50 clinical strains of E. coli and 37 clinical strains of Salmonella spp. In 99% of the cases the results of resistance determinant detection by the microarray technique coincided with those of PCR carried out in the form of several multiplex reactions. In E. coli strains many more resistance genes were found (13 resistance determinants were revealed in over 30% of the samples) compared to Salmonella spp. (seven resistance determinants were found). As to beta-lactamase identification, the method only allowed determination of beta-lactamase TEM and SHV gene type. Genes of CTX-M and OXA type ESBL were revealed only at the subcluster level (CTX-M-1, CTX-M-2, CTX-M-9; OXA-1, OXA-2, OXA-7, and OXA-9).
A microarray was designed for simultaneous determination of 65 genes responsible for development of resistance to macrolides in several types of Gram-negative and Gram-positive bacteria [110]. A panel of 100 probes of 40-60 bases in length was used for gene typing. The probes were applied onto a glass microchip. Genomic DNA was isolated from bacterial cultures, fluorescent label was introduced into it, and it was hybridized on the microchip. Good specificity of the method was demonstrated, and it was possible to reveal for the first time the presence of one of the msr(SA) genes responsible for the efflux system in the Gram-negative microorganism B. fragilis.
Microarrays for SNP Determination
Hybridization analysis on microarrays allows both identification of genes and recognition of single structural distinctions in the probe and in the hybridized sequence, which is necessary for SNP determination. For this aim the method of allele-specific hybridization is most often used.
Allele-specific hybridization on a glass microarray was used to determine fluoroquinolone-resistant E. coli strains [111]. Oligonucleotide structures were proposed for nucleotide polymorphism identification in the gyrA gene, encoding amino acid substitutions in positions 83 and 87. Oligonucleotides were composed of 19 bases, and the detected mutation was in the central position. A group of four oligonucleotides differing in the base at central position was used for detection of one mutation. Optimization of oligonucleotide structures provided high specificity of determination: in all studied cases the result of complementary hybridization exceeded that for non-complementary hybridization by 4-13-fold. In the work an attempt was made to design universal probes using inosine, but this resulted in significant lowering of the analytical signals.
The method of hybridization analysis on microarray was described for detection of several types of ESBLs (TEM, SHV, CTX-M-3, CTX-M-9, MOX, CMY, DHA, ACC, FOX, MIR-1, and ACT-1) and plasmid-encoded AmpC beta-lactamase, including detection of six point mutations in the SHV beta-lactamase gene [112]. Oligonucleotide probes of 19-23 bases in length were immobilized on a glass microarray. Two oligonucleotides with base sequence complementary to the wild type and mutant enzymes were used for detection of point mutations. An asymmetric two-step PCR technique was used for amplification. In the first step a mixture of specific primers was used for amplification of the studied gene fragments. The second step was carried out at a higher temperature of annealing with universal fluorescence-labeled primers in higher concentrations. Optimization of conditions provided mainly single-stranded molecules, which made it possible to exclude the stage of DNA denaturation before hybridization. Rather good correlation was established between data of typing on the microarray and data of phenotyping and sequencing.
For identification of nine resistance determinants and one mutation in the grlA gene encoding the α subunit of DNA topoisomerase in clinical sample of St. aureus, a microarray was designed on the basis of glass-immobilized oligonucleotides [113]. The oligonucleotides were 17-24 bases long. A set of four oligonucleotides was used for detection of point mutation. A fluorescent label was introduced into the DNA after amplification. Hybridization on the microarray was carried out for 4 h at 42°C. The microarray enables simultaneous testing of bacterial resistance to beta-lactams, tetracyclines, gentamycin, macrolides, streptogramin A compounds, and fluoroquinolones. The detection limit was 100 pg DNA, which corresponds to 104 bacterial cells.
An original technology of gel biological chips for gene and SNP identification was designed at the Engelhardt Institute of Molecular Biology [114]. In this method oligonucleotide probes are immobilized on the microarray surface in the form of a hemispherical hydrogel of cells from 100 µm to 1 mm in diameter. Immobilization is carried out with formation of covalent bonds upon ultraviolet irradiation. The high-porosity gel makes possible immobilization of oligonucleotides of up to 200 bases. A fluorescent dye is introduced into the analyzed DNA. Electric fields transverse to the microarray surface introduces hydrodynamic flows that accelerate the kinetics of the labeled DNA hybridization with oligonucleotides. The probe concentration in the gel microarrays exceeds almost 103-fold that in the case of planar microarrays, which significantly increases the sensitivity of the method. This approach was used for detection of antibiotic-resistant forms of Mycobacterium tuberculosis [115]. For this aim SNPs that define resistance to rifampicin were determined in 28 positions of the rpoB gene as well as SNPs responsible for resistance to isoniazid were determined in 11 positions of the katG gene, in five positions of the promoter of the inhA gene, and in five positions of the regulatory region of gene ahpC-oxyR. Fluorescent label was introduced into PCR primers, and the analysis time was 12 h.
