REVIEW: Nanocolonies and Diagnostics of Oncological Diseases Associated with Chromosomal Translocations
E. V. Chetverina* and A. B. Chetverin
Institute of Protein Research, Russian Academy of Sciences, 142290 Pushchino, Moscow Region, Russia; E-mail: helena@vega.protres.ru* To whom correspondence should be addressed.
Received May 26, 2010; Revision received June 16, 2010
This paper reviews chromosomal abnormalities observed in oncological diseases, the history of discovery of chromosomal translocations (a widespread type of chromosomal abnormalities), and statistical data showing a correlation between translocations and emergence of oncological diseases (in particular leukemia). The importance of detection of minimal residual disease (MRD) in treatment of leukemia associated with translocations is discussed along with methods of MRD diagnosis, followed by description of a novel diagnostic procedure for the detection of single chimeric mRNA molecules serving as MRD markers. This procedure includes a number of improvements, of which the most important is the use of a PCR version of the method of nanocolonies (other names are molecular colonies, polonies) that provides for the determination of the absolute titer of RNA tumor markers, excludes false positive results in the detection of chimeric molecules, and significantly exceeds other methods in the sensitivity of MRD detection.
KEY WORDS: chromosomal translocation, leukemia, minimal residual disease, RT-PCR, nanocolonies, molecular colonies, polonies, PCR coloniesDOI: 10.1134/S0006297910130109
Abbreviations: ALL, acute lymphoid leukemia; AML, acute myeloid leukemia; AML1-ETO, chimeric sequence consisting of parts of AML1 and ETO genes; CLL, chronic lymphoid leukemia; CML, chronic myeloid leukemia; FRET, fluorescence resonance energy transfer; MRD, minimal residual disease; PCR, polymerase chain reaction; RCA, rolling circle amplification; RQ-PCR (as well as qPCR and qRT-PCR), real time PCR; RT, reverse transcription; RT-PCR, reverse transcription with following PCR; SNP, single nucleotide polymorphism; Taq DNA polymerase, DNA-dependent DNA polymerase from Thermus aquaticus.
Many malignant diseases are associated with chromosome abnormalities
arising as a result of chromosome translocations. Chromosome
translocations alter gene expression and/or produce fused (chimeric)
genes expressing chimeric products (RNA and proteins) that can serve
both as diagnostic markers and as therapeutic targets. The most
sensitive diagnostics is based on the detection of a chimeric marker
RNA using polymerase chain reaction (PCR) [1, 2]. First, a DNA copy of the chimeric RNA is
synthesized by reverse transcription (RT), and then PCR is applied for
amplification of the fused gene joint region in chimeric cDNA using
primers complementary to sequences localized on different sides of the
junction point [3-5].
Theoretically, PCR allows a single target molecule to be detected. However, in practice the sensitivity of assay of clinical samples containing large amounts of non-target nucleic acids is 100-1000 times lower [6]. Most of the problems that reduce the sensitivity and accuracy of PCR analysis and impede the quantification of RNA targets can be overcome by the method of nanocolonies (other name: molecular colonies or polonies) invented by us [7, 8].
Nanocolonies are formed when template nanomolecules are amplified in a medium whose polymeric matrix forms a three-dimensional network with pore size in the nanometer range [9-16]. Thus, one can “grow” RNA colonies in agarose containing the RNA-dependent RNA polymerase of bacteriophage Qβ [17, 18] or DNA colonies in polyacrylamide gel containing a thermostable DNA polymerase [19, 20]. In this case, on one hand, each colony consists of identical molecules, which in fact allows the original molecule to be analyzed, while on the other hand, each original target molecule forms a colony, which allows the target titer to be directly determined by counting the colonies.
Various applications of nanocolonies are based on their unique ability of spatial separation (compartmentalization) of amplification and expression of individual DNA or RNA molecules. The method of nanocolonies was first used in 1991 for the detection of single airborne molecules of replicating templates, which served as the most important evidence against the hypothesis of spontaneous RNA synthesis [17]. The ability of nanocolonies to detect products of reactions between single molecules allowed RNA self-recombination to be discovered [21] and revealed the diversity of mechanisms of RNA recombination performed by different RNA-dependent RNA polymerases [22, 23]. Since nanocolonies are the progeny of individual template molecules (molecular clones), they provide for the cloning of pure genetic material and in situ gene screening, including by the function of the encoded proteins [24, 25].
The diagnostic potential of nanocolonies is multifaceted. Using viral targets, it was shown that nanocolonies provide for reliable detection of a single DNA molecule or two RNA molecules in a blood sample containing a trillion-fold greater amount of human nucleic acids [19, 20]. Spatial separation of amplification of different nucleic acid molecules present in clinical samples eliminates interference from nonspecific synthesis (that in the case of amplification in liquid is the main factor limiting sensitivity of target assay), excludes competition among simultaneously amplified templates in multiplex analysis [19, 20], and ensures unsurpassed sensitivity of detection of minor drug-resistant malignant cell clones [26]. Finally, the ability of identifying cis-elements makes nanocolonies a unique tool for chimeric RNA detection [27], gene mapping and haplotype determination [28], elucidation of RNA exon composition [29], and in situ sequencing [30].
In this review we discuss the role of chromosomal translocations in the genesis and diagnostics of cancer and describe a complete procedure for the nanocolony-based diagnostics of a chimeric RNA-associated disease as exemplified by the detection of the minimal residual disease (MRD) in an acute myeloid leukemia.
CHROMOSOMAL TRANSLOCATIONS
Chromosomal Abnormalities in Oncological Diseases
Chromosomal abnormalities are characteristic of cancer cells; they are found in all major types of malignant tumors. Today there are 57,709 types of chromosomal abnormalities (listed in the Mitelman database of the National Cancer Institute of the USA [31]), and new types continue to be revealed at an increasing rate due to development of new techniques in molecular cytogenetics and other fields of science.
The World Health Organization Classification of Tumors (a WHO project aimed at providing an international standard for classification of tumors, published as a series of “WHO Blue Books” [32]) recommends that a number of chromosomal abnormalities be used as disease characteristics for diagnosing, choice of treatment tactics, as well as a prognostic marker.
Chromosomal abnormalities are divided into two classes—balanced chromosomal rearrangements and chromosomal unbalance [33]. In the case of chromosomal unbalance the amount of chromosomal material decreases or increases, which is revealed as complete or partial trisomy, monosomy, deletions, and intra- or extrachromosomal amplifications. Balanced chromosomal rearrangements do not change the amount of chromosomal material.
Many of the abnormalities are due to translocations (transfer of a part of a chromosome) that can result in either balanced or unbalanced chromosomal rearrangements. In a narrower sense translocations (also known as reciprocal translocations) comprise the exchange of segments between two nonhomologous chromosomes resulting in a balanced chromatin rearrangement [34]. Such translocations can have two consequences. In some cases, a rearrangement affects regulatory elements of genes without changing their coding regions and alters the expression of a normal gene. In other cases, translocation involves the coding region and results in the formation of a fusion (chimeric) gene. The expression of a chimeric gene results in the appearance of chimeric products, chimeric mRNA and chimeric protein, which, on one hand, exhibit altered properties compared to the original gene products, while on the other hand, can serve as markers of malignant cells [33].
In 1978, the International System for Human Cytogenetic Nomenclature (ISCN) [35] was developed for systematization of cytogenetic abnormalities; it has been repeatedly updated [36, 37], the last changes and additions being introduced in 2009 [38].
According to this nomenclature, chromosome aberrations are denoted by symbols of Latin letters. Here are some of them: t, translocation; del, deletion; ins, insertion; inv, inversion; dic, dicentric chromosome. The aberration resulting in the formation of Philadelphia chromosome (Ph), whose discovery will be described below, is designated as follows: t(9;22)(q34;q11), where t means translocation, the first parentheses indicate numbers of the chromosomes between which it has occurred, and the second parentheses indicate the respective chromosome arms (p or q) along with numbers of the cytogenetic bands containing the breakpoints.
The same gene can be involved in both balanced and unbalanced rearrangements, and it can take part in recombinations with different partners. The ETV6 gene can serve as an example: unbalanced aberration dic(9;12)(p13;p13) involves genes PAX5 and ETV6, and balanced aberrations t(12;21)(p13;q22) and t(12;22)(p13;q12) involve, respectively, gene pairs ETV6/RUNX1 and ETV6/MN1 [31]. All these chromosomal abnormalities are associated with leukemia [31, 39].
The same abnormality can be associated with different diseases. Thus, chromosomal translocation t(12;22)(p13;q12) involving genes ATF1 (synonym CHOP) and EWSR1 (synonym EWS) is found in sarcomas and melanomas [40]. And, vice versa, there are chromosomal translocations whose detection indicates a particular disease. Thus, chromosomal translocation t(11;22)(q24;q12) is detected only in the case of Ewing’s sarcoma and is responsible for 90-95% cases of this disease [41]. In the case of this translocation, genes FLI1 and EWSR1 are fused. The remaining cases of Ewing’s sarcoma are associated with other translocations, the most frequent of which (5-10% cases) t(21;22)(q22;q12) results in the fusion of genes ERG and EWSR1 [42].
The above examples show that formally the same notation of a chromosome aberration (in this example, t(12;22)(p13;q12)) may relate to quite different gene pairs and to different diseases. Sometimes, to avoid confusion, a more precise location of breakpoints in the cytogenetic bands is indicated [33, 34]. Thus, the GeneCards database [43] differently designates translocations involving genes ETV6/MN1 and ATF1/EWS: t(12;22)(p13.2;q12.1) and t(12;22)(p13.12;q12.2), respectively. Also, instead of or in addition to the localization of breakpoints in the chromosomes, the pair of genes involved in translocation can be indicated [44, 45], such as t(12;22)ETV6-MN1 and t(12;22)ATF1-EWS.
Reciprocal (balanced) translocations result in the formation of two chimeric chromosomes and, respectively, two chimeric genes. In other words, if translocation involves genes A and B, it results in a reciprocal pair of chimeric genes A-B (containing the 5′ part of gene A and the 3′ part of gene B) and B-A (containing the 5′ part of gene B and the 3′ part of gene A). Usually in diagnostics the expression product of only one member of a chimeric gene pair is assayed [46].
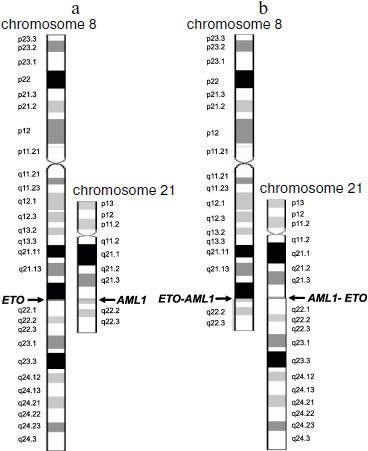
For example, translocation t(8;21)(q22;q22) results in formation of two chimeric genes AML1-ETO and ETO-AML1 (Fig. 1). However, in patients with leukemia associated with this translocation, only a single chimeric transcript type (of two possible) is usually assayed, AML1-ETO mRNA [3, 47].
Translocation t(15;17)(q22;q21) results in the appearance of chimeric PML-RARA mRNA [48], but in some patients chimeric RARA-PML mRNA is detected in addition to the PML-RARA RNA [49].Fig. 1. Schemes of chromosomes 8 and 21 before (a) and after (b) translocation t(8;21)(q22;q22). Schemes of normal chromosomes are from the site of the GeneCards database [43]. Arrows indicate breakpoints.
The emergence of the second chimeric product or its high level may be indicative of a more severe disease, like that in the case of leukemia associated with t(12;21)(p13;q22). In 76% of such patients in addition to the main chimeric product TEL-AML1 RNA, the reciprocal RNA variant AML1-TEL is detected. High level of the latter is associated with unfavorable prognosis [50].
There is an interesting situation concerning translocation t(9;22)(q34;q11) occurring between chromosomes 9 and 22 [51]. There are two main breakpoints in the BCR gene on chromosome 22, M-bcr (major) between exons 12 and 16 and m-bcr (minor) in the first intron. Gene ABL on chromosome 9 contains only a single break site in the first intron [46]. Translocation at M-bcr results in synthesis of chimeric protein BCR-ABL, p210BCR-ABL, characteristic of chronic myeloid leukemia (CML) [52, 53], while translocation at m-bcr results in appearance of chimeric protein BCR-ABL (appearing in the literature as p185BCR-ABL [54] or p190BCR-ABL [55]) that is specific for acute lymphoid leukemia (ALL) [54, 55]. In 65% of CML patients a short chimeric ABL-BCR transcript [56] encoding p40ABL-BCR protein is found, while a long chimeric transcript ABL-BCR encoding protein p96ABL-BCR is detected in 100% of patients with ALL associated with this translocation [57]. The mechanism of leukemogenesis is also different. Thus, p210BCR-ABL is able by itself, i.e. in the absence of p40ABL-BCR, to induce and maintain leukemia, while in the case of ALL both the chimeric proteins p96ABL-BCR and p185BCR-ABL/p190BCR-ABL are involved in the development of the disease [58]. Besides, in the case of this translocation normal genes are expressed along with abnormal ones [59, 60].
It was shown in some cases that only a chimeric allele is expressed, while expression of the normal allele is inhibited. Thus, in the case of Burkitt’s lymphoma, 90% of which are associated with translocations between chromosomes 8 and 14 [61], the chimeric allele is activated, whereas the MYC gene normal allele on chromosome 8 is not transcribed [61, 62]. The chimeric gene products can participate in inhibition of normal gene expression; for example, chimeric proteins AML1-ETO and PML-RARA repress transcription of normal genes AML1 and RARA due to formation of transcriptional corepressor complex containing histone deacetylase [63].
On the History of Discovery of Chromosomal Translocations
For a long time the role of genetic alterations in emergence of oncological diseases was a subject of debate. Back in 1890, David Paul von Hansemann observed asymmetric nucleus fission during mitosis in human cancer cells. He supposed that the malignant transformation of a normal cell is caused by the chromosomal abnormalities occurring at cell division [64].
Only 70 years later, in 1960, Peter Nowell and David Hungerford [65] studied leukocytes of CML patients and detected shortened chromosome 22. Later this chromosome was named Philadelphia (after the city in which it was discovered) [66]. This was the first example of association between a particular chromosomal rearrangement and a certain type of oncological disease. Thereby it was proved that malignant tumors could result from chromosomal alterations in somatic cells. For a long time before that it was assumed, despite Hansemann’s hypothesis, that emergence of numerous chromosomal abnormalities in malignant cells is a manifestation of genome instability1 in such cells, and chromosomal abnormalities are only a consequence rather than the cause of malignant transformation [66]. The subsequent search for the exact location of breakpoints resulted in the discovery of genes involved in translocation and in elucidation of functions of the encoded proteins, including chimeric ones. This, in turn, led to the discovery of inhibitors of those proteins and ensured progress in the treatment of cancer.
1 Genome instability is indeed an intrinsic property and a hallmark of malignant cells. It comprises increased likelihood and inheritance in a number of cell generations of various alterations to the genome [67]. There was an entire epoch, both historical and scientific, between the discoveries of Hansemann and of Nowell. Listed below are just some milestones of this period that in one way or another contributed to the discovery of translocations and revealed their role in emergence of malignancies. Some of these discoveries, although not directly related to translocations, are so global that it is impossible not to mention, as they are historical landmarks in the development of genetics and molecular biology.
Hansemann formulated his hypothesis just two years after the introduction of the term “chromosome” (from Greek “chroma”, stained, and “soma”, body) by Heinrich Waldeyer in 1888 [68] for designation of structures involved in the process comprised of a series of intranuclear changes and termed “karyomitosis” (thread-like metamorphosis) by Walter Flemming in 1878 [69, 70].
In 1909 Frans Janssens discovered crossover between sister chromosomes during meiosis in the gametes of salamanders [71].
In the 1910s Thomas Hunt Morgan et al. carried out intensive studies of drosophila chromosomes and formulated and confirmed experimentally the chromosomal theory of heredity [72, 73].
In 1914 Theodor Boveri published a chromosomal theory of cancer [74]. Today’s discoveries have confirmed the validity of his theory. The main provisions of Boveri’s theory are the assumptions that cancer is a cell’s problem, that a malignant tumor originates from a single cell having an aberrant chromatin complex, and that chromosome abnormalities are inherited by all daughter cells and cause the unlimited proliferation of tumor cells [75]. For a long time Boveri’s theory did not attract attention, like the laws of heredity discovered by Gregor Mendel in 1865 [76] and rediscovered by other researchers in 1900 [77].
In 1921 Alfred Sturtevant, a student of T. Morgan, found that chromosomes can exchange not only with homologous, but also with nonhomologous segments [78]. John Belling and Alfred Blakeslee proposed the term “segmental interchange” for similar chromosome rearrangements they observed in thorn apple (Datura stramonium) cells [79]. In the 1920s another student of T. Morgan, Hermann J. Muller, in experiments with drosophila, and Lewis Stadler with barley and maize, found that X-ray radiation causes a variety of chromosomal distortions [80, 81]. The abundance of mutations gave new material for investigations.
