REVIEW: Activated Leukemic Oncogenes AML1-ETO and c-kit: Role in Development of Acute Myeloid Leukemia and Current Approaches for Their Inhibition
A. V. Rulina*, P. V. Spirin, and V. S. Prassolov
Engelhardt Institute of Molecular Biology, Russian Academy of Sciences, ul. Vavilova 32, 119991 Moscow, Russia; E-mail: rulinaura@mail.ru* To whom correspondence should be addressed.
Received June 3, 2010; Revision received July 24, 2010
Acute myeloid leukemia (AML) is a malignant blood disease caused by different mutations that enhance the proliferative activity and survival of blood cells and affect their differentiation and apoptosis. The most frequent disorders in AML are translocations between chromosomes 21 and 8 leading to production of a chimeric oncogene, AML1-ETO, and hyperexpression of the receptor tyrosine kinase KIT. Mutations in these genes often occur jointly. The presence in cells of two activated oncogenes is likely to trigger their malignization. The current approaches for treatment of oncologic diseases (bone marrow transplantation, radiotherapy, and chemotherapy) have significant shortcomings, and thus many laboratories are intensively developing new approaches against leukemias. Inhibiting expression of activated leukemic oncogenes based on the principle of RNA interference seems to be a promising approach in this field.
KEY WORDS: acute myeloid leukemia (AML), leukemic oncogenes, AML1-ETO, c-kit, RNA interferenceDOI: 10.1134/S0006297910130092
Abbreviations: Akt, a family of proteins playing an important role in intracellular signal transmission in mammals; AML, acute myeloid leukemia; AML1, gene of Acute Myeloid Leukemia 1 encoding hemopoiesis-regulating protein; AML1-ETO, chimeric gene Acute Myeloid Leukemia 1–Eight-Twenty One; casiRNA, cis-acting siRNAs characteristic for plants; CBF, core-binding factor (heterodimeric transcription complex); CBF AML, AML associated with disturbance in CBF function; Cbl, ubiquitin E3 ligase; dsRNA, double-stranded RNA; ERK, extracellular-signal-regulated kinase; ETO, gene Eight-Twenty One; FAB, French-American-British classification of AML; FLT-3, fms-like receptor tyrosine kinase-3; GIST, gastrointestinal stromal tumor; HDAC, histone deacetylase; HZ4-FeSV, feline sarcoma virus; IL, interleukin; JAK, Janus tyrosine kinase; JM-domain, juxtamembrane domain of receptor tyrosine kinase KIT; M-CSF, macrophage colony-stimulating factor; MDS, myelodysplastic syndrome; miRNA, microRNA; MPD, myeloproliferative diseases; mRNA, messenger RNA; N-CoR, nuclear corepressor of transcription; NHR, Nervy protein homologous region; PDGF, platelet-derived growth factor; PI3K, phosphatidylinositol-3 kinase; piRNA, RNAs interacting with PIWI proteins; PIWI, proteins of Ago family (P-element Induced WImpy testis); PLZF, promyelocytic leukemia zinc finger (a transcription repressor); PTGS, posttranscriptional gene silencing; Ras, family of small GTPases involved in intracellular signal transmission; rasiRNA, repeat-associated small interfering RNAs; RdRP, RNA-dependent RNA polymerase; RHD, Runt protein homologous domain; RISC, RNA-induced silencing complex; RLC, RISC-loading complex; SCF, stem cell factor; SFK, Src family kinase; SH2, Src homology 2 domain; SHP-2 and SHP-3, Src homology region 2- and 3-domain phosphatase; shRNA, small hairpin RNA; Sin3, transcription corepressor; siRNA, small interfering RNA; STAT, signal transducers and activator of transcription; VEGFR, vascular endothelial growth factor receptor; VSV-G, vesicular stomatitis virus G protein.
According to predictions of the World Health Organization (WHO),
oncologic diseases in the near future may take the first place, leaving
behind cardiovascular diseases. At present more than 5000 oncologic
diseases are known that are caused by structural–functional
disorders of various genes, including activation of protooncogenes
resulting in cell malignization. The situation is especially
complicated because in every case different genes are involved in the
development of malignancy. This makes difficult the choice of target
genes and their protein products for directed treatment of tumors.
Leukemias are widespread oncologic diseases that are characterized by an increased content of morphologically immature blood cells, blasts, which on one hand do not perform normal functions of mature cells and on the other hand exclude normal precursors because of the active proliferation.
Acute myeloid leukemias (AML) constitute a significant fraction of leukemias. Tumor cells of such patients often contain mutant forms of certain oncogenes, and therefore mutations in these genes are believed to be responsible for malignization of the hematopoietic system cells.
In 20% of patients with AML, leukemic cells carry a translocation between chromosomes 21 and 8 resulting in formation of the chimeric oncogene AML1-ETO producing a fused protein AML1-ETO, which has the activity of a transcription factor. Furthermore, in 70% of patients with AML the receptor tyrosine kinase KIT is hyperexpressed. Mutations of this enzyme in patients with AML do not occur very often (in ~5% of patients), but in the presence of the translocation t(8;21) the mutation frequency increases to 30%. The correlation between these two oncogenes was noted long ago. It was supposed that the presence in the cells of two activated oncogenes (e.g. the transcription factor AML1-ETO and tyrosine kinase c-kit) should trigger their malignization. However, based on recent results it is supposed that in some rare cases even one activated oncogene can induce an appearance of leukemic cells [1].
Bone marrow transplantation, radiotherapy, and chemotherapy are still the main approaches in the treatment of leukemias. Each of these approaches has significant shortcomings, including suppression of immune system, severe radiation intoxications, and appearance of secondary tumors much more resistant to all treatments. Many laboratories are now actively developing new approaches against leukemias. Inhibiting the expression of activated leukemic oncogenes based on the principle of RNA interference seems to be a promising approach that is likely to get wide distribution.
The purpose of this review is to systematize the available knowledge about two major leukemic oncogenes, existing drugs, and also about RNA interference as the most modern approach for suppression of gene activity. The structure and functions of the protein AML1-ETO and its involvement in oncogenesis were considered in detail in a review by D. Baskaran [2], therefore, we have limited it to a shorter description.
ACUTE MYELOID LEUKEMIA
Leukemia is a clonal malignant (neoplastic) family of diseases of the hematopoietic system that comprises many diseases with different etiology. Leukemias are characterized by rapid dissemination of tumor cells throughout the whole hematopoietic system. According to annual statistics of the USA, leukemias are the most frequent cause of death among young people (younger than 40 years old) [3].
By clinical course leukemias are subdivided as follows.
1. Acute leukemias start acutely, display rapid progression, and without treatment lead to death within a few months. In the blood of such patients there is usually a large number of blast cells.
2. Chronic leukemias start gradually and develop slowly, and even without treatment the patients can live for several years. Immature cells but with a tendency for maturation are usually detected in the blood.
It should be noted that the terms “acute” and “chronic” leukemias are used only for convenience – they never change one into the other. The significance of these terms in hematology is different from their significance in other medical disciplines.
Acute leukemias constitute a heterogenous group of malignant diseases of the hematopoietic system with specific damage to blood marrow by blast cells. Later (or from the very beginning) various tissues and organs can be infiltrated by blast cells. Tumor transformation in acute leukemia occurs in the stages of differentiation of pre-parental hematopoietic cells; therefore, they are subdivided based on this trait. Leukemias are subclassified into lymphoblastic ones, i.e. related to the lymphopoiesis precursors, which present 15% of all acute leukemias, and myeloid leukemias related to myelopoiesis precursors, which represent the bulk of acute leukemias in adults [4].
Acute myeloid leukemia (AML) is a malignant disease of blood arising as a result of cancer transformation and disorders in differentiation of hematopoietic cells on the level of myeloid cell precursor cells [5]. Acute myeloid leukemia is difficult to treat, and it causes 1.6% of cancer-caused deaths in the USA [3]. Although chemotherapy results in a complete remission in 65% of patients, recurrences are observed in 70% of cases within 5 years [6].
Clinical and Hematological Characterization of Patients with AML
The initial stage of appearance and development of leukemias is usually asymptomatic: the patients feel healthy until the general dissemination of tumor cells throughout the hematopoietic system. Diagnosis of acute leukemia can be established only morphologically by the presence of blast tumor cells in the blood or in the bone marrow. A decrease in numbers of blood cells because of replacement of normal hematopoietic stem cells by neoplastic cells results in anemia, thrombocytopenia, and neutropenia, which manifests as frequent infectious diseases and ulcerations on the mucosa [4].
In peripheral blood leukocytosis is observed with the presence of blasts and a so-called “leukemic gap”, i.e. a sharp increase in the number of blast cells and the presence of unique mature elements along with the absence of transitional maturing forms. In some cases the leukocyte number is not increased, but blasts are always present. If blast cells are still unable to leave the bone marrow, their number in it is rather high [4]. The diagnosis of AML can be established based on the presence of more than 30% myeloblasts in the bone marrow [5].
Classification of AML
There are two main classifications of AML: the initial FAB classification and the modern WHO classification.
In 1976 French, American, and British (FAB) hematologists developed a classification of acute leukemias based on morphological and cytochemical characteristics of the cells [4, 7]. The FAB classification subdivides AML into eight subclasses, M0-M7. Depending on the subclass, the patients have different prognoses and obtain different treatment [7]. Currently the classification looks as follows (Table 1).
Table 1. FAB classification of acute myeloid
leukemias [7]

The WHO classification of acute myeloid leukemia was developed considering the FAB system, but it is more convenient for clinical application because it takes into account the most significant prognostic signs of the disease (Table 2). Mutations described for the first group are located in the genes highly sensitive to damage caused by some chemical preparations and, thus, can be involved in the appearance of the third group AML [8].
Table 2. Subspecies of acute myeloid
leukemias according to the WHO classification [8]

Molecular Genetics of Acute Leukemia
AML appears as a result of separate cooperating mutations of different genes that increase the proliferative ability and survival and affect the normal differentiation and apoptosis of cell precursors of myeloid, erythroid, megakaryocytic, and monocytic series. All acute leukemias are clonal, i.e. originate from a single mutant hematopoietic cell, which can be either a very early one or a partially differentiated (committed) cell precursor towards different hematopoietic series. The relation of blast cells a particular hematopoietic series and the level of their differentiation then to determine the clinical course of acute leukemia, the program of therapy, and its effectiveness. Based on the major mutations and prognosis of the disease, patients with AML can be divided into three risk groups [9].