METHODS OF MULTIPARAMETRIC HYBRIDIZATION ANALYSIS FOR
IDENTIFICATION OF EXTENDED SPECTRUM BETA-LACTAMASES
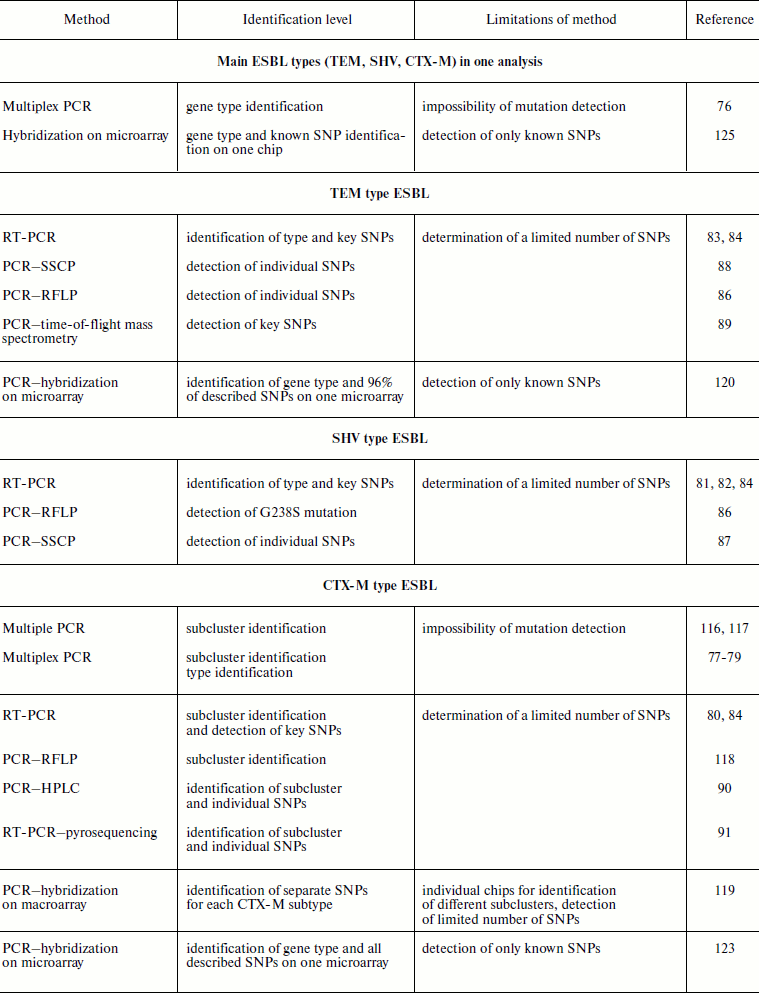
The variety of beta-lactamase types and variants makes rather complicated the problem of their identification for clinical laboratory diagnosis and epidemiological monitoring of the emergence and spreading of infection. The existence of several molecular classes of enzymes as well as their variability makes it necessary to use methods able to provide for simultaneous detection both of several gene types and mutations in them. At first sight it may seem that the problem of ESBL identification is more highly specialized, because formally ESBL are a small part of the beta-lactamase family and belong to one functional subgroup of 12 already described. However this subgroup is one of most numerous concerning the detected and described gene variants and point mutations in them. Therefore ESBL gene typing, i.e. determination of genotype of an enzyme produced by the bacterial strain under study, suggests simultaneous determination of several gene types and several hundreds of mutations in them. Data on the use of various molecular-genetic methods for detection of the main ESBL types that are based on PCR modifications and analysis of amplification products by different methods are given in Table 3. The use of methods based on PCR and the combination of PCR with methods of product analysis based on gel electrophoresis, chromatography, and mini-sequencing usually allows determination of only a limited number of genes and SNPs in a single analysis. Therefore, these methods cannot be used when identification of enzyme genotype is required. Among the total variety of described molecular methods, methods of multiparametric hybridization analysis on microarrays, considered in this section, seem to be the only adequate technologies for ESBL multiparametric gene typing.
Table 3. Molecular-genetic methods of ESBL
identification
The method of hybridization analysis with biotin label revealed by the streptavidin–peroxidase conjugate with subsequent chemiluminescent enzyme detection was proposed for identification of the most widespread variants of CTX-M type beta-lactamase genes [119]. Biotin was introduced into reverse primers used for multiplex PCR. Primer structure was selected so that amplified fragments of the CTX-M beta-lactamase gene belonging to four different genetic subclusters differed in size. Enzymes of CTX-M-8 and CTX-M-25 types were combined in a single subcluster. On the membrane macroarray oligonucleotides were immobilized as bands in an immunoblotter. For hybridization, the array was turned through 90° relative to the initial orientation, and the immunoblotter channels were filled with the amplified DNA solutions in hybridization buffer. The peculiarity of this method is hybridization performed in kinetic regime; to increase specificity of analysis, the sample was washed at enhanced temperature. After washing, the amount of label in DNA duplexes was determined on the support by peroxidase activity in the chemiluminescence reaction.
Differences in Tm of selected oligonucleotide panels did not allow selection of the same conditions for hybridization analysis for all four genetic subclusters. Because of this, a separate membrane array and different temperatures of washing buffer were used after hybridization (from 58 to 65°C) for detection of mutations within each subcluster. To determine which array is necessary for typing of any sample, the subcluster type was determined by the size of amplicon obtained in multiplex PCR. The selected oligonucleotides enabled identification of the presently most widespread gene variants of the CTX-M type beta-lactamases: CTX-M-1, -2, -3, -9, -14, and -15. High productivity of this method (43 samples can be tested in parallel), rather short time of analysis (about 7 h), and inexpensive equipment can be considered as its advantages. Limitations of this method are both unusual conditions of hybridization analysis for different subclusters of CTX-M beta-lactamase genes and a limited number of determined SNPs (four of each SNP for subclusters CTX-M-1, CTX-M-2, and CTX-M-8 and five for subcluster CTX-M-9).
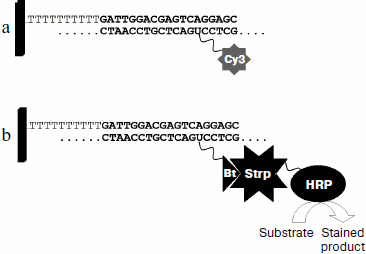
The method of hybridization analysis on glass microarrays was proposed for TEM type beta-lactamase gene typing on the basis of determination of all SNPs [120]. The fluorescent label used in this method was introduced into the gene during PCR in the form of modified deoxyribocytosine triphosphates (dCTP-Cy5). The scheme of the method of hybridization analysis with fluorescent detection is shown in Fig. 4a. Amplified labeled DNA was fragmented before hybridization by DNase for obtaining approximately 50-base-long oligonucleotide strands. It was shown earlier that this increases hybridization efficiency [121]. Hybridization was followed by scanning of fluorescence activity on the microarray surface. For one SNP determination a set of four oligonucleotides with unique base sequence corresponding to the TEM beta-lactamase gene structure in a given region and differing from each other by just a single nucleotide in the central position, which could be one of four nucleotides A, G, C, or T, was used (Fig. 5a). The most stable duplex is formed in the case of the labeled fragment hybridization with a fully complementary oligonucleotide, and correspondingly, in this case a higher analytical signal (PM, perfect match) is detected (Fig. 5b). If duplexes are formed during hybridization with three other oligonucleotides or with only some of them, then the observed signal (MM, mismatch) is indicative of the level of unspecific hybridization.