In 1931 Barbara McClintock discovered mobile genes (transposons) able to migrate within the genome [82]. In 1944 Oswald T. Avery, Colin Macleod, and Maclyn McCarty discovered in their experiments on bacterial transformation that DNA is the hereditary material [83]. In 1953 James D. Watson and Francis H. Crick deciphered the structure of DNA [84]. In 1956 Jo H. Tjio and Albert Levan determined the exact number of human chromosomes [85]. They found that the number of human chromosomes is 46 rather than 48, as was thought before. This event was later called the birth of human cytogenetics [86]. In 1960 Peter Nowell and David Hungerford discovered the Philadelphia chromosome [65]. In 1968 Torbjorn Caspersson et al. stained plant chromosomes with acrichine (quinacrine) and observed banded chromosomes [87] with alternate dark and light bands of varied intensities (a schematic image of such chromosomes is shown in Fig. 1). This made it possible to easily differentiate individual chromosomes and their regions. This method was later used for staining human chromosomes [88].
In 1971 at a meeting in Paris, cytogeneticists adopted a nomenclature for banded chromosomes according to which chromosomes were numbered in order of decreasing size, and chromosomal bands were enumerated beginning from centromeres. The only exception was the triple chromosome observed in Down’s syndrome that was already called the 21th. To escape confusion, number 21 was reserved for this chromosome, and therefore chromosome 22 is longer than chromosome 21. Also accepted was the convention to designate the short and long chromosome arms by letters “p” and “q”, respectively, as established in Chicago in 1966 [89] (Fig. 1). In 1972 Janet D. Rowley [90] discovered a translocation between chromosomes 8 and 21, the first translocation associated with a particular disease, acute myeloid leukemia. In 1973 she found that the Philadelphia chromosome is a result of translocation between chromosomes 9 and 22 [91]. In 1976 translocation between chromosomes 8 and 14 associated with Burkitt’s lymphoma [92] was detected, for which the participating genes were identified earlier than for other translocations; they included gene MYC and the variable region of genes for immunoglobulin heavy chains [93-95]. In 1982-1984 the fused genes of the Philadelphia chromosome were identified as oncogenes ABL (chr9) and BCR (chr22) [51, 96]. In 1991 the genes involved in a translocation between chromosomes 8 and 21 were identified as AML1 and ETO [97, 98].
Since then many chromosome translocations associated with malignancies and the involved genes have been discovered [31, 66].
Leukemia-Associated Chromosome Translocations
Leukemia. Leukemia is a malignant tumor of the hemopoietic system emerging in bone marrow. Blood cells in leukemia are able to proliferate at later stages of hemopoiesis compared to the norm. Incompletely differentiated cells proliferate in acute leukemia, whereas mature or nearly mature cells proliferate in chronic leukemia [99]. Leukemic cells are not able to function like normal blood cells. A decreased cell number and loss of the functions characteristic of normal cells lead to anemia, suppression of the immune system (and, consequently, to infectious disease), bleeding, adverse metabolic effects of increased cellular turnover, while circulation in the peripheral blood of undifferentiated cells results in appearance of leukemic infiltrates in various organs [100]. Acute leukemia is an aggressive disease, and without treatment it very quickly (in weeks or months) results in death. In the case of a chronic leukemia, patients can live without therapy for several months or years [101].
In 2001 WHO together with the Hematopathology Society and European Association of Hematopathology published “Classification of Tumors of Hemopoietic and Lymphoid Tissues” in the frame of the third edition of the series “WHO Classification of Tumors” [102]. This classification makes use of all available information (morphological, cytochemical, immunophenotypic, genetic, and clinical) for identification of clinically important symptoms. There can be myeloid or lymphoid leukemia depending on the type of progenitor cells whose differentiation is impaired [102]. Impairing the differentiation of lymphocytes leads to lymphoid leukemia; if other blood cells are affected, then myeloid leukemia develops. In the WHO classification the term “myeloid” relates to all cells belonging to granulocytic, monocytic, erythroid, and megalocytic lineages [45].
Thus, four main leukemia types are distinguished: AML (acute myeloid leukemia), ALL (acute lymphoid leukemia), CML (chronic myeloid leukemia), and CLL (chronic lymphoid leukemia) [103]. Myeloid leukemia is also termed myeloleukemia [104], acute myeloid leukemia – myeloblast leukemia [105, 106], lymphoid leukemia – lympholeukemia [104, 105], and acute lymphoid leukemia – lymphoblast leukemia [105, 106]. The approximate frequencies and other features of the different types of leukemia are shown in Table 1.
Table 1. Different leukemia types in USA
(data of the National Cancer Institute of the USA [103])
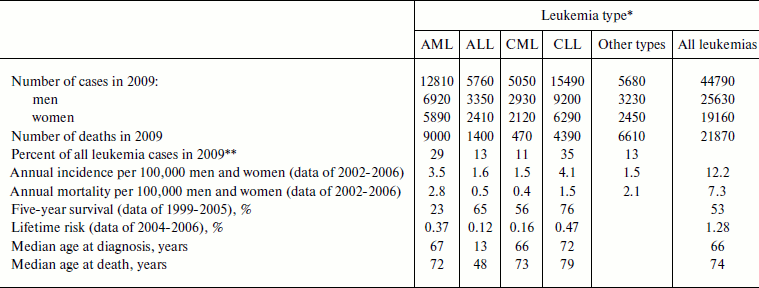
*AML, acute myeloid leukemia; ALL, acute lymphoid leukemia;
CML, chronic myeloid leukemia; CLL, chronic lymphoid leukemia.
**From data shown in the first line of the table.
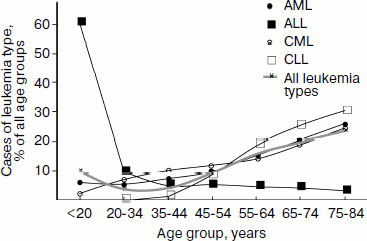
Epidemiology. In 2006 in Russia 33% of boys and 31% of girls were diagnosed with leukemia among those children of age 0-14 who developed cancer, which, depending on age, constituted 5.1 (age, 0-4), 3.7 (5-9), and 2.6 (10-14) cases per 100,000 children [107]. Among all age groups, 2.4% of men and 2.1% of women were diagnosed with leukemia of those who developed malignant tumors, which constituted 7.5 cases per 100,000 men and women [104]. Although these data show that on average adults are diagnosed with leukemia even more frequently than children, however approximately two-fold decrease in the leukemia incidence is observed for people 20-45 years old compared to those younger than 20 years old (Fig. 2, thicker gray curve). Frequency of the individual leukemia types also changes with age: chronic leukemia is rare in children, with CLL being virtually absent from this group, while it prevails in adults (Fig. 2).
In Russia in 2006, 34.4% of boys and 29.4% of girls who died of all malignant tumors died of leukemia [108], which, depending on age, constituted 1.9 (age 0-4), 1.2 (5-9), and 1.1 (10-14) deaths per 100,000 children [107]. Among adults, leukemias caused death of 5.4 of 100,000 people, in men being 2.5% and in women 2.9% in the structure of mortality caused by malignancy [108].Fig. 2. Distribution of leukemia types among age groups in 2002-2006 in the USA. Based on data of the National Cancer Institute of the USA [103].
In the USA leukemias are among the ten most widespread malignancies, and as of January 1, 2006 there were 231,586 patients suffering from leukemia or in the stage of remission [102]. Among all children of age 0-14 diagnosed with cancer in 2006, 30.6% were diagnosed with leukemia (altogether 2779 children, 2180 with ALL and 373 with AML), which constituted 4.7 patients per 100,000 children. Leukemia caused death of 28.7% of children (altogether 417 children, 188 with ALL and 137 with AML) who died of malignancies or 0.7 deaths per 100,000 children [109]. Other statistical data for individual leukemia types are given in Table 1.
Leukemia-associated chromosomal translocations. It is not accidental that translocations were discovered in leukemia patients: 40-50% of AML cases are associated with chromosomal translocations and 95% of CML cases are associated with Philadelphia chromosome formed upon t(9;22)(q34;q11.2) translocation (Table 2 and [110]). At present, about 500 leukemia-associated chromosomal translocations are known [111].
Table 2. Most frequent chromosomal
abnormalities in leukemia
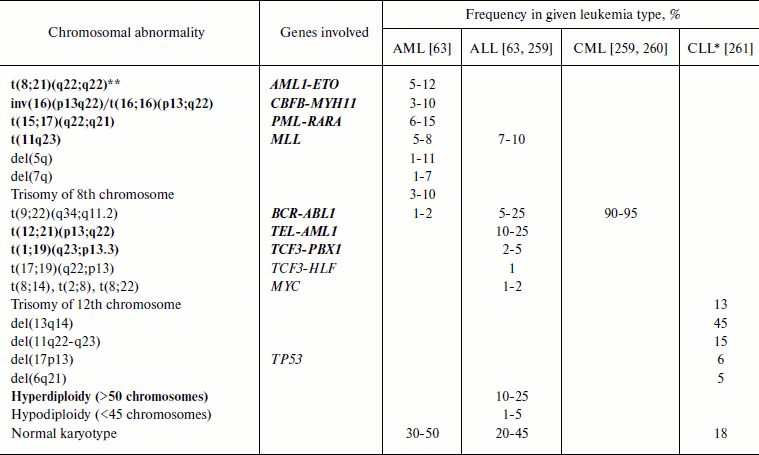
Note: AML, acute myeloid leukemia; ALL, acute lymphoid leukemia; CML,
chronic myeloid leukemia; CLL, chronic lymphoid leukemia.
Gene abbreviations (synonyms are given in parentheses in accordance with
databases Genatlas [113] and GeneCards [43]; underlined is the main gene symbol according to
the HGNC nomenclature [262]):
AML1 (RUNX1, PEBP2A2, CBFA2,
AMLCR1, RUN1, EVI13, TSG21A,
AML1-EVI-1, EVI-1), acute myeloid leukemia 1;
BCR (ALL, CML, PHL, D22S11,
D22S662, BCR-ABL1, BCR1, FLJ16453),
breakpoint cluster region;
CBFB (PEBP2B), core-binding factor, beta
subunit;
ETO (RUNX1T1, MTG8, AM1L1T1, CDR,
CBFA2T1, MGC2796, MTG8b, ZMYND2), eight twenty
one protein gene (ETO) = myeloid translocation gene on 8q22
(MTG8);
HLF − hepatic leukemia factor;
MLL (EC 2.1.1.43, ALL1, CDK6/MLL fusion
protein, CXXC7, FLJ11783, HRX, HRX1,
HTRX, HTRX1, KMT2A, MLL/GAS7, MLL1A,
MLL-AF4 der(11) fusion protein, TET1-MLL,
TRX1), myeloid/lymphoid or mixed-lineage leukemia;
MYH11 (SMHC, SMMHC, AAT4, FAA4,
FLJ35232, MGC126726, MGC32963, KIAA0866),
myosin, heavy polypeptide 11, smooth muscle;
MYC (c-MYC, bHLHe39, MRTL), v-myc
myelocytomatosis viral oncogene homolog (avian);
PBX1 (HOXP, PMX1, DKFZp686B09108,
MGC126627, PRL), pre-B cell leukemia homeobox 1;
PML (MYL, TRIM19, PP8675, RNF71),
promyelocytic leukemia;
RARA (NR1B1, C13A4, RAR, RAR-alpha),
retinoic acid receptor, alpha;
TCF3 (E2A, ITF1, TCFE2A, EVI104,
TFE2, MGC129647, MGC129648, bHLHb21,
VDIR), transcription factor 3 (E2A immunoglobulin enhancer binding
factors E12/E47);
TEL (ETV6, TEL/ABL, TEL1), TEL
oncogene;
TP53 (LFS1, TRP53, p53, FLJ92943),
tumor protein p53.
*Data were recalculated as percent of all CLL cases (in original
article they were given as percent of all chromosomal abnormalities in
CLL).
**Recurrent abnormalities according to WHO classification are shown
in bold.
Frequencies of the most frequent of widespread chromosome abnormalities in different types of leukemia are given in Table 2. It should be noted that the frequency of a particular chromosome abnormality is strongly dependent on the patient’s age. Thus, in ALL patients frequency of t(9;22) increases with age (3% in children and 25% in adults) while that of t(12;21) decreases (22% in children and 2% in adults) [112]. In AML patients less than 1 year old translocations of chromosome 11 are major ones (more than 30% of all AML cases), with the most frequent of them being t(4;11), while in all other age groups these translocations appear in 1-3% of AML cases [89]. In children under one year old the translocation t(8;21) is virtually absent, but then it becomes the most frequent at the age of 1-19 years, constituting 15% of AML cases in this age group [89].
Search for the chromosomal translocation breakpoints is one of the most efficient approaches for revealing new genes involved in cell growth regulation and induction of malignant transformation. Thus, the discovery of chromosomal translocation t(8;21) revealed the AML1 gene located on chromosome 21 and encoding the hemopoiesis transcription factor of the same name [97].
Many genes were named after the type of leukemia in which they were detected, for example the name of gene AML1 originates from the type of leukemia that emerges upon distortions of this gene, acute myeloid leukemia (different examples of this kind of names are given in the legend to Table 2). Gene BCR (for “breakpoint cluster region”) involved in t(9;22) translocation acquired its name upon identification of breakpoints in chromosome 22 at the site of fusion of this gene with gene ABL [96].
Later it was found that many genes discovered along this way including those discussed here encode tyrosine (ABL) or serine (BCR) kinases or key transcription factors (AML1, MLL, PML, ETO, PBX1) [33, 66, 113, 114]. Clearly, changes in such genes should affect a number of processes in the organism.
Chromosomal translocations associated with acute myeloid leukemia. Acute myeloid leukemia (AML) is an aggressive type of leukemia that is most frequent in early childhood and in old age [115].
According to the WHO Classification of 2001, recurrent chromosomal abnormalities in AML include t(8;21)(q22;q22) (AML1-ETO), inv(16)(p13q22) or t(16;16)(p13;q22) (CBF-MYH11), t(15;17)(q22;q12) (PML-RARA), and t11q23 (MLL) that altogether are associated with 30% of all AML cases [116] and are characterized by favorable prognosis [117]. In 2008, translocations t(6;9)(p23;q34) (DEK-NUP214), inv(3)(q21q26.2) or t(3;3)(q21;q26.2) (RPN1-EVI1), t(1;22)(p13;q13) (RBM15-MKL1) were added to the list of recurrent chromosomal abnormalities in AML [45].
Chromosomal translocation t(8;21)(q22;q22). A nanocolony-based diagnostics for oncological diseases associated with chromosomal translocations has been developed by diagnosing leukemia associated with translocation t(8;21)(q22;q22) as an example. This variant of leukemia comprises up to 12% of all cases of myeloid leukemia (Table 2) and is characterized by favorable prognosis [118]. The five-year survival rate for patients under 55 years old with this chromosomal abnormality is from 60 to 70% [119].
This translocation leads to the fusion of genes AML1 (located on chromosome 21, GenBank number D43969) and ETO (located on chromosome 8, GenBank number D14289) and hence to the formation of a chimeric gene AML1-ETO [98] whose expression results in the synthesis of mRNA AML1-ETO, which serves as a marker of this type of leukemia [3, 46]. In accordance with the HGNC nomenclature, this mRNA is named RUNX1-RUNX1T1 (Table 2).
The 150 kb-long AML1 gene includes nine exons [3] and encodes a key hemopoiesis transcription factor regulating expression of an entire group of genes [120-122]. Besides t(8;21)(q22;q22), gene AML1 is also involved in at least 31 translocations, three of which, like AML1-ETO, involve chromosome 8 [123]. Gene ETO of 87 kb in length includes 13 exons [3] and encodes a transcription corepressor interacting with other corepressors, including histone deacetylases [124, 125].
Breakpoints in t(8;21)(q22;q22) occur within introns, and although several regions of break and fusion points were detected [126], in this translocation only a single type of chimeric transcript (AML1-ETO) is formed in which exon 5 of the AML1 gene is fused to exon 2 of the ETO gene. The fusion points in all patients with this leukemia type coincide with single nucleotide accuracy [46, 127, 128].
In the 1990s there were reports on the existence of alternative splicing variants with a shifted open reading frame, harboring insertions of 46-82 nucleotides in length between the fifth exon of AML1 and the second exon of ETO [129, 130]. However, in all cases such variants were minor compared to the main variant [129, 130] and might have been generated by retrovirus polymerases during reverse transcription [131] and/or by DNA polymerases during PCR [2, 132, 133].
For AML1-ETO mRNA, alternative splicing variants encoding shorter chimeric proteins but containing the same sequence around the fusion point as the main variant were found [134, 135].
