REVIEW: Constitutive and Induced Functions of the p53 Gene
A. O. Zheltukhin and P. M. Chumakov*
Engelhardt Institute of Molecular Biology, Russian Academy of Sciences, ul. Vavilova 32, 119991 Moscow, Russia; E-mail: peter@chumakov.com* To whom correspondence should be addressed.
Received April 27, 2010; Revision received July 25, 2010
The p53 tumor suppressor serves to secure genetic stability of multicellular organisms. It suppresses the accumulation of mutations in somatic cells and substantially decreases the probability of malignant diseases. The p53 gene acts highly selectively through multiple mechanisms. Under relatively favorable conditions, p53 helps to maintain intracellular homeostasis by balancing anabolic and catabolic processes and by timely elimination of reactive oxygen species. These functions of p53 facilitate maximal efficiency and survival of cells under conditions of physiological stresses. In the case of grave molecular damage caused by severe stress, a significant amount of highly active p53 is induced leading to irreversible growth arrest and programmed cell death. The induced functions of p53 secure the timely elimination from the organism of damaged and potentially dangerous cells. Collectively, the functions of p53 contribute to the prevention of malignant and other diseases and decelerate the aging process.
KEY WORDS: p53 tumor suppressor, cancer prevention, signal transduction, molecular damage, aging, apoptosis, autophagy, cell cycle control, oncogenes, tumor suppressor genes.DOI: 10.1134/S0006297910130110
Abbreviations: Akt, protein kinase B and its gene; AMPK, AMP-dependent kinase; GSK3β, glycogen synthase kinase encoding gene; IGF-1, insulin-like growth factor; iPS, induced pluripotent stem cells; clonogenic index, number of clones formed upon plating 100 cells per dish; LKB1, gene encoding AMPK-activating PK and several different kinases; MDM2, gene encoding Mdm2 protein, ubiquitin E3 class ligase involved in protein p53 stability regulation; mTOR, mammalian and human TOR homolog; NA, nucleic acids; nutlins (from Nutley, a small town in the USA where a Hoffman-LaRoche company facility is located), small molecules able to destroy complex of p53 and Mdm2 proteins; PIP2 and PIP3, phosphatidylinositol bis-phosphate and phosphatidylinositol triphosphate; PK, protein kinase; pRB, product of retinoblastoma suppressor gene; PTEN, tumor suppressor encoding “tensin-homologous protein phosphatase”; ROS, reactive oxygen species; TOR, protein kinase target of rapamycin; TORC1, first TOR-containing kinase complex; TORC2, second TOR-containing kinase complex; TSC1, tuberin, product of TSC1 suppressor gene damaged in hereditary tuberous sclerosis; TSC2, hamartin, product of TSC2 suppressor gene damaged in hereditary tuberous sclerosis.
Thirty years have passed since the discovery of p53. During this time
the exceptional importance of this protein within an organism has been
demonstrated, and its encoding gene is now one of the most studied
human genes. A few times new data on p53 forced fundamental revisions
in concepts concerning the role of p53 within the organism. Each new
turn in studies uncovered additional aspects in p53 function and opened
new perspectives for application of the new knowledge in prevention and
cure of diseases. It is now clear that the role of p53 is not limited
to the emergency response to severe stresses and to cell damage, but it
is also important for maintaining homeostasis in the organism under
normal conditions. Recent studies have also revealed a somewhat
controversial role of p53 in pathologies. Being an important component
of the system that clears the organism from damaged cells and a factor
contributing to balancing of metabolic processes, p53 contributes to
prevention of malignancy. However, the functions of p53 that are
directed toward protection may themselves produce collateral damage or
create negative background for normal cells and tissues, thereby
contributing to chronic pathologies and premature aging. As functions
of p53 are finely balanced between “good and evil”, deeper
understanding of p53-dependent processes would contribute to
development of optimal preventative measures.
p53 was first discovered as a “tumor antigen”. The level of p53 is low in normal cells, but it accumulates upon transformation by some viruses and in tumors [1-8]. After molecular cloning of the p53 gene, it became possible to express p53 ectopically in different cells. As p53 was found capable of transforming and immortalizing cells, it was assigned to the family of oncogenes [9-15]. However, 10 years later it has became clear that oncogenic properties of p53 represent an artifact owing to the fact that the first cloned p53 sequences were obtained from transformed cells that contained mutations in the protein-coding region of the p53 gene [16-18]. Quite opposite, the p53 gene isolated from normal cells behaved as a tumor suppressor upon introduction into cells. It had practically no influence on the function of normal cells, but was capable of stopping proliferation of transformed and tumor cells [19-21]. Depending on tissue origin, introduction of the wild-type p53 into tumor cells resulted in three types of responses: arrest of cell division at the cell cycle checkpoints [22, 23], induction of cell death (apoptosis) [24], or irreversible arrest of cell divisions accompanied by hallmarks of senescence [25-27]. Functionally, p53 meets the main criteria of a tumor suppressor: (a) its function is affected in many tumors, (b) wild-type p53 inhibits growth of tumor cells both in cell culture and in laboratory animals, (c) the p53 gene is mutated in the hereditary Li-Fraumeni syndrome [28] resulting in early development of multiple malignancies, and (d) mice with genomic knockout of the p53 gene die early of tumors [29]. In tumors the p53 gene often contains point mutations affecting the protein structure [30] leading to intracellular accumulation of faulty p53, which inhibits the function of the product of the second undamaged gene copy, i.e. it exhibits the dominant negative effect. Besides, mutant p53 protein acquires a number of novel activities not characteristic of normal p53, and these activities contribute to oncogenic properties of tumor cells [31]. Being a tumor suppressor in the norm, the p53 gene is converted to a dominant oncogene by mutations.
The level of p53 protein and its activity are low in normal cells, but DNA damaging stresses (such as UV or ionizing irradiation) induce accumulation and functional activation of p53. The induction of p53 can also result from some other damaging effects (hyperthermia, hypoxia) or by disturbances in cell physiology, such as disrupted mitotic spindle, errors in chromosome segregation upon mitoses, faults in actin cytoskeleton, cell contacts, extracellular matrix, etc. Viral infection or oncogene activation also induces p53 and signal toward removal of the damaged cells [32]. As a variety of conditions threatening cell genome integrity can induce the p53 response, David Lane has defined p53 as a “guardian of the genome” [33]. p53 prevents proliferation of defective cells. If a stress such as irradiation results in a DNA damage, then subsequent DNA replication may convert the damage to an inheritable mutation. If a cell begins the next division before completion of chromosome segregation, then the daughter cells may inherit incorrect chromosome number. p53 prevents such events by postponing cell divisions until the damage is repaired or by killing the damaged cell before the division occurs.
The above model laid the foundation for a new concept explaining how genetic stability of multicellular organisms is maintained, and further studies have brought its verification. As animals evolve to be more and more complex, systems supervising the accuracy of developmental programs specified by the genome acquire particular importance. An error at any step of a complex processes may result in substantial distortions at the downstream steps and results in the development of pathology. To prevent such situations, each individual cell of the organism should have a feedback signaling that reports about stepping outside the permitted genetically defined limits. In these regards, functions of p53 prevent catastrophes analogous to the emergency brake in a train. By supervising over correct execution of the genetic program in each separate cell, p53 carries an important function of preventing pathologies through clearing the organism of potentially dangerous cells.
Emerging new data force rethinking of the view that p53 is only an induced protein. The strong effects produced by p53 in response to damaging factors put into the shade other aspects of p53 function [34]. Only a few studies focused on the possible role of p53 in normal cells and its potential role under conditions of physiological stresses had been generally out of consideration. It was commonly assumed that p53 activity is absent in normal cells, and that it is required either to restrain propagation of defective cells or to accelerate damage repair. A strong argument in favor of such view was the apparently normal appearance of p53 knockout mice. The early death of phenotypically normal p53 knockout mice caused by lymphomas could be easily explained in light of the tumor suppressor role of p53 through timely removal of defective cells.
Recently several new activities of p53 have been revealed under physiological conditions in cells that are not exposed to extreme stresses [35, 36]. Along with its function in emergencies, p53 participates in milder adaptive processes by modulation of metabolism, increasing activity of antioxidant defense and detoxification, affecting the rate of protein biosynthesis, supervising the autophagy process that is directed to repair of non-dividing cells, and stimulation of reproductive functions [37-39]. Although these activities of p53 are not so visible, they also contribute to protection of genome integrity even without substantial induction of p53 levels. p53 also functions in embryos by protecting against defects of development. In response to emerging damage, p53 stops pluripotency of embryonic stem cells by repressing the inhibitor of differentiation Nanog and stops further cell divisions, thereby preventing their participation in the formation of the organism [40]. This same function of p53 protects genetic stability of the organism by preventing reverse differentiation (reprogramming) of specialized cells. Deliberate reprogramming of cells is a promising approach to artificial regeneration of tissues and organs. Overcoming a natural barrier to obtaining fully compatible stem cells from the patient’s own body by development of methods for temporal suspension of p53 function is important in disease control and in life-extending approaches.
Unlike the p53 function preventing propagation of defective cells, the ability of p53 to control homeostasis in normal cells [41] contributes to prevention of malignancy and of such widespread diseases as atherosclerosis, metabolic syndrome, obesity, diabetes, neurodegenerative diseases, and premature aging.
However, there is also a “dark side” of p53, as its excessive activity can itself lead to pathologies. Chronic stresses and local inflammation can persistently stimulate p53, thus leading to apoptosis in separate cells and release by dying cells of reactive oxygen species (ROS). This collateral stress, in turn, results in changes in intercellular matrix and launching further pathological events. Due to the pathologic feedbacks, the p53 related mechanisms contribute to diseases. Another undesirable effect of p53 is observed during chemo- and radiotherapy of cancer. Although the therapy provide more or less selective killing of tumor cells, the associated stresses induce p53 and contribute to toxic effects in normal tissue. Understanding mechanisms of p53 induction and considering measures for its temporary suppression would undoubtedly help to prevent these undesired complications.