Two types of mutations are considered in AML. Class II mutations affect genes of transcription factors and lead to changes in their functions or activity and influence cell differentiation. The most frequent mutations of class II in AML are the translocation t(8;21)(q22;q22) and inversion inv(16)(p13q22). On the molecular level the translocation t(8;21) and the inversion inv(16) result in appearance of fused proteins AML1-ETO and CBFβ/MYH11, which are transcription factors [10]. Such mutations are necessary but insufficient for tumor transformation. To acquire the tumor phenotype, secondary genetic rearrangements are required that are called class I mutations. Mutations of class I occur in the genes of tyrosine kinases, lead to their constitutive activation, and influence cell proliferation and survival. In AML mutations most often occur in the genes of tyrosine kinases KIT and FLT3 [11].
ACTIVATED LEUKEMIC ONCOGENES
AML1-ETO
Involvement of CBF in appearance and progression of acute myeloid leukemias. CBF is a heterodimeric transcription factor consisting of subunits CBFα (AML1) and CBFβ. CBF regulates expression of a number of genes involved in hematopoiesis [12]. The AML CBF group includes leukemias specified by mutations in the genes encoding subunits of this complex. These mutations include the above-mentioned translocations between the 8th and 21st chromosomes [t(8;21)(q22;q22)] and the pericentric inversion in chromosome 16 [inv(16)(p13q22)]. In adults with de novo developed AML, the translocation t(8;21) is observed in 7% of cases and the inversion inv(16) in 8% of cases [10]. Chimeric genes produced as a result of cytogenetic anomalies contribute to AML progression [13].
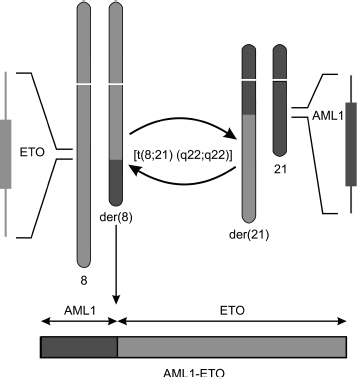
The translocation [t(8;21)(q22;q22)] affects gene AML1 (or RUNX1, CBFα) on the 21st chromosome and gene ETO (or MTG8) on the 8th chromosome. This translocation results in formation of a chimeric gene that encodes the fused protein AML1-ETO (Fig. 1).
Transcription factor AML1. AML1 as a component of CBF complex. The AML1 gene codes the DNA-binding α-subunit of transcription complex CBF. The conservative Runt Homology Domain (RHD) localized on the N-terminus of AML1 protein is necessary for binding with DNA and cofactor CBFβ [14]. The C-terminus of protein AML1 contains trans-activating and repressor domains. The heterodimeric complex AML1–CBFβ binds with DNA at a definite site with consensus sequence PyGPyGGTPy (where Py is the pyrimidine base cytosine or thymine). Protein CBFβ strengthens AML1 binding with DNA and also stabilizes AML1, protecting it against ubiquitin-dependent proteolysis [15]. CBF regulates transcription of various genes required for differentiation of cells of the myeloid and lymphoid series through binding with other transcription factors depending on the promoter context [14].Fig. 1. Scheme of translocation t(8;21)(q22;q22) resulting in formation of fused gene AML1-ETO. One copy of gene alleles remains unaffected, which must be taken into account on studies of functions and action mechanism of AML1-ETO protein.
Interactions of AML1 with other proteins. 1) AML1 as a transcription activator. The majority of activated genes are associated with hematopoiesis: genes of T- and B-cell receptors, interleukin-3 (IL-3), macrophage colony-stimulating factor receptor (M-CSFR), and genes of neutrophil elastase, myeloperoxidase, and granzyme B. During the transcription activated via the CBF complex a crucial role is played by protein–protein interactions with involvement of the C-terminus of the full-size protein AML1. CBF is a scaffolding protein responsible for assemblage of transcription factors in promoter regions of genes, although it is unable to activate transcription. For operating, it needs the contribution of other transcription factors such as AP-1, Myb, C/EBPβ, and/or proteins of the Ets family [16]. The complex of transcription factors assembled on CBF attracts additional cofactors such as p300, CREB-binding protein, and the “yes”-associated protein-1 (yap1), the binding with which is also controlled by AML1. These cofactors in turn acetylate histones and uncoil condensed DNA making the gene promoter regions available for RNA polymerase, which acquires the ability to effectively initiate transcription [17].
2) Protein AML1 as a transcription repressor. The AML1 protein is a context-dependent transcription repressor. In particular, it inhibits transcription of genes CD4, p21WAF-1, HERF1, MRP14, Pim-2, Stefin-3, and the gene of uridine phosphorylase and of bone sialoprotein [18]. The best known functioning of AML1 as a transcription repressor is inhibition of the expression of gene CD4 in CD4+CD8+ thymocytes during T-cell differentiation. Binding with the silencer region of the gene CD4, the protein AML1 attracts to this locus the chromatin-remodeling complex BAF [19]. In other cases the repression by AML1 is due to binding with transcription corepressors that form complexes with histone deacetylases (HDAC). These enzymes suppress the gene expression due to deacetylation of histones that promotes condensation of chromatin. Some of HDAC proteins can directly interact with AML1 protein and thus repress transcription [16].
3) Protein AML1 in epigenetic silencing. In drosophila and mice the protein AML1 is also involved in epigenetic gene silencing [16]. Protein SUV39H1 is a specific methyltransferase of histone H3 lysine 9. Methylation of histone H3 lysine 9 is a posttranslational modification that allows histone H3 to bind with protein HP1 and causes gene silencing. Co-immunoprecipitation in cell line Cos7 revealed an association of proteins AML1 and SUV39H1 [20].
Transcription factor ETO family. Protein structure. The protein family (Eight-Twenty One) ETO comprises: 1) ETO (MTG8); 2) ETO2 (MTG16, MTGR2), and 3) MTGR1. All members of the ETO family, similarly to the AML family members, are involved in activation of transcription and are scaffolding proteins [21]. These proteins are characterized by the presence of four evolutionarily conservative Nervi Homology Region domains (NHR 1-4) [22]. The domain NHR1 is responsible for protein–protein interactions and is a homolog to the TATA-box of drosophila. Domain NHR2 contains heptade repeats of hydrophobic amino acids and is necessary for homo- and heterodimerization of the ETO family proteins. Domain NHR3 has coiled-coil structure and seems to be involved in interaction with some corepressors together with NHR4. Domain NHR4 contains two zinc finger motifs on the C-terminus (CxxCCxxC and CxxCHxxC) that are involved in protein–protein interactions. It was recently shown that nonconservative regions of the ETO protein (NHR outsiders) are also involved in its interactions with corepressors of transcription [23].
Main interactions of ETO with other proteins. a) Histone deacetylases. Deacetylation of histones influences their interaction with key regulators of translation. Consequently, the HDAC proteins can regulate gene expression and arrange the so-called “histone code”.
ETO recruits HDAC proteins by directly interacting with them (mainly due to the NHR2 and NHR4 domains) or binding with components of complexes, which include HDAC. The HDAC proteins are components of multiprotein complexes such as N-CoR, SMRT, or Sin3. Both N-CoR and SMRT can locally deacetylate histones “labeled” by DNA-binding transcription regulators. Sin3 seems to be involved in global genome deacetylation. HDAC is an enzymatic component of the complex, whereas other proteins seem to regulate or direct the HDAC proteins [23].
b) Transcription repressors PLZF and Gfi-1. According to the corepressor model, protein ETO interacts with transcription receptors that are specific to the nucleotide sequence. By now two such repressors are known, PLZF and Gfi-1. ETO interacts with PLZF directly [24]. PLZF is found in hematopoietic cells where it functions as a growth suppressor, and its expression decreases during myeloid differentiation [25].
In both in vivo and in vitro experiments protein ETO has been shown to interact with transcription repressor Gfi-1, which is also expressed in myeloid cells. Gfi-1 is supposed to regulate hematopoiesis and influence the viability of hematopoietic cells [26].
c) ETO–ETO interactions. On interacting with each other, proteins of the ETO family can form high molecular weight oligomers. Oligomerization of ETO increases its affinity for proteins N-CoR, SMRT, and other corepressors. Homooligomerization seems to be a prerequisite for functioning of the fused oncoprotein AML1-ETO [27].
Fused protein AML1-ETO. The chimeric gene AML1-ETO is produced as a result of the translocation t(8;21) that leads to fusion of gene AML1 on chromosome 21q22 with the gene ETO on chromosome 8q22. Breakpoints in this molecular rearrangement are constant and occur in exon 5 of the AML1 gene and in exon 2 of the ETO gene [11]. The gene AML1-ETO encodes the fused protein, which is a transcription factor. Protein AML1-ETO contains the N-terminal region of AML1 protein, which includes the DNA- and CBFβ-binding RHD domain, whereas the C-terminal part belongs to ETO protein including its four NHR domains [28].
The role of protein AML1-ETO in the progress of leukemia is explained by the dominant negative influence of protein AML1. The “knock in” phenotype of mice expressing the fused gene AML1-ETO is identical to the phenotype of mice with homozygous knockout of the AML1 gene (mice with disorders in hematopoiesis) [28]. The fused protein AML1-ETO is shown to inhibit the differentiation of cells of the myeloid and erythroid series [1].
AML1-ETO as a repressor of transcription. AML1 activates transcription promoting differentiation of granulocytes due to transactivation of series-specific target genes. Formation of the fused protein AML1-ETO results in the replacement of the AML1 activation domains by the ETO repressor domains. The fused protein AML1-ETO binds with DNA via the RHD domain from the AML1 moiety. Then the fused protein via the NHR domains of ETO effectively recruits corepressors, which in turn recruit HDAC. Due to activity of HDAC proteins the gene transcription is suppressed. As a result, the fused protein inhibits the expression AML1 target genes instead of activating them [21].
AML1-ETO as an activator of transcription. Among 24 target genes of AML1-ETO only 10 genes were targets for AML1 [29]. This means that protein AML1-ETO can influence expression of genes that under normal conditions are not controlled by AML1 protein. Later AML1-ETO was shown to activate and repress the expression of nearly the same numbers of genes. The trans-activated target genes are exemplified by: 1) Jagged1, which is a Notch-ligand responsible for proliferation of stem cells; 2) Plakoglobin, which acts as a mediator in the Wnt-signaling pathway; 3) β-catenin, which is also involved in Wnt-signaling; 4) receptor of nerve growth factor RKA in human CD34+ hematopoietic precursor cells; 5) antiapoptotic gene Bcl-2; 6) C/EBPe gene, the product of which trans-activates gene G-CSFR expression via interaction with C/EBP binding site in regulatory regions of DNA [1, 21]. All target genes in the regulatory regions have binding sites with AML1. Many genes whose expression is activated by protein AML1-ETO are involved in the self-renovation of stem cells. Due to transactivation of such genes, the fused protein AML1-ETO provides for the expansion of multipotent precursor cells.