The consequences of translocation are numerous [125]. Analysis of the effects of translocations, including that of t(8;21), has shown that in AML the expression profile of over 1000 genes involved in transcription, cell cycle, protein synthesis, and apoptosis is altered [136]. Recent investigations revealed the ability of chimeric protein AML1-ETO to directly inhibit a number hemopoiesis transcription factors that are suppressors of malignant tumors in several key points of differentiation [137].
Is chromosomal translocation sufficient for emergence of disease? Chromosomal translocations are frequent and mostly prenatal, early genetic events [138]. The study of leukemia cases in monozygous twins and retrospective investigation of archival neonatal blood spots have shown that often (but not always) chromosomal translocations associated with child leukemias emerge in the womb [139-141], and that latent period in some cases can exceed 10 years [142]. Prenatal transmission of a leukemic clone from mother to child is also possible [143, 144].
Chromosomal translocation does not necessarily result in cancer. Thus, translocations t(8;21) and t(12;21) are detected in the cord blood of newborns at a frequency 100 times exceeding the disease rate in children with the respective leukemia type [145]. In most cases additional genetic alterations are needed to trigger the development of disease [146, 147], that is, a “second hit” is required [146-149]. Thus, leukemia was shown to emerge in situations when the ETO part of the chimeric protein loses its ability to bind histone deacetylase complex [150]. The loss of this ability may be caused by a number of factors, such as the absence of a binding domain in the ETO portion due to early translation termination (for example, due to occurrence of an early stop codon because of alternative splicing [135, 151]) or proteolytic cleavage, as well as due to mutations in the histone deacetylase complex itself [152]. Various factors, including abnormal immune response to infection [153], can contribute to genetic changes and malignant transformation of a pre-leukemia clone [115].
The requirement of additional genetic changes for disease development does not diminish the therapeutic and diagnostic value of ascertaining the type of translocation upon emergence of the disease and monitoring the chimeric product during the treatment process. Once emerged, the disease remains firmly associated with the presence of a malignant cell clone carrying the primary chromosomal translocation and with the presence in clinical samples of the related chimeric RNA marker [110, 154].
MINIMAL RESIDUAL DISEASE
Definition of Minimal Residual Disease
An important characteristic of cancer including leukemia is minimal residual disease (MRD), a post-treatment condition of the patient’s body still retaining a small number of tumor cells that cannot be revealed during standard cytological investigation of blood and bone marrow but are detectable by more sensitive methods [110] (see “Methods for Diagnosis of Minimal Residual Disease”).
Recently, because of the availability of efficient antileukemia drugs and sensitive methods for the detection of leukemia molecular markers, it has become possible to distinguish between the clinical-hematological remission in the absence of clinical symptoms [4, 155] and molecular remission. There is some disagreement on defining the term “molecular remission”. Some authors consider molecular remission as a “persistent long-term absence of a specific RNA marker or preservation of its low level in blood and bone marrow” [156]. Other researchers define molecular remission as the complete absence of a specific RNA marker in blood and bone marrow as determined by the currently most sensitive PCR assay [157]. The same discord is observed with the definition of “molecular relapse”. As a consensus, molecular relapse can be defined as “the appearance of RNA marker or increase in its amount during hematological remission” [156].
Probably the reason for the lack of agreement on how molecular remission and molecular relapse should be defined lies in the fact that it is unknown what amount of a marker RNA can be regarded as “low” or is contained in the blood and bone marrow when it is not detected, and how long should its “persistent long-term absence” be to make sure that there is no risk of relapse. The uncertainty of these parameters is due to limitations of the methods used to determine MRD, in particular, to problems of conventional (solution) PCR [8].
Importance of Detection of Minimal Residual Disease
Investigation of the molecular mechanisms of leukemogenesis stimulated the discovery of molecular targets for therapeutic intervention [63, 158]. The use of new chemical preparations has resulted in significantly better outcomes of the treatment of different types of leukemia [119, 159-161].
When acute myeloid leukemia is just diagnosed, more than 20% of the blood and bone marrow nucleated cells are blast cells, whereas these under-differentiated cells are virtually absent from peripheral blood in normal conditions and in clinical and hematological remission and make up less than 5% of bone marrow cells [45, 110]. When modern intensive chemotherapy is used, remission occurs in 60-80% of AML patients [154].
However, without the use of adjuvant therapies, in nearly all AML cases a relapse (resumption of clinical symptoms, often starting with the appearance of leukemic infiltration foci [162]) occurs after a few weeks or months of remission [154]. Leukemic cells that survived treatment can become resistant to antileukemic drugs, thus resulting in an increased MRD level, despite the fact that cytomorphologically the patient remains in remission [154]. In most cases, the leukemic cells resume to proliferate resulting in a clinical relapse. It is from relapse, which is observed in 30-50% of cases of leukemia associated with t(8; 21) (q22; q22), [163, 164], that patients eventually die.
Quantitative diagnosis of MRD is important at nearly all stages of treatment of the disease [165]. With the use of modern drugs at the early stages of treatment, the number of leukemic cells rapidly drops to a cytologically undetectable level, and determination of MRD at this stage has prognostic value that exceeds all known classical prognostic factors [166]. Clinical studies have shown that patients with undetectable MRD have an excellent prognosis and are good candidates for reduced treatment intensity, or at least should not be subjected to further intensification of treatment, unlike patients with high MRD level who need urgent intensification of treatment and even addition of new therapeutic approaches [110, 166, 167]. Prerequisites for successful bone marrow transplantation are maximal reduction of MRD before transplantation [166, 168] and favorable results of monitoring the MRD kinetics after transplantation [169]. It is particularly important to monitor MRD during remission for early detection and prevention of impending relapse [154, 170].
Keeping all this in mind, one can conclude that diagnosing MRD is important not only for monitoring the course and prognosis of the diseases, but also for choosing the most effective and lowest-risk treatment strategy [154]; therefore, it is necessary to include MRD determination into all treatment protocols to ascertain the effectiveness of a selected treatment procedure for a given patient [166].
Methods for Diagnosing Minimal Residual Disease
Methods used for MRD detection should meet the following requirements: high sensitivity, specificity, accuracy in quantification of the marker, technical practicability (ease of standardization and speed of the assay) for clinical use, as well as intra- and inter-laboratory reproducibility [166].
MRD is detected using different approaches, such as fluorescent in situ hybridization (FISH), flow cytometry, and PCR [44]. Sensitivity of MRD diagnostics is expressed by the inverse of the number of normal cells per detected leukemic cell.
Fluorescence in situ hybridization. Fluorescence in situ hybridization (FISH) comprises hybridization of interphase or metaphase chromosomes with gene-specific fluorescent probes and counting the labeled chromosome using a fluorescence microscope.
The sensitivity of FISH depends on the probe type and the number of examined cells [44] and is in the range 10–1-10–3; in this case translocations are detected at a lower sensitivity than the loss or increase in the number of chromosomes [110].
In FISH variants known as SKY (spectral karyotyping) and M-FISH (multiplex FISH) sets of differently colored probes specific for various chromosomes [44] are used. Co-hybridization of different probes with metaphase chromosomes reveals a number of genetic disorders, including translocations [171]. The method includes preparation of metaphase chromosomes, their treatment for hybridization, hybridization itself, scanning, and image development, and it takes about 6 days [171].
FISH is applicable to 40-60% of cases, and besides the inability to analyze a large number of cells, its sensitivity is limited by false positive results due to random signal colocalization [110].
Flow cytometry. This method is based on abnormal immunophenotype of leukemic cells (LAIP, leukemia-associated aberrant immunophenotypes). The procedure of MRD detection by flow cytometry is as follows: nucleated blood or bone marrow cells are isolated, incubated with fluorescently labeled leukemic cell surface antigen-specific monoclonal antibodies, and passed through a flow cytometer to register the fluorescence produced by the antibody-binding cells [44]. The most accurate results are obtained using multiparametric flow cytometry and up to six antibodies labeled with different fluorophores [172-175]. Among advantages of this method are its applicability in the absence of known genetic markers (chimeric genes or other genetic abnormalities), relatively low cost, and speed (1-2 days) [175].
Theoretically, the sensitivity of the method can be as high as 10–6, but in practice it is much lower [175]. The following factors decrease the sensitivity of flow cytometry: (1) insufficient difference in antigenic profiles of tumor and normal cells, (2) existence of several leukemic cell subpopulations, some of which as minor clones that are difficult to identify, (3) inability to track immunophenotypic switch between diagnosis and relapse, (4) need to count a large number of cells and need for technical expertise [175], (5) loss of cells depending on their type and the isolation procedure [176].
If the immunophenotype of leukemic cells is sufficiently different from the immunophenotype of normal cells, then the sensitivity is 10–4. In other cases, when immunophenotypes of the leukemic and normal cells partially overlap, detection sensitivity decreases to 10–3 [177].
For achieving higher specificity, it was suggested to determine individual immunophenotype of leukemic cells for each patient using a large set of antibodies and to compare this immunophenotype with that of normal bone marrow cells of the same patient to find the maximal difference [154]. However, it was concluded at a conference in Bethesda in 2006 that even with this approach reliable results could not be obtained with flow cytometry in 50% of cases. To reduce cost, the 2006 Bethesda International Consensus recommended minimizing initial investigations and directing efforts to more intense analysis of samples of those patients in whom initial investigations revealed a pronounced aberrant immunophenotype, and even in this case it is suggested to use about 30 types of antibodies for initial screening [178].
The switch of leukemic cell immunophenotype in the period between diagnosis and recurrence, which is observed in 90% of patients [173], is an even more serious problem, often resulting in impossibility of MRD detection and probably emerging due to instability of malignant cell genomes or due to expansion during relapse of a small subpopulation of leukemic cells phenotypically different from the main one and undetectable at the time of diagnosis [154, 166]. Probably in future it will become possible to reveal typical changes in leukemic cell immunophenotypes by correlating these changes with recurrent genetic abnormalities specific for a given type of leukemia, as was done in the case of chromosomal translocation t(15;17) (PML-RARA) [179].
Polymerase chain reaction. If leukemia is associated with the formation of a chimeric RNA, then the most sensitive and specific method is the detection of the marker RNA using reverse transcription (RT) followed by polymerase chain reaction (PCR) [154]. The method includes isolation from a clinical sample of total RNA, cDNA synthesis using reverse transcription, and then cDNA amplification using PCR. The region of a chimeric cDNA that includes the fusion point of sequences of the contributing genes is amplified in PCR using primers complementary to sequences located on different sides of the fusion point. Since only in the given type of leukemia these sequences occur within the same molecule, DNA synthesis during RT-PCR should ideally take place only when the sample contains the appropriate chimeric RNA.
Most popular is a variant of the method that allows the PCR product to be detected during synthesis, in “real time”. This variant is designated RQ-PCR [173] or qPCR (qRT-PCR) [180]. The PCR is carried out in the presence of fluorescent reporter molecules (fluorescent intercalating dyes and/or fluorescently labeled oligonucleotide probes complementary to the PCR product) whose fluorescence is enhanced upon interaction with amplified DNA. The amount of target is estimated by the number of PCR cycles at which the fluorescent signal begins to exceed the background. The higher the starting amount of the target, the earlier this occurs.
The sensitivity of RT-PCR for the detection of chimeric transcript is 10–4-10–6 [173]. In routine use it usually does not exceed 10–5 [181].
Among factors restricting the sensitivity of detection of an RNA target is the susceptibility of RNA to degradation and variability (non-reproducibility) of the reverse transcription yield [177], limited primer specificity, decrease in sensitivity of detection in the presence of non-target nucleic acids [180], need for signal normalization using external and internal standards, inconsistency of data from different laboratories [154], and non-reproducibility of the measurements of small amounts of the target [180].
If leukemia is not associated with the formation of a chimeric transcript, then other targets, such as aberrantly expressed genes, should be used for MRD detection by RT-PCR. Thus, the Wilms’ tumor gene (WT1) is highly expressed in 70% of patients with the diagnosis of acute leukemia, and detection of the respective mRNA in blood or bone marrow is indicative of the presence, continuation, or relapse of the disease [182]. Limitation in this approach is the high background from RNAs of normal bone marrow cells, which often decreases the reliability of determination [177].
Other potential targets for MRD detection include the mutant nucleophosmin gene NPM1 whose expression sharply increases in AML [183]. Specific targets (both for MRD detection using QRT-PCR and for therapy) can be miRNAs whose expression profile correlates with the leukemia type [63].
It is probably not worth arguing which of the abovementioned methods is better. Each is good in its own way in a particular situation. At the time of diagnosis, when the number of leukemic cells in the blood is high and high sensitivity of analysis is not required, but there is a need to identify the type of genetic abnormality and possible markers, it is logical to use FISH and other cytological methods.
For MRD diagnostics, if the disease is associated with translocation and, hence, with the emergence of a chimeric RNA, it is preferable to use quantitative RT-PCR as the most sensitive method capable of detecting the target directly associated with leukemogenesis [110, 154, 166]. However, if a given type of leukemia is not associated with translocation and there is no chimeric marker, then multiparametric flow cytometry can be used for detecting leukemic cells (in this case the immunophenotype of the leukemic cells has to be first determined). Alternatively, quantitative RT-PCR can be used to assay a disease marker that is either associated with another type of genetic abnormality or the product of a hyperexpressed gene.
USE OF NANOCOLONIES FOR DIAGNOSING MINIMAL RESIDUAL
DISEASE
Nanocolonies
Principle of nanocolonies. Nanocolonies (or molecular colonies) are formed when exponential amplification of nanomolecules, such as RNA and DNA, is carried out in a solid medium with nanometer-sized pores [9, 12]. The key moment in this case is immobilization of the medium. There is no convection in such a medium, and therefore the progeny of each original molecule does not spread over the whole reaction volume as occurs in a liquid medium, but it is concentrated around the parental molecule and forms a colony. Thus, each colony represents the progeny of a single molecule, i.e. it is a molecular clone and consists of identical molecules, which makes it possible to detect, count, and analyze single DNA and RNA molecules.
Any enzymatic system for nucleic acid amplification can be used for growing nanocolonies [6]. A variant of this method is DNA amplification in a polyacrylamide gel with the use of polymerase chain reaction [9, 12, 19, 184]. When PCR in carried out in a thin gel layer, nanocolonies are arranged in a single plane and produce a two-dimensional pattern. Simple counting of nanocolonies allows the titer of the initial DNA and RNA molecules in a sample to be directly determined.
Growing DNA colonies. Since PCR requires temperature cycling over a broad range of temperatures, including heating to 92-94°C for melting a double-stranded DNA, a thermostable medium such as a polyacrylamide gel is used for growing DNA colonies using this reaction [9, 12]. The gel is prepared in a well in a microscope slide [7, 19]. It is cast, washed, and dried.
To analyze RNA, RT is first carried out to convert RNA into the DNA form. Directly before experiment the dry gels are impregnated with the complete reaction mixture containing deoxynucleotides, oligonucleotide primers, a thermostable DNA-dependent DNA polymerase, and the analyzed sample, DNA or cDNA, and the wells are sealed. Then slides with gels are placed in a thermocycler with flat-bed heater (such as commonly used for in situ PCR) and temperature cycling (PCR itself) is started.
For the assays of chimeric AML1-ETO mRNA, the marker of leukemia associated with translocation t(8;21), we used the RT and PCR primers recommended for MRD detection by the Europe Against Cancer Program (an institution which, in particular, develops standards for leukemia diagnostics) [3].
Detection of chimeric nanocolonies. Nanocolonies can be detected by a number of means [18, 28, 185, 186]. We have elaborated two methods that use fluorescence to detect chimeric colonies (consisting of molecules carrying fused sequences of AML1 and ETO gene fragments). Both are based on the hybridization of amplified DNA with specific probes complementary to an internal region of the amplified target.
One method includes hybridization of nanocolonies on nylon membrane [185, 187]: after PCR, colonies are transferred from gel by blotting onto the membrane, which is then hybridized with a mixture of two fluorescent probes. One probe, specific for the AML part, is labeled by a red fluorophore (cyanine-5, Cy5), the other specific of the ETO part and is labeled by a green fluorophore (cyanine-3, Cy3) (see scheme in Fig. 6a below). Then the membrane is analyzed using a fluorescence scanner at two wavelengths, optimal for red and green fluorophores, respectively. The ability of a colony for hybridization with both the probes specific for the two genes shows that this colony is formed by the chimeric molecule.
The other method of detection comprises hybridization of colonies directly in gel, in “real time” (Fig. 3). To this end, a detection system was developed which is based on the principle of FRET (fluorescence resonance energy transfer) [188] and comprises a pair of adjacently hybridizing probes carrying at proximal termini two fluorophores, donor and acceptor (see scheme in Fig. 6a below). For efficient FRET the distance between the hybridized probes should not exceed a few nucleotides [189]. The gel is scanned during PCR, with excitation of the donor and detecting fluorescence of the acceptor; in this case, resonance transfer of energy from the donor to the acceptor occurs. The probes are complementary to the AML1-ETO cDNA around the point of fusion of the AML1 and ETO sequences, so that one probe is complementary to the AML sequence while the other is complementary to the ETO sequence.