Evolution adjusts and aligns intracellular processes in such a way that under physiologically normal conditions they are in the state of homeostasis. In an ideal organism, when all genes are fully functional and are in homozygous state, such equilibrium is possible. However, individual organisms are not ideal—they contain many defective alleles and therefore even without strong environmental influences the homeostasis could be compromised by launching pathogenic mechanisms and chronic diseases. Practically all pathologies are associated with stresses in tissues, and the p53 mechanisms contribute to these processes.
Thus, p53 plays a dual role—by securing the genome stability and protecting the organism from diseases it may incidentally complicate and aggravate some pathological processes. At the level of individual cells it can either induce their death if the cells are hopelessly damaged or, help the cells to resume normal function if the damage is manageable. These seemingly exclusive potentials of a single gene stem from the complex regulation of p53 activity. Many examples of the regulation have been considered in our recent review [32]. Here we would like to consider two scenarios of p53 function, one occurring under conditions of extreme damage and the other – under conditions of everyday life. Each of the activities contributes to overall preventive functions of p53 and has tremendous value for medical practice.
MODULATION OF p53 ACTIVITY
It has long been assumed that p53 activity is practically absent in normal cells under non-stressful conditions. At the transcription level the p53 gene is expressed at approximately the same intensity in most tissues despite the fact that the upstream region of the p53 gene contains multiple recognition sites for transcription factors AP1, YY1, NFkB, HOXA5, and PAX. Therefore, it is possible that some regulation at the transcription level may indeed occur, although the question requires additional studies. With the exception of HOXA5, the mentioned transcription factors were shown to repress p53 gene transcription [42]. Certain modulation towards increase in the transcription level is also possible due to the effect of poorly studied transcript Wrap53 corresponding to the opposite DNA strand of the p53 gene [43, 44]. Some data point to regulation of mRNA level through binding to 3′-untranslated region of stabilizing protein HuR; it is possible that the mechanism contributes to p53 induction upon stresses [45, 46].
The structure of p53 mRNA specifies an additional mechanism of regulation, which is due to translation initiation from two internal ribosome entry sites (IRES) responsible for CAP-independent translation initiation [47, 48]. Possibly, this regulatory mechanism is related to the expression of multiple minor alternatively spliced p53 mRNA isoforms [49]. In particular, an isoform specifying a protein lacking forty N-terminal amino acids, although a minor one, is translated more actively than the full-sized mRNA, so that the full-sized and truncated proteins are synthesized at approximately the same levels [50]. The functional significance of multiple p53 isoforms is poorly understood, although there are data implicating one of the isoforms in DNA repair [51].
Under normal physiological conditions, the intracellular level of p53 is very low because the newly synthesized protein is rapidly degraded in proteasomes. Most newly synthesized p53 protein is immediately directed to 20S proteasomes without prior ubiquitin coupling. The 20S proteasomes are involved in accelerated destruction of proteins having unfolded structure [52] or PEST motifs. Near the N-terminal segment of p53 there is an unstructured site responsible for the destruction of p53 in 20S proteasomes. The rate of p53 entry into 20S proteasomes can be modulated by two oxidoreductases, NQO1 and NQO2, which bind to unstructured regions of p53 and prevent its entry into 20S proteasomes. This activity of NQO1 and NQO2 seems not to be related to the reductase activity of the proteins. The NQO1 and NQO2 activity can be modulated depending on the physiological condition of the cell, in particular, by elevated ROS or by disrupted pyrimidine nucleotide biosyntheses.
Many details of regulation of p53 degradation in 20S proteasomes are still not clear, although it is quite possible that the modulation of p53 level by this mechanism plays an important role in prevention of some pathologies. NQO2 is known to be the main target for action of resveratrol [53, 54], which can counteract the aging processes, and, similarly to caloric restriction exerts a rejuvenating effect under high calorie diet [55, 56].
The ubiquitin-dependent pathway for p53 degradation in 26S proteasomes is better studied. Inhibition of the ubiquitin-mediated degradation results in massive accumulation of p53 protein during stresses and various disturbances in cell physiology. Due to activity of class E3 ubiquitin ligases, p53 undergoes polyubiquitination that is required for its entry into 26S proteasomes. The process is tightly regulated, and there are feedbacks that induce destabilization of p53 as its activity increases. Mdm2 protein is the main E3 ligase for p53 [57-59]. Mdm2 binds at the N-terminal region of p53, inhibits its transcription activity, and simultaneously stimulates export of the protein from the cell nucleus into the cytoplasm. However, the main function of Mdm2 is polyubiquitination of p53 at several C-terminal lysines. The MDM2 gene contains a strong p53-responsive element that specifies its activation upon increase in p53 protein level, thus stimulating its further destruction, which is important for restoration of low p53 level after the termination of stress [60]. The Mdm2 homologous protein MdmX is not an E3 ligase, but it can bind to Mdm2 and regulate the process of p53 degradation [61-63]. It is interesting that the Mdm2–MdmX complex also binds to the p53-induced cell cycle inhibitor p21 and contributes to its degradation in the 20S proteasomes [64]. A single-nucleotide polymorphism site SNP309 is located near the MDM2 gene promoter. A single nucleotide change in this site results in higher expression of Mdm2 and hence a decreased level of p53 and predisposition to malignancies [63]. In addition to Mdm2 protein, p53 is also ubiquitinated by several E3 ligases including Pirh2 [65] and COP1 [66], which like Mdm2 are products of p53 stimulated genes. By analogy with Mdm2, the Pirh2 protein can bind to other proteins (p27Kip1 and pRB) and stimulate their ubiquitin-independent degradation in 20S proteasomes [64, 67, 68]. Thus, the p53-regulated protein Pirh2 contributes to the release of the cell cycle block caused by p27Kip1 and pRB, which enhances p53-dependent apoptosis. The list of E3 ligases known to contribute to p53 degradation is expanding and includes CARP1/2 [69], TOPORS [70], synoviolin [71], and TRIM24 [72, 73].
By the influence of stresses, the process of p53 degradation is slowed down, leading to its accumulation. Numerous mechanisms are involved in stabilization of p53. A product of an alternative splice isoform from the CDKN2 (p16) gene p19ARF is able to bind to Mdm2 and to drive it to the nucleolus, thus retarding p53 export into the cytoplasm where it is destroyed in 26S proteasomes [74]. In normal cells the level of p19ARF protein is low, but it increases substantially after activation of oncogenes, thus leading to accumulation of transcription-active nuclear p53 [75]. The interaction of p19ARF and Mdm2 is in turn regulated by E3 ligase ARF-BP/MULE, which simultaneously binds to p19ARF and p53. The binding prevents p19ARF-mediated inhibition of Mdm2, while ARF-BP/MULE independently ubiquitinates p53 and contributes to its degradation [42, 76]. This mechanism probably helps to restore low level of p53 after ending of a stress.
Stabilization of p53 upon ribosomal stress can follow a different mechanism: ribosomal proteins L5, L11, and L23 bind to Mdm2 protein and prevent its interaction with p53 [77-80]. An additional mechanism for p53 stability regulation involves the deubiquitinase HAUSP, which removes ubiquitin from p53 and thus contributes to its accumulation [81]. Besides, HAUSP prevents ubiquitination of Mdm2 [82, 83], and the process is enhanced by DAXX protein [84]. It is interesting that the product of tumor suppressor RASSF1A disrupts the Mdm2–DAXX–HAUSP protein complex and causes accumulation of p53 by promoting degradation of Mdm2 [85].
The p53 protein is also stabilized and activated by modifications that protect it from E3 ligases and qualitatively change parameters of p53 activity. It should be emphasized that p53 is a protein that is subject to a great variety of posttranslational modifications. At least 36 various positions have been described in the amino acid sequence at which modifications of p53 can be detected [86, 87]. Most often modifications leading to p53 stabilization are associated with phosphorylation of N-terminal serines 15 and 20 by kinases ATM/ATR/DNA-PK, Chk1, and Chk2. This same site in the p53 molecule is responsible for binding to Mdm2 protein. Owing to phosphorylation at this site the interaction of p53 with Mdm2 is prevented, leading to stabilization of transcriptionally active p53. The phosphorylation can be detected soon after application of stresses including the DNA damage [88-90]. p53 can also be stabilized by alternative mechanisms, and the role of protein modifications can vary in different tissues [91, 92].
p53 KILLS OR ARRESTS DIVISION OF DAMAGED AND GENETICALLY MODIFIED
CELLS, THUS DECREASING PROBABILITY OF CANCER
The ability of p53 to arrest cell proliferation has been known for a long time, which justifies its definition as a tumor suppressor gene. When introduced into tumor cells, the intact p53 gene was found to inhibit cell proliferation and colony formation in cell culture, while no such effect was observed in normal fibroblasts. It was found later that the cessation of tumor cell proliferation is associated with several tissue-specific processes launched by p53. The main factor responsible for selective suppression of tumor cells is because they are abnormal. As during multistep carcinogenesis, a cancer cell acquires numerous mutations, and p53 introduced into such cells receives multiple feedbacks signaling about the abnormalities, which results in immediate p53 response against the abnormal cell.
The p53 tumor suppressor acts mainly as a transcription factor, and the character of its effect on a cell is defined by the set of products of p53-regulated genes. Besides p53 transcription function, p53 can also be transported to mitochondria, where it directly induces cytochrome c release and apoptosis by interaction with proteins BclXL/Bcl2 and Bak [32, 93-97].
Acting as a specific transcription factor, p53 induces expression of numerous genes, and the list of p53-induced genes is continuously expanding. In different cell models the set of p53-induced genes may greatly differ The p53-induced gene spectra may also differ depending on the character of inducing factors and on the existing cell environment. The most extreme result of p53 activation is induction of p53-dependent cell death, which results in removal of an irreversibly damaged cell from the organism.