Fig. 4. Scheme of DNA hybridization analysis on microarrays with different labels: a) fluorescent dye; b) biotin (Bt). Strp, streptavidin; HRP, horseradish peroxidase.
Molecular design of oligonucleotide structures for 41 SNPs in TEM beta-lactamase genes was carried out in this work. The following rules of oligonucleotide structure selection for SNP recognition were observed [122]: (i) oligonucleotides should be 17-26 bases long; (ii) the G + C content should be in the range of 35-70%; (iii) the probability of dimer and cyclic structure formation should be minimal; (iv) Tm differences for different oligonucleotide groups should not exceed 5-10°C.Fig. 5. a) Scheme of point mutation detection in gene by allele-specific hybridization. b) The result of point mutation detection in a gene by hybridization analysis on microchip. c) Representation of results of hybridization analysis for detection of a single mutation in a gene in relative units.
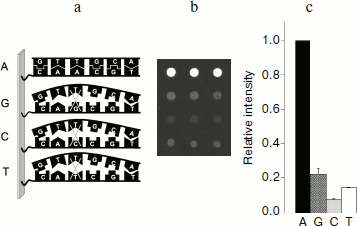
The principle of internal signal normalization was used for nucleotide type determination in the analyzed gene position. For this aim all results of hybridization within the set of four oligonucleotides were normalized to the signal of complementary hybridization. In this way hybridization signals were obtained in relative units: signal of complementary hybridization became equal to 1, whereas the other three signals of non-complementary hybridization were expressed in its parts (Fig. 5c):
RIPM = IPM/IPM = 1,
RIMM = IMM/IPM,
where IPM is intensity of signal of hybridization with fully complementary oligonucleotide, IMM is intensity of signal of hybridization with the other three oligonucleotides, RIPM is fraction of complementary hybridization, RIMM is fraction of noncomplementary (unspecific) hybridization.
Parameter of unspecific hybridization fraction RIMM = IMM/IPM was used for estimation of SNP detection specificity. If the fraction of unspecific hybridization exceeded 0.7, then the SNP detection was considered as unspecific. Molecular design of oligonucleotide structures for SNP detection in TEM type beta-lactamases allowed highly specific detection of mutations: over 99% of RIMM values did not exceed 0.4, and only a single group of oligonucleotides was characterized by RIMM = 0.52. The authors of this work tried to reduce significantly the hybridization time from three hours to 15 min in order to elaborate a method for express analysis of resistant strains that could be used in clinical laboratories. It was found that in the case of 1-h-long hybridization reaction, in 84% of cases RIMM values are not higher than 0.4, 14% of RIMM values are not higher than 0.6, while in the case of a single mutation an RIMM value close to 0.7 is observed. Reduction of hybridization time to 30 min revealed similar specificity with lower reproducibility. Further reduction of hybridization time to 15 min resulted in significant lowering of the detection specificity: six positions were identified incorrectly, while 21% of RIMM values exceeded 0.7. Thus, it was found that hybridization time could be reduced to 30 min. In this case total time of analysis was 3.5 h, which is a significant improvement compared to standard phenotypic tests taking 2-3 days.
We have used the same approach for elaboration of hybridization analysis on microarrays for gene typing of CTX-M type beta lactamases [123] that differ from other class A ESBL by lower homology between the same type genes. Because of this, oligonucleotide structures for genetic subcluster identification were also selected along with oligonucleotides for identification of point mutations in genes.
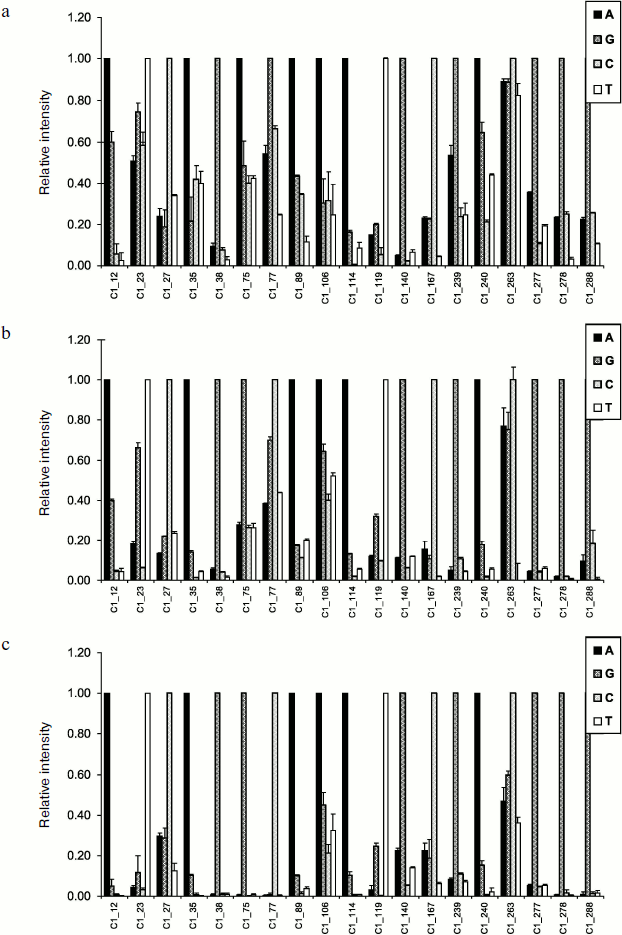
On the whole, the CTX-M type beta lactamases differ from TEM and SHV type beta-lactamases by higher G–C content, which resulted in a higher level of unspecific hybridization compared to previous data [120]. Thorough molecular design of specific oligonucleotide structures was carried out for lowering the level of unspecific hybridization. Introduction of artificial substitutions into oligonucleotide structure was used to increase specificity of the central oligonucleotide detection. Results of testing of the CTX-M-3 beta-lactamase-producing control strain by hybridization analysis on microarrays using fluorescent dye Cy3 as DNA label are shown in Fig. 6a. Specificity of separate SNP detection during hybridization at 45°C was not high enough: 16% of RIMM values did not exceed 0.7. On increase of hybridization temperature to 47°C the specificity of SNP identification was improved insignificantly: the fraction of RIMM values below 0.4 increased, but 10% of RIMM values exceeded 0.7 (Fig. 6b).