In clinical practice, chimeric RNAs should be better assayed with the second method. First, the gel does not need to be opened after amplification of the target, which simplifies the procedure and minimizes the risk of cross contamination between samples. Second, the gel can be scanned at a single wavelength only and chimeric colonies are directly revealed; there is no need to compare the scans obtained for different fluorophores.Fig. 3. Detection of AML1-ETO chimeric cDNA in real time. Colonies of AML1-ETO cDNA were obtained using asymmetric PCR in a gel in the presence of FRET probes. Above the gel image the PCR cycle number is indicated, after which the image was obtained by scanning the gel using a blue laser (488 nm) and red emission filter (670 nm). The length of the amplified PCR product was 260 bp.
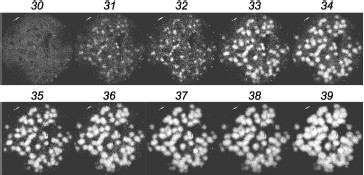
Instead of FRET probes, a single probe complementary to the fusion site of the chimeric cDNA components, a molecular beacon [190], or a combination of an intercalating dye and a probe labeled with appropriate fluorophore can be used. We have demonstrated the ability of such probes to reveal nanocolonies [186]. It is possible to detect nanocolonies formed by different targets during multiplex PCR using several such probes labeled with different fluorophores.
Some translocations, whose break and fusion points differ in different patients, result in the formation of dissimilar variants of the chimeric RNA whose sequences around the fusion point are different. This is observed for translocations t(9;22) (BCR-ABL1), t(4;11) (MLL-AF4), t(15;17) (PML-RARA), and inv(16) (CBFB-MYH11) [46]. Therefore, if the variant of a chimeric RNA is not known, chimeric colonies can be detected, along with identification of the chimeric RNA variant, in the real time format by using a set of molecular beacons labeled with different fluorophores and complementary to different parts of the chimeric sequence or to different variants of the fusion sequence.
Sensitivity of DNA detection using nanocolonies. It was shown that, within statistical scatter, the number of nanocolonies was equal to the number of DNA molecules added to the gel [19, 20, 24]. Thus, nanocolonies can reveal 100% of DNA molecules. In other words, if the sample introduced into the gel contains at least one DNA target molecule, the target will be detected, i.e. the sensitivity of the detection of a DNA target is one molecule.
The sensitivity of target detection using nanocolonies does not decrease in the case of multiplex PCR (even if the targets differ in concentration more than 106-fold), or when PCR is carried out in the presence of a huge excess of non-target DNA [19].
Diagnostic Procedure and Its Development
The optimal amount of an analyzed sample is a prerequisite for obtaining reliable quantitative data [191, 192]; therefore, the RNA yield and integrity after a series of manipulations (sample collection, preservation and transportation, as well as RNA isolation) are critical parameters for successful RT-PCR analysis [180].
When we developed the MRD diagnostic procedure, we paid attention to recommendations of the Europe Against Cancer Program for MRD monitoring by RT-PCR [3, 5, 46, 193] as these recommendations are the best currently available in the field of MRD diagnostics.
The material for MRD diagnostics using RT-PCR is an RNA preparation isolated from the patient’s blood or bone marrow. However, such clinical samples contain a huge amount of ribonucleases, which makes the isolation of intact RNA a difficult problem. Also, blood and bone marrow contain many different RT and PCR inhibitors (heme, iron ions, polyphenols, polysaccharides, etc.). The maximal detection sensitivity is only achieved if RNA is extracted from nucleated cells that are first isolated from the blood or bone marrow [173] (such a protocol is recommended by the Europe Against Cancer Program [193]). This approach eliminates the bulk of RT and PCR inhibitors but has a number of disadvantages that can result both in false positive results (detecting a chimeric RNA when it is not contained in the sample) and false negative results (failure to detect chimeric RNA present in the sample). Isolation of nucleated cells is too expensive, time-consuming, and technically complicated a procedure for routine use in clinical laboratories, and there is a high risk of cross contamination of the samples and hence, of a false positive result. In addition, in this case 40-60% of cells are lost [194], which complicates the quantitative assessment and increases the risk of RNA degradation [195] and, hence, of false negative results.
Therefore, we developed a method for RNA isolation directly from whole blood and bone marrow rather than from fractionated clinical sample. Also, all stages of the diagnostic procedure were optimized and characterized quantitatively. The detection of the target itself was carried out using nanocolonies.
Conservation of clinical samples. During the work we confronted the need to develop a method for conservation of blood and bone marrow samples that would provide for complete preservation of nucleic acids, particularly RNA, prior to their isolation. In 1979 J. Chirgwin et al. suggested lysing animal tissues with a solution containing guanidine thiocyanate (a powerful chaotropic agent), sarcosyl (detergent), 2-mercaptoethanol (S–S-bond reducer), EDTA (polyvalent metal chelator), and citrate (buffer) [196]. Such combination of reagents efficiently deproteinates nucleic acids and inhibits ribonucleases. By mixing the whole blood with such a lysing solution we showed that RNA and DNA are preserved in the lysate without detectable changes for at least a fortnight at 4°C [197, 198]. If necessary, guanidine thiocyanate lysates of whole blood can be stored at –20°C for over one year with preservation of both DNA and RNA. For convenience of sample transportation, RNA preservation can be also provided at room temperature (at least up to two weeks) after precipitating nucleic acids by the addition of two volumes of isopropanol to the lysate. The lysate composition is completely compatible with the most widely used procedures for nucleic acid isolation, including RNA isolation using extraction by acidic phenol [199] and RNA and DNA isolation by adsorption on silica or glass beads or filters [200] used, for example, in the NucliSens extractor (Organon Teknika, The Netherlands). The lysate with added isopropanol is completely compatible with our method of simultaneous isolation of RNA and DNA [20] and with RNA isolation using extraction by acidic phenol; it is only necessary to dissolve the isopropanol pellet in the lysing solution before the extraction (our unpublished data). Actually, the process of sample conservation replaces the step of obtaining lysate in the abovementioned methods. Thus, the method of sample preservation comprises mixing the sample, immediately upon its collection, with lysing solution containing the abovementioned components.
RNA isolation. Our protocol for RNA isolation is based on the well known method of Chomczynski proposed for RNA isolation from animal tissues and cells [199]. The method includes phenol extraction of guanidine thiocyanate lysates at pH 4.0, in which only RNA remains in the aqueous phase, while DNA in complex with proteins is transferred into the organic phase. Using internal controls we showed that when used for the extraction of whole blood lysates, this method yields 85-90% of high molecular weight RNAs [20].
However, it turned out that the resulting preparation contains contaminations that inhibit both RT and PCR. To remove the inhibitors, the isolation procedure was supplemented by RNA precipitation with acetone–ethanol mixture and gel filtration through a spun column [27].
Optimization of reverse transcription. The Europe Against Cancer Program recommends that the RT step is carried out using a mixture of random hexanucleotides as primer, 200 units [3] or 100 units [46] of RNase H¯ MMLV reverse transcriptase, and that 10% of the RT product is used in PCR.
For the synthesis of cDNA we used a sequence-specific reverse primer. Although the use of random hexanucleotides to prime RT gives the highest yield of cDNA, it is not good for quantitative RNA determinations for a number of reasons [201]. One reason is that the RT yield is overestimated, because RT may start from many points, resulting in more than one cDNA molecule being synthesized from one RNA template. In this case the number of cDNA copies per RNA template molecule is not fixed and depends both on the RNA length and structure and on the extent of its degradation. Another reason is that upon random priming on RNA isolated from clinical samples the bulk of cDNA is synthesized by copying the ribosomal RNAs, which constitute over 90% of the preparation, and these cDNA can compete with the amplification of a specific target present in the sample at low concentration. Therefore, the use in RT of a specific primer instead of random hexanucleotides may result in a higher yield of the specific PCR product [202].
The recommendation to use in PCR only 10% of the cDNA obtained at the RT step artificially decreases the assay sensitivity and seems to be due to the fact that some low molecular weight components of the RT reaction, such as dithiothreitol, may inhibit PCR. We found that gel filtration of the RT products completely eliminates inhibition and makes it possible to use in PCR all the synthesized cDNA.
It turned out that the use of the recommended amount of reverse transcriptase inhibits colony growth even if a pre-synthesized cDNA is used as a PCR template [27]. In other words, if a mock sample containing the RNA preparation isolated from a donor blood and no chimeric RNA was subjected to RT and combined with the AML1-ETO cDNA just before PCR, the chimeric DNA colonies grew to a lesser intensity compared with controls, in which no RT product was added or the RNA preparation was added to the PCR mixture omitting the RT step. This means that if RT is first carried out on donor blood free of chimeric RNA, and then, before PCR, the AML1-ETO cDNA is added to the RT product, the chimeric DNA colonies grow less intensively than the controls free of added RT product, or if the RNA preparation was added into the gel immediately before PCR without an RT stage. Probably the reason for the inhibition of colony growth is a competition from the nonspecific cDNA synthesized on the blood RNA during reverse transcription. Inhibition can be completely eliminated if the amount of reverse transcriptase is reduced a hundred-fold [27]. Reducing the amount of reverse transcriptase also increases the yield of reverse transcription, i.e. the number of colonies increases up to 50% of the number of chimeric RNA molecules subjected to RT [27].
Quantitative evaluation of the diagnostic procedure. Quantitative control of each step of the developed diagnostic procedure permitted the absolute titer of a target in the initial clinical sample to be accurately calculated without determining control (reference) RNAs.
Quantitative evaluation of each step of the procedure gave the following results: the method used for collection and conservation of clinical samples excluded any RNA degradation and preserved 100% of the initial RNA amount [197]; the yield of the improved RNA isolation procedure was close to 100% [27]; the yield of a reverse transcription of the AML1-ETO mRNA was reproducibly 50% [27], and finally, the sensitive and quantitative PCR colony format, also resistant to the huge excess of RNA from non-leukemic cells, reveals 100% of cDNA molecules [19, 20, 27]. Hence, the overall yield of the diagnostic procedure is 50% of the AML1-ETO mRNA molecules present in a clinical sample; in other words, every second molecule of RNA target is detected [27].
Thus, our diagnostic procedure eliminates most of the problems associated with the use of RT-PCR for MRD detection.
MRD Detection Using Nanocolonies
Detection of chimeric AML1-ETO mRNA in clinical samples. The developed diagnostic procedure was tested by the determination of the absolute titer of chimeric AML1-ETO mRNA in blood and bone marrow of patients suffering from leukemia associated with chromosomal translocation t(8;21)(q22;q22) after a course of chemotherapy [27].
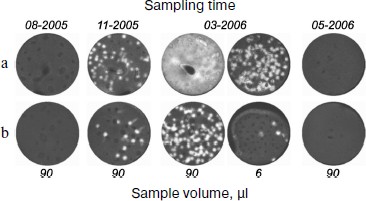
Figure 4 shows the titer dynamics for one of the patients. The first sample was negative. However, the chimeric RNA was detected in a sample taken five months before the clinical relapse accompanied by a burst of the titer. Although in all cases its content in bone marrow was approximately one order of magnitude higher than in blood, the analysis of blood, whose sampling is less traumatic for a patient, also makes it possible to detect the appearance of the chimeric RNA. The repeated course of chemotherapy after clinical relapse again decreased the titer of this RNA to zero.
Thus, the detection of AML1-ETO mRNA with the use of nanocolonies allows MRD to be diagnosed several months prior to clinical relapse.Fig. 4. Retrospective analysis of dynamics of AML-ETO mRNA titer in blood and bone marrow of a patient with leukemia associated with chromosomal translocation t(8;21)(q22;q22). The figure is reproduced from [27]. The AML1-ETO mRNA in the indicated volume of bone marrow (a) and blood (b) taken from a patient on specified dates was determined using the developed diagnostic procedure comprising reverse transcription of the RNA isolated from the clinical samples followed by real time in-gel PCR.
Reliability of negative result of assay of clinical samples. The RNA preparations isolated from clinical samples were tested for the RNA yield and integrity by spectrophotometric and electrophoretic analysis, respectively. Electrophoretic pattern showed that the modified method of RNA isolation allows the intact RNA to be reproducibly isolated (Fig. 5a, [27]). The yield of RNA from bone marrow was higher than that from the same volume of blood, which correlates with a higher content of nucleated cells in bone marrow [203]. Even almost 5000-nucleotides-long 28S RNA was preserved, which ensures the virtually complete intactness of the shorter 260-nucleotides-long AML1-ETO mRNA fragment amplified in PCR.
Spectrophotometric analysis was carried out by recording a spectrum rather than by mere measuring the sample absorption at 260 nm (as recommended by the Europe Against Cancer Program [193]) because in addition to RNA, the absorption at 260 nm can be affected by the sample light scattering, as well as by the presence of light-absorbing substances of non-nucleotide nature contained both in the blood and the reagents used for isolation, such as polyphenols, phenol, and mercaptoethanol. In calculating the RNA marker titer the number of detected molecules was normalized to the volume of clinical sample because the content of nucleated cells varies greatly between samples. To standardize the RNA yield, an exact (and hence necessarily incomplete) volume of the aqueous phase was collected during extractions.Fig. 5. Elimination of false negative results in the detection of chimeric AML1-ETO mRNA using nanocolonies. a) Quantitative and qualitative characterization by electrophoresis in 1% agarose of RNA preparations isolated from clinical samples. The amount of RNA in a lane corresponds to a 20-µl aliquot of bone marrow (BM) or whole blood (blood). Arrows point to the positions of tRNA and ribosomal 18S and 28S RNAs. b) Reliability of negative results (upper row) is confirmed by the appearance of cDNA colonies in samples to which 135 molecules of the AML1-ETO mRNA was added (bottom row). DNA colonies were revealed by in-gel hybridization with FRET probes. c) Lack of interference from a large amount of non-target RNAs to the nanocolony determination of a chimeric RNA target. The non-target RNAs present in an RNA preparation isolated from clinical samples significantly reduce the amount of the specific product of a solution PCR (on the right) but do not interfere with the growth of nanocolonies (on the left). The products of reverse transcription of AML1-ETO RNA mixed with total RNA isolated from the indicated blood aliquots were amplified by in-gel PCR (on the left; the AML1-ETO cDNA colonies were detected by hybridization with a fluorescent probe) and by a standard, i.e. solution PCR (on the right; PCR products were analyzed by electrophoresis through a polyacrylamide gel and staining with ethidium bromide).
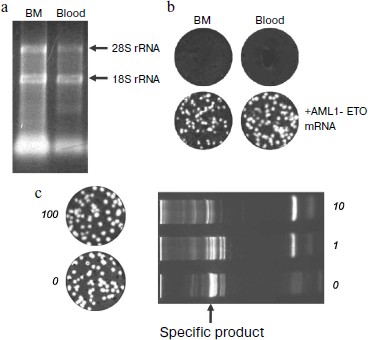
The reliability of negative results was confirmed by running control experiments in which the AML1-ETO RNA was added to RNA preparations that showed negative results. The appearance of colonies accounting for 50% of the added RNA target molecules was indicative of the absence of RT and PCR inhibitors in the analyzed samples (Fig. 5b). Hence, if RNA preparations isolated from blood and bone marrow contained at least two molecules of chimeric RNA they would have been detected.
Overcoming false negative results characteristic of solution PCR. The main reason for false negative results is nonspecific synthesis [8] that inevitably occurs due to a limited specificity of primers and hence their erroneous hybridization with non-target RNAs always present in the preparations isolated from clinical samples. The problem of nonspecific synthesis is aggravated when a chimeric RNA is detected because of the presence of a large excess of the homologous mRNAs from normal (non-leukemic) cells in which both the genes contributing to the chimera are expressed. One of the used primers is perfectly complementary to its respective gene; hence selectivity of amplification of the chimeric cDNA is entirely determined by the other primer. Nonspecific incorporation of any of the two primers may result in the formation of non-chimeric products, including those of a high molecular weight, whose amplification will compete with that of the assayed target. As a result, the real sensitivity of diagnosis, including real-time PCR, defined as the minimum detectable fraction of the leukemic cells of total number of nucleated cells, is usually no better than 10–4 [4, 5]. As the number of nucleated cells is ≈107 in 1 ml of blood and ≈108 in 1 ml of bone marrow, it follows that leukemic cells can only be detected if their amount is 103 or 104 in 1 ml of blood or bone marrow, respectively. Thus, due to the limited sensitivity of solution RT-PCR the MRD often remains undetected, and because of the difficulty of quantifying the chimeric mRNA it is often not possible to monitor the dynamics of disease relapse.
A different situation is observed when nanocolonies are used. Since nanocolonies formed by the specific and nonspecific templates are spatially separated, they do not interfere with each other during amplification. For example, the products of reverse transcription of RNA isolated from 100 µl of blood do not change the number of nanocolonies formed during in-gel PCR, which allows the titer of the target RNA to be reliably determined (Fig. 5c, left), but result in a decreased yield of the specific product and in the accumulation of nonspecific products of both low and high molecular weight, even if present at an order of magnitude lower amount (Fig. 5c, right).