The p53-controlled genes are involved in the induction of cell death by several alternative mechanisms. p53 induces transcription of proapoptotic genes that regulate permeability of mitochondrial pores, such as PUMA [98, 99], NOXA [100], BAX [101], and OKL38 [102], represses antiapoptotic genes Bcl2 [103] and ARC [104], and activates gene APAF1 [105-107], which results in activation of the mitochondrial apoptosis: release into the cytosol of cytochrome c, assembly of apoptosomes, and launch of proteolytic cascade initiated by caspase 9 [108]. The above pathway of apoptosis is a predominant p53-dependent response to severe stresses in many cell types [96].
p53 is also able to activate the extrinsic pathway of apoptosis that uses death receptors localized on the cell surface and a proteolytic cascade initiated by caspase 8 [108]. In some cell types the p53 induction activates transcription of genes FAS (APO1) [109] and KILLER/DR5 genes [110], thus increasing cell sensitivity to external ligands of these death receptors [111], and induces death receptor ligands TRAIL [112] and Fas [113].
In addition, p53 can initiate cell death by inducing a number of different genes, including caspase 10 [114], PERP [115], PIDD [116], WIP1 [117], Scotin [118], p53AIP1 [119], GML, STAG1, p53CABC1, and p53RDL1 [120], which are involved in apoptosis or in other forms of programmed cell death. It can also stimulate a number of genes whose products stimulate production and release of ROS (PIG3, PIG8 [121], FDXR [122]) and contribute to apoptosis [121].
Finally, p53 can control synthesis of non-coding micro RNAs (miRNAs) that are involved in regulation of still more sets of genes [123-127]. Forceful inhibition of p53-activated miR34 compromises the p53-dependent apoptosis in response to genotoxic stresses, which suggests the contribution of this mechanism to the suppressor activity of p53.
Along with the induction of cell death, p53 is able to arrest cell divisions by affecting the transitions of the cell cycle. Cyclin-dependent kinase (CDK2/4) inhibitor p21, the product of the CDKN1A gene, was the first identified p53 transcription target [128]. Besides, p53 induces a number of other genes, such as 14-3-3σ, GADD45, BTG2, B99/GTSE-1, REPRIMO, HZF, and MCG10, that can delay or arrest cell cycle progression [129-133].
The induction of p21 occurs even upon moderate increase in p53 activity, which results in transient delay of cell cycle progression in the late G1 phase, prior to initiation of DNA synthesis. The p21-mediated cell cycle delay gives the cell time to repair small DNA damage, after which the p53 level returns to the norm and the cell can resume further divisions.
Some cell types do not undergo apoptosis even in response to severe DNA damage, but instead they irreversibly stop dividing. The state of irreversible cell cycle arrest is defined as cell senescence. Sometimes the state is called replicative senescence because a similar condition is observed in the case of critical telomere shortening upon long-term culturing of normal cells. The p53-dependent replicative senescence plays a decisive role in carcinoma and sarcoma suppression upon pharmacological reactivation of p53 or upon the reintroduction of wild-type p53 with different vectors [134, 135]. Unlike the transient cell cycle arrest, replicative senescence has some characteristic features such as expression of acid beta-galactosidase, increased intracellular ROS level, and induction of CDK inhibitor p16 [136, 137].
The classical state of replicative senescence is observed upon exceeding the allowable limits of cell divisions in culture. It is associated with critical telomere shortening, which initiates p53 induction [138, 139]. Telomerase activity is generally absent in most normal cells. Therefore, the replicative capacity of a cell depends on telomere length. The telomerase gene (hTERT) is repressed by p53, although the mechanism of repression is not quite clear. According to one model, p53 binds to transcription factor Sp1, thus preventing activation of the hTERT gene promoter [140, 141]. Another model suggests a leading role of the p53-regulated product of the CDKN1 gene (p21), which is involved in assembly on the hTERT gene promoter of a repressor complex containing pRB bound to E2F [142, 143]. In any case, loss of p53 activity is accompanied by activation of hTERT gene transcription. Active telomerase restores proper length of telomeres and maintain further unlimited divisions of the cell [144]. Therefore, loss of p53 activity results in cell immortalization, while stress activation of p53 leads to replicative senescence.
Prolonged induction of p53-dependent p21 plays a pivotal role in the establishment of replicative senescence. A similar effect is observed upon deliberate expression of p21 protein even in p53-negative cells [145]. Different p53-regulated genes, including the gene PAI-1, also contribute to the establishment of p53-dependent replicative senescence [146, 147], although the p21 protein is obligatory for the process. This was demonstrated in mice expressing a p53 mutant defective in proapoptotic function, but competent in induction of p21. Such mutant protects mice from tumors with strict dependence on the expression of p21 [148-150]. Nevertheless, deletion of the p21 gene in mice per se does not result in noticeable increase in tumor frequency [151].
Thus, it was found by exclusion that abrogation of neither the p53-induced apoptosis nor the p53-dependent replicative senescence and cell cycle arrest, when applied alone, are sufficient to disrupt the tumor suppressor function of p53. It raises question of what activity of p53 is the most important for the prevention of tumor formation? At first glance, the existence of numerous pathways of p53-mediated cell death may suggest the possible key role of apoptosis and other forms of programmed cell death. In support, a p53 mutant with disrupted ability to arrest the cell cycle but with preserved apoptosis function is still able to efficiently protect transgenic mice against the development of spontaneous tumors [152]. However, the antitumor activity of p53 is not defined exclusively by its ability to induce apoptosis. In mice the deletion of the PUMA gene, which is required for p53-dependent apoptosis of various cell types [153], was not associated with increase in tumor frequency [154]. Besides, a p53 mutant lacking apoptosis function but able to arrest the cell cycle still protects mice well against the development of tumors [155]. The results of these experiments clearly point to the existence of additional p53-dependent mechanisms contributing to protection against cancer.
CONTROL BY p53 OF TISSUE AND ORGANISMAL RESPONSE TO DAMAGED AND
MODIFIED CELLS
It still needs to be found what would be the phenotype of mice with combined homozygous deletions of genes encoding PUMA and p21, which abrogates p53-dependent apoptosis and cell cycle arrest together, and whether they retain the ability to resist the development of tumors. Taking into account the great variety of functions provided by different p53-induced genes, one can speculate that the antitumor function of p53 is not confined to mere prevention of propagation of defective cells. A significant group of p53-regulated genes encodes secreted factors that may affect the tissue environment of damaged cells and prevent their survival and spreading. By secreting p53-induced factors defective cells signal to the environment about the emerging danger, which then mobilizes protective mechanisms within the tissue and the organism. p53 can induce the inhibitor of plasminogen activator PAI-1 [156], matrix metalloproteinase 2 (collagenase IV), MMP2 [157], and maspin [158], a serpin class protease inhibitor that influences the dynamics of extracellular matrix and serves as a separate tumor suppressor [159]. It also controls the suppressor of metastasis gene KAI1/CD82 [160] encoding a tetraspanin family protein. The product of KAI1 is localized on the cell surface where it interacts with other tetraspanins, integrins, and chemokines and influences cell migration, cell adhesion, and signal transduction. The KAI1 protein is used by certain viruses to facilitate their penetration into the cell. The increase in KAI1 expression results in endocytosis of receptors of epidermal growth factor and slower cell migration and invasiveness [161].
Activation of p53, in particular by irradiation, induces production of several secretory factors whose function inhibits cell proliferation [162]. Treatments that induce p53 activation, such as anticancer therapy, may cause collateral damage through the effects that p53-induced secreted factors produce to normal tissue. The induction of p53 may also affect hormone regulation of tissue metabolism, as one of the commonly induced p53-regulated genes IGF-BP3 [163-165] encodes a protein that binds to insulin-like growth factor. Most likely this mechanism directly affects the IGF-BP3-secreting damaged cell, as it was shown that IGF-BP3 decreases tumor cell survival and increases their sensitivity to apoptosis [166]. However, this factor may also influence the surrounding normal cells, causing a decrease in glucose uptake that may contribute to restriction of damaged cell spreading.
p53 influences neoangiogenesis. Among p53-induced genes there are several genes encoding angiogenesis inhibitors. Thrombospondin (TSP-1) [167], a matrix glycoprotein that interacts with numerous tissue protein factors, also inhibits fibroblast growth factor 2, which is required for blood vessel invasion [168]. Besides, p53 induces the thrombospondin-related angiogenesis inhibitor BAI1 that is expressed in brain [169] and GD-AIF, a poorly characterized angiogenesis inhibitor secreted by glioblastoma cells [170]. Through the action of these and other factors, as soon as a cell encounters a p53-inducing damage, the surrounding tissues receive signals that prevent penetration into this area of new vessels, which limits further proliferation and spreading of genetically modified cells.
Recently the ability of p53 to modify endosome dynamics and exosome secretion was identify, which may play an important role in suppression of tumor development [171-173]. Endosomes are involved in processes of intracellular signal transduction though regulation of ligand interactions with appropriate membrane receptors [174]. Internalization of ligand-bound membrane receptors into endosomal vesicles serves their further delivery into different cell structures. Receptor complexes are modified during the transport; some receptors are then destroyed, others return to the cell surface. Modulation of endosomal transport can influence signal transduction. In addition to the classical pathway for protein secretion through endoplasmic reticulum and Golgi apparatus, secretion through exosomes represents a separate pathway, which involves multivesicular particles (MVP). The process of exosomal secretion proceeds by membrane budding into the endosome opening, during which a part of the cytoplasm is engulfed by endosomes that are surrounded by an additional membrane. Such MVP are then delivered to the cell surface, fused with the outer membrane, and the endosomes with their content are released to form exosomes. It is worth noting that the exosomal vesicles can interact with dendritic cells, which contributes to immunization of the organism against their content [175]. Exosomes can fuse with other cells, thus transmitting signals between cells, or release their content into intracellular matrix, thereby changing its composition [164, 175].