Protein AML1-ETO is a suppressor of cell differentiation. It was recently supposed that the progress of acute leukemias could be caused by the loss of function of series-specific transcription factors. Under the complete loss of the function the cells die, but a partial loss results in disorders of homeostasis of hematopoietic stem cells sufficient for development of leukemias [30]. In particular, disorders in the series-specific determination of hematopoietic stem cells cause expansion of the compartment of self-renovated precursor cells, which can change to tumor cells on the appearance of additional mutations. Via direct protein–protein interactions (through RHD or ETO domains) the fused protein AML1-ETO can suppress transcription factors (E-proteins, PU.1, C/EBPα, and GATA-1) that play a key role in differentiation of cells of the hematopoietic series [21]. AML1-ETO is also shown to inhibit expression of genes involved in excision repair of DNA and thus, possibly, to increase genetic instability within the cell population. It seems that the suppression by protein AML1-ETO of differentiation of hematopoietic cells is not absolute, but the continuous expansion of precursor cells on the background of genetic instability can serve a basis for additional secondary mutations [31].
Functions of AML1-ETO protein. Studies on functions of fused proteins containing the protein moiety AML1 or CBFβ have shown the involvement of these proteins in appearance of populations of preleukemic cells, which manifests itself by accumulation of early cell precursors with a decreased ability for differentiation but without a pronounced leukemia. Studies on mice have confirmed that mutations in gene AML1 are initiating events in development of acute leukemias, although for their progress secondary mutations are required [14, 33].
The fused protein AML1-ETO is a multifunctional protein that plays an important role in the regulation of various cell programs such as differentiation, proliferation, apoptosis, and self-renovation in both in vitro and in vivo models. This protein regulates target genes of AML1 and also other target genes because it interacts with different transcription regulators. Moreover, for development of AML additional mutations are required. An unanswered question is what genes are crucial targets for the fused protein and what molecular pathways jointly with AML1-ETO are involved in the neoplastic transformation of hematopoietic cells. A huge amount of new information about the human and mouse genome and also new technologies are promising for a significant simplification of studies on these interesting problems [1].
Receptor Tyrosine Kinase KIT
SCF is allelic with SL-locus, and c-kit is allelic with W-locus. Many years ago it was noted that mutations in the W-locus (the dominant white spotting locus) on the fifth mouse chromosome or in the Steel (Sl) locus on the tenth chromosome are manifested similarly: the mice are characterized by an affected color of the fur, macrocytic anemia, and sterility. Similar phenotypic manifestations of mutations in different loci led to the conclusion that products encoded by these regions of the chromosomes must perform common functions together [33]. Later this hypothesis was confirmed. In 1986 a viral acute transforming oncogene v-kit was discovered, which is expressed by the Hardy–Zuckerman 4 feline sarcoma virus [34]. A year later its cellular homolog c-kit was found encoding protein KIT, which is a receptor tyrosine kinase. In 1988 c-kit was mapped to the W-locus of mice, and then a KIT ligand, the stem cell factor (SCF) encoded by the Sl locus, was found [35]. These pioneer studies proved that the SCF/KIT system influences melanogenesis, gametogenesis, and hematopoiesis [34].
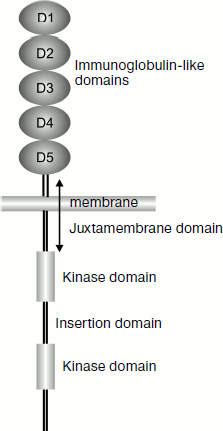
KIT as a type III receptor tyrosine kinase. Gene c-kit encodes the protein KIT (or CD117 with molecular weight of 145 kDa), which is a cytokine receptor and is expressed on the surface of hematopoietic stem cells and of many other cells. KIT is a type III receptor tyrosine kinase of the monomeric receptor family. This group also includes platelet growth factor receptor (PDGF-R), macrophage colony-stimulating factor receptor (M-CSF-R), and also fms-like tyrosine kinase-3/embryonic liver kinase-2 (Flt-3/Flt-2). This class of kinases (also called the PDGF-R family) plays a key role in the regulation of hematopoiesis and embryogenesis [34]. The class III tyrosine kinases are characterized by the presence of five extracellular immunoglobulin-like domains responsible for binding with the ligand (Fig. 2). These kinases are discriminated from other types of tyrosine kinases by the presence of an insertion consisting of 70-100 amino acids in the kinase domain (80 amino acids in the case of KIT). The insertion domain can be phosphorylated and serve a binding site for some important signaling molecules [36].
Structure of c-kit gene. The gene c-kit is located in locus q11-q12 of the human fourth chromosome [37]. In mice it was mapped in the W-locus of the fifth chromosome [38]. The gene consists of 21 exons and 20 introns. The exons are small in size – about 100 bp, except for exon 21 that has a size of 2407 bp. As a result, no more than 6% of sequences of mRNA containing more than 89 kb can code proteins [39]. The first exon contains the 5′-untranslated region (5′-UTR), the initiation site, and a signal for translocation across the membrane. The extracellular domain is coded in exons 2-9 and in the 5′-end of exon 10. The transmembrane domain is located in exon 10. The cytoplasmic domain is coded in the 3′-end of exon 10 and in exons 11-20. They also contain kinase domains separated by the insertion domain, which includes the 3′-end of exon 14, exon 15, and the 5′-end of exon 16. Exon 12 contains an ATP-binding site (Gly-X-Gly-X-X-Gly). Exon 21 contains the site of translation termination and 3′-UTR [40].Fig. 2. Scheme of location of protein KIT domains.
The promoter region of the c-kit gene includes no typical TATA-box specific for many eukaryotic promoters and has about 70% G/C-composition similarly to the housekeeping gene promoters and to genes of some tyrosine kinases such as EGFR, c-fms, and insulin receptor [41]. The promoter regions of human and mouse are highly homologous (about 75% of the sequences). The binding sites of transcription factors are also conservative [42].
Known forms of KIT and alternative splicing. In humans there are four protein isoforms that are produced due to alternative splicing. Two of them are discriminated by the presence (or absence) of the tetrapeptide GNNK in the extracellular part of the juxtamembrane domain. Two other forms are characterized by the presence or absence of a single serine residue within the insertion domain of the KIT protein. In embryonic cells of mouse testes a shortened form of c-kit (truncated c-kit, tr-kit) is expressed from the cryptic promoter [43]. Such a shortened KIT is unable to phosphorylate proteins but can transmit a signal. Microinjections of tr-kit into mouse oocytes trigger in them the transition from metaphase to anaphase [44].
A soluble form of KIT is also found that seems to be a result of proteolysis, although the protease itself involved in the detachment of the extracellular domain has not been identified. The contribution to this process of protein kinase C [45], tumor necrosis factor α-converting enzyme (TACE) [46], and matrix metalloproteinase-9 (MMP-9) [47] was shown using indirect approaches. The soluble form of the receptor, similarly to its membrane-bound form, is glycosylated and binds with SCF [48, 49], and this leads to its competition with the membrane-bound KIT. This results in suppression of SCF-dependent growth and, possibly, in mobilization of colony-forming blood cells and their departure from the bone marrow [47, 50].
Signal transmission involving KIT. Interactions between stem cell factor (SCF) and receptor tyrosine kinase KIT. The binding with SCF occurs with involvement of three extracellular domains of KIT (D1, D2, and D3) with a spatial position arranged like the letter “Γ”. This structure is rigid, with strictly determined angles that are supported by large interdomain interactions. Amino acid residues involved in these interactions are conservative in mammals, and therefore the spatial structure of the domains and their mutual orientation also remain conservative. This results in consolidation of the spatial orientation of the D1 domain loop C′ involved in the interaction with SCF [34].
The region of interaction between SCF and KIT includes three areas (D1/SCF, D2/SCF, and D3/SCF) that are supported by a large number of salt bridges and hydrogen bonds. These interactions provide for SCF⋅KIT dimer formation. This is associated with significant conformational changes in SCF—unstructured or poorly structured areas become clearly visible. Then the dimers dimerize with production of tetrameric complexes 2⋅(SCF⋅KIT). Only SCF molecules contribute to production of tetramers, the molecules being joined “head-to-head” [34].
The receptor dimerization induces its enzymatic activity. Autophosphorylation results in modification of some tyrosine residues (especially Tyr568 and Tyr570) that are outside the kinase domains and act as binding sites for signal-transmitting molecules containing Src homology 2 (SH2) or phosphotyrosine binding (PTB) domains [51].
Regulation of KIT activity and signal termination. Upon phosphorylation the receptor can be ubiquitinated by ubiquitin E3 ligases, among which protein Cbl is the most important. This protein binds with activated KIT using adaptor proteins and is phosphorylated by SFK. It seems that just the SFK-dependent phosphorylation of Cbl and the subsequent ubiquitination of KIT lead to internalization of the receptor and its degradation in lysosomes [35].
SHP-1 and SHP-2 are the main tyrosine phosphatases regulating KIT activity. KIT is inactivated under the influence of SHP-1, whereas SHP-2 stimulates the signal transmission. Other phosphatases are also involved in regulation of KIT activity but their role is still unclear [35].
The juxtamembrane (JM) domain functions as an intramolecular inhibitor. This domain of KIT has a stable secondary structure and is tightly bound with the ATP-binding region of the receptor. In this position KIT is inhibited and cannot phosphorylate proteins. Phosphorylation of the JM domain changes its conformation and abolishes the inhibition of the receptor [52].
Signaling pathways triggered by KIT. The activation of PI3K under the influence of KIT is associated with cell division, differentiation, adhesion, secretion, survival, and reorganization of the cytoskeleton. KIT stimulates cell survival via the PI3K-dependent activation of Akt and phosphorylation of Bad, which is a proapoptotic protein. On SCF stimulation the levels of antiapoptotic proteins Bcl-2 and Bcl-xL increase and the amount of proapoptotic factor Bax decreases [53]. The physiological role of the KIT-dependent activation of PI3K was shown by two independent studies using transgenic mice expressing a mutant c-kit. The loss of PI3K kinase activity appeared to be critical for gametogenesis—the mice were sterile [35].
The ligand-dependent activation of KIT leads to rapid activation of Src family kinases (SFK). In the cell this family of kinases is responsible for survival, chemotaxis, adhesion, proliferation, and migration. The major site of SFK binding with KIT is tyrosine Y568. In 2004 experiments were performed on mice expressing KIT with tyrosine in position 568 substituted by phenylalanine (Y568F). In these mice T- and B-cell differentiation was inhibited. The involvement of SFK in the development of lymphocytes could be supposed if Y568 was not also a binding site for other signaling molecules [35].
The stimulation of SCF is associated with activation of the signaling cascade Ras/Erk, which plays an important role in cell division, survival, and tumor transformation. KIT also triggers other signaling pathways such as the JAK/STAT pathway, activates phospholipase C-γ, and also interacts with various adaptor proteins [34].