Comparison of results obtained with the same cDNA preparations using nanocolonies and the standard (solution) real time PCR has shown that the two methods give similar results when the titer of AML1-ETO RNA in a patient’s blood or bone marrow is high [204]. Such a high titer is observed either before treatment or in a relapse. However, during remission, when the titer of RNA target is low, the sensitivity of standard PCR may be not sufficient for the detection of MRD [204].
Overcoming false positive results. Nonspecific synthesis can result in false positive results as well. As shown by the Europe Against Cancer Program, false positive results occur at a 10% rate when clinical samples are assayed for AML1-ETO RNA [46]. And this occurs in the best (reference) laboratories.
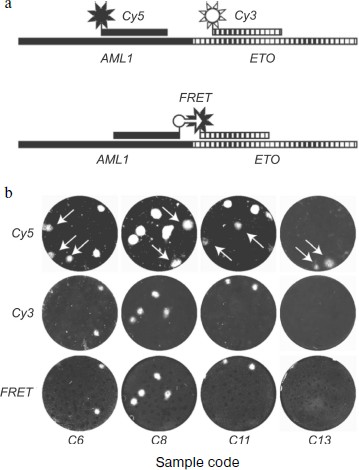
Figure 6 illustrates both the problem of false positive background and the means for its solution. Arrows point to DNA colonies hybridized with the AML-specific probe but not hybridized with the ETO-specific probe. These colonies are formed by non-chimeric molecules containing only the AML1 gene sequence. The fact that mRNA AML1 forms false positive colonies more frequently than does mRNA ETO is not accidental, because the expression level of the AML1 gene in normal tissues greatly exceeds that of the ETO gene [205].
The use of FRET probes also provides for discrimination between colonies formed by chimeric and non-chimeric molecules. Since resonance energy transfer is only possible when fluorophores are close to each other, luminescence of colonies serves as evidence that they consist of chimeric sequences capable of hybridization with both probes.Fig. 6. Elimination of false positive results in detection of chimeric AML1-ETO RNA using nanocolonies. The figure is reproduced from [27]. a) Schemes of AML1-ETO-chimera hybridization with lone probes labeled by red (Cy5) or green (Cy3) fluorophores and with a pair of probes labeled with fluorophores capable of FRET (FAM is the donor and Cy5 is the acceptor). b) Hybridization of nanocolonies grown during RT-PCR of RNA isolated from clinical samples with probes labeled by Cy5 (upper row), Cy3 (middle row), and with FRET probes (bottom row). Arrows point to colonies hybridized with the Cy5-AML probe but not hybridized with the Cy3-ETO and FRET probes.
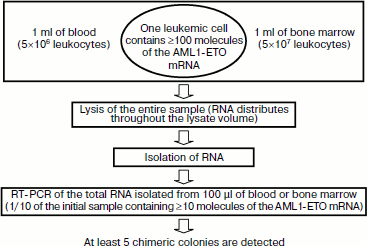
Sensitivity of MRD detection using nanocolonies. A single leukemic cell can be found among 107-108 normal cells using the above-described diagnostic procedure employing the PCR version of nanocolonies (Fig. 7). The calculation of this value was based on two assumptions: (i) 1 ml of patient’s blood and bone marrow contains a standard number of leukocytes (5×106 and 5×107, respectively [203]) and (ii) as follows from diverse data in the literature, a single leukemic cell contains at least 100 molecules of AML1-ETO mRNA [27].
One of the features of our procedure is that the clinical sample is lysed immediately after sampling. This releases RNA molecules from cells. These RNA are distributed throughout the entire volume of the lysate. If 1 ml of the sample contains a single leukemic cell, then ≥100 chimeric RNA molecules will be present in the lysate. The portion of total RNA subjected to reverse transcription and PCR corresponds to 100 µl of the initial sample (or 1/10 portion). This portion is expected to contain ≥10 chimeric RNA molecules that will give on average five colonies (because of the 50% yield of RT), i.e. the single leukemic cell present in the lysate will be detected. It is possible to achieve in blood analysis the same sensitivity as in the analysis of bone marrow if 10 ml of blood is lysed and RNA equivalent to 1 ml blood is taken in RT-PCR. This corresponds to diagnostics sensitivity of 10–7-10–8, which exceeds by several orders of magnitude the maximal sensitivity achieved by other methods.Fig. 7. Sensitivity of minimal residual disease (MRD) diagnostics using nanocolonies. If 1 ml of analyzed sample (blood or bone marrow) contains at least one leukemia cell, then it will be detected. This corresponds to the sensitivity of MRD diagnostics of 10–7-10–8.
Advantages of using nanocolonies for MRD detection. In addition to unsurpassed sensitivity, tolerance to the interference from nonspecific synthesis, and the possibility of discriminating between chimeric and non-chimeric PCR products, nanocolonies have other advantages over standard (solution) PCR.
Spatial separation of nanocolonies not only significantly decreases the interference from nonspecific synthesis, but also eliminates the competition between targets [19], which makes it possible to carry out multiplex PCR in a gel and to monitor MRD both by detecting a single chimeric RNA and a combination of markers complicating the course of leukemia of a given type [117]. Spatial separation of targets also prevents the recombination of templates that is observed in solution PCR [2, 132, 133] and is able to produce artifactual chimeric sequences and hence false positive results.
Counting the colonies permits the absolute titer of RNA target in clinical samples to be determined. The nanocolony technique is digital; it does not rely on measurement of the signal intensity; it is only needed that a colony gets “matured” to the detectable level. The problems characteristic of standard PCR, such as irreproducibility of reaction conditions, dissimilar amplification rates of different targets in multiplex PCR, inaccuracy of signal measurements [8], may affect the size but not the number of nanocolonies. Thus, the nanocolony technique is truly digital and more precise and reliable than the “analog” methods based on the measurement of a signal level, including standard (solution) PCR.
Technically, the only difference between the nanocolony method and standard PCR is that reaction mixture is introduced into a well coated with a film of dry gel, rather than into a test tube. Manufacture of disposable cassettes (a prototype of such cassette was described [206, 207]) carrying a gel ready for soaking the PCR mixture would make a nanocolony-based assay no more labor-consuming than the standard PCR.
By the cost, PCR in nanocolonies is comparable to standard PCR. The gel volume in the discussed experiments was only 65 µl, i.e. consumption of reagents was nearly the same as in a standard 50-µl PCR reaction. Thinner gels can also be used [184], which would reduce the consumption of reagents. However, in fact the nanocolony technology is cheaper than standard PCR because it allows the number of processed samples to be greatly reduced.
OTHER APPLICATIONS OF NANOCOLONIES FOR DIAGNOSTICS OF ONCOLOGICAL
DISEASES
The opportunities provided by nanocolonies are much broader than the detection and quantification of a chimeric RNA or another marker. Some of the opportunities were successfully demonstrated by our foreign colleagues, who employ nanocolonies under names “polonies” (for “polymerase colony”) [184] or “PCR-colonies” [208, 209].
Detection of Genetic Alterations
Sequencing in nanocolonies. Often there is a need not only to detect, but also to determine the nucleotide sequence of a chimeric mRNA [210]. Nanocolonies provide such an opportunity [15].
Church et al. [211] have shown that nanocolonies can be used for rapid and inexpensive sequencing using at least two approaches: sequencing by oligonucleotide ligation [211] and by primer extension [30]. Both approaches are based on the fact that each colony contains identical multiple copies of a single fragment of a longer sequence, and sequencing of all colonies (in other words, “reading” the sequence of all the fragments) occurs simultaneously.
Sequencing by oligonucleotide ligation includes template-dependent in situ ligation of a hybridized primer with a fluorescently labeled oligonucleotide. A single sequencing round comprises hybridization of the analyzed sequence with the primer and a number of pools of fluorescently labeled degenerate oligonucleotide nanomers (the fluorophore color in each pool corresponds to one of four possible bases at a certain nanomer position), ligation of the hybridized primer and nanomers, washing off unincorporated nanomers, scanning the gel at four wavelengths, hybrid denaturing, and washing off the ligation products. In the next round, hybridization and ligation are carried out using another pool of nanomers carrying the fluorophore-labeled bases at a different position on the nanomers.
Sequencing by primer extension is based on a method developed by Affymetrix for the detection of SNPs (single nucleotide polymorphisms) [212], and its improved variant consists of hybridization of an immobilized DNA with a primer complementary to a flanking region whose sequence is known, followed by reiteration of the following steps: extension of the primer, using DNA polymerase with nucleotides whose 3′ hydroxyl is reversibly blocked with a fluorescent group (a unique fluorophore corresponds to each base type), washing off unincorporated nucleotide, scanning, and unblocking the 3′-hydroxyls along with removal of the fluorophore.
Since many thousands of colonies can be resolved in one gel [184, 213], in situ sequencing has a very high sequencing capacity, up to sequencing of the whole genome. Therefore, sequencing in nanocolonies can reveal the whole spectrum of genetic alterations specific for the tumor of a particular patient and define all possible molecular diagnostic markers and targets, thus enabling a personalized therapy.
In essence, sequencing in nanocolonies is nothing but the method of a total large-scale sequencing [214, 215]. The large-scale sequencing of individual human genomes is now widely used under various names, in particular “massively parallel sequencing” [215], to search for chromosomal translocations and various genetic abnormalities associated with malignant tumors of epithelial origin, the most common types of cancer [216, 217].
Detection of SNP and haplotype analysis. SNPs (single nucleotide polymorphisms) can be detected similarly to sequencing in colonies. To this end, just a single cycle of elongation of a primer hybridized next to the supposed SNP by the fluorescence-labeled nucleotide is sufficient.
In many cases the effect of an SNP on gene expression or on the encoded protein function is caused not by a single substitution but rather by multiple substitutions, i.e. by the haplotype [218]. Haplotype determination is of important diagnostic and prognostic value in malignancy, in particular in leukemia [219], but it is a difficult problem due to the need to separate sister chromosomes.
Nanocolonies help to overcome this problem because in-gel amplification of two or more segments of the same DNA molecule (such as different loci of the same chromosome) results in the formation of overlapping colonies. On the contrary, separate colonies form when segments belonging to different DNA molecules (different chromosomes) are amplified. During subsequent SNP analysis using a primer extension by fluorescently labeled nucleotides, either the same colony will be labeled by different fluorophores (if substitutions belong to the same chromosome) or different colonies will be labeled (if substitutions are localized on different chromosomes). As a result, nanocolonies allow the haplotype to be determined at a minimum cost and rather easily and quickly [28].
Church et al. recently mapped SNPs on a region of human chromosome 7 of 153×106 bp length [220]. According to these authors, the use of such a long-distance haplotyping can make it possible to detect and characterize recombinations emerging in single cells during meiosis and even more rare somatic recombinations emerging at mitosis, as well as chromosomal translocations resulting in the transformation of a normal cell to a malignant one [220].
Determination of allele copy number. The course of malignant diseases including those associated with chromosomal translocations is complicated in the case of loss of a whole chromosome or its part carrying a normal (non-chimeric) gene or tumor suppressor genes [221, 222], i.e. when loss of heterozygosity (LOH) occurs.
Since nanocolonies can be directly counted, they are ideal for any quantitative assays of DNA and RNA targets including LOH detection. Edwards et al. used nanocolonies to analyze pancreatic cells (Panc-1) and found that the number of K-ras2 gene colonies was almost twice the number of p53 gene colonies, which points to LOH in the region of the p53 gene [223].
Determination of drug resistance. Resistance of leukemic cells to treatment in relapse is explained by emergence and rapid proliferation of the pool of leukemic cells carrying mutations conferring resistance to the drug used [224, 225]. Information concerning existence of such mutations is necessary to choose an adequate scheme of treatment for a particular patient.
Over 90% of cases of resistance to kinase inhibitor imatinib, widely used for chemotherapy of t(9;22)(q34;q11.2)-associated CML, are caused by nine mutations in the kinase domain of the BCR-ABL gene [226]. The use of nanocolonies allows a single imatinib-resistant cell to be detected among 104 drug-sensitive cells [26].
Transcriptome Analysis
Alternative splicing. Inherited and acquired genetic changes may also result in oncological diseases via the alternative splicing mechanism. A genome-wide analysis of alternative splicing in 2 million human ESTs (expressed sequence tags, short cDNA fragments that are tags of mRNAs) revealed cancer-specific splice variants in 316 genes and identified three types of cancer-specific splicing shifts. The first type, accounting for more than half of the shifts, comprised a loss of one splice isoform, the second type constituted a complete shift from one splice form in norm to another in tumors, and the third consisted of an increased amount of one isoform in tumors. It is believed that many of the shifts act by disrupting a tumor suppressor function [29]. A similar approach of other authors revealed over 15 thousand tumor-specific alternatively spliced isoforms in almost 10,000 genes from 27 types of cancer, and in some cases a correlation between the type of cancer and a variant of alternative splicing was detected [227].
Similarly to ascertaining whether nanocolonies are chimeric, it is possible to search and quantitatively measure differently spliced variants. After reverse transcription and subsequent in-gel PCR with a pair of primers matching the invariable terminal exons (or carrying out multiplex PCR with primers matching each possible exon), colonies could be revealed by hybridization with exon-specific fluorescently labeled probes. The combination of probes hybridizing with a colony (or an overlap of colonies in multiplex PCR) would reveal the exons contained in the same mRNA molecule. Colonies can also be detected by primer extension with a labeled nucleotide as in the case of SNP detection [228].
Nanocolonies have enabled detection of new isoforms and quantitative assessment of all alternatively spliced isoforms, including minor ones [228-230]. The use of nanocolonies for searching for all spliced mRNAs of the CD44 gene that includes 10 variable exons and is theoretically capable of producing over 1000 combinations, has revealed 69 mRNA isoforms, which more than twice exceeds the number of isoforms previously detected by other methods [228]. Quantitative analysis of the exon distribution in normal and transformed cells showed that the proportion of major isoforms is practically unchanged. At the same time, there are statistically reliable changes in the number and proportions of minor isoforms.
Gene expression profile. Since nanocolonies enable high throughput sequencing, they can be used for establishing gene expression profiles. Thus, Garraway et al. [231] used massively parallel sequencing and discovered 11 new chimeric transcripts in melanoma.
Total mRNA sequencing has a number of advantages compared to hybridization on microchips, in particular because it allows somatic mutations to be detected and allele-specific gene expression to be precisely measured. Life Technologies Inc. together with the Mayo Clinic used mRNA sequencing to compare gene expression profiles in tumor and normal tissues in three patients with squamous cell carcinoma of the oral cavity [232]. The study resulted in the detection of a tumor-associated allelic imbalance leading to a suppression of the expression of several genes, and in identification of specific genes known for other types of cancer but not previously implicated in this type of cancer.
The ability of nanocolonies to precisely measure the expression of different alleles of the same gene was demonstrated by measuring the expression level of protein kinase D2 in eight heterozygous patients with SNP 4208G/A, and the results were compared with published data [233]. Allele variants of mRNA were detected in the same way as in SNP analysis, by extension with labeled nucleotides of a primer hybridized with DNA colonies. Mere counting the colonies labeled with different fluorophores allowed the ratio of the allele activities to be exactly determined. The ratio of alleles themselves, determined as 1 : 1, was used as a control. Edwards et al. concluded that nanocolonies have at least two indisputable advantages: they provide for a digital assay (more precise than an “analog” one) and absolute measurement of allele expression levels.
Detection of Epigenetic Changes
Aberrant methylation. Diagnostics and treatment of malignant diseases are aimed at the targets caused both by both genetic alterations and epigenetic events. One such epigenetic event is aberrant DNA methylation (an enzymatic methylation of cytosine residues in DNA not observed in the norm) [63, 234].
Analysis of methylation of the genome of AML patients revealed 16 subgroups of AML, five of which being newly detected [235, 236]. Also, a correlation was found between the gene methylation profile and the genetic abnormality (chromosomal translocations AML1-ETO, CBFb-MYH11, and PML-RARA are associated with specific methylation profiles) and clinical outcome of the disease.
Methylation of promoter-associated CpG-rich regions (CpG islands) can result in inactivation of tumor gene suppressors and progress of the disease [237]. Testing the DNA methyltransferase inhibitor 5-azacytidine, which decreases the promoter methylation level and thus activates suppressors of tumor genes [238], has shown that this drug was effective in 48% of AML patients [239].
Nanocolonies provide for highly sensitive detection and quantitative assessment of aberrant methylation. Thus, nanocolonies generated by the isothermal amplification system RCA (rolling circle amplification) were used for quantitative analysis of methylation of the gene of tumor suppressor p16 in stomach tumor cells [240]. The analysis is based on the fact that DNA pre-treatment with bisulfite results in a conversion of unmodified cytosines to uracils without affecting the methylated cytosines. The substitutions of U for C by such treatment are detected by hybridization with fluorescent probes. This approach makes it possible to detect a single methylated sequence among a million unmethylated ones and to determine the proportion at which the sites belonging to the same molecule are methylated (if several methylation sites are analyzed).