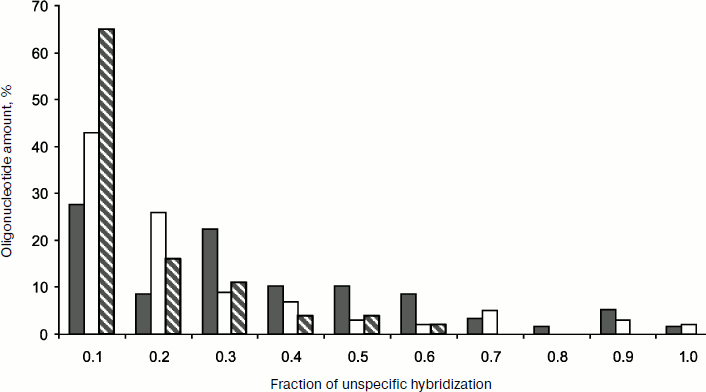
Since hybridization efficiency and specificity are defined by the oligonucleotide strand secondary structure that can be influenced by the introduced label, we studied the effect of an alternative biotin label having significantly smaller size compared to fluorescent labels of the cyanin group [124]. The scheme of hybridization analysis with biotin label is shown in Fig. 4b. Biotin was introduced into the beta-lactamase gene during PCR (in the form of dUTP-biotin). The introduced biotin was revealed using the streptavidin–horseradish peroxidase (HRP) conjugate and HRP colorimetric detection with substrates whose oxidation is accompanied by formation of intensely stained product adsorbed on the glass surface near the enzyme molecules. Figure 6c shows results of testing of the CTX-M-3 beta-lactamase-producing control strain by hybridization analysis on microarrays using biotin label. The specificity of SNP revealed by hybridization analysis with biotin label was significantly higher even at 45°C. Data in Fig. 7 compare specificity in revealing 19 different SNPs described for the CTX-M-1 subcluster beta-lactamases upon hybridization analysis with fluorescent and colorimetric detection. The use of biotin label provided the best specificity in detection of point mutations: 94% of RIMM values were below 0.4. All in all, oligonucleotide probe structures were selected for detection of 67 SNPs in CTX-M type beta-lactamase genes. The elaborated method was proven using a collection of 94 clinical microbial strains of the Enterobacteriaceae family. A full 100% coincidence was obtained between gene typing by hybridization analysis on the microarray and complete gene sequencing.Fig. 6. Intensity profiles of hybridization signals of labeled DNA amplified from control strain producing CTX-M-3 beta-lactamase. Mutation numbers in CTX-M-1 beta-lactamase sequence are marked on the abscissa. a) Cy3 label, hybridization conditions 45°C, 1 h; b) Cy3 label, hybridization conditions 47°C, 1 h; c) biotin label, hybridization conditions 45°C, 2 h.
Thus experiments on multiparametric SNP typing in TEM and CTX-M type beta-lactamase genes by allele-specific hybridization on microarrays revealed the good potential of this method. The peculiarity of the considered approach is its sufficient universality and the possibility of extension of the type of identified genes and mutations.Fig. 7. Relative specificity of point mutation detection in CTX-M type beta-lactamase genes by hybridization analysis on microarrays with fluorescence and colorimetric detection. Black columns: Cy3 DNA label, hybridization conditions 45°C, 1 h; white columns: Cy3 DNA label, hybridization conditions 47°C, 1 h; hatched columns: biotin label, hybridization conditions 45°C, 2 h.
Later this was confirmed during elaboration of integrated technique for simultaneous gene typing for the three most clinically important ESBL (TEM, SHV, and CTX-M types) on the basis of SNP detection in encoding genes [125]. Two multiplex PCRs were elaborated for gene amplification: in one of them genes of TEM and SHV type beta-lactamases were amplified, while the other was used for amplification of the CTX-M type beta-lactamase two gene fragments belonging to all four subclusters and comprising in aggregate the full-size gene (open reading frame). The fluorescent label Cy3 introduced into the gene during PCR was used as the DNA label. A set of four oligonucleotides differing in nucleotide type in the central position or in that close to central was used for one SNP detection. It was proposed to place oligonucleotide groups on the microchip in the form of modules, each of which contains oligonucleotide groups for detection of mutations in the same type beta-lactamase genes. This will make possible in future to add new oligonucleotide groups for revealing new mutations in analyzed gene types as well as new oligonucleotide modules for revealing new beta-lactamase types.
The main problem of integration of the earlier developed oligonucleotide panels for gene typing of TEM, SHV, and CTX-M type beta-lactamases into a single microarray is how to optimize the conditions providing the efficient hybridization of structurally different oligonucleotides with retention of high sensitivity and specificity of analysis. Due to structural differences between genes of studied ESBL types and higher G–C content in CTX-M type genes, hybridization temperature was increased to 47°C instead of that chosen earlier for gene typing of TEM type beta-lactamases to increase the specificity of the analysis. Hybridization was carried out for 1 h under automatic conditions in a hybridization tube. Artificial substitutions were introduced into some oligonucleotide sequences to lower the probability of formation of dimer and other stable secondary structure elements. Molecular design of specific oligonucleotide structures provided very high specificity of SNP determination: in 94% of oligonucleotides the fraction of unspecific hybridization was below 0.4. The developed method allows simultaneous detection of over 150 SNP, which is much higher than the number of parameters simultaneously determined by other methods.