KIT interacts with tyrosine phosphatases of SHP-1 and SHP-2 proteins. The activation of SHP-2 is necessary for full activation of the Ras/Erk pathway. The role of SHP-2 is established in differentiation of embryonic cells and hematopoiesis. KIT is also shown to interact with transcription factors Mitf and Slug [35, 54, 55].
Expression and role of c-kit in normal hematopoiesis. The KIT/SCF system is involved in hematopoiesis. KIT is expressed in hematopoietic stem cells in the bone marrow of adults (in ~8% of all bone marrow cells) and in peripheral blood (in 0.1% of cells) [56, 57]. The c-kit gene is also expressed in more committed precursors: erythroid, myeloid, and also in precursors of megakaryocytes [58], mast cells [59], and natural killers. Moreover, KIT is involved in the early development of T- and B-lymphocytes [60, 61].
Studies in vitro have shown that SCF supports the viability of stem cells, but additional factors are required for proliferation. Together with other cytokines such as erythropoietin (Epo), IL-1, IL-3, IL-6, IL-7, GM-CSF, G-CSF, etc., SCF promotes the amplification of committed cell precursors. It seems that differentiation of cell precursors into a particular cell species depends not only on SCF but also on additional factors. Thus, SCF together with EPO lead to an increase in the population of erythroid precursors, and SCF together with IL-7 or GM-CSF lead to expansion of myeloid precursors [62].
c-kit and AML. Expression and mutations of c-kit in tumor cells. KIT is involved in such processes important for tumor progression as stimulation of cell proliferation, decrease in sensitivity to apoptotic signals, and cell migration and adhesion. Hyperexpression or mutations of KIT are generally observed in the majority of patients with AML, MDS/MPD, gastrointestinal stromal tumor (GIST), and mastocytosis. It must be taken into account that the decrease in sensitivity to apoptosis promotes the appearance of drug resistance [63].
The role of KIT in oncogenesis is twofold. On one hand, in many tumors KIT is constitutively activated [64], and on the other hand the loss of KIT functions is associated with development of thyroid gland carcinoma [65].
The constitutive activation of the receptor can occur either by means of an activating mutation, through an autocrine loop, or via a paracrine activation. The autocrine loop implicates the concurrent expression by the cells of KIT and SCF. Such mutations were found in small cell pulmonary cancer [66] and in GIST [67].
Activating mutations of KIT in tumors can occur in the juxtamembrane (JM) and kinase domains. As mentioned, the JM domain in wild type KIT inhibits the receptor due to interaction with the ATP-binding region. Mutations in the JM domain of KIT diminish the time required for the activation of KIT. But the substrate specificity of the receptor and its binding constant for ATP are retained [23]. Mutations in the JM domain of KIT are observed in different tumors such as in AML and GIST [68, 69].
Molecular modeling showed that mutations in the kinase domain of KIT and Flt-3 (the structurally related RTK) failed to directly stabilize the active conformation of the enzyme. The balance was shifted towards the activated form of the phosphotransferase domain due to a significant destabilization of the inactive conformation of the kinase [70]. Such mutations were shown for mastocytosis [68, 71, 72], AML [68, 72], and GIST [68, 69].
Expression of c-kit in AML. Normally KIT is expressed only in non-differentiated or in poorly differentiated hematopoietic cells. But in patients with AML the c-kit gene product is expressed on the surface of fully differentiated cells [73]. Studies in vitro revealed that different cell lines of AML carried products of the c-kit gene. In these cells the degree of receptor phosphorylation correlated with the proliferation level [74, 75]. Introduction of the c-kit gene into cells enhanced their proliferative potential [76]. These data suggest that the activation of KIT in tumor cells should play a role in the excess proliferation and disorders of blood cell differentiation during the progress of AML.
In 63-85% of cases mature cells of patients with AML are KIT-positive [68, 77-83]. Most often expression of c-kit is observed in the cells of patients with AML of the M0, M1, and M2 types. In type M5 and M7 the expression of KIT is less frequent [77-79, 81, 82]. Patients with KIT expression in tumor cells suffer higher mortality [8, 84].
Mutations of the c-kit gene in acute myeloid leukemia. An increased proliferation of cells during carcinogenesis appears either as a result of mutations in the c-kit gene leading to constitutive activation of the receptor or as a result of hyperexpression of the receptor. Mutations of the c-kit gene are relatively infrequent in patients with AML (6% of all patients with AML) [85, 86] and are observed mainly in AML cases with presence of t(8,21) or inv(16) (30% of patients with CBF-AML) [11, 87].
The most frequent mutation is substitution of aspartic acid in position 816 of the KIT kinase domain (exon 17) by D816V, D816I, D816P, D816N, D816Y, and D816H [8, 88, 89]. Mutation of this amino acid leads to constitutive activation of the receptor [49]. Another activating mutation N822K was recently found in exon 17 [51, 74]. Mutations leading to deletions and insertions in exon 8 of the c-kit gene are also frequent [87]. Mutations in exon 8 affect immunoglobulin domains of the receptor. They manifest themselves in hyperactivation of the receptor in response to the action of stem cell factor (SCF) [90]. Tandem repeats in exons 11 and 12 (the receptor juxtamembrane domain) also are mutations leading to constitutive activation of KIT [91].
Drugs used for suppression of KIT in AML. The KIT protein is involved in such processes important for tumor development as cell proliferation, loss of sensitivity to apoptotic signals, migration, and adhesion. Hyperexpression or KIT mutations are constantly observed in the majority of patients with AML, MDS/MPD, GIST, and mastocytosis. It must be taken into account that the decrease in sensitivity to apoptosis promotes the appearance of drug resistance [66]. All these findings have stimulated development of various chemical preparations for inhibition of the activity of this protein.
Gleevec® (STI571) is an inhibitor of tyrosine kinases specific to BCR-ABL, PDGFR, and KIT [92]. Studies on model cell lines indicated a significant decrease in the proliferation and suppression of antiapoptotic effect caused by the activation of KIT. On fresh blood samples taken from patients these effects were noticeably worse [93, 94]. Clinical testing did not show therapeutic effect of STI571 in KIT+ patients with AML [95-99].
Studies on crystalline structure of the receptor and inhibitor have shown that STI571 cannot be an ideal inhibitor of KIT. It acts as a competitive inhibitor by binding with the ATP-binding site and prevents the activation of the receptor. But the molecule of STI571 is too large and destroys inside the receptor the bonds that are responsible for the inactivated conformation [50].
SU6668 and SU5416 are low molecular weight inhibitors of tyrosine kinases VEGFR-1, VEGFR-2 (KDR), VEGFR-3, KIT, and FLT3 [100-102]. A study on a model cell line and on blood cells from patients with AML has shown the efficiency of these inhibitors—the proliferation decreased and the sensitivity to apoptosis increased [103]. In experiments on mice SU5416 and SU6668 inhibited metastasizing, microvascularization, and cell proliferation [104]. Clinical testing was also successful [105, 106], but it is still unclear whether these positive results are due just to inhibition of KIT or are caused by inhibition of vascular endothelium growth factor receptor (VEGFR).
Dasatinib (BMS-354825) is a specific inhibitor of Src and BCR/ABL kinases that displayed good results in clinical testing on patients with chronic myeloid leukemia [107]. In cell lines Dasatinib inhibited the ligand-dependent phosphorylation of KIT and cell proliferation [108, 109]. Clinical testing on four patients with KIT+ AML was also promising: in all patients the blood picture significantly improved and in one patient a complete remission was achieved upon addition of other preparations [110].
Sorafenib was initially developed as an inhibitor of tyrosine kinases C-RAF and B-RAF, but later it was shown to also inhibit other tyrosine kinases including KIT, FLT3, PDGFR, and VEGFR. On model cell lines Sorafenib inhibited the ligand-dependent phosphorylation of KIT and cell proliferation [111]. This drug is now used for treatment of kidney and liver cancers; promising results were also obtained in the treatment of thyroid gland carcinoma [112].
Sunitinib (SU11248) is a low molecular weight inhibitor of tyrosine kinases including FLT3, VEGFR, and PDGFR [113]. On model cell lines SU11248 inhibited the ligand-dependent phosphorylation of KIT and cell proliferation [111]; in patients with AML Sunitinib induced a short-term remission [113, 114].
Other inhibitors of tyrosine kinases such as Ki11502, PD180970 and MLN518, PKC412, and ABT-869 that have a particular influence on KIT are now being subjected to preclinical testing [115-118].
RNA interference based on suppressing gene expression on the posttranscriptional level using short oligonucleotides seems to be a promising modern approach for the treatment of tumors.
RNA interference is now the most popular approach for gene knockdown, the approach being easy to perform, inexpensive, relatively nontoxic, and highly specific. During recent years siRNA technology has been successfully used in genomics to study functions of genes and their interactions and also to search for new pharmaceutical preparations. RNA interference is thought promising as a basis for new biomedical approaches against various diseases including tumors. The therapeutic use of interfering RNAs is mainly prevented by difficulties in their delivery into target cells. Systems with lentiviral vectors are now used for gene delivery and expression in cells of higher animals and humans both in vitro and in vivo. Such vectors carry as a target gene a precursor of small interfering RNAs (shRNA), which are converted into siRNA under the influence of intracellular mechanisms.
INTERFERENCE
History of the Problem
The phenomenon of RNA interference was discovered in 1990 in an attempt to create a new petunia cultivar with brighter flowers. The researchers introduced into petunia cells a construction that encoded the gene chs. The enzyme chalcone synthase produced by this gene is responsible for synthesis of a violet pigment. However, instead of the desired enhancement of the color, flowers of the transgenic plants were either entirely white or had white spots or sectors. The cells of these plants contained a decreased concentration of mRNA of both endogenous and exogenous chs genes. This phenomenon was called co-suppression [119]. In 1994 another group of researchers showed on the example of the petunia chs gene expression that, although the cytoplasm of these plants lacks the mRNA of the chs gene, the transcription of the gene in the nucleus is retained at the former level [120]. Thus, it was concluded that the co-suppression should occur at the posttranscriptional level. After many cases of posttranscriptional degradation of RNA in plants were detected as a result of injections of plant, bacterial, and viral transgenes, co-suppression was re-termed posttranscriptional gene silencing (PTGS).
In 1999 small RNAs were shown to be involved in PTGS: in plant cells with PTGS induced by injection of dsRNA short RNA molecules were formed (about 25 nucleotides in length) with sense and antisense polarity, whereas no such molecules were observed in the control plants [121]. Then similar molecules were detected in drosophila upon initiation of PTGS by long dsRNAs [122]. Small RNAs involved in PTGS were called small interfering RNAs (siRNAs).
The discovery of interference induced great interest and stimulated intensive studies in this field. Rather soon it was found that the silencing phenomenon occurred not only in plants, but also in fungi and animals [123, 124]. During recent years the molecular mechanism of RNA interference has been studied, and it was shown to be a fundamental and evolutionarily conservative mechanism of regulation of the gene expression and became widely used in molecular biology as a tool for analyzing gene functions.