CONCLUSION
In 2006 almost half a million (475,432) new cancer patients were diagnosed in Russia [104]. As in many other countries, this disease is the second mortality factor after cardiovascular diseases. Altogether, in 2006 in Russia 286,232 deaths, or 333 per 100,000 women and men, were caused by malignant neoplasms [108]. According to the United States National Cancer Institute data for the period of 2004-2006, the probability of getting cancer during one’s lifetime is 40% [103].
It was assumed for a long time that chromosomal translocations are mainly associated with malignant tumors of hemopoietic and lymphatic tissues. However, by 2004, sixty-four chimeric genes associated with solid tumors were revealed with the most widespread types of malignancies being among them [241], including breast cancers [242]. It was found in 2005 that 80% of cases of prostate cancer are associated with rearrangements of the ERG and ETV1 genes of the ETS family of transcription factors, including chromosomal translocations involving either of these genes and gene TMPRSS2 [243]. Chimeric TMPRSS2-ERG mRNA was revealed in 40-60% of cases of prostate cancer [243-245], and a correlation is observed between its emergence (including certain isoforms) and clinical outcome of the disease [244, 245]. In 2007 it was found in Japan that 6.7% of cases on non-small-cell lung carcinoma are associated with chromosomal inversion inv(2)(p21;p23), resulting in the appearance of chimeric EML4-ALK RNA [246]. The same chimeric RNA was detected in 2.4% of cases of breast cancers, 2.4% of cases of colorectal cancers, and 11.3% of cases of non-small-cell lung carcinoma in the USA [247].
It was found by 2007 that 20% of all oncological diseases are associated with chromosomal translocations [248]. Now chromosomal translocations are considered as the main factor responsible for the emergence of many types of malignant tumors [34, 249, 250]. Today there is an intensive search for new chromosomal translocations resulting in the emergence of chimeric RNA and proteins able to serve both as diagnostic markers of a disease and as targets of its treatment [251, 252].
The high-throughput sequencing of individual human genomes enables revealing the whole spectrum of chromosomal abnormalities and stimulates detection of new translocations that cannot be revealed by other methods [216, 217]. Clearly, the next step should be the ascertaining which of the chromosomal abnormalities cause a malignant transformation of the cells and which are nonspecific and by themselves do not result in disease, or as it is usual to say now, which abnormalities are “drivers” and which are “passengers” of carcinogenesis [253-255].
In April 2008, the International Cancer Genome Consortium (ICGC) was launched to coordinate the building of a catalog of genetic abnormalities (somatic mutations) in 50 different types of malignancies that are of clinical and societal importance around the globe [256]. In April 2010 the ICGC announced its plans to sequence the genomes of 25,000 malignant tumors (500 tumors of each malignancy type) [257], which look quite realistic with the next generation of sequencing techniques. The aim of such a global study is to comprehensively describe the genomic, transcriptomic, and epigenomic changes in 50 different tumor types and/or subtypes to reveal the repertoire of these changes (i.e. to identify the disease “drivers”) and to make the collected data available to researchers around the world as soon as they are posted at the open access site of the Consortium [257]. Eleven countries have contributed to this project [256].
In fact, the treatment of malignant disease has shifted to the next level, the “personalized medicine”. “We are moving into an era where the prescription for cancer treatment should be based on the genetics of each patient’s tumor” according to a press release of Eric Lander, an ICGC member and the president and director of the Broad Institute of Harvard University and the Massachusetts Institute of Technology [258].
Progress in medicine and other sciences is so fast that there is every reason to believe that cancer, instead of being frightening with fatal outcome, will soon become an “ordinary” treatable disease. We hope that nanocolonies will substantially contribute to the solution of this problem through the formation of a basis for efficient methods for screening and early diagnosis, evaluation of the efficacy of treatment of each individual patient, identification of drug-resistant malignant cell clones in order to select the optimal treatment regimen, detection of the minimal residual disease, and early prediction of relapse, as well as large-scale genome sequencing to detect the full range of chromosomal abnormalities.
This work was supported by the program of Molecular and Cell Biology of the Russian Academy of Sciences, the Russian Foundation for Basic Research, and the Federal Targeted Program “Research and Development in Priority Areas of Science and Technology”.
REFERENCES
1.Saiki, R. K., Scharf, S., Faloona, F., Mullis, K.
B., Horn, G. T., Erlich, H. A., and Arnheim, N. (1985) Science,
230, 1350-1354.
2.Saiki, R. K., Gelfand, D. H., Stoffel, S., Scharf,
S. J., Higuchi, R., Horn, G. T., Mullis, K. B., and Erlich, H. A.
(1988) Science, 239, 487-491.
3.Van Dongen, J. J., Macintyre, E. A., Gabert, J. A.,
Delabesse, E., Rossi, V., Saglio, G., Gottardi, E., Rambaldi, A.,
Dotti, G., Griesinger, F., Parreira, A., Gameiro, P., Diaz, M. G.,
Malec, M., Langerak, A. W., San Miguel, J. F., and Biondi, A. (1999)
Leukemia, 13, 1901-1928.
4.Baer, M. R. (2002) Curr. Oncol. Rep.,
4, 398-402.
5.Van der Velden, V. H., Hochhaus, A., Cazzaniga, G.,
Szczepanski, T., Gabert, J., and van Dongen, J. J. (2003)
Leukemia, 17, 1013-1034.
6.LeDuc, J. W., Damon, I., Meegan, J. M., Relman, D.
A., Huggins, J., and Jahrling, P. B. (2002) Emerg. Infect. Dis.,
8, 743-745.
7.Chetverin, A. B., and Chetverina, H. V. (2002)
Mol. Biol. (Moscow), 36, 320-327.
8.Chetverin, A. B., and Chetverina, H. V. (2003)
Mol. Med. (Moscow), 2, 30-40.
9.Chetverin, A. B., and Chetverina, H. V. (1995) RF
Patent No. 2048522.
10.Chetverin, A. B., and Chetverina, H. V. (1998) RF
Patent No. 2114175.
11.Chetverin, A. B., and Chetverina, H. V. (1998) RF
Patent No. 2114915.
12.Chetverin, A. B., and Chetverina, H. V. (1997)
U.S. Patent 5,616,478.
13.Chetverin, A. B., and Chetverina, H. V. (1999)
U.S. Patent 5,958,698.
14.Chetverin, A. B., and Chetverina, H. V. (1999)
U.S. Patent 6,001,568.
15.Chetverina, H. V., and Chetverin, A. B.
(2008) Biochemistry (Moscow), 73, 1361-1387.
16.Chetverin, A. B., and Chetverina, H. V. (2008)
Prog. Nucleic Acid Res. Mol. Biol., 82,
219-255.
17.Chetverin, A. B., Chetverina, H. V., and
Munishkin, A. V. (1991) J. Mol. Biol., 222, 3-9.
18.Chetverina, H. V., and Chetverin, A. B. (1993)
Nucleic Acids Res., 21, 2349-2353.
19.Chetverina, H. V., Samatov, T. R., Ugarov, V. I.,
and Chetverin, A. B. (2002) BioTechniques, 33,
150-156.
20.Chetverina, H. V., Falaleeva, M. V., and
Chetverin, A. B. (2004) Anal. Biochem., 334, 376-381.
21.Chetverina, H. V., Demidenko, A. A., Ugarov, V.
I., and Chetverin, A. B. (1999) FEBS Lett., 450,
89-94.
22.Chetverin, A. B., Chetverina, H. V., Demidenko,
A. A., and Ugarov, V. I. (1997) Cell, 88,
503-513.
23.Chetverin, A. B., Kopein, D. S., Chetverina, H.
V., Demidenko, A. A., and Ugarov, V. I. (2005) J. Biol. Chem.,
280, 8748-8755.
24.Samatov, T. R., Chetverina, H. V., and Chetverin,
A. B. (2005) Nucleic Acids Res., 33, e145.
25.Chetverin, A. B., Samatov, T. R., and Chetverina,
H. V. (2008) Cell-Free Protein Synthesis: Methods and Protocols
(Spirin, A. S., and Swartz, J. R., eds.) Wiley-VCH, Weinheim, pp.
191-206.
26.Nardi, V., Raz, T., Cao, X., Wu, C. J., Stone, R.
M., Cortes, J., Deininger, M. W., Church, G., Zhu, J., and Daley, G. Q.
(2008) Oncogene, 27, 775-782.
27.Falaleeva, M. V., Chetverina, H. V., Kravchenko,
A. V., and Chetverin, A. B (2009) Mol. Biol. (Moscow),
43, 180-189.
28.Mitra, R. D., Butty, V. L., Shendure, J.,
Williams, B. R., Housman, D. E., and Church, G. M. (2003) Proc.
Natl. Acad. Sci. USA, 100, 5926-5931.
29.Xu, Q., and Lee, C. (2003) Nucleic Acids
Res., 31, 5635-5643.
30.Mitra, R. D., Shendure, J., Olejnik, J.,
Krzymanska-Olejnik, E., and Church, G. M. (2003) Anal. Biochem.,
320, 55-65.
31.Mitelman, F., Johansson, B., and Mertens, F.
(eds.) (2010) Mitelman Database of Chromosome Aberrations in
Cancer, http://cgap.nci.nih.gov/Chromosomes/Mitelman, data
on February 18, 2010.
32.Kleihues, P., and Sobin, L. H. (2000) World
Health Organization Classification of Tumors. Cancer, 88,
2887.
33.Frohling, S., and Dohner, H. (2008) N. Engl.
J. Med., 359, 722-734.
34.Nambiar, M., Kari, V., and Raghavan, S. C. (2008)
Biochim. Biophys. Acta, 1786, 139-152.
35.Bergsma, D. (ed.) (1978) An International
System for Human Cytogenetic Nomenclature (1978), ISCN. Report of
the Standing Commitee on Human Cytogenetic Nomenclature, S. Karger,
Basel.
36.Mitelman, F. (ed.) (1995) ISCN 1995: An
International System for Human Cytogenetic Nomenclature, S. Karger,
Basel.
37.ISCN 2005: An International System for Human
Cytogenetic Nomenclature (Cytogenetic and Genome Research)
(2005), S. Karger, Basel.
38.Brothman, A. R., Persons, D. L., and Shaffer, L.
G. (2010) Cytogenet. Genome Res., 127, 1-4.
39.Belloni, E., Trubia, M., Mancini, M., Derme, V.,
Nanni, M., Lahortiga, I., Riccioni, R., Confalonieri, S., Lo-Coco, F.,
Di Fiore, P. P., and Pelicci, P. G. (2004) Genes Chromosomes
Cancer, 41, 272-277.
40.Kim, J., and Pelletier, J. (1999) Physiol.
Genom., 1, 127-138.
41.Ladanyi, M. (2002) Cancer Biol. Ther.,
1, 330-336.
42.Sorensen, P. H. B., Lessnick, S. L.,
Lopez-Terrada, D., Liu, X. F., Triche, T. J., and Denny, C. T. (1994)
Nat. Genet., 6, 146-151.
43.Weizmann Institute of Science, Israel, http://www.genecards.org/
44.Gulley, M. L., Shea, T. C., and Fedoriw, Y.
(2010) J. Mol. Diagn., 12, 3-16.
45.Vardiman, J. W., Thiele, J., Arber, D. A.,
Brunning, R. D., Borowitz, M. J., Porwit, A., Harris, N. L., Le Beau,
M. M., Hellstrom-Lindberg, E., Tefferi, A., and Bloomfield, C. D.
(2009) Blood, 114, 937-951.
46.Gabert, J., Beillard, E., van der Velden, V. H.,
Bi, W., Grimwade, D., Pallisgaard, N., Barbany, G., Cazzaniga, G.,
Cayuela, J. M., Cave, H., Pane, F., Aerts, J. L., De Micheli, D.,
Thirion, X., Pradel, V., Gonzalez, M., Viehmann, S., Malec, M., Saglio,
G., and van Dongen, J. J. (2003) Leukemia, 17,
2318-2357.
47.Peterson, L. F., Boyapati, A., Ahn, E. Y., Biggs,
J. R., Okumura, A. J., Lo, M. C., Yan, M., and Zhang, D. E. (2007)
Blood, 110, 799-805.
48.Kakizuka, A., Miller, W. H., Jr., Umesono, K.,
Warrell, R. P., Jr, Frankel, S. R., Murty, V. V., Dmitrovsky, E., and
Evans, R. M. (1991) Cell, 66, 663-674.
49.Alcalay, M., Zangrilli, D., Fagioli, M.,
Pandolfi, P. P., Mencarelli, A., Lo Coco, F., Biondi, A., Grignani, F.,
and Pelicci, P. G. (1992) Proc. Natl. Acad. Sci. USA, 89,
4840-4844.
50.Stams, W. A., den Boer, M. L., Beverloo, H. B.,
Meijerink, J. P., van Wering, E. R., Janka-Schaub, G. E., and Pieters,
R. (2005) Clin. Cancer Res., 11, 2974-2980.
51.De Klein, A., van Kessel, A. G., Grosveld, G.,
Bartram, C. R., Hagemeijer, A., Bootsma, D., Spurr, N. K., Heisterkamp,
N., Groffen, J., and Stephenson, J. R. (1982) Nature,
300, 765-767.
52.Ben-Neriah, Y., Daley, G. Q., Mes-Masson, A. M.,
Witte, O. N., and Baltimore, D. (1986) Science, 233,
212-214.
53.Faderl, S., Talpaz, M., Estrov, Z.,
O’Brien, S., Kurzrock, R., and Kantarjian, H. M. (1999) N.
Engl. J. Med., 341, 164-172.
54.Clark, S. S., McLaughlin, J., Crist, W. M.,
Champlin, R., and Witte, O. N. (1987) Science, 235,
85-88.
55.Hermans, A., Heisterkamp, N., von Linden, M., van
Baal, S., Meijer, D., van der Plas, D., Wiedemann, L. M., Groffen, J.,
Bootsma, D., and Grosveld, G. (1987) Cell, 51, 33-40.
56.Melo, J. V., Gordon, D. E., Cross, N. C., and
Goldman, J. M. (1993) Blood, 81, 158-165.
57.Melo, J. V., Gordon, D. E., Tuszynski, A., Dhut,
S., Young, B. D., and Goldman, J. M. (1993) Blood, 81,
2488-2491.
58.Zheng, X., Oancea, C., Henschler, R., Moore, M.
A., and Ruthardt, M. (2009) PLoS One, 4, e7661.
59.Diamond, J., Goldman, J. M., and Melo, J. V.
(1995) Blood, 85, 2171-2175.
60.Zheng, X., Guller, S., Beissert, T., Puccetti,
E., and Ruthardt, M. (2006) BMC Cancer, 6, 262.
61.Ar-Rushdi, A., Nishikura, K., Erikson, J., Watt,
R., Rovera, G., and Croce, C. M. (1983) Science, 222,
390-393.
62.Nishikura, K., Ar-Rushdi, A., Erikson, J., Watt,
R., Rovera, G., and Croce, C. M. (1983) Proc. Natl. Acad. Sci.
USA, 80, 4822-4826.
63.Chen, J., Odenike, O., and Rowley, J. D. (2010)
Nat. Rev. Cancer, 10, 23-36.
64.Birnold, L. P., Coghlan, B., and Jersmann, H.
(2009) Cell Oncol., 31, 61.
65.Nowell, P. C., and Hungerford, D. A. (1960)
Science, 132, 1497.
66.Rowley, J. D. (2001) Nat. Rev. Cancer,
1, 245-250.
67.Kopnin, B. P. (2007) Mol. Biol. (Moscow),
41, 369-380.
68.Winkelmann, A. (2007) Clin. Anat.,
20, 231-234.
69.Flemming, W. J. (1965) Cell Biol.,
25, 3-69.
70.Paweletz, N. (2001) Nature Rev. Cell
Biol., 2, 72-75.
71.Stent, G. S., and Calendar, R.
(1981) Molecular Genetics. An Introductory Narrative, 2nd
Edn., W. H. Freeman and Company, San Francisco.
72.Sturtevant, A. H., Bridges, C. B., and Morgan, T.
H. (1919) Proc. Natl. Acad. Sci. USA, 5, 168-173.
73.Morgan, T. H., Sturtevant, A. H., and Bridge, C.
B. (1920) Proc. Natl. Acad. Sci. USA, 6, 162-164.
74.Boveri, T. (2008) J. Cell Sci.,
121, Suppl. 1, 1-84.
75.McKusick, V. A. (1985) J. Med. Genet.,
22, 431-440.
76.Mendel, G. (1986) Am. Zool., 26,
749-752.
77.Watson, J. D. (1978) Molecular Biology of the
Gene, 3nd Edn., Harvard University and Cold Spring Harbor
Laboratory, Cold Spring Harbor, NY.
78.Sturtevant, A. H. (1921) Proc. Natl. Acad.
Sci. USA, 7, 235-237.
79.Belling, J., and Blakeslee, A. F. (1926) Proc.
Natl. Acad. Sci. USA, 12, 7-11.