Recently it has been found that p53 regulates transcription of several genes (TSAP6, Caveolin-1, and Chmp4C) that are somehow involved in dynamics of endosomes [171-173]. The activation of TSAP6 stimulates exosomal secretion [176]. Caveolin-1, a structural component of membrane vesicle caveolae, participates in the internalization of membrane-bound receptors including those of epidermal growth factor and TGF-β 1, 1. The process is stimulated by p53, resulting in limited accessibility of growth factor receptors and retardation of proliferation [173]. Chmp4C is a component of MVP that binds to transported cargo [179], and its activation stimulates exosomal secretion. The role of p53 in dynamics of endosomal transport requires further detailed study. There is an interesting hypothesis concerning participation of endosomes in p53-induced immunization of the organism against tumor cells [180]. It suggests that p53 may inhibit tumor development at the organismal level, and that damage in an individual cell may provide the signal for mobilizing protective systems of the organism.
p53 AND DNA REPAIR
Errors occurring during DNA replication may lead to accumulation of mutations and contribute to the development of malignancy. One of the measures against mutation could be transient arrest of cell divisions, and another complementary measure is activation of DNA repair. p53 is involved in DNA repair at several levels and not only as a transcription factor. It was found that p53 can directly recognize DNA damage and bind to the damaged sites. Also, p53 can bind to single-stranded DNA regions and to non-protected DNA termini that originate from double-stranded DNA breaks; it is able to recognize and bind to unpaired DNA regions [181-186]. Through the binding p53 participates in damage recognition, which then serves as a signal for DNA repair activation or induction of apoptosis. It has also been found that p53 is directly involved in the excision of a damaged DNA region due to its own 3′→5′ exonuclease activity and ability to catalyze topological rearrangement in an altered DNA region [181, 187, 188]. Owing to its own exonuclease activity, p53 plays a proofreading role by correcting errors made by DNA polymerase due to insufficiency of its exonuclease [189] or primase [190]. Finally, p53 is directly involved in increasing reading accuracy of reverse transcriptase [191-194]. The enzymic activities of p53 toward DNA probably represent its most ancient functions that enforce genetic stability [195].
The next level of p53 participation in DNA repair includes its interaction with numerous repair proteins such as RAD51, 53BP1, BRCA1/2, BARD1, MDC1, HMG1, BLM, WRN, MRE1, RPA1, HEF-1, ERCC6, SNF5, DNApolα, and mtDNApolγ 1-204]. Interaction with p53 either modifies activities of repair systems or serves to assist DNA damage signal transduction, which subsequently results in p53 activation via corresponding systems. Upon binding to the site of DNA damage, p53 becomes accessible for activating modifications by DNA-dependent protein kinase (DNA-PK) and ATM and ATR kinases.
p53 influences DNA repair also due to its transcription activity. Members of the global genome repair (GGR) system, XPE (DDB2) and XPC were revealed among transcriptional targets of p53 [133, 205, 206]. Deficiency in the GGR system and prevalence of transcription-coupled repair (TCR) were observed in tumor cells upon inhibition of p53 activity [132]. p53 stimulates transcription of the DNA polymerase η gene and thus enhances DNA repair near the replication fork [207]. Factors of unpaired nucleotide repair MSH2, PCNA, MLH1, and PMS2 are also induced by p53 [208-211]. Finally, p53 protein induces a homolog of the ribonucleotide reductase p53R2 β-subunit gene [212]. The main β-subunit is required for nuclear DNA replication during the S phase, but p53R2 is involved in replication of mitochondrial genome and in mtDNA repair [213, 214]. This partially explains why in p53-deficient cells the mitochondrial genome is unstable, leading to impaired mitochondrial function [215].
Due to the ability of p53 to stimulate DNA repair systems, during p53-mediated transient cell cycle arrest the cells may accelerate the DNA damage repair process.
FUNCTIONS OF p53 UNDER PHYSIOLOGICAL CONDITIONS
Since p53 is a stress-induced protein, it produces rather strong effects in cells subjected to stresses. The diversity of the p53 induction pathways in response to various stresses and the brightness of the observed effects have kept researchers busy over many years. The strong effects produced by stresses have been leaving in the shade some other activities of p53 that are not associated with such a strong manifestations but perhaps play no lesser role in the mission of p53 of providing stability of the genome, securing precise execution of genome-specified developmental programs. Only recently accumulated information has forced revision of the dogma that without stress p53 activity is practically absent.
Studying of complex biological processes tend to follow a reductionist approach, which acknowledge the brightest effects and plot on their basis provisional schemes that often play a restraining role in obtaining a wide and comprehensive picture. Eventually, accumulated data started to suggest that p53 function is not limited to measures of stopping damaged cells from propagation and spreading. The activity of p53 can be revealed in everyday life, even in the absence of any extreme damaging influences, but rather under light and moderate physiological loads and stresses. As organisms became more and more complex the p53 gene probably acquired new functions related to control over social behavior of cells, forming additional connections with components of different pathways and developing even more sophisticated mechanisms regulating its function.
Activities of p53 under normal conditions become most obvious when considering systems where p53 is either completely absent (animals with homozygous p53 gene deletion) or in which its activity is very much decreased due to RNA interference or other factors that negatively control its functions. From studying such systems, it has become clear that p53 participates in regulation of daily homeostasis, takes part in normal physiological processes, and helps cells to balance metabolic processes. p53 may participate in aligning concerted actions of different signaling pathways, timely mobilize antioxidant defenses, and intervene with tissue renewal and stem cell mobilization. It looks very probable that through its ability to intervene with these processes, p53 affects the pace of organism aging. This new view places p53 in the center of adaptation mechanisms that either help individual cells to cope with daily stresses or make the decision for their elimination from the organism if the stress exposure is excessive.
ANTIOXIDANT FUNCTION OF p53
Irradiation (gamma, X-ray, and UV) and DNA-modifying chemicals are among the most commonly considered unfavorable factors that contribute to mutagenesis. Therefore, these factors have been in the center of studies on mechanisms of p53 activation in response to stresses. However, endogenous ROS that are constantly produced in the organism are the most common factor that contributes to modification of DNA and acquisition of mutations [216]. Every day about 20 thousand bases are oxidized in each cell [217].
The perception of ROS has significantly changed during recent years. Earlier ROS were considered as clearly harmful compounds, byproducts of aerobic metabolism produced mainly due to electron leakage from the mitochondrial electron transport chain (ETC). Leaking electrons, instead of participating in the formation of water molecules, are transferred to molecular oxygen and form superoxide anionic radicals (O2•–) [3, 218, 219] that gives rise to different ROS, such as hydrogen peroxide (H2O2), hydroxyl radicals (•OH, OH–), and alcoxyl/peroxyl radicals (RO•/ROO•). The bulk of superoxide anion is formed in ETC complexes I and III, and due to their strong charge they easily penetrate the internal mitochondrial membrane and are then released into the matrix [219, 220]. Superoxide anion is rapidly transformed by superoxide dismutase (SOD) to produce H2O2, or through interaction with transition metals it can also give rise to highly reactive hydroxyl radical (the Fenton reaction). In addition to mitochondria, ROS are produced in peroxisomes, where they participate in the oxidation of fatty acids; they are also formed as byproducts of detoxification reactions mediated by cytochrome P450.
Because of their ability to oxidize biological molecules, ROS are highly damaging to lipids, proteins, and nucleic acids, which contribute to mutagenic load on the genome and to development of pathologies. However, more recently it was found that besides their undesirable properties, ROS play important physiological roles in the organism. Hydrogen peroxide is a signal molecule in numerous regulatory processes and is also used by leukocytes for protection against microbes [221]. During leukocyte activation the superoxide anion is produced by membrane-bound NADPH-dependent oxidase, which contributes to killing of invading bacteria. A similar mechanism is used in signaling pathways where the activity of NADPH oxidase transfers an electron to an oxygen molecule with formation of superoxide anion, which is rapidly converted into H2O2. Hydrogen peroxide then modifies redox-sensitive components of signaling pathways (protein phosphatases, proteinases, some transcription factors) and in this way participates in signal transduction mechanisms. Thus, most intercellular communications as well as interaction of the cells with external protein ligands are accompanied by transient bursts in the level of hydrogen peroxide. Therefore, systems need to be in place that eliminate the excess of ROS in such a way that the antioxidant mechanisms do not interfere with the signaling processes.
ROS homeostasis is maintained by a number of antioxidant systems. Catalase remove excessive H2O2 with high efficiency, although the source of the H2O2 being removed by catalase is mainly of exogenous origin. Glutathione peroxidase catalyzes the reduction of H2O2 and lipid peroxides using glutathione, which is oxidized during the reaction and is then regenerated by glutathione reductase [222]. However, the role of these highly efficient enzymes is still not quite clear. Genetic approaches with knockout animals show that neither catalase nor glutathione peroxidase play a substantial role in protection against endogenous ROS. However, the endogenously produced ROS play a leading role in pathologies and aging [222-225]. The endogenous H2O2 is preferentially removed by peroxiredoxins, thiol antioxidant enzymes that are oxidized during catalysis and then are re-reduced using the thioredoxin–thioredoxin reductase system [226].
Changes in levels of intracellular ROS take place in a number of physiological and pathological conditions. Responses to the changes depend both on the cell type and the ROS level [227]. When exceeding certain limits, an oxidative stress condition occurs that results in induction of p53. Activation of p53 results in inhibition of proliferation or development of replicative senescence or apoptosis [227, 228]. p53 prevents proliferation of cells with oxidative DNA damage, and thus plays a role in prevention of pathology.
It is remarkable that one third of all genes induced by H2O2 treatment are transcription targets of p53 [229]; at the transcription level p53 is able to induce numerous genes (such as PIG3, PIG6, and FDXR) whose products are either involved in ROS generation or increase cell sensitivity to oxidative stress [121, 230, 231]. This indicates that p53 activities may aggravate oxidative stress conditions and contribute to induction of apoptosis through oxidative destruction of mitochondrial structures [121]. Some other p53-regulated genes (PUMA, Bax, etc.) may elevate ROS through direct induction of apoptosis, which is accompanied by a substantial release of mitochondrial ROS.
However, among transcription targets of p53 there are also several genes with apparently antioxidant function. For example, p53 regulates aldehyde dehydrogenase 4 ALDH12 [232], a microsomal homolog of glutathione transferase PIG12 [121], glutathione peroxidase GPX1, superoxide dismutase SOD2 [233], catalase [234], two members of the sestrin family SESN1 and SESN2 [235, 236], TIGAR [237], glutaminase 2 GLP2 [238], as well as p53INP1 [239]. It may look paradoxical that p53 is able to induce simultaneously both pro- and antioxidant genes. To resolve the paradox, p53 functions were switched off in cell cultures. It was found that in many cell types abrogation of p53 activity either by RNA interference or by expression of p53-inhibiting proteins (Mdm2, papilloma virus E6 protein, dominant-negative p53 mutants) results in significant increase in intracellular ROS levels [35]. It is noteworthy that in cultures obtained from p53-knockout mice as well as in tissues of these mice the ROS level was also increased, along with substantially reduced expression level of several p53-regulated antioxidant genes. Thus, even in the absence of stresses, under normal physiological conditions p53 activity is required for maintaining sufficient expression of antioxidant genes. The data indicate that in normal cells p53 has antioxidant function, and that the basal non-induced expression level of p53 is sufficient to carry out this function [35, 240].