Mechanism of RNA Interference
RNA interference is initiated by appearance in the cell of exogenous or endogenous dsRNA. The interference efficiency correlates with the length of dsRNA: the longer is dsRNA, the greater amount of siRNA is produced and the greater number of target sites will be recognized on the mRNA molecule. The minimal size of dsRNA sufficient for inducing interference is 26 bp. Most likely this limitation prevents degradation of the cell’s own mRNA with short intramolecular self-complementary structures [125].
The dsRNA is recognized and cleaved by the enzyme Dicer from the family of type III RNases [126]. This protein is conservative in protozoans, plants, fungi, and animals; Dicer cleaves long dsRNAs from both ends [127]. This produces short dsRNAs of 21-28 nucleotides in length (a species-specific trait) [128]. These fragments contain on the 3′-ends two projected unpaired nucleotides. The 3′- and 5′-ends carry, respectively, hydroxyl and phosphate groups. And just such a structure of the enzyme is required for involvement in the further stages of the process leading to the RNA silencing [129].
In the next stage a RISC-loading complex (RLC) is produced. To realize this, to the enzyme Dicer containing a fragment of dsRNA a special protein (TRBP in human and R2D2 in Drosophila) is joined. RLC is involved in two very important processes: the choice of the chain for participation in the subsequent events and also the transfer of this chain onto the RISC complex.
In the RLC complex RNA has a specific position: Dicer binds the end of dsRNA with the lower melting temperature and R2D2 binds the opposite end. The location of the RNA fragment in RLC seems to predetermine its location in the protein Argonaut and what chain will be a guide and what chain will be destroyed without participation in the subsequent events (passenger chain). The chain with the lower melting temperature of the 5′-end becomes a guide [130].
The RLC transfers these double-stranded siRNAs onto one protein of the Argonaut (Ago) family, which is the major protein of the RISC complex (Fig. 3). One of the Ago protein domains cleaves the passenger chain between the ninth and tenth nucleotides from the 5′-end of the guide, and after this the passenger chain leaves the complex, and the guide remains within the functionally active RISC complex [131].
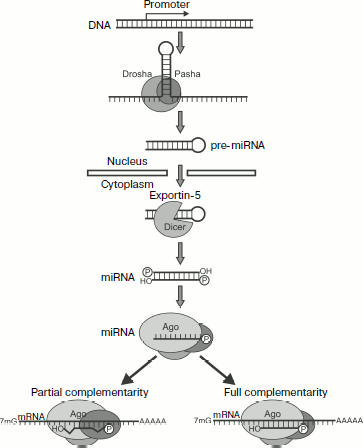
Types of Small RNAsFig. 3. Biogenesis and action mechanism of miRNA [132]. The first stage of miRNA processing occurs in the nucleus. The transcription of a gene results in primary transcripts that contain hairpin-producing self-complementary regions. These transcripts are cut by a microprocessor complex (Pasha and Drosha proteins) producing pre-miRNA of ~70 nucleotides. Then pre-miRNAs under the influence of exportin-5 are transferred into the cytoplasm to be processed by Dicer (which cuts off the hairpin loop) and Ago enzymes. As a result, a mature RISC complex is produced that contains one chain of miRNA. The further development of events depends on the degree of homology between miRNA and the target mRNA. In the majority of animal miRNAs studied there is no complete correlation to the nucleotide sequence of the target mRNA; they usually bind with the 3′-UTR of mRNA and act as translation inhibitors. In plants miRNAs usually display greater homology with mRNA, and therefore they more frequently act via induction of PTGS.
These molecules are virtually indistinguishable biochemically and functionally, and thus they are classified based on their precursors.
siRNAs are small interfering RNAs of 21 nucleotides length formed from long double-stranded RNA molecules (dsRNA).
miRNAs are small interfering RNAs of ~22 nucleotides length formed from intramolecular double-stranded structures (hairpins) of RNA precursors [132, 133].
piRNAs are small one-stranded RNAs of 24-31 nucleotides length specific for sex cells; they are produced without the involvement of Dicer [133].
miRNAs. miRNAs are found in both plant and animal kingdoms. The genes of miRNAs are usually combined in clusters and transcribed as total polycistronic units (TU), but there are exceptions when the miRNA gene possesses its own promoter [134]. Biogenesis of these molecules is the most completely studied in animals (Fig. 3). The first stage of miRNA processing occurs in the nucleus. The transcription of the gene results in primary transcripts that contain self-complementary regions producing hairpins. These transcripts remaining inside the nucleus are cleaved by a microprocessor complex with production of pre-miRNA of ~70 nucleotides.
The microprocessor complex includes two proteins: 1) Drosha, which like Dicer is a member of the type III family of RNases [135]. This protein cleaves the primary transcript; 2) Pasha (for D. melanogaster and C. elegans; its human analog is DGCR8), which contains two dsRNA-binding domains. DGCR8 binds with transcript at the hairpin base and helps the enzyme Drosha to cleave the stalk at the distance of 11 bp from the base. This results in production of pre-miRNAs (Fig. 3). Most likely the cleaving occurs co-transcriptionally and is coupled with splicing. In addition to classic miRNAs the so-called “mirtrons”, or small introns, are described, which form hairpins cut as a result of splicing; in fact, they already are pre-miRNAs and do not require Drosha for being cut [134].
Then pre-miRNAs are transferred by exportin-5 into the cytoplasm where they are processed by Dicer (which cuts off the hairpin loop) and Ago by the above-described mechanism. As a result, a mature RISC complex is produced that contains one chain of miRNA (Fig. 3) [136]. It should be remembered that the majority of miRNA duplexes contain noncomplementary nucleotides that prevents their cutting by the enzyme Ago. In this case the passenger chain is believed to be removed by RNA helicase [134].
The further development of events depends on the degree of homology between miRNA and the target mRNA. In the majority of animal miRNAs studied there is no complete correlation with the nucleotide sequence of the target mRNA; they usually bind with the 3′-UTR of mRNA and act as translation inhibitors [133, 137]. In plants miRNAs are usually more homologous to mRNA, and therefore they more frequently act via induction of PTGS [138].
siRNAs. Endogenous siRNAs are most frequently produced from complementary pairs of transcripts encoded by transposons, repeated elements of the genome, or, in the case of incomplete complementarity between transcripts of mRNAs of different proteins. Another type of siRNA precursors is one-stranded self-complementary transcripts, which form long hairpin structures (for miRNA the hairpin stalk length is markedly less). All the above-listed double-stranded precursors of small RNAs are produced in the nucleus, and upon leaving it they are subjected to processing with involvement of the Dicer and Ago proteins, and afterword remain within the RISC complex where they perform their functions in PTGS [133].
In plants many different siRNAs are found as well as proteins involved in their production. Thus, Arabidopsis thaliana possesses four Dicer-like (DCL) proteins and 10 proteins of the Argonaut family, each performing unique functions. Most likely this diversity is associated with the necessity for immovable organisms to struggle against biotic and abiotic stresses. Plant siRNAs are also characterized by methylation of the 3′-end by the enzyme HEN1. Moreover, the RNA-dependent RNA polymerase (RdRP) is involved in production of endogenous siRNAs. Plant siRNAs are classified as follows:
1) casiRNAs are produced from transposons and tandem repeats; they contribute to chromatin reorganization;
2) tasiRNAs are produced in association with processing of miRNAs;
3) natsiRNAs are synthesized in response to stress. They are produced from a pair of partially complementary transcripts, one of which is usually expressed constitutively and the other only under stress conditions;
4) lsiRNAs (“long” siRNAs) similarly to natsiRNAs are produced from complementary pairs of transcripts, and their synthesis is induced by stress, but they are noticeably longer and include 30-40 nucleotides [139].
The genomes of mammals and drosophila do not encode RNA-dependent RNA polymerases (RdRP); therefore, the discovery in them of endogenous siRNAs was unexpected. The first mammalian siRNAs found were directed against a mobile element of the genome, LINE1. This element has promoters for the sense and antisense chains; complementary transcripts form dsRNAs, which initiate RNA interference. Later siRNAs involved in silencing of transposons were also detected in the nematode Caenorhabditis elegans and in drosophila [139].
siRNAs produced during viral infections. In plants and fungi RNA interference is involved in the struggle against viruses using siRNAs produced from dsRNA, which appears on the replication of the virus. These siRNAs are completely complementary to the viral RNA and within the RISC complex promote its degradation or suppression of translation [140].
Mammals and C. elegans have one type of Dicer enzyme, whereas Drosophila has two types: Dcr-1 for producing miRNA and Dcr-2 for producing siRNA. The matter is that Drosophila actively uses RNA interference for both regulating its genes and protecting against viruses, and because of the Dicer specialization the competition for the enzyme between pre-miRNAs and viral dsRNAs is decreased. Note that the genes Dcr-2 and Ago-2 belong to the most rapidly evolving genes, which can be due to the evolutionary pressure on PTGS from viruses. For mammals viral gene silencing is not very important because during evolution they have acquired an effective protein-based immune system [140].
piRNAs. piRNAs (24-29 nucleotides) are mainly expressed in sex cells. Their generation depends on PIWI proteins (a subfamily of Ago proteins), which are also characteristic for sex cells. The majority of piRNAs are coded by repeated inter-gene elements of DNA, including mobile elements [141]. Unlike other small RNAs, the generation of piRNAs does not depend on Dicer because they never form stable duplexes [142].
Technology of RNA Interference
From the time of discovery of RNA interference researchers have predicted the great importance of this phenomenon for practice. During recent years methods for regulation of gene expression based on features of small RNAs have been under extremely rapid development. RNA interference is now the most popular approach for gene knockdown (selective inactivation of gene expression) because it is rather easy, inexpensive, highly specific, and relatively nontoxic. For some years siRNA technology has been successfully applied in genomics for studies of gene functions and interactions and also in searches for new pharmaceuticals. It seems that this approach will also be used in medicine, e.g. against viral and parasitic diseases, in the treatment of cancer, etc. Methodical aspects of siRNA technology and some problems associated with its use are described below.
Synthesis of siRNA in vitro. Gene functions in plants, worms, and drosophila are usually studied using long molecules of dsRNA. But they cannot be used for the same purpose in mammals because dsRNAs longer than 30 bp activate the mammalian interferon system. This is associated with initiation of expression of more than 100 different genes including the gene of dsRNA-binding protein kinase R responsible for nonspecific suppression of translation and degradation of RNA [143]. Therefore, to induce RNA interference in mammalian cells synthetic double-stranded molecules of siRNA 21-22 bp in length are used [144].