80.Muller, H. J. (1928) Proc. Natl. Acad. Sci.
USA, 14, 714-726.
81.Stadler, L. J. (1928) Science, 68,
186-187.
82.McClintock, B. (1930) Proc. Natl. Acad. Sci.
USA, 16, 791-796.
83.Avery, O. T., Macleod, C. M., and McCarty, M.
(1944) J. Exp. Med., 79, 137-158.
84.Watson, J. D., and Crick, F. H. (1953)
Nature, 171, 737-738.
85.Tjio, J. H., and Levan, A. (1956)
Hereditas, 42, 1-6.
86.Garcia-Sagredo, J. M. (2008) Biochim. Biophys.
Acta, 1779, 363-375.
87.Caspersson, T., Zech, L., Modest, E. J., Foley,
G. E., Wagh, U., and Simonsson, E. (1969) Exp. Cell. Res.,
58, 128-140.
88.Caspersson, T., Lomakka, G., and Zech, L. (1971)
Hereditas, 67, 89-102.
89.Rowley, J. D. (2008) Blood, 112,
2183-2189.
90.Rowley, J. D. (1973) Ann. Genet.,
16, 109-112.
91.Rowley, J. D. (1973) Nature, 243,
290-293.
92.Zech, L., Haglund, U., Nilsson, K., and Klein, G.
(1976) Int. J. Cancer, 17, 47-56.
93.Taub, R., Kirsch, I., Morton, C., Lenoir, G.,
Swan, D., Tronick, S., Aaronson, S., and Leder, P. (1982) Proc.
Natl. Acad. Sci. USA, 79, 7837-7841.
94.Dalla-Favera, R., Bregni, M., Erikson, J.,
Patterson, D., Gallo, R. C., and Croce, C. M. (1982) Proc. Natl.
Acad. Sci. USA, 79, 7824-7827.
95.Erikson, J., Finan, J., Nowell, P. C., and Croce,
C. M. (1982) Proc. Natl. Acad. Sci. USA, 79,
5611-5615.
96.Groffen, J., Stephenson, J. R., Heisterkamp, N.,
de Klein, A., Bartram, C. R., and Grosveld, G. (1984) Cell,
36, 93-99.
97.Miyoshi, H., Shimizu, K., Kozu, T., Maseki, N.,
Kaneko, Y., and Ohki, M. (1991) Proc. Natl. Acad. Sci. USA,
88, 10431-10434.
98.Erickson, P., Gao, J., Chang, K. S., Look, T.,
Whisenant, E., Raimondi, S., Lasher, R., Trujillo, J., Rowley, J., and
Drabkin, H. (1992) Blood, 80, 1825-1831.
99.Boyle, P., and Levin, B. (eds.) (2008) World
Cancer Report 2008, International Agency for Research on Cancer,
Lyon.
100.Henderson, E. S., and Han, T. (1986) CA
Cancer J. Clin., 36, 322-350.
101.Denisov, I. N., and Ulumbekov, E. G. (eds.)
(1999) Reference Guide-Book of Medical Practitioner. 2000 Diseases
from A to Z [in Russian], GEOTAR-MED, Moscow.
102.Jaffe, E. S., Harris, N. L., Stein, H., et
al. (eds.) (2001) World Health Organization Classification of
Tumors. Pathology and Genetics of Tumors of Hematopoietic and Lymphoid
Tissues, IARC, Lyon, France.
103.Horner, M. J., Ries, L. A. G., Krapcho, M., et
al. (eds.) SEER Cancer Statistics Review (1975-2006), National
Cancer Institute, Bethesda, MD (http://seer.cancer.gov/csr/1975_2006/, based on
November 2008 SEER data submission, posted to the SEER web site,
2009).
104.Davydov, M. I., and Axel, E. M. (2008) Bull.
Blokhin Res. Center of Oncol. RAMS, 19, 52-90.
105.Koziney, G. I., Sarycheva, T. G., Lugovskaya,
S. A., Dyagileva, O. A., Pogorelov, V. M., and Protsenko, D. D. (2009)
Hematological Atlas: Handbook of Medical Laboratory Assistant
[in Russian], Prakticheskaya Meditsina, Moscow.
106.Mentkevich, G. L., and Mayakova, S. A. (eds.)
(2009) Leukemia in Children [in Russian], Prakticheskaya
Meditsina, Moscow.
107.Axel, E. M., and Gorbacheva, I. A. (2008)
Bull. Blockhin Russ. Center of Oncol. RAMS, 19,
135-152.
108.Davydov, M. I., and Axel, E. M. (2008) Bull.
Blokhin Res. Center of Oncol. RAMS, 19, 91-119.
109.U.S. Cancer Statistics Working Group. United
States Cancer Statistics (USCS): 1999-2006 Incidence and Mortality
Web-Based Report (2010) U.S. Department of Health and Human
Services, Centers for Disease Control and Prevention and National
Cancer Institute, Atlanta (available at www.cdc.gov/uscs).
110.Raanani, P., and Ben-Bassat, I. (2004) Acta
Haematol., 112, 40-54.
111.Atlas of Genetics and Cytogenetics in
Oncology and Haematology (http://atlasgeneticsoncology.org//index.html).
112.Pui, C. H., Relling, M. V., and Downing, J. R.
(2004) N. Engl. J. Med., 350, 1535-1548.
113.GENE DATAbase: http://genatlas.medecine.univ-paris5.fr/
114.Zhang, Y., and Rowley, J. D. (2006) DNA
Repair, 5, 1282-1297.
115.Deschler, B., and Lubbert, M. (2006)
Cancer, 107, 2099-2107.
116.Vardiman, J. W., Harris, N. L., and Brunning R.
D. (2002) Blood, 100, 2292-2302.
117.Marcucci, G. (2009) Clin. Adv. Hematol.
Oncol., 7, 448-451.
118.Mrozek, K., and Bloomfield, C. D. (2006)
Hematol. Am. Soc. Hematol. Educ. Progr., 169-177.
119.Ravandi, F., Burnett, A. K., Agura, E. D., and
Kantarjian, H. M. (2007) Cancer, 110, 1900-1910.
120.Lutterbach, B., and Hiebert, S. W. (2000)
Gene, 245, 223-235.
121.Wang, L., Huang, G., Zhao, X., Hatlen, M. A.,
Vu, L., Liu, F., and Nimer, S. D. (2009) Blood Cells Mol. Dis.,
43, 30-34.
122.Kumano, K., and Kurokawa, M. (2010) J. Cell
Physiol., 222, 282-285.
123.De Braekeleer, E., Ferec, C., and de
Braekeleer, M. (2009) Anticancer Res., 29, 1031-1037.
124.Hug, B. A., and Lazar, M. A. (2004)
Oncogene, 23, 4270-4274.
125.Davis, J. N., McGhee, L., and Meyers, S. (2003)
Gene, 303, 1-10.
126.Peterson, L. F., and Zhang, D. E. (2004)
Oncogene, 23, 4255-4262.
127.Downing, J. R., Head, D. R., Curcio-Brint, A.
M., Hulshof, M. G., Motroni, T. A., Raimondi, S. C., Carroll, A. J.,
Drabkin, H. A., Willman, C., Theil, K. S., Civin, C. I., and Erickson,
P. (1993) Blood, 81, 2860-2865.
128.Kozu, T., Miyoshi, H., Shimizu, K., Maseki, N.,
Kaneko, Y., Asou, H., Kamada, N., and Ohki, M. (1993) Blood,
82, 1270-1276.
129.Tighe, J. E., and Calabi, F. (1994)
Blood, 84, 2115-2121.
130.Van de Locht, L. T., Smetsers, T. F., Wittebol,
S., Raymakers, R. A., and Mensink, E. J. (1994) Leukemia,
8, 1780-1784.
131.Goodrich, D. W., and Duesberg, P. H. (1990)
Proc. Natl. Acad. Sci. USA, 87, 2052-2056.
132.Meyerhans, A., Vartanian, J. P., and
Wain-Hobson, S. (1990) Nucleic Acids Res., 18,
1687-1691.
133.Paabo, S., Irwin, D. M., and Wilson, A. C.
(1990) J. Biol. Chem., 265, 4718-4721.
134.Era, T., Asou, N., Kunisada, T., Yamasaki, H.,
Asou, H., Kamada, N., Nishikawa, S., Yamaguchi, K., and Takatsuki, K.
(1995) Genes Chromosomes Cancer, 13, 25-33.
135.Yan, M., Kanbe, E., Peterson, L. F., Boyapati,
A., Miao, Y., Wang, Y., Chen, I. M., Chen, Z., Rowley, J. D., Willman,
C. L., and Zhang, D. E. (2006) Nat. Med., 12,
945-949.
136.Lee, S., Chen, J., Zhou, G., Shi, R. Z.,
Bouffard, G. G., Kocherginsky, M., Ge, X., Sun, M., Jayathilaka, N.,
Kim, Y. C., Emmanuel, N., Bohlander, S. K., Minden, M., Kline, J.,
Ozer, O., Larson, R. A., LeBeau, M. M., Green, E. D., Trent, J.,
Karrison, T., Liu, P. P., Wang, S. M., and Rowley, J. D. (2006)
Proc. Natl. Acad. Sci. USA, 103, 1030-1035.
137.Elagib, K. E., and Goldfarb, A. N. (2007)
Cancer Lett., 251, 179-186.
138.Bateman, C. M., Colman, S. M., Chaplin, T.,
Young, B. D., Eden, T. O., Bhakta, M., Gratias, E. J., van Wering, E.
R., Cazzaniga, G., Harrison, C. J., Hain, R., Ancliff, P., Ford, A. M.,
Kearney, L., and Greaves, M. (2010) Blood, 115,
3553-3558.
139.Ford, A. M., Ridge, S. A., Cabrera, M. E.,
Mahmoud, H., Steel, C. M., Chan, L. C., and Greaves, M. (1993)
Nature, 363, 358-360.
140.Ford, A. M., Bennett, C. A., Price, C. M.,
Bruin, M. C., van Wering, E. R., and Greaves, M. (1998) Proc. Natl.
Acad. Sci. USA, 95, 4584-4588.
141.Maia, A. T., Koechling, J., Corbett, R.,
Metzler, M., Wiemels, J. L., and Greaves, M. (2004) Genes
Chromosomes Cancer, 39, 335-340.
142.McHale, C. M., and Smith, M. T. (2004) Am.
J. Hematol., 75, 254-257.
143.Osada, S., Horibe, K., Oiwa, K., Yoshida, J.,
Iwamura, H., Matsuoka, H., Adachi, K., Morishima, Y., Ohno, R., Ueda,
R., et al. (1990) Cancer, 65, 1146-1149.
144.Isoda, T., Ford, A. M., Tomizawa, D., van
Delft, F. W., de Castro, D. G., Mitsuiki, N., Score, J., Taki, T.,
Morio, T., Takagi, M., Saji, H., Greaves, M., and Mizutani, S. (2009)
Proc. Natl. Acad. Sci. USA, 106, 17882-17885.
145.Mori, H., Colman, S. M., Xiao, Z., Ford, A. M.,
Healy, L. E., Donaldson, C., Hows, J. M., Navarrete, C., and Greaves,
M. (2002) Proc. Natl. Acad. Sci. USA, 99, 8242-8247.
146.Yuan, Y., Zhou, L., Miyamoto, T., Iwasaki, H.,
Harakawa, N., Hetherington, C. J., Burel, S. A., Lagasse, E., Weissman,
I. L., Akashi, K., and Zhang, D. E. (2001) Proc. Natl. Acad. Sci.
USA, 98, 10398-10403.
147.Armitage, P., and Doll, R. (1957) Br. J.
Cancer, 11, 161-169.
148.Knudson, A. G. (2001) Nat. Rev. Cancer,
1, 157-162.
149.Kern, S. E. (2002) Cancer Biol. Ther.,
1, 571-581.
150.Amann, J. M., Nip, J., Strom, D. K.,
Lutterbach, B., Harada, H., Lenny, N., Downing, J. R., Meyers, S., and
Hiebert, S. W. (2001) Mol. Cell Biol., 21, 6470-6483.
151.Wolford, J. K., and Prochazka, M. (1998)
Gene, 212, 103-109.
152.Hess, J. L., and Hug, B. A. (2004) Proc.
Natl. Acad. Sci. USA, 101, 16985-16986.
153.Ford, A. M., Palmi, C., Bueno, C., Hong, D.,
Cardus, P., Knight, D., Cazzaniga, G., Enver, T., and Greaves, M.
(2009) J. Clin. Invest., 119, 826-836.
154.Kern, W., Schoch, C., Haferlach, T., and
Schnittger, S. (2005) Crit. Rev. Oncol. Hematol., 56,
283-309.
155.Ferrara, F., Palmieri, S., and Mele, G. (2004)
Haematologica, 89, 998-1008.
156.Baturina, Yu. A., Popa, A. V., Sokova, O. I.,
Kirichenko, O. P., Konstantinova, L. N., Kulagina, O. E., Shamansky, S.
V., Mayakova, S. A., Maschan, A. A., and Fleishman, E. V. (2002)
Gematol. Transfuziol., 47, 10-14.
157.Ito, Y., and Miyamura, K. (1994) Leuk.
Lymphoma, 16, 57-64.
158.Fathi, A. T., Grant, S., and Karp, J. E. (2010)
Cancer Treat. Rev., 36, 142-150.
159.Faderl, S., O’Brien, S., Pui, C. H.,
Stock, W., Wetzler, M., Hoelzer, D., and Kantarjian, H. M. (2010)
Cancer, 116, 1165-1176.
160.Fausel, C. (2007) J. Manag. Care Pharm.,
13 (8 Suppl. A), 8-12.
161.Hamilton, A., Gallipoli, P., Nicholson, E., and
Holyoake, T. L. (2010) J. Pathol., 220, 404-418.
162.Baranov, A. A., Bazhenova, L. K., Bayandina, G.
N., Geppe, N. A., Gorelov, A. V., Zinov’eva, G. A., Kaganov, B.
S., Kapranova, E. I., Lyskina, G. A., Podchernyaeva, N. S.,
Shakhbazyan, I. E., Shishov, A. Ya., and Erdes, S. I. (2002)
Childhood Diseases. A Textbook for Higher Medical School [in
Russian], GEOTAR-MED, Moscow.
163.Marcucci, G., Caligiuri, M. A., and Bloomfield,
C. D. (2003) Eur. J. Haematol., 71, 143-154.
164.Leroy, H., de Botton, S., Grardel-Duflos, N.,
Darre, S., Leleu, X., Roumier, C., Morschhauser, F., Lai, J. L.,
Bauters, F., Fenaux, P., and Preudhomme, C. (2005) Leukemia,
19, 367-372.
165.Campana, D. (2009) Semin. Hematol.,
46, 100-106.
166.Szczepanski, T. (2007) Leukemia,
21, 622-626.
167.Stow, P., Key, L., Chen, X., Pan, Q., Neale, G.
A., Coustan-Smith, E., Mullighan, C. G., Zhou, Y., Pui, C. H., and
Campana, D. (2010) Blood, 115, 4657-4663.
168.Bader, P., Kreyenberg, H., Henze, G. H.,
Eckert, C., Reising, M., Willasch, A., Barth, A., Borkhardt, A.,
Peters, C., Handgretinger, R., Sykora, K. W., Holter, W., Kabisch, H.,
Klingebiel, T., and von Stackelberg, A., ALL-REZ BFM Study Group (2009)
J. Clin. Oncol., 27, 377-384.
169.Spinelli, O., Peruta, B., Tosi, M., Guerini,
V., Salvi, A., Zanotti, M. C., Oldani, E., Grassi, A., Intermesoli, T.,
Mico, C., Rossi, G., Fabris, P., Lambertenghi-Deliliers, G., Angelucci,
E., Barbui, T., Bassan, R., and Rambaldi, A. (2007)
Haematologica, 92, 612-618.
170.Bassan, R., Spinelli, O., Oldani, E.,
Intermesoli, T., Tosi, M., Peruta, B., Rossi, G., Borlenghi, E.,
Pogliani, E. M., Terruzzi, E., Fabris, P., Cassibba, V.,
Lambertenghi-Deliliers, G., Cortelezzi, A., Bosi, A., Gianfaldoni, G.,
Ciceri, F., Bernardi, M., Gallamini, A., Mattei, D., di Bona, E.,
Romani, C., Scattolin, A. M., Barbui, T., and Rambaldi, A. (2009)
Blood, 113, 4153-4162.
171.Padilla-Nash, H. M., Barenboim-Stapleton, L.,
Difilippantonio, M. J., and Ried, T. (2006) Nat. Protoc.,
1, 3129-3142.
172.Lucio, P., Gaipa, G., van Lochem, E. G., van
Wering, E. R., Porwit-MacDonald, A., Faria, T., Bjorklund, E., Biondi,
A., van den Beemd, M. W., Baars, E., Vidriales, B., Parreira, A., van
Dongen, J. J., San Miguel, J. F., and Orfao, A.; BIOMED-I (2001)
Leukemia, 15, 1185-1192.