The important question is what factors affect the decision of p53 to selectively activate either pro-, or antioxidant genes? The answer was found when testing different expression levels of p53. Under low levels of active p53 in cells that are maintained either without stresses, or are subjected to moderate physiological stress, p53 activates a set of antioxidant genes such as SESN1/2, GPX1 [35], TIGAR [237], catalase [234], along with CDK inhibitor p21 [35]. These genes are sensitive to even low levels of p53. It is quite appropriate that p21 is among the most sensitive targets as it can fine tune proliferation by its delay when the ROS level reaches a certain limit. Quite opposite, high levels of p53 that are observed under severe stresses result in activation of proapoptotic and prooxidant genes [35]. Thus, the capacity of p53 to induce different functional sets of genes and to produce quite opposite effects is defined by the severity of the stress and by the expression level and activity of p53. Under normal conditions, or during physiological stresses, p53 acts as a survival factor that helps the cell to cope with its working load. After exceeding a certain limit of stress or achieving damage level that cannot be efficiently repaired, p53 acts as a “terminator” inducing genetic death of a damaged cell. These scenarios are depicted in Fig. 1.
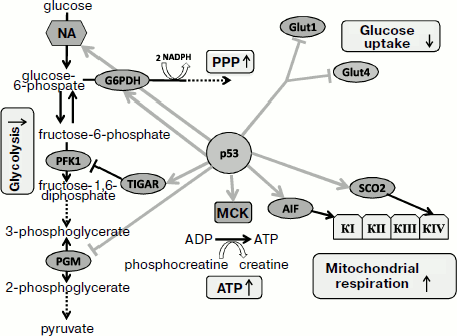
The role of antioxidant activity of p53 in enforcing genome stability as well as its contribution to tumor suppression have been demonstrated both in cell culture and in the p53-knockout mouse model [35]. The absence of p53 was associated not only with increased intracellular ROS levels but also with acceleration of mutagenesis. However, addition of antioxidant to the medium efficiently decreased the mutation frequency. Feeding antioxidant to p53-knockout mice was sufficient to abrogate the development of malignant lymphomas, which suggests a leading role of increased ROS as the cause for tumorigenic phenotype in p53-knockout mice [35].Fig. 1. Switching between different functions of p53 protein depending on the severity of stress and damage. Under physiological conditions or during mild stress, low p53 levels stimulate transcription of genes that maintain intracellular homeostasis. The functions aim to help optimal function of the cell and its survival. Under conditions of severe stress that produce serious damage, much higher levels of transcription-active p53 is induced, which results in activation of a different sets of genes whose function result in either arrest of cell divisions or programmed cell death. NA, nucleic acid; PPP, pentose-phosphate pathway; MCK, muscle creatine kinase.
The antioxidant activity of p53 was revealed only recently, and therefore its mechanisms require additional study. While the consequences of the induction of such enzymes as catalase and glutathione peroxidase are quite clear, mechanisms associated with the induction of other p53-regulated antioxidant genes seem more complicated. Sestrins are members of a small family consisting of three closely related genes (SESN1-3). The products of these genes are ubiquitously expressed in many cell types. Analysis of protein structure of sestrins reveals a region of high homology with the product of the bacterial AhpD gene [241], a thioredoxin-like protein playing a role in regeneration of bacterial peroxiredoxin AhpC [242].
Unlike bacterial peroxiredoxins, which are robust enzymes, animal and human peroxiredoxins, which are also involved in decomposition of H2O2, are prone to inactivation by excessive concentrations of H2O2 due to the oxidation of their catalytic cysteine to sulfinate Cys-SO2. Unlike Cys-SO, the sulfinic acid derivative of cysteine in peroxiredoxins cannot be regenerated by a thioredoxin molecule, as it cannot establish with the latter an intermolecular disulfide bond [243-246]. Such mode of regulation of peroxidase activity of peroxiredoxins is characteristic only to organisms that utilize H2O2 as a signaling molecule. When a peroxide burst is being conveyed, the antioxidant defense firewall is switched off, which allows modification of redox-sensitive targets in signaling pathways. However, to prevent the development of oxidative stress the peroxiredoxin activity then needs to be restored. The latter process is controlled by sulfiredoxin. It reduces Cys-SO2 to Cys-SO, which can be then reduced by the thioredoxin regeneration system [247]. Sestrins are also involved in the regeneration of peroxiredoxin Cys-SO2 [241]. They form a complex with sulfiredoxin and apparently control of the rate of this process, although details of the participation of sestrin require additional study.
Sestrins are also negative regulators of the mTORC1 pathway, which will be described in more detail in following sections. The inhibition of mTORC1 can also reduce ROS levels due to retardation of anabolic processes. Therefore the contribution of the two activities of sestrins in the overall antioxidant function needs further clarification.
The genes that are controlled by physiological levels of p53 and affect cell metabolism can also contribute to antioxidant activity. Functions of these genes will be described in more detail below. The p53-induced regulator of glycolysis and apoptosis TIGAR is a distant structural and functional homolog of the bis-phosphatase domain of the bifunctional enzyme 6-phosphofructo-2-kinase/fructo-2,6-bisphosphatase [237]. Stimulation of TIGAR expression results in decrease in intracellular level of fructo-2,6-bisphosphate, a powerful positive allosteric effector of 6-phosphofructo-1-kinase. The latter enzyme serves as a potent stimulator of glycolysis. Besides, fructose-2,6-bisphospate is an allosteric inhibitor of fructose-1,6-bisphosphatase, which stimulates gluconeogenesis [248]. Thus, activation of TIGAR results in inhibition of glycolysis and accumulation of fructose-6-phosphate that is then isomerized to glucose-6-phosphate, which serves as a substrate for the pentose-phosphate pathway. The pathway is involved in generation of NADPH and thus stimulates accumulation of reduced glutathione, thereby lowering intracellular ROS level. Consistent with this, lower level of ROS is observed in cells with accelerated TIGAR expression, while the opposite effect is observed upon endogenous inhibition of TIGAR by RNA interference [249]. Changes in redox balance of the cell during increased expression of TIGAR result in increased resistance to apoptosis following H2O2 treatment, which is consistent with the contribution of TIGAR to the antioxidant function of p53. TIGAR belongs to the group of p53-regulated genes that are most sensitive even to a slight increase in p53, although it is expressed to some extent even in p53-deficient cells [237].
Since the mitochondrial ETC significantly contributes to total cellular ROS level, change in oxidative phosphorylation should affect the ROS level. Low levels of p53 in unstressed cells are sufficient for the expression of the SCO2 gene, which is involved in the assembly of cytochrome c oxidase complex in mitochondria, the most important site for O2 consumption [250]. In cells devoid of p53 activity the processes of mitochondrial respiration are substantially compromised [250, 251], which theoretically can be accompanied by lower production of mitochondrial superoxide and decreased ROS level [252]. An interesting model is proposed for p53-induced proapoptotic gene AIF under physiological conditions [253]. The product of the AIF gene exhibits NADPH oxidase activity [254]; it is localized in intermembrane mitochondrial space and somehow participates in the function of ETC complex I [255]. Deficiency in this gene results in defects in oxidative phosphorylation, and in particular, the Harlequin mouse strain with an insertion within the AIF gene is characterized by chronic oxidative stress and progressive neurodegeneration [256]. AIF also displays an apoptogenic activity: in response to stresses, AIF, like cytochrome c, is released through the mitochondrial pore, but then it is transported into the cell nucleus where it induces a caspase-independent apoptosis [257, 258]. Development of this form of apoptosis requires activation of poly(ADP)ribose polymerase (PARP-1) [259] and formation PAR-polymer contributing to release of AIF from mitochondria [260, 261]. After entering nuclei, AIF induces chromosome fragmentation [262] and condensation [263] due to its direct interaction with DNA [262]. The ability of AIF to induce apoptosis does not require its NADPH oxidase activity [264], which is necessary exclusively for its mitochondrial function [265]. It is interesting that p53 causes AIF induction at the transcription level in normal non-stressed cells. In this case p53 activation by stresses does not result in further increase in AIF expression [253]. Thus, by activating AIF under physiological conditions, p53 adjusts oxidative phosphorylation function and simultaneously increases sensitivity of the cell to apoptosis under stress conditions. The activation of the ETC can somewhat increase production of ROS, but AIF deficiency results in impaired mitochondrial functions, which also favors ROS release.
Phosphoglycerate mutase (PGM), which regulates the balance between mitochondrial respiration and glycolysis, is also controlled by p53. This p53-dependent gene exhibits strictly tissue-specific regulation: in fibroblasts p53 serves as a posttranscriptional repressor of PGM [266] leading to slower glycolysis and stimulated respiration and ROS production, while in muscle cells p53 activates PGM at transcriptional level [267], which stimulates glycolysis and exerts an antioxidant effect [252, 268]. Another p53-regulated gene, GLS2, encoding glutaminase 2, activates glutamine metabolism and contributes to enhancement of mitochondrial respiration. The increase in GLS2 expression stimulates glutathione synthesis, which contributes to antioxidant defense [238].
In addition to the its role as transcription factor in antioxidant function of p53 under physiological stresses, some role in the process can be also attributed to the fraction of p53 delivered to mitochondria. It has been mentioned that a fraction of p53 can induce apoptosis through direct interaction with proteins of the Bcl2 family in mitochondria [93, 95]. However, p53 apparently has an additional function upon delivery to mitochondria, as in the absence of severe stresses it can even contribute to cell survival by increasing the mitochondrial genome stability [269]. p53 binds to mitochondrial DNA polymerase γ and contributes to mitochondrial DNA replication [270]. Thus, protein p53 can exert diverse effects on mitochondrial physiology, which explains why in p53-knockout cells there a substantial depletion of mitochondrial DNA, reduction in mitochondrial mass, and lowered ratio of the mitochondrial superoxide to H2O2 is observed [215].