Note that the in vitro synthesized siRNA must be fully complementary to mRNA of the target because even one nucleotide difference can abolish the activity of this siRNA molecule. The efficiency of suppression of the gene expression significantly depends on the choice of a target region on the mRNA molecule with which the complementary siRNA must form a duplex, because the secondary structure of the target mRNA or proteins bound with it can prevent its availability for siRNA. The first 75-100 nucleotides of mRNA are not recommended for use as target sites because they are very likely to contain protein-binding regulatory sequences. Thus, in every case the choice of a target sequence for siRNA is individual and is found by trial-and-error. Usually three or four different siRNAs are synthesized complementary to different regions of the mRNA molecule, and by experiment the most efficient siRNA is selected [145].
A sequence of 21 nucleotides is thought to be optimal for the target region and, correspondingly, for siRNA. Moreover, the target sequence must begin with two adenine residues. The effect of RNA interference is the highest if the synthetic double-stranded siRNA has 19 paired bases and two protruding uridines on the 3′-ends of each of the chains. Sometimes the unpaired uridine residues are replaced by deoxythymidine, which makes the molecule more stable inside the cell. The 5′-ends must be phosphorylated for siRNA functioning, but the phosphorylation is not significant for design of the molecule because is can occur directly inside the cell [132].
The in vitro synthesized siRNA is usually delivered into the cells by transfection by means of lipophilic agents or by electroporation. If electroporation is chosen, one has to take into account a significant (>50%) death of the cells during the procedure. The effect of RNA interference does not develop immediately after the cell transfection because siRNA provides for degradation only of mRNA but not of the protein. The effect usually develops during ~18 h, but in the case of stable proteins this period can be longer. The main shortcoming of the approach is that the action of siRNA is transient and lasts only during three to five cell divisions. Therefore, in the case of stable proteins or if more time is required for manifestation of the cell phenotype, either additional transfections are performed or constructions are used which promote the expression of siRNA directly inside the cells [132].
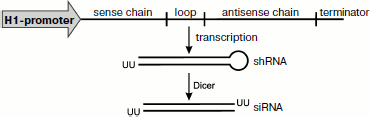
Expression of small RNAs inside the cell. To obtain a prolonged effect of RNA interference, DNA constructs are used with sequences encoding the spacer-separated sense and antisense chains of siRNA located between the promoter and terminator of RNA polymerase III (pol III) (Fig. 4). It is very convenient to use just pol III because in this case the transcription is terminated by a sequence consisting of four thymidines, which promotes production of short RNAs with 1-4 uridines on the 3′-end. Transcription of such matrix leads to production of RNA hairpins (shRNAs) similar to pre-miRNA. The optimal expression of such a transcript is obtained with a spacer consisting of nine bases. Upon transcription the loop linking two complementary sequences of shRNA is cut off by Dicer, and this results in production of siRNA. The efficiency of RNA interference increases significantly when the sequence encoding the human pre-miRNA loop is used instead of a random spacer [132].
At first, to obtain the shRNA expression inside the cells, plasmids were used with an inserted selective marker. Later more convenient viral vectors began to be used, and retroviral vectors are now most popular.Fig. 4. Expression of shRNA. For expression of small RNAs inside the cell DNA constructs are used with sequences encoding spacer-separated sense and antisense chains of siRNA located between the promoter and terminator of RNA polymerase III (pol III). Transcription of such matrix leads to production of RNA hairpins (shRNAs) similar to pre-miRNA. Upon transcription, the loop linking two complementary sequences of shRNA is cut off by Dicer, and this results in production of siRNA.
Acute myeloid leukemia (AML) is a malignant blood disease caused by different mutations that enhance the proliferative activity and survival of blood cells and affect their differentiation and apoptosis. In every case the progress of malignancy is contributed by various genes. This affects choice of target genes and their protein products for directed therapy of tumors. The situation is even more difficult because in the tumor development not one but several genes are usually involved. It was suggested that the presence in cells of a pair of activated oncogenes (in particular, transcription factor and tyrosinase) should trigger their malignization. The oncogene pair AML1-ETO and c-kit is found most frequently. The current approaches for treatment of oncological diseases (bone marrow transplantation, radio- and chemotherapy) have significant shortcomings such as suppression of immunity, severe radio intoxications, and appearance of secondary tumors that are significantly more resistant to all treatments. New approaches for the struggle against leukemia are under development in many laboratories. Inhibition of expression of activated leukemic oncogenes based on the principle of RNA interference seems to be a promising approach.
REFERENCES
1.Peterson, L. F., and Zhang, D. E. (2004)
Oncogene, 23, 4255-4262.
2.Baskaran, D., Spirin, P. V., and Prassolov, V. S.
(2010) Mol. Biol. (Moscow), 44, 1-13.
3.Jemal, A., Siegel, R., Ward, E., Hao, Y., Xu, J.,
and Thun, M. J. (2009) C. A. Cancer J. Clin., 59,
225-249.
4.Vorob’ev, A. I. (2002) Guide in
Hematology [in Russian], Vol. 1, Newdiamed, Moscow.
5.Rubnitz, J. E., Gibson, B., and Smith, F. O. (2008)
Pediatr. Clin. North. Am., 55, 21-51.
6.Advani, A. S. (2006) Curr. Hematol. Malig.
Rep., 1, 101-107.
7.Bennett, J. M., Catovsky, D., Daniel, M. T.,
Flandrin, G., Galton, D. A., Gralnick, H. R., and Sultan, C. (1976)
Br. J. Haematol., 33, 451-458.
8.Vardiman, J. W., Harris, N. L., and Brunning, R. D.
(2002) Blood, 100, 2292-2302.
9.Grimwade, D., Walker, H., Oliver, F., Wheatley, K.,
Harrison, C., Harrison, G., Rees, J., Hann, I., Stevens, R., Burnett,
A., and Goldstone, A. (1998) Blood, 92, 2322-2333.
10.Lasa, A., Carricondo, M. T., Carnicer, M. J.,
Perea, G., Aventin, A., and Nomdedeu, J. F. (2006)
Haematologica, 91, 1283-1284.
11.Kelly, L. M., and Gilliland, D. G. (2002)
Annu. Rev. Genom. Hum. Genet., 3, 179-198.
12.Gilliland, D. G. (1998) Leukemia,
12, Suppl. 1, 7-12.
13.Paschka, P., Marcucci, G., Ruppert, A. S.,
Mrozek, K., Chen, H., Kittles, R. A., Vukosavljevic, T., Perrotti, D.,
Vardiman, J. W., Carroll, A. J., Kolitz, J. E., Larson, R. A., and
Bloomfield, C. D. (2006) J. Clin. Oncol., 24,
3904-3911.
14.Niebuhr, B., Fischer, M., Tager,
M., Cammenga, J., and Stocking, C. (2008) Blood Cells Mol.
Dis., 40, 211-218.
15.Huang, G., Shigesada, K., Ito, K., Wee, H.
J., Yokomizo, T., and Ito, Y. (2001) EMBO J., 20,
723-733.
16.Perry, C., Eldor, A., and Soreq, H.
(2002) Leuk. Res., 26, 221-228.
17.Kitabayashi, I., Yokoyama, A., Shimizu, K., and
Ohki, M. (1998) EMBO J., 17, 2994-3004.
18.Durst, K. L., and Hiebert, S. W. (2004)
Oncogene, 23, 4220-4224.
19.Taniuchi, I., Osato, M., Egawa,
T., Sunshine, M. J., Bae, S. C., Komori, T., Ito,
Y., and Littman, D. R. (2002) Cell, 111,
621-633.
20.Durst, K. L., Lutterbach, B., Kummalue,
T., Friedman, A. D., and Hiebert, S. W. (2003) Mol. Cell.
Biol., 23, 607-619.
21.Elagib, K. E., and Goldfarb, A. N. (2007)
Cancer Lett., 251, 179-186.
22.Feinstein, P. G., Kornfeld,
K., Hogness, D. S., and Mann, R. S. (1995) Genetics,
140, 573-586.
23.Hug, B. A., and Lazar, M. A. (2004)
Oncogene, 23, 4270-4274.
24.Costoya, J. A., and Pandolfi, P. P. (2001)
Curr. Opin. Hematol., 8, 212-217.
25.Melnick, A. M., Westendorf, J. J., Polinger,
A., Carlile, G. W., Arai, S., Ball, H. J., Lutterbach,
B., Hiebert, S. W., and Licht, J. D. (2000) Mol. Cell.
Biol., 20, 2075-2086.
26.McGhee, L., Bryan, J., Elliott,
L., Grimes, H. L., Kazanjian, A., Davis, J. N., and
Meyers, S. (2003) J. Cell Biochem., 89, 1005-1018.
27.Minucci, S., Maccarana, M., Cioce, M., de Luca,
P., Gelmetti, V., Segalla, S., di Croce, L., Giavara, S., Matteucci,
C., Gobbi, A., Bianchini, A., Colombo, E., Schiavoni, I., Badaracco,
G., Hu, X., Lazar, M. A., Landsberger, N., Nervi, C., and Pelicci, P.
G. (2000) Mol. Cell, 5, 811-820.
28.Downing, J. R. (2003) Curr. Opin. Genet.
Dev., 13, 48-54.
29.Shimada, H., Ichikawa, H., Nakamura, S., Katsu,
R., Iwasa, M., Kitabayashi, I., and Ohki, M. (2000) Blood,
96, 655-663.
30.Rosenbauer, F., Koschmieder, S., Steidl, U., and
Tenen, D. G. (2005) Blood, 106, 1519-1524.
31.Frosina, G. (2000) Eur. J.
Biochem., 267, 2135-2149.
32.Basecke, J., Schwieger, M., Griesinger, F.,
Schiedlmeier, B., Wulf, G., Trumper, L., and Stocking, C. (2005)
Leuk. Lymphoma, 46, 265-272.
33.Russell, E. S. (1979) Adv. Gen.,
20, 357-459.
34.Liu, H., Chen, X., Focia, P., and He, X. (2007)
EMBO J., 26, 891-901.
35.Lennartsson, J., Voytyuk, O., Heiss, E.,
Sundberg, C., Sun, J., and Ronnstrand, L. (2005) Cancer Ther.,
3, 5-28.
36.Roskoski, R., Jr. (2005) Biochem. Biophys.
Res. Commun., 338, 1307-1315.
37.D’Auriol, L., Mattei, M. G., Andre, C., and
Galibert, F. (1998) Hum. Genet., 78, 374-376.
38.Geissler, E. N., Cheng, S. V., Gusella, J. F.,
and Housman, D. E. (1988) Proc. Natl. Acad. Sci. USA, 85,
9635-9639.
39.Vandenbark, G. R., Chen, Y., Friday, E., Pavlik,
K., Anthony, B., deCastro, C., and Kaufman, R. E. (1996) Cell.
Growth Differ., 7, 1383-1392.