The integrated microchip was tested using 60 samples already characterized by standard phenotyping tests for strain sensitivity to panels of antibiotics and their combinations with inhibitors. Hybridization analysis on the microarray revealed 93% sensitivity and 100% specificity of this method. ESBLs of one or several types were found in 54 samples. For four samples phenotypically identified resistance could not be explained by ESBL production. Successful identification of different gene type mixtures (like TEM-1 and SHV-12; TEM-1, SHV-1, and CTX-M-15) and mixture of two genes belonging to the same gene type (like CTX-M-15 and CTX-M-14b; SHV-1 and SHV-14) can be considered as advantages of the developed method. These results were confirmed by sequencing. It should be noted that detection of a mixture of two genes on a microarray is more clear and reliable, especially for genes of the same type, whereas in the case of sequencing detection of two chromatographic peaks causes difficulties especially for automatic data processing.
The variety of beta-lactamases and extremely rapid spreading of known resistance determinants and emergence of new determinants and new combinations of previously described resistance determinants make necessary elaboration of adequate methods for their clinical diagnostics for choosing the correct course of therapy and monitoring of spreading of infectious diseases. Molecular-biological methods of gene structure analysis are indispensable for solution of these problems. Amplification and hybridization technologies have already provided a qualitatively new level of diagnosis of many infectious and genetic diseases.
Antibiotic resistance of infectious pathogens was tested at the “micro” level when resistance is determined in a single clinic or in a separate group of patients, as well as at “macro” level in inter-center, national, and international studies when hundreds, thousands, and tens of thousands of pathogen strains collected in different clinics are analyzed. For large-scale investigations it is desirable to have methods allowing detection of numerous parameters in the same reaction that are characterized by sufficient sensitivity and specificity along with high productivity.
Broad introduction of sequencing for these aims is difficult due to complication and laboriousness of the method, the necessity of specialized expensive equipment, and to rather high cost of analysis. Multiparametric analysis on microarrays seems to be a convenient alternative to sequencing. Multiparametric determination of genes and mutations in them by hybridization analysis on microarrays has great potential for use in investigation of molecular mechanisms of infection resistance and spreading.
This work was supported by Russian Federal Program “National Technological Resources 2007-2011” (State contract GP/07/442/NTB/K) and a grant of the German Federal Ministry of Education and Science within the framework of the GenoMik (Genome Research on Microorganisms) project.
REFERENCES
1.Livermore, D. M. (1995) Clin. Microbiol.
Rev., 8, 557-584.
2.Pitout, J. D., and Laupland, K. B. (2008) Lancet
Infect. Dis., 8, 159-166.
3.Perez, F., Endimiani, A., Hujer, K. M., and Bonomo,
R. A. (2000) Curr. Opin Pharmacol., 7, 459-469.
4.Schwaber, M. J., and Carmeli, Y. (2007) J.
Antimicrob. Chemother., 60, 913-9205.
5.Schwaber, M. J., Navon-Venezia, S., Kaye, K. S.,
Ben Ami, R., Schwartz, D., and Carmeli, Y. (2006) Antimicrob. Agents
Chemother., 50, 1257-1262.
6.Bradford, P. (2001) Clin. Microbiol. Rev.,
14, 933-951.
7.Babic, M., Hujer, A. M., and Bonomo, R. A. (2006)
Drug Resistance Updates, 9, 142-156.
8.Paterson, D. L., and Bonomo, R. A. (2005) Clin.
Microbiol. Rev., 18, 657-686.
9.Sidorenko, S. V., and Tishkov, V. I. (2004)
Uspekhi Biol. Khim., 44, 263-306.
10.Page, M. I. (1987) Advan. Phys. Org.
Chem., 23, 165-170.
11.Massova, I., and Mobashery, S. (1998)
Antimicrob. Agents Chemother., 48, 1-17.
12.Nukaga, M., Haruta, S., Tanimoto, K., Kogure, K.,
Taniguchi, K., Tamaki, M., and Sawai, T. (1995) J. Biol. Chem.,
270, 5729-5735.
13.Ghuysen, J. M. (1994) Trends Microbiol.,
2, 372-380.
14.James, P. A., and Reeves, D. S. (1996)
J. Chemother., 8, Suppl. 2, 37-47.
15.Chambers, H. F. (1997) Clin. Microbiol. Rev.
J., 10, 781-791.
16.Heritier, C., Poirel, L., Lambert, T., and
Nordmann, P. (2005) Antimicrob. Agents Chemother., 49,
3198-3202.
17.Heritier, C., Poirel, L., Fournier, P. E.,
Claverie, J. M., Rauolt, D., and Nordmann, P. (2005) Antimicrob.
Agents Chemother., 49, 4174-4179.
18.Livermore, D. M., and Pearson, A. (2007) Clin.
Microbiol. Infect., 13, Suppl. 2, 7-16.
19.Harada, S., Yoshikazu, I., and Keizo, Y. (2008)
Korean J. Lab. Med., 28, 401-412.
20.Woodford, N., and Ellington, M. J. (2007)
Clin. Microbiol. Infect., 13, 5-18.
21.Medeiros, A. A. (1997) Clin. Infect. Dis.,
24, Suppl. 1, S19-S45.
22.Poirel, L., Naas, T., and Nordmann, P. (2008)
Clin. Microbiol. Infect., 14, 75-81.
23.Weldhagen, G. F. (2004) Int. J. Antimicrob.
Agents, 23, 556-562.
24.Fleming, P. C., Goldner, M., and Glass, D. G.
(1963) Lancet, 1, 1399-1401.
25.Richmond, M. H., and Sykes, R. B. (1973) Adv.
Microb. Physiol., 9, 31-38.
26.Sykes, R. B., and Matthew, M. (1976) J.
Antimicrob. Chemother., 2, 115-157.
27.Bush, K., Jacoby, G. A., and Medeiros, A. A.
(1995) Antimicrob. Agents Chemother., 39, 1211-1233.
28.Ambler, R. P. (1980) Philos. Trans. R. Soc.
Lond. B. Biol. Sci., 289, 321-331.
29.Bush, K. (2001) Clin. Infect. Des.,
32, 1085-1089.
30.Philippon, A., Arlet, G., and Jacoby, G. A.
(2002) Antimicrob. Agents Chemother., 46, 1-11.