173.Van der Velden, V. H., Boeckx, N., van Wering,
E. R., and van Dongen, J. J. (2004) J. Biol. Regul. Homeost.
Agents, 18, 146-154.
174.Al-Mawali, A., Gillis, D., Hissaria, P., and
Lewis, I. (2008) Am. J. Clin. Pathol., 129, 934-945.
175.Al-Mawali, A., Gillis, D., and Lewis, I. (2009)
Am. J. Clin. Pathol., 131, 16-26.
176.Craig, F. E., and Foon, K. A. (2008)
Blood, 111, 3941-3967.
177.Shook, D., Coustan-Smith, E., Ribeiro, R. C.,
Rubnitz, J. E., and Campana, D. (2009) Clin. Lymphoma Myeloma,
9, Suppl. 3, S281-285.
178.Wood, B. L., Arroz, M., Barnett, D.,
DiGiuseppe, J., Greig, B., Kussick, S. J., Oldaker, T., Shenkin, M.,
Stone, E., and Wallace, P. (2007) Cytometry. B. Clin. Cytom.,
72, Suppl. 1, S14-22.
179.Dimov, N. D., Medeiros, L. J., Ravandi, F., and
Bueso-Ramos, C. E. (2010) Am. J. Clin. Pathol., 133,
484-490.
180.Bustin, S. A., and Mueller, R. (2005) Clin.
Sci. (Lond.), 109, 365-379.
181.Sessions, J. (2007) J. Manag. Care
Pharm., 13 (8 Suppl. A), 4-7.
182.Yang, L., Han, Y., Suarez Saiz, F., and Minden,
M. D. (2007) Leukemia, 21, 868-876.
183.Barragan, E., Pajuelo, J. C., Ballester, S.,
Fuster, O., Cervera, J., Moscardo, F., Senent, L., Such, E., Sanz, M.
A., and Bolufer, P. (2008) Clin. Chim. Acta, 395,
120-123.
184.Mitra, R. D., and Church, G. M. (1999)
Nucleic Acids Res., 27, e34.
185.Chetverina, H. V., Kravchenko, A. V.,
Falaleeva, M. V., and Chetverin, A. B. (2007) Bioorg. Khim.,
33, 456-463.
186.Samatov, T. R., Chetverina, H. V., and
Chetverin, A. B. (2006) Anal. Biochem., 356, 300-302.
187.Chetverina, H. V., Chetverin, A. B., and
Kravchenko, A. V. (2008) RF Patent No. 2338787 C2.
188.Forster, T. (1948) Ann. Physik,
2, 55-75.
189.Cardullo, R. A., Agrawal, S., Flores, C.,
Zamecnik, P. C., and Wolf, D. E. (1988) Proc. Natl. Acad. Sci.
USA, 85, 8790-8794.
190.Tyagi, S., Landegren, U., Tazi, M., Lizardi, P.
M., and Kramer, F. R. (1996) Proc. Natl. Acad. Sci. USA,
93, 5395-5400.
191.Muller, M. C., Hordt, T., Paschka, P., Merx K.,
la Rosee, P., Hehlmann, R., and Hochhaus, A. (2004) Acta
Haematol., 112, 30-33.
192.Nucleic Acid Sample Preparation for
Downstream Analyses. Principles and Methods (2010) Handbooks from
GE Healthcare, GE Healthcare Bio-Sciences AB.
193.Beillard, E., Pallisgaard, N., van der Velden,
V. H., Bi, W., Dee, R., van der Schoot, E., Delabesse, E., Macintyre,
E., Gottardi, E., Saglio, G., Watzinger, F., Lion, T., van Dongen, J.
J., Hokland, P., and Gabert, J. (2003) Leukemia, 17,
2474-2486.
194.Boyum, A. (1964) Nature, 204,
793-794.
195.Van der Velden, V. H., Boeckx, N., Gonzalez,
M., Malec, M., Barbany, G., Lion, T., Gottardi, E., Pallisgaard, N.,
Beillard, E., Hop, W. C., Hoogeveen, P. G., Gabert, J., and van Dongen,
J. J., Europe Against Cancer Program (2004) Leukemia, 18,
884-886.
196.Chirgwin, J. M., Przybyla, A. E., MacDonald, R.
J., and Rutter, W. J. (1979) Biochemistry, 18,
5294-5299.
197.Kravchenko, A. V., Chetverina, H. V., and
Chetverin, A. B. (2006) Bioorg. Khim., 32,
609-614.
198.Chetverina, H. V., Chetverin, A. B., and
Kravchenko, A. V. (2008) RF Patent No. 2322058 C2.
199.Chomczynski, P., and Sacchi, N. (1987) Anal.
Biochem., 162, 156-159.
200.Boom, R., Sol, C. J., Salimans, M. M., Jansen,
C. L., Wertheim-van Dillen, P. M., and van der Noordaa, J. (1990) J.
Clin. Microbiol., 28, 495-503.
201.Bustin, S. A., and Mueller, R. (2006) Mol.
Aspects Med., 27, 192-223.
202.Leparc, G. G., and Mitra, R. D. (2007)
Nucleic Acids Res., 35, e146.
203.Alberts, B., Bray, D., Lewis, J., Raff, M.,
Roberts, K., and Watson, J. D. (1994) Molecular Biology of the
Cell, 3rd Edn., Garland Publishing, NY.
204.Falaleeva, M. V., Chetverina, H. V.,
Kravchenko, A. V., Zabollotneva, Yu. A., Samatov, T. R., Bobrynina, V.
O., Maschan, M. A., and Chetverin, A. B. (2006) Reports of
Research-Practical Conference “Living Systems” (within the
Framework of Federal Program “Investigations and Developments on
Priority Trends in the Science and Technology in 2002-2006”),
pp. 238-243.
205.Okumura, A. J., Peterson, L. F., Lo, M. C., and
Zhang, D. E. (2007) Exp. Hematol., 35, 978-988.
206.Chetverin, A. B., Samatov, T. R., and
Chetverina, H. V. (2006) RF Patent pending No. 2006109271.
207.Chetverin, A. B., Samatov, T. R., and
Chetverina, H. V. (2007) PCT Publication WO2007111639.
208.Merritt, J., Butz, J. A., Ogunnaike, B. A., and
Edwards, J. S. (2005) Biotechnol. Bioeng., 92,
519-531.
209.Merritt, J., Roberts, K. G., Butz, J. A., and
Edwards, J. S. (2007) Genom. Med., 1, 113-124.
210.Luthra, R., Sanchez-Vega, B., and Medeiros, L.
J. (2004) Mod. Pathol., 17, 96-103.
211.Shendure, J., Porreca, G. J., Reppas, N. B.,
Lin, X., McCutcheon, J. P., Rosenbaum, A. M., Wang, M. D., Zhang, K.,
Mitra, R. D., and Church, G. M. (2005) Science, 309,
1728-1732.
212.Shapero, M. H., Leuther, K. K., Nguyen, A.,
Scott, M., and Jones, K. W. (2001) Genome Res.,
11, 1926-1934.
213.Cai, F., Chen, H., Hicks, C. B., Bartlett, J.
A., Zhu, J., and Gao, F. (2007) Nat. Meth., 4,
123-125.
214.Bentley, D. R. (2006) Curr. Opin. Genet.
Dev., 16, 545-552.
215.Ten Bosch, J. R., and Grody, W. W. (2008) J.
Mol. Diagn., 10, 484-492.
216.Campbell, P. J., Stephens, P. J., Pleasance, E.
D., O’Meara, S., Li, H., Santarius, T., Stebbings, L. A., Leroy,
C., Edkins, S., Hardy, C., Teague, J. W., Menzies, A., Goodhead, I.,
Turner, D. J., Clee, C. M., Quail, M. A., Cox, A., Brown, C., Durbin,
R., Hurles, M. E., Edwards, P. A., Bignell, G. R., Stratton, M. R., and
Futreal, P. A. (2008) Nat. Genet., 40, 722-729.
217.Newman, S., and Edwards, P. A. (2010) Genome
Med., 2, 19.
218.Davidson, S. (2000) Nat. Biotechnol.,
18, 1134-1135.
219.Eklund, E. A. (2010) Curr. Opin.
Hematol., 17, 75-78.
220.Zhang, K., Zhu, J., Shendure, J., Porreca, G.
J., Aach, J. D., Mitra, R. D., and Church, G. M. (2006) Nat.
Genet., 38, 382-387.
221.Reeve, A. E., Sih, S. A., Raizis, A. M., and
Feinberg, A. P. (1989) Mol. Cell Biol., 9, 1799-1803.
222.Silva, F. P., Morolli, B., Storlazzi, C. T.,
Anelli, L., Wessels, H., Bezrookove, V., Kluin-Nelemans, H. C., and
Giphart-Gassler, M. (2003) Oncogene, 22, 538-547.
223.Butz, J., Wickstrom, E., and Edwards, J. S.
(2003) BMC Biotechnol., 3, 11.
224.Warner, J. K., Wang, J. C., Hope, K. J., Jin,
L., and Dick, J. E. (2004) Oncogene, 23, 7164-7177.
225.Wang, J. C., and Dick, J. E. (2005) Trends
Cell Biol., 15, 494-501.
226.Hughes, T., Deininger, M., Hochhaus, A.,
Branford, S., Radich, J., Kaeda, J., Baccarani, M., Cortes, J., Cross,
N. C., Druker, B. J., Gabert, J., Grimwade, D., Hehlmann, R.,
Kamel-Reid, S., Lipton, J. H., Longtine, J., Martinelli, G., Saglio,
G., Soverini, S., Stock, W., and Goldman, J. M. (2006) Blood,
108, 28-37.
227.He, C., Zhou, F., Zuo, Z., Cheng, H., and Zhou,
R. (2009) PLoS One, 4, e4732.
228.Zhu, J., Shendure, J., Mitra, R. D., and
Church, G. M. (2003) Science, 301, 836-838.
229.Conrad, C., Zhu, J., Conrad, C., Schoenfeld,
D., Fang, Z., Ingelsson, M., Stamm, S., Church, G., and Hyman, B. T.
(2007) J. Neurochem., 103, 1228-1236.
230.Butz, J. A., Roberts, K. G., and Edwards, J. S.
(2004) Biotechnol. Prog., 20, 1836-1839.
231.Berger, M. F., Levin, J. Z., Vijayendran, K.,
Sivachenko, A., Adiconis, X., Maguire, J., Johnson, L. A., Robinson,
J., Verhaak, R. G., Sougnez, C., Onofrio, R. C., Ziaugra, L.,
Cibulskis, K., Laine, E., Barretina, J., Winckler, W., Fisher, D. E.,
Getz, G., Meyerson, M., Jaffe, D. B., Gabriel, S. B., Lander, E. S.,
Dummer, R., Gnirke, A., Nusbaum, C., and Garraway, L. A. (2010)
Genome Res., February 23.
232.Tuch, B. B., Laborde, R. R., Xu, X., Gu, J.,
Chung, C. B., Monighetti, C. K., Stanley, S. J., Olsen, K. D.,
Kasperbauer, J. L., Moore, E. J., Broomer, A. J., Tan, R., Brzoska, P.
M., Muller, M. W., Siddiqui, A. S., Asmann, Y. W., Sun, Y., Kuersten,
S., Barker, M. A., De La Vega, F. M., and Smith, D. I. (2010) PLoS
One, 5, e9317.
233.Butz, J. A., Yan, H., Mikkilineni, V., and
Edwards, J. S. (2004) BMC Genet., 5, 3.
234.Sharma, S., Kelly, T. K., and Jones, P. A.
(2010) Carcinogenesis, 31, 27-36.
235.Figueroa, M. E., Lugthart, S., Li, Y.,
Erpelinck-Verschueren, C., Deng, X., Christos, P. J., Schifano, E.,
Booth, J., van Putten, W., Skrabanek, L., Campagne, F., Mazumdar, M.,
Greally, J. M., Valk, P. J., Lowenberg, B., Delwel, R., and Melnick, A.
(2010) Cancer Cell, 17, 13-27.
236.Bullinger, L., and Armstrong, S. A. (2010)
Cancer Cell, 17, 1-3.
237.Toyota, M., Kopecky, K. J., Toyota, M. O.,
Jair, K. W., Willman, C. L., and Issa, J. P. (2001) Blood,
97, 2823-2829.
238.Santini, V., Kantarjian, H. M., and Issa, J. P.
(2001) Ann. Int. Med., 134, 573-586.
239.Silverman, L. R., McKenzie, D. R., Peterson, B.
L., et al. (2006) J. Clin. Oncol., 24, 3895-3903.
240.Zhou, D., Zhang, R., Fang, R., Cheng, L., Xiao,
P., and Lu, Z. (2008) Electrophoresis, 29, 626-633.
241.Mitelman, F., Johansson, B., and Mertens, F.
(2004) Nat. Genet., 36, 331-334.
242.Tognon, C., Knezevich, S. R., Huntsman, D.,
Roskelley, C. D., Melnyk, N., Mathers, J. A., Becker, L., Carneiro, F.,
MacPherson, N., Horsman, D., Poremba, C., and Sorensen, P. H. (2002)
Cancer Cell, 2, 367-376.
243.Tomlins, S. A., Rhodes, D. R., Perner, S.,
Dhanasekaran, S. M., Mehra, R., Sun, X. W., Varambally, S., Cao, X.,
Tchinda, J., Kuefer, R., Lee, C., Montie, J. E., Shah, R. B., Pienta,
K. J., Rubin, M. A., and Chinnaiyan, A. M. (2005) Science,
310, 644-648.
244.Wang, J., Cai, Y., Ren, C., and Ittmann, M.
(2006) Cancer Res., 66, 8347-8351.
245.Winnes, M., Lissbrant, E., Damber, J. E., and
Stenman, G. (2007) Oncol. Rep., 17, 1033-1036.
246.Soda, M., Choi, Y. L., Enomoto, M., Takada, S.,
Yamashita, Y., Ishikawa, S., Fujiwara, S., Watanabe, H., Kurashina, K.,
Hatanaka, H., Bando, M., Ohno, S., Ishikawa, Y., Aburatani, H., Niki,
T., Sohara, Y., Sugiyama, Y., and Mano, H. (2007) Nature,
448, 561-566.
247.Lin, E., Li, L., Guan, Y., Soriano, R., Rivers,
C. S., Mohan, S., Pandita, A., Tang, J., and Modrusan, Z. (2009)
Mol. Cancer Res., 7, 1466-1476.
248.Mitelman, F., Johansson, B., and Mertens, F.
(2007) Nat. Rev. Cancer, 7, 233-245.
249.Brenner, J. C., and Chinnaiyan, A. M. (2009)
Biochim. Biophys. Acta, 1796, 201-215.
250.Edwards, P. A. (2010) J. Pathol.,
220, 244-254.
251.Morris, D. S., Tomlins, S. A., Montie, J. E.,
and Chinnaiyan, A. M. (2008) BJU Int., 102, 276-282.
252.Howarth, K. D., Blood, K. A., Ng, B. L.,
Beavis, J. C., Chua, Y., Cooke, S. L., Raby, S., Ichimura, K., Collins,
V. P., Carter, N. P., and Edwards, P. A. (2008) Oncogene,
27, 3345-3359.
253.Greenman, C., et al. (2007) Nature,
446, 153-158.
254.Campbell, P. J., Pleasance, E. D., Stephens, P.
J., Dicks, E., Rance, R., Goodhead, I., Follows, G. A., Green, A. R.,
Futreal, P. A., and Stratton, M. R. (2008) Proc. Natl. Acad. Sci.
USA, 105, 13081-13086.
255.Wang, X. S., Prensner, J. R., Chen, G., Cao,
Q., Han, B., Dhanasekaran, S. M., Ponnala, R., Cao, X., Varambally, S.,
Thomas, D. G., Giordano, T. J., Beer, D. G., Palanisamy, N., Sartor, M.
A., Omenn, G. S., and Chinnaiyan, A. M. (2009) Nat. Biotechnol.,
27, 1005-1011.
256.http://www.icgc.org/files/icgc/ICGC_April_29_2008_en.pdf
257.International Network of Cancer Genome Projects
(2010) Nature, 15, 464, 993-998.
258.Thomas, U. G. (2010) International Consortium
Plans to Sequence 25,000 Cancer Tumors, BioTechnique, 48,
News.
259.Gollin, S. M. (2007) Semin. Cancer
Biol., 17, 74-79.
260.PCR Testing for BCR-ABL Gene Rearrangement in
CML (2003) MSAC reference 9a (i) Assessment report, Canberra (http://nzhta.chmeds.ac.nz/publications/poly04.pdf).
261.Dohner, H., Stilgenbauer, S., Benner, A.,
Leupolt, E., Krober, A., Bullinger, L., Dohner, K., Bentz, M., and
Lichter, P. (2000) N. Engl. J. Med., 343, 1910-1916.
262.HUGO Gene Nomenclature Committee at the
European Bioinformatics Institute (http://www.genenames.org).