40.Vandenbark, G. R., deCastro, C. M., Taylor, H.,
Dew-Knight, S., and Kaufman, R. E. (1992) Oncogene, 7,
1259-1266.
41.Yamamoto, K., Tojo, A., Aoki, N., and Shibuya, M.
(1993) Jpn. J. Cancer Res., 84, 1136-1144.
42.Yasuda, H., Galli, S. J., and Geissler, E. N.
(1993) Biochem. Biophys. Res. Commun., 191, 893-901.
43.Albanesi, C., Geremia, R., Giorgio, M., Dolci,
S., Sette, C., and Rossi, P. (1996) Development, 122,
1291-1302.
44.Paronetto, M. P., Farini, D., Sammarco, I.,
Maturo, G., Vespasiani, G., Geremia, R., Rossi, P., and Sette, C.
(2004) Am. J. Pathol., 164, 1243-1251.
45.Yee, N. S., Langen, H., and Besmer, P. (1993)
J. Biol. Chem., 268, 14189-14201.
46.Cruz, A. C., Frank, B. T., Edwards, S. T., Dazin,
P. F., Peschon, J. J., and Fang, K. C. (2004) J. Biol. Chem.,
279, 5612-5620.
47.Heissig, B., Hattori, K., Dias, S., Friedrich,
M., Ferris, B., Hackett, N. R., Crystal, R. G., Besmer, P., Lyden, D.,
Moore, M. A. S., Werb, Z., and Rafii, S. (2002) Cell,
109, 625-637.
48.Wypych, J., Bennett, L. G., Schwartz, M. G.,
Clogston, C. L., Lu, H. S., Broudy, V. C., Bartley, T. D., Parker, V.
P., and Langley, K. E. (1995) Blood, 85, 66-73.
49.Turner, A. M., Bennett, L. G., Lin, N. L.,
Wypych, J., Bartley, T. D., Hunt, R. W., Atkins, H. L., Langley, K. E.,
Parker, V., and Martin, F. (1995) Blood, 85,
2052-2058.
50.Nakamura, Y., Tajima, F., Ishiga, K., Yamazaki,
H., Oshimura, M., Shiota, G., and Murawaki, Y. (2004) Exp.
Hematol., 32, 390-396.
51.Mol, C. D., Dougan, D. R., Schneider, T. R.,
Skene, R. J., Kraus, M. L., Scheibe, D. N., Snell, G. P., Zou, H.,
Sang, B. C., and Wilson, K. P. (2004) J. Biol. Chem.,
279, 31655-31663.
52.Chan, P. M., Ilangumaran, S., La Rose, J.,
Chakrabartty, A., and Rottapel, R. (2003) Mol. Cell. Biol.,
23, 3067-3078.
53.Wandzioch, E., Edling, C., Pulmer, R., Carisson,
L., and Hallberg, B. (2004) Blood, 104, 51-57.
54.Perez-Losada, J., Sanchez-Martin, M., Perez-Caro,
M., Perez-Mancera, P., and Sanchez-Garcia, I. (2003) Oncogene,
22, 4205-4211.
55.Perez-Losada, J., Sanchez-Martin, M.,
Rodriguez-Garcia, A., Sanchez, M., Orfao, A., Flores, T., and
Sanchez-Garcia, I. (2002) Blood, 100, 1274-1286.
56.Hassan, H. T., and El-Sheemy, M. (2004) J.
Roy. Soc. Med., 97, 465-471.
57.Ogawa, M., Matsuzaki, Y., Nishikawa, S., Hayashi,
S., Kunisada, T., Sudo, T., Kina, T., Nakauchi, H., and Nishikawa, S.
(1991) J. Exp. Med., 174, 63-71.
58.Andre, C., d’Auriol, L., Lacombe, C.,
Gisselbrecht, S., and Galibert, F. (1989) Oncogene, 4,
1047-1049.
59.Galli, M. C., Giardina, P. J., Migliaccio, A. R.,
and Migliaccio, G. (1993) Int. J. Clin. Lab. Res., 23,
70-77.
60.DeCastro, C. M., Denning, S. M., Langdon, S.,
Vandenbark, G. R., Kurtzberg, J., Scearce, R., Haynes, B. F., and
Kaufman, R. E. (1994) Exp. Hematol., 22, 1025-1033.
61.Lennartsson, J., Voytyuk, O., Heiss, E.,
Sundberg, C., Sun, J., and Ronnstrand, L. (2005) Cancer Ther.,
3, 5-28.
62.Galli, S. J., Tsai, M., and Wershil, B. K. (1993)
Am. J. Pathol., 142, 965-974.
63.Hassan, H. T., and Zander, A. (1996) Acta
Haematol., 95, 257-262.
64.Ulivi, P., Zoli, W., Medri, L., Amadori, D.,
Saragoni, L., Barbanti, F., Calistri, D., and Silvestrini, R. (2004)
Breast Cancer Res. Treat., 83, 33-42.
65.Tanaka, T., Umeki, K., Yamamoto, I., Kotani, T.,
Sakamoto, F., Noguchi, S., and Ohtaki, S. (1995) Endocr. J.,
42, 723-728.
66.Krystal, G. W., Hines, S. J., and Organ, C. P.
(1996) Cancer Res., 56, 370-376.
67.Theou-Anton, N., Tabone, S., Brouty-Boye, D.,
Saffroy, R., Ronnstrand, L., Lemoine, A., and Emile, J. F. (2006)
Br. J. Cancer, 94, 1180-1185.
68.Kitayama, H., Kanakura, Y., Furitsu, T.,
Tsujimura, T., Oritani, K., Ikeda, H., Sugahara, H., Mitsui, H.,
Kanayama, Y., and Kitamura, Y. (1995) Blood, 85,
790-798.
69.Feng, F., Liu, X. H., Xie, Q., Liu, W. Q., Bai,
C. G., and Ma, D. L. (2003) World J. Gastroenterol., 9,
2548-2551.
70.Torrent, M., Rickert, K., Pan, B. S., and
Sepp-Lorenzino, L. (2004) J. Mol. Graph. Model., 23,
153-165.
71.Longley, B. J., Reguera, M. J., and Ma, Y. (2001)
Leuk. Res., 25, 571-576.
72.Boissan, M., Feger, F., Guillosson, J. J., and
Arock, M. (2000) J. Leukoc. Biol., 67, 135-148.
73.Ikeda, H., Kanakura, Y., Tamaki, T., Kuriu, A.,
Kitayama, H., Ishikawa, J., Kanayama, Y., Yonezawa, T., Tarui, S., and
Griffin, J. D. (1991) Blood, 78, 2962-2968.
74.Kuriu, A., Ikeda, H., Kanakura, Y., Griffin, J.
D., Druker, B., Yagura, H., Kitayama, H., Ishikawa, J., Nishiura, T.,
and Kanayama, Y. (1991) Blood, 78, 2834-2840.
75.Beghini, A., Magnani, I., Ripamonti, C. B., and
Larizza, L. (2002) Hematol. J., 3, 157-163.
76.Hu, Q., Trevisan, M., Xu, Y., Dong, W., Berger,
S. A., Lyman, S. D., and Minden, M. D. (1995) J. Clin. Invest.,
95, 2530-2538.
77.Bene, M. C., Bernier, M., Casasnovas, R. O.,
Castoldi, G., Knapp, W., Lanza, F., Ludwig, W. D., Matutes, E., Orfao,
A., Sperling, C., and van’t Veer, M. B. (1998) Blood,
92, 596-599.
78.Cascavilla, N., Musto, P., D’Arena, G.,
Melillo, L., Carella, A. M., Petrilli, M. P., Sanpaolo, G., and
Carotenuto, M. (1998) Haematol. J., 83, 392-397.
79.Hans, C. P., Finn, W. G., Singleton, T. P.,
Schnitzer, B., and Ross, C. W. (2002) Am. J. Clin. Pathol.,
117, 301-305.
80.Omar, S., Abdel-Kader, M., Matar, M., and Meabed,
M. (2001) J. Egypt. Nat. Cancer Inst., 13, 191-201.
81.Schwartz, S., Heinecke, A., Zimmermann, M.,
Creutzig, U., Schoch, C., Harbott, J., Fonatsch, C., Lffler, H.,
Bchner, T., Ludwig, W. D., and Thiel, E. (1999) Leuk. Lymphoma,
34, 85-94.
82.Tsao, A. S., Kantarjian, H., Thomas, D., Giles,
F., Cortes, J., Garcia-Manero, G., Huh, Y., Yang, Y., Shen, Y.,
Albitar, M., and Estey, E. (2004) Leuk. Res., 28,
373-378.
83.Valverde, L. R., Matutes, E., Farahat, N.,
Heffernan, A., Owusu-Ankomah, K., Morilla, R., and Catovsky, D. (1996)
Ann. Hematol., 72, 11-15.
84.Ashman, L. K., Roberts, M. M., Gadd, S. J.,
Cooper, S. J., and Juttner, C. A. (1988) Leuk. Res., 12,
923-928.
85.Longley, B. J., Jr., Metcalfe, D. D., Tharp, M.,
Wang, X., Tyrrell, L., Lu, S.-Z., Heitjan, D., and Ma, Y. (1999)
Proc. Natl. Acad. Sci. USA, 96, 1609-1614.
86.Zaker, F., Mohammadzadeh, M., and Mohammadi, M.
(2012) Arch. Iran. Med., 13, 21-25.
87.Gari, M., Goodeve, A., Wilson, G., Winship, P.,
Langabeer, S., Linch, D., Vandenberghe, E., Peake, I., and Reilly, J.
(1999) Br. J. Haematol., 105, 894-900.
88.Bacher, U., Haferlach, T., Kern,
W., Haferlach, C., and Schnittger, S. (2007) Haematol.
J., 92, 744-752.
89.Sun, J., Pedersen, M., and Ronnstrand, L. (2009)
J. Biol. Chem., 284, 11039-11047.
90.Kohl, T. M., Schnittger, S., Ellwart, J. W.,
Hiddemann, W., and Spiekermann, K. (2005) Blood, 105,
3319-3321.
91.Corbacioglu, S., Kilic, M., Westhoff, M. A.,
Reinhardt, D., Fulda, S., and Debatin, K. M. (2006) Blood,
108, 3504-3513.
92.Kindler, T., Breitenbuecher, F., Marx, A., Hess,
G., Gschaidmeier, H., Gamm, H., Kirkpatrick, C. J., Huber, C., and
Fischer, T. (2003) Blood, 101, 2960-2962.
93.Heinrich, M. C., Griffith, D. J., Druker, B. J.,
Wait, C. L., Ott, K. A., and Zigler, A. J. (2000) Blood,
96, 925-932.
94.Scappini, B., Onida, F., Kantarjian, H. M., Dong,
L., Verstovsek, S., Keating, M. J., and Beran, M. (2001) Clin.