31.Maltezou, H. C. (2009) Int. J. Antimicrob.
Agents, 33, e1-e7.
32.Tamilselvi, A., and Govindasamy, M. (2008) J.
Biol. Inorg. Chem., 13, 1039-1053.
33.Pitout, J. D., and Laupland, K. B. (2008)
Lancet Infect. Dis., 8, 159-166.
34.Livermore, D. M. (2008)
Clin. Microbiol. Infect., 14, Suppl. 1, 3-10.
35.Empel, J., Baraniak, A., Literacka, E., Mrowka,
A., Fiett, J., Sadowy, E., et al. (2008) Antimicrob. Agents
Chemother., 52, 2449-2454.
36.Rodriguez-Bano, J., Navarro, M. D., Romero, L.,
Martinez-Martinez, L., Muniain, M. A., Perea, E. J., Perez-Cano,
R., and Pascual, A. (2004) J. Clin. Microb., 42,
1089-1094.
37.Paterson, D. L., Hujer, K. M., Hujer, A. M.,
Yeiser, B., Bonomo, M. D., Rice, L. B., et al. (2003) Antimicrob.
Agents Chemother., 47, 3554-3560.
38.Bush, K. (2008)
Clin. Microbiol. Infect., 14, Suppl. 1, 134-143.
39.Quinteros, M., Radice, M., Gardella, N.,
Rodriguez, M. M., Costa, N., Korbenfeld, Couto, D., and Gutkind, G.
(2003) Antimicrob. Agents Chemother., 47, 2864-2867.
40.Mulvey, M. R., Bryce, E., Boyd, D.,
Ofner-Agostini, M., Christianson, S., Simor, A. E., and Paton, Sh.
(2004) Antimicrob. Agents Chemother., 48, 1204-1214.
41.
Hawkey, P. M. (2008)
Clin. Microbiol. Infect., 14, Suppl. 1, 159-165.
42.Wu, T. L., Chia, J. H., Su, L. H., Kuo, A. J.,
Chu, C., and Chiu, C. H. (2003) J. Clin. Microbiol., 41,
4836-4838.
43.Schlesinger, J., Navon-Venezia, S., Chmelnitsky,
I., Hammer-Munz, O., Leavitt, A., and Gold, H. S. (2005) Antimicrob.
Agents Chemother., 49, 1150-1156.
44.Livermore, D. M., Canton, R., Gniadkowski, M.,
Nordmann, P., Rossolini, G. M., Arlet, G., Ayala, J., Coque, T. M.,
Kern-Zdanowicz, I., Luzzaro, F., Poirel, L., and Woodford, N. (2007)
J. Antimicrob. Chemother., 59, 165-174.
45.Livermore, D. M., and Hawkey, P. M. (2005) J.
Antimicrob. Chemother., 56, 451-454.
46.Lartigue, M. F., Zinsius, C., Wenger, A., Bille,
J., Poirel, L., and Nordmann, P. (2007) Antimicrob. Agents
Chemother., 51, 2855-2860.
47.Mugnaioli, C., Luzzaro, F., de Luca, F.,
Brigante, G., Perilli, M., Amicosante, G., Stefani, S., Toniolo, A.,
and Rossolini, G. M. (2006) Antimicrob. Agents Chemother.,
50, 2700-2706.
48.Pagani, L., Dell’Amico, E., Migliavacca,
R., D’Andrea, M. M., Giacobone, E., Amicosante, G., Romero, E.,
and Rossolini, G. M. (2003) J. Clin. Microbiol., 41,
4264-4269.
49.Wang, H., Kelkar, S., Wu, W., Chen, M., and
Quinn, J. P. (2003) Antimicrob. Agents Chemother., 47,
790-793.
50.Canton, R., and Coque, T. M. (2006) Curr.
Opin. Microbiol., 9, 466-475.
51.Jones, J. L., Muccioli, C., Belfort, R., Holland,
G. N., Roberts, J. M., and Silveira, C. (2007) Emerging Infect.
Dis., 13, 513-514.
52.Mudrak, D. E., Ikryannikova, L. N., Sidorenko, S.
V., and Il’ina, E. N. (2007) Antibiot. Khimoter.,
52, 10-16.
53.Egorova, S., Kaftyreva, L., Grimont, P. A., and
Weill, F. X. (2007) Microb. Drug Resist., 13,
102-107.
54.Ivanov, D. V., and Egorov, A. M. (2008)
Biomed. Khim., 54, 104-113.
55.Sidorenko, S. V., Berezin, A. G., and Ivanov, D.
V. (2004) Antibiot. Khimoter., 49, 6-15.
56.Edelstein, M., Pimkin, M., Palagin, I.,
Edelstein, I., and Stratchounski, L. (2003)
Antimicrob. Agents Chemother., 47, 3724-3732.
57.Datta, N., and Kontomichalou, P. (1965)
Nature, 208, 239-241.
58.Du Bois, S. K., Marriott, M. S., and Amyes, S. G.
(1995) J. Antimicrob. Chemother., 35, 7-22.
59.Mabilat, C., Lourencao-Vital, J., Goussard, S.,
and Courvalin, P. (1992) Mol. Gen. Genet., 235,
113-121.
60.Chaibi, E. B., Sirot, D., Paul, G., and Labia, R.
(1999) J. Antimicrob. Chemother., 43, 447-458.
61.Strynadka, N. C., Adachi, H., Jensen, S. E.,
Johns, K., Sielecki, A., Betzel, C., Suton, K., and James, M. N. (1992)
Nature, 359, 700-705.
62.Petrosino, J. F., and Palzkill, T. (1996) J.
Bacteriol., 178, 1821-1828.
63.Hayes, F., Hallet, B., and Cao, Y. (1997) J.
Biol. Chem., 272, 28833-28836.
64.Knothe H., Shah, P., Kremery, V., Antal, M., and
Mitsuhasi, S. (1983) Infection, 11, 315-317.
65.Bauernfield, A., Grimm, H., and Shweighart, S.
(1990) Infection, 18, 294-298.
66.Bonnet, R. (2004)
Antimicrob. Agents Chemother., 48, 1-14.