It is still not clear whether the antioxidant activity of p53 is induced in response to mild physiological stresses, or it is constitutive. Increased ROS levels result in activation of p53 by several mechanisms including the introduction of covalent modifications to the p53 molecule. However, most of these modifications are not ROS-specific. The p53 molecule contains two clusters of redox-sensitive cysteines, the oxidation of which influences its DNA-binding activity [252, 271]. Oxidation of these cysteines induces S-glutathionylation and lowers DNA-binding activity of p53 [272, 273]. However, oxidation of certain cysteines can change the spectrum of p53 specificity towards DNA elements of different p53-regulated genes [274]. This, in turn, can be important for selective expression of certain sets of genes in response to oxidation. It was also shown that a number of redox-active enzymes (thioredoxin, thioredoxin reductase, APE/Ref-1) can modulate p53 activity [199, 275, 276], which suggests the possibility of antioxidant activity regulation in response to physiological stresses [277].
Another p53-regulated antioxidant gene, TP53INP1, is induced in response to stresses, and in cells deficient in expression of this gene the ROS levels are high [239]. Forced expression of TP53INP1 in p53-deficient cells results in lower levels of ROS, which points to p53-independent character of its antioxidant activity. When TP53INP1 is switched off in p53-expressing cells, there was abnormal induction of some of the p53-dependent genes, including the proapoptotic genes PUMA and Bax, antioxidant sestrins, CDKN1a (p21), and TA-p73. The mechanism by which TP53INP1 affects the p53 pathways is in its ability to interact with protein kinases HIPK2 and PKCδ and participate in phosphorylation of p53 at Ser46 [278-280]. Previously Ser46 phosphorylation was considered as being particularly important for the induction of p53-dependnt apoptosis [281]; now it becomes clear that it is also important for other activities of p53, including the antioxidant function.
The p53-independent function of TP53INP1, which is responsible for lowering ROS level, is less understandable. TP53INP1-deficient mice are susceptible to the development of colon cancer on the background of chronic inflammatory process that is accompanied by increased levels of ROS [282]. This suggests a role of the TP53INP1 gene as an independent tumor suppressor. Some p53-independent functions can be mediated by the closely related p73 gene, because TP53INP1 can also be induced in response to TA-p73 [283]. An alternative explanation can be associated with a recently identified closely related gene TP53INP2, which together with TP53INP1 arose in evolution due to duplication of a common ancestral gene. In response to starvation or upon treatment with the mTOR inhibitor rapamycin and the PI3K inhibitor wortmannin, TP53INP2 is transported from the nucleus into autophagosomal structures where it interacts with Atg8, an LC3-like protein, and with autophagosomal protein VMP1 [284]. Thus, TP53INP2 is involved in the mechanism of autophagy induction; particularly, autophagy is completely inhibited in cells deficient in the product of the TP53INP2 gene [285]. In light of the role of TP53INP2, the antioxidant activity of TP53INP1 can be also associated with this process of autophagy, although so far the hypothesis has no experimental confirmation.
ROLE OF p53 IN REGULATION OF METABOLISM
Studies of cancer cell metabolism reveal specific features associated by the absence of p53 activity. However, it should be kept in mind that cancer is a lethal pathology, and therefore the carcinogenesis process has not been subject to evolution. Mechanisms of antitumor activity of p53 should therefore be considered in lights of normal processes that the p53 gene controls. Until recently, studies on p53 were mainly focused on its involvement with processes regulating cell divisions and cell death, partly because the main cause of cancer is often considered a result of distorted regulation in these processes. At the turn of the centuries, six major hallmarks of cancer cells had been described [286]. They include (1) self-sufficiency of growth signals; (2) insensitivity to anti-growth signals; (3) evading apoptosis; (4) limitless replicative potential; (5) sustained angiogenesis; (6) tissue invasion and metastasis. It becomes clear from previous sections that p53, a universal tumor suppressor, counteracts each of the above hallmarks of cancer. However, the list of cancer hallmarks should also be supplemented by the seventh not least important property, metabolic transformation, without which the cell cannot rapidly divide under conditions of limited resources provided by surrounding normal tissues.
Metabolic transformation of cancer cells to a significant extent is also associated with the loss of p53 activity. We will consider this in more detail below, but first we would like to describe the role of p53 in normal tissue metabolism.
p53 CONTROLS OPTIMAL BALANCE BETWEEN GLYCOLYSIS AND AEROBIC
RESPIRATION
Energy consumptions of cells differ substantially depending on the tissue affiliation, physiological condition, proliferation status, etc. In normal cells glucose serves as the main external source of energy that is later transformed to the energy of ATP through the processes of glycolysis and subsequent oxidative phosphorylation of acetyl-CoA. Glycolysis is the ancient anaerobic process in the cytoplasm in which one molecule of glucose gives rise to two pyruvates, two ATP molecules, and one NADH molecule. The next aerobic stage in mitochondria completes the oxidation of glucose, giving rise to approximately 30 molecules of ATP. Despite the high efficiency of aerobic respiration, it is a rather slow process, while glycolysis allows rapid ATP synthesis. In addition to its important energy-supplying function, the mitochondrial tricarboxylic acid cycle serves as the main source of metabolites used in anabolic processes. In the case of continuous expenditure of metabolites removed from the tricarboxylic acid cycle, the latter is stopped until the pools are somehow replenished [287]. Therefore, despite obviously high energetic efficiency of aerobic respiration, ATP production by glycolysis can be advantageous in situations when rapid release of energy is required, such as in the case of intensive contraction of muscle fibers [288] or when massive formation of cell structures (membranes, organelles) is required in rapidly proliferating cells [289]. This is why the supervision of the balance between glycolysis and the mitochondrial branch of metabolism needs to be strictly regulated [290].
p53 is able to coordinate processes of energy metabolism depending on the cell proliferative condition [228, 251], and this function of p53 can be carried out in the absence of stresses, under normal physiological conditions. Loss of p53 activity is associated with impaired aerobic respiration and increase in the cell’s dependence on glycolysis [291-293]. By modulating the activity of p53, glycolysis can be partial switched to the pentose-phosphate shunt pathway [34, 228, 294, 295].
p53 AND GLYCOLYSIS
Functions of p53 affect glucose metabolism at several levels. In some cell types p53 can inhibit glucose transport through the plasma membrane by repressing the transcription of GLUT1 and GLUT4 genes encoding glucose transporters [296]. In other cell types p53, on the contrary, stimulates glucose transport by enhancing transcription of the gene for hexokinase II [297], the enzyme converting glucose to glucose-6-phosphate and thus initiating the process of glycolysis. At first blush, such activity is not consistent with p53 function as tumor suppressor, as the increase in hexokinase II activity is characteristic of many cancer cells [298]. However, if one assumes that in the absence of strong stresses p53 plays the role of a survival factor, it is quite reasonable that the mild p53-dependent increase in enzyme activity can help the cell to escape from the cell cycle arrest due to the lack of energy resources [299] (see also below). p53 also regulates phosphoglycerate mutase (PGM), the enzyme that reversibly converts phosphoglycerate. The p53-dependent increase in PGM gene transcription notably increases glycolytic capacity [266]. The PGM gene contains a p53-responsive element responsible for transcription activation, at least in cardiomyocytes [267]. Since p53 can also repress the PGM gene at posttranscriptional stages [266], this makes the regulation even more complicated.
As mentioned in the section concerning antioxidant function of p53, the TIGAR gene encoding a p53-regulated homolog of the bis-phosphatase domain of 6-phosphofructo-2-kinase, is able to send some glycolysis metabolites along the pentose-phosphate pathway, which is required for the syntheses of NADPH and glutathione, as well as synthesis of ribose necessary for the synthesis of nucleotides [237]. It is interesting that p53 can also activate glucose-6-phosphate dehydrogenase (G6PD) [300], which catalyzes an important step in the pentose-phosphate pathway [301]. Inhibition of glycolysis and activation of the pentose-phosphate pathway probably contribute to cell survival, helping cells to recover from minor damages [228].
The muscle creatine kinase (MCK) gene was among the first identified p53 transcription targets [302]. A functional p53-responsive element was also found in the gene encoding a creatine kinase isoform expressed in brain [303]. The creatine kinase restored exhausted ATP resources by phosphorylation of ADP and consumption of phosphocreatine as the energy resource in tissues capable of sharp increase in ATP consumption, such as skeletal muscle, brain, or smooth muscle. Thus, restoration of ATP level in response to p53 helps to maintain intracellular homeostasis and contributes to cell survival.
STIMULATION OF MITOCHONDRIAL RESPIRATION
Inhibition of p53 results in noticeable impairment of mitochondrial biogenesis [291], lower oxygen consumption [250], stimulation of glycolysis, and increase in lactic acid secretion. The overall level of ATP production in p53-knockout cells does not decrease. However, while in p53-proficient mouse fibroblasts the proportion of ATP produced by glycolysis to that produced in mitochondria is 1 : 3, in p53-knockout fibroblasts it increases to 3 : 1 [250], which points to the role of p53 in production of mitochondrial ATP. The SCO2 gene is responsible for this role, and it is regulated by basal p53 levels [250]. The SCO2 gene encodes a copper chaperon that participates in the assembly of mitochondrial complex IV (cytochrome c oxidase) [304]. The decreased aerobic respiration in p53-deficient cells can be restored by introduction of genetic constructs expressing SCO2 to the physiological level. The decrease in ATP production by mitochondria and enhancement of glycolysis are also observed upon deletion of one allele of the SCO2 gene. Collectively, the results confirm the role of p53 in controlling the SCO2 gene even in the absence of stresses, although they do not exclude potential involvement of other p53-regulated genes in mitochondrial energetics. One such gene could be the p53-regulated AIF gene, which is involved in the assembly of complex I of the mitochondrial ETC [255], the other could be the glutaminase 2 gene GLS2 that is also regulated by p53 [238]. Glutaminase stimulates mitochondrial respiration and ATP synthesis at the expense of increased production of glutamate and α-ketoglutarate.