Cancer Res., 7, 3884-3893.
95.Kindler, T., Breitenbuecher, F., Marx, A., Beck,
J., Hess, G., Weinkauf, B., Duyster, J., Peschel, C., Kirkpatrick, C.
J., Theobald, M., Gschaidmeier, H., Huber, C., and Fischer, T. (2004)
Blood, 103, 3644-3654.
96.Cortes, J., Giles, F., O’Brien, S., Thomas,
D., Albitar, M., Rios, M. B., Talpaz, M., Garcia-Manero, G., Faderl,
S., Letvak, L., Salvado, A., and Kantarjian, H. (2003) Cancer,
97, 2760-2766.
97.Cairoli, R., Beghini, A., Morello, E., Grillo,
G., Montillo, M., Larizza, L., and Morra, E. (2005) Leuk. Res.,
29, 397-400.
98.Heidel, F., Cortes, J., Rucker, F. G., Aulitzky,
W., Letvak, L., Kindler, T., Huber, C., Dohner, H., Kantarjian, H., and
Fischer, T. (2007) Cancer, 109, 907-914.
99.Piccaluga, P. P., Malagola, M., Rondoni, M.,
Arpinati, M., Paolini, S., Candoni, A., Fanin, R., Messa, E., Pirrotta,
M. T., Lauria, F., Visani, G., Alberti, D., Rancati, F., Vinaccia, V.,
Russo, D., Saglio, G., Baccarani, M., and Martinelli, G. (2007)
Haematol. J., 92, 1721-1722.
100.Fong, T. A., Shawver, L. K., Sun, L., Tang, C.,
App, H., Powell, T. J., Kim, Y. H., Schreck, R., Wang, X., Risau, W.,
Ullrich, A., Hirth, K. P., and McMahon, G. (1999) Cancer Res.,
59, 99-106.
101.O’Farrell, A., Yuen, H., Smolich, B.,
Hannah, A., Louie, S., Hong, W., Stopeck, A., Silverman, L., Lancet,
J., and Karp, J. (2004) Leuk. Res., 8, 679-689.
102.Loges, S., Tinnefeld, H., Metzner, A., Jucker,
M., Butzal, M., Bruweleit, M., Fischer, U., Draab, E., Schuch, G.,
O’Farrell, A. M., Hossfeld, D. K., Bokemeyer, C., and Fiedler, W.
(2005) Blood (ASH Annual Meeting Abstracts), 106,
Abstract 2793.
103.Smolich, B. D., Yuen, H. A., West, K. A.,
Giles, F. J., Albitar, M., and Cherrington, J. M. (2001) Blood,
97, 1413-1421.
104.Shaheen, R. M., Davis, D. W., Liu, W.,
Zebrowski, B. K., Wilson, M. R., Bucana, C. D., McConkey, D. J.,
McMahon, G., and Ellis, L. M. (1999) Cancer Res., 59,
5412-5416.
105.Giles, F. J., Stopeck, A. T., Silverman, L. R.,
Lancet, J. E., Cooper, M. A., Hannah, A. L., Cherrington, J. M.,
O’Farrell, A. M., Yuen, H. A., Louie, S. G., Hong, W., Cortes, J.
E., Verstovsek, S., Albitar, M., O’Brien, S. M., Kantarjian, H.
M., and Karp, J. E. (2003) Blood, 102, 795-801.
106.Fiedler, W., Serve, H., Dohner, H., Schwittay,
M., Ottmann, O. G., O’Farrell, A. M., Bello, C. L., Allred, R.,
Manning, W. C., Cherrington, J. M., Louie, S. G., Hong, W., Brega, N.
M., Massimini, G., Scigalla, P., Berdel, W. E., and Hossfeld, D. K.
(2005) Blood, 105, 986-993.
107.Talpaz, M., Shah, N. P., Kantarjian, H.,
Donato, N., Nicoll, J., Paquette, R., Cortes, J., O’Brien, S.,
Nicaise, C., Bleickardt, E., Blackwood-Chirchir, M. A., Iyer, V., Chen,
T. T., Huang, F., Decillis, A. P., and Sawyers, C. L. (2006) N.
Engl. J. Med., 354, 2531-2541.
108.Schittenhelm, M. M., Shiraga, S., Schroeder,
A., Corbin, A. S., Griffith, D., Lee, F. Y., Bokemeyer, C., Deininger,
M., Druker, B. J., and Heinrich, M. C. (2006) Cancer Res.,
66, 473-481.
109.Kolb, E. A., Gorlick, R., Houghton, P. J.,
Morton, C. L., Lock, R. B., Tajbakhsh, M., Reynolds, C. P., Maris, J.
M., Keir, S. T., Billups, C. A., and Smith, M. A. (2008) Pediatr.
Blood Cancer, 50, 1198-1206.
110.Cioch, M., Dmoszynska, A., and Wach, M. (2008)
Haematol. J., 93, 480, Abs. 1236.
111.Hu, S., Niu, H., Minkin, P., Orwick, S.,
Shimada, A., Inaba, H., Dahl, G. V., Rubnitz, J., and Baker, S. D.
(2008) Mol. Cancer Ther., 7, 1110-1120.
112.Hasskarl, J. (2010) Recent Results
Cancer Res., 184, 61-70.
113.Fiedler, W., Serve, H., Dohner, H., Schwittay,
M., Ottmann, O. G., O’Farrell, A. M., Bello, C. L., Allred, R.,
Manning, W. C., Cherrington, J. M., Louie, S. G., Hong, W., Brega, N.
M., Massimini, G., Scigalla, P., Berdel, W. E., and Hossfeld, D. K.
(2005) Blood, 105, 986-993.
114.Ikezoe, T., Nishioka, C., Tasaka, T., Yang, Y.,
Komatsu, N., Togitani, K., Koeffler, H. P., and Taguchi, H. (2006)
Mol. Cancer Ther., 5, 2522-2530.
115.Corbin, A. S., Griswold, I. J., La Rosee, P.,
Yee, K. W., Heinrich, M. C., Reimer, C. L., Druker, B. J., and
Deininger, M. W. (2004) Blood, 104, 3754-3757.
116.Growney, J. D., Clark, J. J., Adelsperger, J.,
Stone, R., Fabbro, D., Griffin, J. D., and Gilliland, D. G. (2005)
Blood, 106, 721-724.
117.Shankar, D. B., Chang, J. C., Parcells, B.,
Sandoval, S., Li, J., Wei, R.-Q., Tapang, P., Davidsen, S. K., Albert,
D. H., Glaser, K. B., Moore, T. B., and Sakamoto, K. M. (2005)
Blood (ASH Annual Meeting Abstracts), 106,
Abstract 616.
118.Nishioka, C., Ikezoe, T., Yang, J., Miwa, A.,
Tasaka, T., Kuwayama, Y., Togitani, K., Koeffler, H. P., and Yokoyama,
A. (2008) Blood, 111, 5086-5092.
119.Napoli, C., Lemieux, C., and Jorgensen, R.
(1990) Plant Cell, 2, 279-289.
120.Van Blokland, R., van der Geest, N., Mol, J. N.
M., and Kooter, J. M. (1994) Plant J., 6, 861-877.
121.Hamilton, A. J., and Baulcombe, D. C. (1999)
Science, 286, 950-952.
122.Tuschl, T., Zamore, P. D., Lehmann, R., Bartel,
D. P., and Sharp, P. A. (1999) Genes Dev., 13,
3191-3197.
123.Cogoni, C., and Macino, G. (1997) Trends
Plant Sci., 2, 438-443.
124.Fire, A., Xu, S., Montgomery, M. K., Kostas, S.
A., Driver, S. E., and Mello, C. C. (1998) Nature, 391,
806-811.
125.Elbashir, S. M., Harborth, J., Weber, K., and
Tuschl, T. (2002) Methods, 26, 199-213.
126.Bernstein, E., Caudy, A. A., Hammond, S. M.,
and Hannon, G. J. (2001) Nature, 409, 363-366.
127.Lima, W. F., Murray, H., Nichols, J. G., Wu,
H., Sun, H., Prakash, T. P., Berdeja, A. R., Gaus, H. J., and Crooke,
S. T. (2009) J. Biol. Chem., 284, 2535-2548.
128.Chiu, Y. L., and Rana, T. M. (2002) Mol.
Cell, 10, 549-561.
129.Tomari, Y., Matranga, C., Haley, B., Martinez,
N., and Zamore, P. D. (2004) Science, 306, 1377-1380.
130.Song, J. J., Smith, S. K., Hannon, G. J., and
Joshua-Tor, L. (2004) Science, 305, 1434-1437.
131.Leuschner, P., Ameres, S. L., Kueng, S., and
Martinez, J. (2006) EMBO Rep., 7, 314-320.
132.Vilgelm, A. E., Chumakov, S. P., and Prassolov,
V. S. (2006) Mol. Biol. (Moscow), 40, 387-403.
133.Kim, V. N., Han, J., and Siomi, M. C. (2009)
Nat. Rev. Mol. Cell Biol., 10, 126-139.
134.Borchert, G. M., Lanier, W., and Davidson, B.
L. (2006) Nat. Struct. Mol. Biol., 13, 1097-1101.
135.Khvorova, A., Reynolds, A., and Jayasena, S. D.
(2003) Cell, 115, 209-216.
136.Grosshans, H., and Slack, F. J. (2002) J.
Cell Biol., 156, 17-21.
137.Llave, C., Kasschau, K. D., Rector, M. A., and
Carrington, J. C. (2002) Plant Cell, 14, 1605-1619.
138.Leung, A. K., and Sharp, P. A. (2007)
Cell, 130, 581-585.
139.Haasnoot, J., Westerhout, E. M., and Berkhout,
B. (2007) Nature Biotech., 25, 1435-1443.
140.Siomi, M. C., Mannen, T., and Siomi, H. (2010)
Genes Dev., 24, 636-646.
141.Li, C., Vagin, V. V., Lee, S., Xu, J., Ma, S.,
Xi, H., Seitz, H., Horwich, M. D., Syrzycka, M., Honda, B. M., Kittler,
E. L., Zapp, M. L., Klattenhoff, C., Schulz, N., Theurkauf, W. E.,
Wenig, Z., and Zamore, P. D. (2009) Cell, 137,
509-521.
142.Gunawardane, L. S., Saito, K., Nishida, K. M.,
Miyoshi, K., Kawamura, Y., Nagami, T., Siomi, H., and Siomi, M. C.
(2007) Science, 315, 1587-1590.
143.Tuschl, T., and Borkhardt, A. (2002) Mol.
Interv., 2, 158-167.
144.Kim, V. N. (2003) J. Korean Med. Sci.,
18, 309-318.
145.Hu, W. S., and Pathak, V. K. (2000)
Pharmacol. Rev., 52, 493-511.