67.Tzouvelekis, L. S., Tzelepi, E., Tassios, P. T.,
and Legakis, N. J. (2000) Int. J. Antimicrob. Agents, 14,
137-143.
68.Bonnet, R., Champs, C. D., Sirot, D., Chanal, C.,
Labia, R., and Sirot, J. (1999) Antimicrob. Agents Chemother.,
43, 2671-2677.
69.Steward, C. D., Rasheed, J. K., Hubert, S. K.,
Biddle, J. W., Raney, P. M., Anderson, G. J., Williams, P. P.,
Brittain, K. L., Oliver, A., McGowan, J. E., Jr., and Tenover, F. C.
(2001) J. Clin. Microbiol., 39, 2864-2872.
70.Weigand, I., Geiss, H. K., Mack, D., Sturenburg,
E., and Seifert, H. (2007) J. Clin. Microbiol., 45,
1167-1174.
71.Robberts, F. J. L., Kohner, P. C., and Patel, R.
(2009) J. Clin. Microbiol., 47, 358-361.
72.Drieux, L., Brossier, F., Sougakoff, W., and
Jarlier, V. (2008) Clin. Microbiol. Infect., 14,
90-103.
73.Wiegand, I., Geiss, H. K., Mack, D., Sturenburg,
E., and Seifert, H. (2007) J. Clin. Microbiol., 45,
1167-1174.
74.Paterson, D. L. (2008) Curr. Opin Infect.
Dis., 14, 697-701.
75.Brookes, A. J. (1999) Gene, 234, 177-186.
76.Monstein, H. J., Ostholm-Balkhed, A., Nilsson, M.
V., Nilsson, M., Dornbusch, K., and Nilsson, L. E. (2007)
APMIS, 115, 1400-1408.
77.Woodford, N., Fagan, E. J., and Ellington, M. J.
(2006) J. Antimicrob. Chemother., 57, 154-155.
78.Xu, L., Ensor, V., Gossain, S., Nye, K., and
Hawkey, P. (2005) J. Med. Microbiol., 54, 1183-1187.
79.Pitout, J. D. D., Hamilton, N., Church, D. L.,
Nordmann, P., and Poirel, L. (2007) Clin. Microbiol. Infect.,
13, 291-297.
80.Birkett, C. I., Ludlam, H. A., Woodford, N.,
Brown, D. F. G., Brown, N. M., Roberts, M. T. M., Milner, N., and
Curran, M. D. (2007) J. Med. Microbiol., 56, 52-55.
81.Szabo, D., Melan, M. A., Hujer, A. M., Bonomo, R.
A., Hujer, K. M., Bethel, C. R., Kristof, K., and Paterson, D. L.
(2005) Antimicrob. Agents Chemother., 49, 4716-4720.
82.
Randegger, C. C., and Hachler, H. (2001)
Antimicrob. Agents Chemother., 45, 1730-1736.
83.Nikulin, A., Alexeev, Y., and Edelstein, M.
(2007) Int. J. Antimicrob. Agents, 29, Suppl. 2, S222.
84.Stepanova, M., Nikulin, A., Sukhorukova, M., and
Edelstein, M. (2007) Abstr. 17th Europ. Congr. Clin. Microbiol.
Infect. Dis./25th Int. Congr. Chemother., March 31-April 3, 2007,
Munich, Germany, 1732-130.
85.Schumm, J. W., Knowlton, R. G., Braman, J. C.,
Barker, D. F., Botstein, D., Akots, G., Brown, V. A., Gravius, T. C.,
Helms, C., and Hsiao, K. (1989) Am. J. Hum. Genet., 42,
143-159.
86.Kalai Blagui, S., Achour, W., Abdeladhim, A., and
Ben Hassen, A. (2009) Pathol. Biol., 57, 420-424.
87.M’Zali, F.-H., Gascoyne-Binzi, D. M.,
Heritage, J., and Hawkey, P. M. (1996) J. Antimicrob.
Chemother., 37, 797-802.
88.Alonso, R., Fernandez-Aranguiz, A., Colom, K.,
and Cisterna, R. (2002) J. Microbiol. Meth., 50,
85-90.
89.Ikryannikova, L. N., Shitikov, E. A., Zhivankova,
D. G., Il’ina, E. N., Edelstein, M. V., and Govorun, V. M. (2008)
J. Microbiol. Meth., 75, 385-391.
90.Xu, L., Evans, J., Ling, T., Nye, K., and Hawkey,
P. (2007) Antimicrob. Agents Chemother., 51,
1446-1454.
91.Naas, T., Oxacelay, C., and Nordmann, P. (2007)
Antimicrob. Agents Chemother., 51, 223-230.
92.Poirel, L., Naas, T., and Nordmann, P. (2006)
J. Clin. Microbiol., 44, 3008-3011.
93.Southern, E. M., Maskos, U., and Elder, J. K.
(1992) Genomics, 13, 1008-1017.
94.Doss, V. A., Parvathi, S., Raju, B. A., and Devi,
N. A. (2004) Dis. Markers, 20, 317-323.
95.Rodriguez, I., Barownick, W., Helmuth, R.,
Mendoza, M. C., Rodicio, M. R., Schroeter, A., and Guerra, B. (2009)
J. Antimicrob. Chemother., 64, 301-309.
96.Schena, M., Shalon, D., Davis, R. W., and Brown,
P. O. (1995) Science, 270, 467-470.
97.Heller, M. J. (2002) Annu. Rev. Biomed.,
4, 129-153.
98.Guckenberger, M., Kurz, S., Aepinus, C., Theiss,
S., Haller, S., Leimbach, T., Panzner, U., Weber, J., Paul, H.,
Unkmeir, A., Frosch, M., and Dietrich, G. (2002) Amer. Soc.
Microbiol., 184, 2546-2551.
99.Chizhikov, V., Rasooly, A., Chumakov, K., and
Levy, D. D. (2001) Appl. Environ. Microbiol., 67,
3258-3263.
100.Cao, X., Wang, Y. F., Zhang, C. F., and Gao, W.