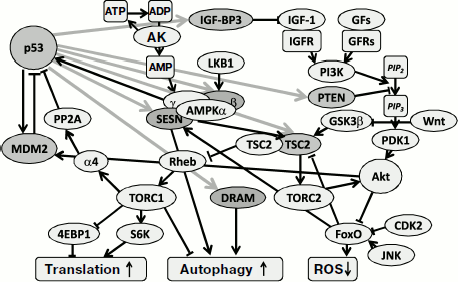
Since the deficiency of mitochondrial function in p53-negative cells is easily compensated by enhanced glycolysis, p53 can coordinate balancing of ATP production. Besides known feedback mechanisms, a wide spectrum of p53-regulated genes is involved in maintaining ATP levels. However, p53-dependent processes in different tissues can vary, which should be considered when analyzing sometimes-contradictory results obtained in different cell models. The role of p53 protein in regulation of glycolysis and aerobic respiration is shown in Fig. 2.
Regulation of p53 functions that maintain homeostasis of metabolic processes proceeds with substantially lower amplitude in its level and activity compared to that during the induction of p53 by severe stresses or lethal damage. Experiments with cell-free systems indirectly suggest that energy status of the cell may affect transcription activity of p53 though its direct interaction with metabolites. In particular, an ADP molecule is able to bind the tetrameric form of p53, thus contributing to its interaction with DNA elements [305], while binding of ATP (or GTP) and NAD+ act in the opposite direction [305, 306]. However, there is still no data concerning the possible regulation of p53 by metabolites in undamaged cells, and this problem clearly requires elucidation.Fig. 2. Effect of p53-induced genes on glycolysis and mitochondrial respiration. Physiological levels of p53 retard glycolysis and activate the pentose-phosphate pathway and the aerobic phase of respiration. In some tissues p53 also contributes to ATP regeneration from phosphocreatine resources.
p53 AND THE WARBURG EFFECT
During malignant transformation cells undergo a number of alterations that are required for their survival and autonomous proliferation within an organism, and these contribute to tumorigenesis. One of most important hallmarks of cancer cells is specific transformation of their metabolism that is characterized by switching of metabolic processes to prevalence of glycolysis as the main source of energy. This property of tumor cells was described in the 1920s by Otto Warburg, who noted the tendency of tumors for “aerobic fermentation”, i.e. to prevalence of the glycolytic pathway even under sufficient supply of oxygen [307-309]. Warburg believed that the process of carcinogenesis is launched when, for some reason, the cells acquire irreversible injury to respiration, which is followed by selection of “less differentiated and primitive cells that succeed in replacing the irreversibly lost respiration by fermentation energy” and which then begin to “grow wildly” [307].
Warburg was the first who suggested that damage to mitochondria and the compensation of oxidative phosphorylation by intensive glycolysis is the basis of cancer. This is indeed characteristic for most tumors, and the property is widely used in the method of cancer diagnosis (positron-emission tomography, PET) that reveals regions of intensive glucose fermentation. The same property is used in the recently suggested very simple method of cancer therapy using dichloroacetic acid (DCA), which inhibits glycolysis by blocking the conversion of pyruvate to lactate [310, 311]. Since regulation of mitochondrial respiration is broken in cancer cells, they cannot live without glycolysis and are forced to die. Although this method has not reached the stage of extended clinical trials (due to the low price of DCA and lack of interest from pharmaceutical industry) [310, 311], work on the development of new generations of combined antitumor drugs that involve DCA as an active group is now under way [312].
The “Warburg effect” is characteristic of cancer cells, but it can also be observed during intensive proliferation in normal cells, such as lymphocytes and hemopoietic and embryonic cells [287, 313]. Although the aerobic pathway is more energy efficient, the glycolytic conversion of glucose to lactic acid is very rapid process [314], and the high rate of energy recovery is particularly important for intensively proliferating cells. Besides, proliferating cells continuously consume metabolites (such as NADPH, citrate, and glycerol for lipid synthesis, ribose for nucleotide synthesis, etc.). Some of these materials are removed from the Krebs cycle, others are withdrawn from the products of glycolysis and the pentose-phosphate pathway. This means that significant metabolic efforts begin to be directed to ATP consumption rather than for its production. The lack of ATP is easily covered by rapid breakdown of glucose, but as the rate of glycolysis exceeds the capacity of further oxidation of pyruvate, its excesses is converted into secreted lactate, which can be used by other cells.
Recent studies [228] show that p53 is involved in optimization of metabolism in normal proliferating cells by redirecting part of the glucose catalysis products to the pentose-phosphate pathway. This pathway is important for the synthesis of metabolites required for biosynthesis of nucleic acids and for stimulating the reduction of NAD+ to NADH. The latter is used for in the conversion of the excess of pyruvate to lactate and for the synthesis of glutathione, thus contributing to the neutralization of ROS. In response to lowering ATP level, p53 induces the expression of the SCO2 gene and thus stimulates oxidative phosphorylation that compensates for the lack of ATP. At the same time, accumulating NAD+ allosterically activates p53, which can stimulate its reduction to NADPH through the p53-dependent induction of the TIGAR gene.
The Warburg effect observed in cancer cells cannot be explained only as a normal process that regulates metabolism in rapidly proliferating cells. Mutations that introduce irreversible changes to different signaling pathways inevitably induce breakdown of the whole regulatory network. The malfunctioning processes emit multiple signals that result in p53 response, restriction of damaged cell proliferation or apoptosis. A mutation could result in the loss of p53 functions, thus allowing uncontrolled proliferation and lifting the ban for competition between altered cells for oxygen and nutrients and for selection of more rapidly growing cells. As tumor cells preferably use glycolysis, they intensively synthesize and secrete lactic acid. An acidic medium is especially harmful for normal cells, whereas cancer cells withstand it well [315]. Taking into account that oxygen supply is often limited in tumors, the acidic environment produced by the cells relying on glycolysis creates additional selective pressure in favor of cells depending on glycolysis.
Mutation of p53 results in deficiency of mitochondrial respiration due to decreased expression of SCO2 and, possibly, of other p53-regulated components of the respiratory chain [253, 281, 293, 303]. The absence of p53 activity increases expression of glucose transporters GLUT1 and GLUT4, phosphoglycerate mutase, and hexokinase, and lowers expression of TIGAR and glutaminase 2. All these changes stimulate glycolysis. The condition of hypoxia that is characteristic of developing tumor is a p53 inducer that not only restrains proliferation, but also inhibits formation of blood vessels [316]. However, when p53-dependent mechanisms are broken, the ban for neoangiogenesis is relieved. Besides, hypoxia induces transcription factor HIF1α that additionally stimulates expression of many enzymes of glycolysis and stimulates angiogenesis. However, despite numerous factors favoring aerobic glycolysis, the contribution of p53 deficiency to this process is the most important because the loss of p53 creates conditions that favor the selection of even more malignant cell variants.
Presently available data on aerobic glycolysis indicate that the Warburg hypothesis linking cancerogenesis with deficiency in mitochondrial physiology and first suggested almost a century ago has now received convincing confirmation.
REGULATION OF BALANCE BETWEEN ANABOLIC AND CATABOLIC
PROCESSES
Until recently, when considering the cell division and cell cycle control processes little attention has been given to their energy support mechanisms. Division of a cell requires its doubling in mass, meaning expenditure of significant resources for building new organelles, membranes, proteins, nucleic acids, etc. The growth in mass and the division process need to be tightly coordinated. By entering a division, the cell should have the resources for the formation of two daughter cells. Likewise, as a non-dividing cell could encounters different conditions, such as variation in functional tasks, changing accessibility of nutrients, growth factors, and hormones, it faces a choice – either to continue its growth in mass, or to use a part of its own mass to compensate for the deficient external supply of energy and building material.
There is a system in cells that integrate signals from growth factors, available nutrients, energy status (AMP to ATP ratio), and accordingly balances anabolic and catabolic processes, The main integrator of incoming signals is the complex formed by products of the TSC1 and TSC2 genes (hamartin and tuberin, respectively). The scheme of processes regulated by products of these genes is shown in Fig. 3. Both genes are tumor suppressors [317, 318] associated with a rare hereditary disease, tuberose sclerosis (TSC), which is characterized by multiple systemic benign tumors (tubers or hamartomas) affecting the brain, internal organs, and skin [319]. These two unrelated proteins of 140 and 200 kDa do not have pronounced homology with other proteins with the exception of the TSC2 C-terminal domain, which is homologous to GTPase-activating protein Rap1GAP. The two proteins form a heterodimeric complex in which TSC1 stabilizes TSC2 against ubiquitination and destruction in proteasomes [320, 321]. The functions of the proteins are interdependent, and therefore inhibition of each of them separately results in the same consequences [322]. The TSC1–TSC2 complex is regulated by phosphorylation by several protein kinases at not less than five sites in TSC1 and eleven sites in TSC2 [323], and while modification by certain protein kinases (AMPK, GSK3β, and REDD) induces activation, other protein kinases (Akt/PKB, CDK1, IKKβ, ERK, RSK1) inhibit TSC1–TSC2 function [323].
TheTSC1–TSC2 complex regulates mTOR (mammalian target of rapamycin) kinase. The mTOR kinase is the most important stimulator of CAP-dependent translation and an autophagy inhibitor [324-326]. Its function stimulates anabolic processes and assists in growth of cell mass. The mTOR protein is a catalytic subunit of two Ser/Thr kinase complexes TORC1 and TORC2 [326]. Only TORC1 is directly involved in regulation of protein synthesis and cell growth [324], while TORC2 is involved in regulation of cell motility and cytoskeleton organization through its control over the PKCα and SGK1 phosphorylation [327, 328] and also influences cell survival through activating phosphorylation of Akt kinase [324, 328]. In addition to mTOR, the TORC1 complex includes as subunits proteins PRAS40, mLST8, and RAPTOR; besides, in the presence of rapamycin the FKBP12/rapamycin complex binds to mTOR and inhibits the TORC1 activity [329]. The TORC2 complex consists of subunits mTOR, mLST8, Sin1, and RICTOR and is relatively resistant to rapamycin [329].Fig. 3. Involvement of p53 in control of anabolic and catabolic processes. p53 regulates some genes that inhibit anabolic processes through downregulation of mTOR kinase. p53-regulated genes also modulate processes controlled by the Akt gene product. Finally, p53 controls autophagy and contributes to mobilization of internal energy and plastic resources to repair non-dividing cells.