J. (2006) Biosens. Bioelectron., 15, 393-398.
101.Ozsoz, M. (2003) Anal. Chem., 75,
2181-2187.
102.Chee, M., Yang, R., Hubbell, E., Berno, A.,
Huang, X. C., Stern, D., Winkler, J., Lockhart, D. J., Morris, M. S.,
and Fodor, S. P. (1996) Science, 274, 610-614.
103.Shogo, M., Shigehiko, U., Suzuki, O., Urano,
A., and Abe, S. (2005) Mar. Biotechnol., 6, 430-434.
104.Michikawa, Y., Fujimoto, K., Kinoshita, K.,
Kawai, S., Sugahara, K., Suga, T., Otsuka, Y., Fujiwara, K., Iwakawa,
M., and Imai, T. (2006) Anal. Sci., 22, 1537-1545.
105.Lee, Y., Lee, C. S., Kim, Y. J., Chun, S.,
Park, S., Kim, Y. S., and Han, B. D. (2002) Mol. Cells,
14, 192-197.
106.Chen, S., Zhao, S., McDermott, P. F.,
Schroeder, C. M., White, D. G., and Meng, J. (2005) Mol. Cell
Probes, 19, 195-201.
107.Cleven, B. E. E., Palka-Santini, M., Gielen, J., Meembor, S.,
Kronke, M., and Krut, O. (2006) J. Clin. Microbiol., 44,
2389-2397.
108.Perreten, V., Vorlet-Fawer, L., Slickers, P.,
Ehricht, R., Kuhnert, P., and Frey, J. (2005) J. Clin.
Microbiol., 43, 2291-2302.
109.Batchelor, M., Hopkins, K. L., Liebana, E.,
Slickers, P., Ehricht, R., Mafura, M., Aarestrup, F., Mevius, D.,
Clifton-Hadley, F. A., Woodward, M. J., Davies, R. H., Threlfall, E.
J., and Anjum, M. F. (2008) Int. J. Antimicrob. Agents,
31, 440-451.
110.Cassone, M., Andrea, M. M. D., Ianelli, F.,
Oggioni, M. R., Rossolini, G. M., and Pozzi, G. (2006) Antimicrob.
Agents Chemother., 50, 2038-2041.
111.Yu, X., Susa, M., Knabbe, C., Schmid, R. D.,
and Bachmann, T. T. (2004) J. Clin. Microbiol., 42,
4083-4091.
112.Zhu, L. X., Zhang, Z. W., Liang, D., Jiang, D.,
Wang, C., Du, N., Zhang, Q., Mitchelson, K., and Cheng, J. (2007)
Antimicrob. Agents Chemother., 51, 3707-3713.
113.Strommenger, B., Schmidt, C., Werner, G.,
Roessle-Lorch, B., Bachmann, T. T., and Witte, W. (2007) Mol. Cell
Probes, 21, 161-170.
114.Rubina, A. Yu., Pan’kov, S. V.,
Dementieva, E. I., Pen’kov, D. N., Butygin, A. V., Vasiliskov, V.
A., Chudinov, A. V., Mikheikin, A. L., Mikhailovich, V. M., and
Mirzabekov, A. D. (2004) Anal. Biochem., 325, 92-106.
115.Gryadunov, D., Mikhailovich, V., Lapa, S.,
Roudinskii, N., Donnikov, M., Pan’kov, S., Markova, O.,
Kuz’ min, A., Chernousova, L., Skotnikova, O., Moroz, A.,
Zasedatelev, A., and Mirzabekov, A. (2005) Clin. Microb.
Infect., 11, 531-539.
116.Shibata, N., Kurokawa, H., Doi, Y., Yagi, T.,
Yamane, K., Wachino, J., Suzuki, S., Kimura, K., Ishikawa, S., Kato,
H., Ozawa, Y., Shibayama, K., Kai, K., Konda, T., and Arakawa, Y.
(2006) Antimicrob. Agents Chemother., 50, 791-795.
117.Pitout, J. D. D., Hossain, A., and Hanson, N.
D. (2004) J. Clin. Microbiol., 42, 5715-5721.
118.Schmitt, J., Jacobs, E., and Schmidt, H. (2007)
J. Med. Microbiol., 56, 241-249.
119.Ensor, V. M., Livermore, D. M., and Hawkey, P.
M. (2007) J. Antimicrob. Chemother., 59, 387-395.
120.Grimm, V., Ezaki, S., Susa, M., Knabbe, C.,
Schmid, R. D., and Bachmann, T. T. (2004) J. Clin. Microbiol.,
42, 3766-3774.
121.Hughes, T. R., Mao, M., Jones, A. R., Burchard,
J., Marton, M. J., Shannon, K. W., Lefkowitz, S. M., Ziman, M.,
Schelter, J. M., Meyer, M. R., Kobayashi, S., Davis, C., Dai, H., He,
Y. D., Stephaniants, S. B., Cavet, G., Walker, W. L., West, A., Coffey,
E., Shoemaker, D. D., Stoughton, R., Blanchard, A. P., Friend, S. H.,
and Linsley, P. S. (2001) Nat. Biotechnol., 19,
342-347.
122.Bodrossy, L. (2003) A Beginner’s Guide
to Microarrays, Kluwer Academic Publishers, New York, pp.
43-92.
123.Rubtsova, M. Yu., Ulyashova, M. M., Ignatenko,
O. V., Edelstein, M. V., Egorov, A. M., Schmid, R. D., and Bachmann, T.
T. (2008) Abstr. 8th Int. Meet. Microb. Epidemiol. Markers,
Zakopane, Poland, May 14-17, 109.
124.Ulyashova, M. M., Rubtsova, M. Yu., Bachmann,
T. T., and Egorov, A. M. (2008) Bull. of Moscow State Univ.,
Ser. 2, Chemistry, 49, 96-101.
125.Leinberger, D. M., Grimm, V., Rubtsova, M.,
Weile, Y., Schroppe, K., Wichelhaus, T. A., Knabbe, C., Schmid, R. D.,
and Bachmann, T. T. (2010) J. Clin. Microbiol., 48,
460-471.