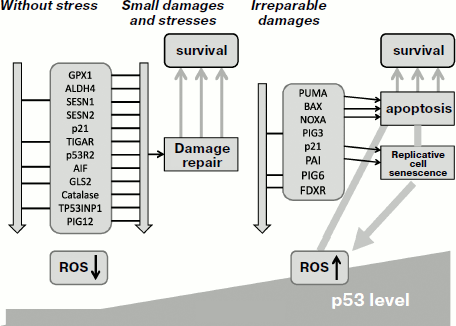
The dissociation of TSC1 and TSC2 and binding of TSC2 to proteins of the 14-3-3 family occur after receiving stimulating signals from growth factors [330]. This results in inhibition of TSC1–TSC2 blocking activity towards TORC1 and following activation of TORC1 protein kinase activity, which phosphorylates its main targets, a p70 protein, the kinase of ribosomal S6 protein (S6K1), and protein binding translation initiation factor eIF4E (4EBP1). The phosphorylation activates S6K1 kinase and inhibits 4EBP1, which stimulates CAP-dependent translation. Phosphorylation of these proteins contributes to assembly of ribosomes and enhances protein translation [329]. Simultaneously TORC1 inhibits autophagy that is launched upon shortage of nutrient substances and exhaustion of the cell energy resources. The process is mediated by phosphorylation of two proteins involved in autophagy initiation, a homolog of Atg1 protein, a product of the UNC-51-like kinase gene ULK1, and of Atg13 protein homolog [331]. Thus, TORC1 activity enhances anabolic processes through activation of protein biosynthesis and inhibits catabolic activity though stopping the process of “self-eating”, the autophagy. The TORC1 activity results in cell mass increase.
If the TSC1–TSC2 complex is in activated state, it blocks the TORC1 kinase activity. Through its GAP domain (that activates GTPases), TSC2 inhibits the activity of small GTPase Rheb that stops the activation of TORC1 [326, 332]. Unlike TORC1, activity of TORC2 is not inhibited but rather is activated by the TSC1–TSC2 complex. Although the activation mechanism is still poorly studied, it has been found that Rheb is not involved in regulation of TORC2. Apparently, TORC2 activation is due to direct binding of the TSC1–TSC2 complex to TORC2 [333]. Activated TORC2 is involved in feedback resulting in inhibition of TSC1–TSC2 complex: TORC2 is a powerful activator of Akt kinase that in turn phosphorylates and inhibits TSC1–TSC2. The mechanism relieves TORC1 repression and appropriately attenuates the TORC2 activity.
Due to switching of mTOR activity there is either activation of anabolic processes and cell mass increase or, on the contrary, inhibition of protein and lipid biosynthesis and activation of autophagy, which provides an important mechanism for adaptation to changing availability of nutrients.
CONTROL OF CELL ENERGY STATUS
As mentioned above, the TSC1–TSC2 complex integrates information on availability of nutrients, signals from external growth factors, and signals monitoring cell energy status, and through the activity of TORC1 regulates the balance of anabolic and catabolic processes. The TSC1–TSC2 complex obtains information from two PK groups acting in opposite directions. Upon exhaustion of ATP resources, the AMP-dependent kinase (AMPK) is activated, and inhibits TORC1 through activation of TSC1–TSC2, whereas in response to growth factors and hormones that signal about sufficient nutrients, Akt or another kinase inhibits the TSC1–TSC2 complex and activates TORC1.
p53 is providing the higher order control by ensuring appropriate order of processes and modulating activity of certain components of the system for optimized function. Specifically, the TSC2 gene is a direct transcription target of p53 [334, 335], which tunes up mTOR by making it more sensitive to factors that inhibit its activity through the TSC1–TSC2 complex. In other words, it contributes to inhibition of mTOR activity, although like in the regulation of many other p53 targets the effect on TSC2 could be tissue-specific [335].
Limited supply of glucose, the main source of energy for the cell, results in depletion of ATP resources. Adenylate kinase quickly converts two ADP molecules to ATP and AMP, thus partially restoring the ATP pool, but increasing AMP level. AMPK is responsible for maintenance of energy balance in the cells and in the whole organism [336]. AMPK carries out its function by phosphorylation of TSC2 and some substrates such as HMG-Co reductase and acetyl-CoA carboxylase (ACC). The latter inhibits cholesterol and fatty acids biosyntheses, protein biosynthesis through the TSC2 phosphorylation, glucose synthesis in liver, and reduces insulin synthesis. AMPK simultaneously stimulates glucose consumption, fatty acid oxidation, and mitochondrial respiration [337].
AMPK consists of catalytic (α) and two regulatory (β and γ) subunits [338]. Weak activation of AMPK occurs upon AMP binding to the γ subunit. However, more powerful activation occurs upon α subunit phosphorylation at Thr172 [339] by Ca2+/calmodulin-sensitive kinase and/or by constitutively active kinase LKB1. It is interesting that the LKB1 kinase gene is a tumor suppressor inhibited in a number of human malignancies [340]. Due to continuous stimulation by LKB1, AMPK is initially active, but its activity is modulated by protein phosphatase 2C (PP2C) that dephosphorylates Thr172. AMP binding modifies AMPK conformation in such a way that it becomes inaccessible for the PP2C phosphatase, which results in increase in its activity [339, 341].
AMPK phosphorylates TSC2 at Ser1345, which contributes to its activation and additionally makes TSC2 accessible for subsequent activating phosphorylation at Ser1345 and Ser1337 by glycogen synthase kinase GSK3β [342], which additionally activates the complex and deeply inhibits TORC1 function. It is interesting that at this stage the pathways of AMPK Wnt signaling intersect. After binding to the Frizzled family receptors, Wnt protein stimulates proliferation. The Wnt signaling pathway inhibits GSK3β activity and thus eliminates the GSK3β super-activating effect on TSC2. As a result, the required for cell proliferation TORC1 is activated [329].
The functions of p53 and AMPK are closely interconnected. The AMPK β subunit serves as an immediate transcription target for p53 [334]. The subunit is a central component of AMPK that serves as a scaffold for further binding of α and γ subunits, and it is responsible for intracellular localization and activity of AMPK [343]. In turn, p53 is controlled by AMPK and LKB1. Kinase LKB1 forms a complex with p53 and directly or through an intermediate phosphorylates it at Ser15 and Ser392 [344]. LKB1 in the complex with p53 binds to regulatory elements within CDKN1 (p21) and other p53-regulated genes, and due to phosphorylation of chromatin and components of the transcriptional machinery it is involved in transcriptional activation of these genes [344]. After activation, AMPK can phosphorylate p53 and additionally it can activate p53 promoter [345], thereby additionally enhancing the p53 effects.
Similar to arresting cell divisions in response to DNA damage, regulation of cell proliferation also depends on availability of nutrients. In low glucose medium normal fibroblasts enter transient p53-dependent arrest at the G1/S boundary, which is also AMPK-dependent and proceeds even with fully active mTOR. The cell cycle arrest represents a checkpoint that restricts further cell divisions under depleted energy resources. In p53 knockout mouse cells there is no metabolic checkpoint. Moreover, although normal p53-proficient fibroblasts when placed in a low-glucose medium are relatively resistant to further complete removal of glucose, p53-deficient fibroblasts are not protected against such treatment [346]. Thus, p53 assists in the adaptation of cells to decreased availability of glucose and increases their survival under conditions of starvation.
The mechanism of metabolic checkpoint activation includes direct phosphorylation of p53 at Ser15 catalyzed by AMPK. The phosphorylation increases transcription activity leading to cell cycle arrest by p53-regulated genes, such as CDKN1-p21 [346]. The mechanism of lowering p53 activity after the end of oxygen starvation involves the mTOR-mediated phosphorylation of protein phosphatase PP2A that dephosphorylates Ser15 on p53 [165]. However, the p53- and AMPK-dependent reactions to decreasing glucose level vary greatly in different cell types. Normal thymocytes and a human osteosarcoma cell line are characterized by induction of apoptosis following phosphorylation of p53 at Ser46 [345]. Apoptosis is also characteristic to oncogene-transformed cells and to cells with checkpoints defective due to inactivation of pRB, which highlights the link between the ability to induce cell cycle arrest and the p53-stimulated cell survival [334]. It should be also noted that survival of cells that can stop the cell cycle can be additionally enhanced by stimulation of p53-dependent autophagy [347] during which the nutrients for viability are obtained by partial digestion of the cytoplasm [348].
As already mentioned, AMPK phosphorylates TSC2 and through the inhibition of TORC1 postpones anabolic processes and stimulates catabolic autophagy. However, TSC2 activation is also controlled by p53. Under hypoxia, p53 and HIF1α induce the REDD1 gene whose product activates TSC2 due to its displacement from inactive complex with 14-3-3 protein [349]. It was also found that p53-dependent sestrins (SESN1 and SESN2), in addition to their antioxidant activity associated with peroxiredoxin regeneration [241], are involved in inhibition of TORC1. Sestrins are able to bind the AMPK α subunit and induce its activation even without participation of protein kinase LKB1 and elevated AMP [350]. Sestrin binding also inhibits dephosphorylation of α subunit of AMPK at Thr172 catalyzed by protein phosphatase PP2C, which maintains high activity of AMPK. Besides, sestrins directly bind to TSC2 and due to their interaction with AMPK contribute to preferable phosphorylation of TSC2. Thus, sestrins contribute to highly efficient inhibition of TORC1 activity, which results in delay of anabolic processes and stimulation of autophagy [351]. Since sestrins increase AMPK activity, by this mechanism they contribute to the AMPK-dependent activation of p53, which results in an additional increase in expression of p53-dependent sestrins. Another positive feedback between AMPK and sestrins forms due to the ability of AMPK to activate transcription factors of the FOXO family [352, 353]. FOXO proteins, in turn, activate sestrins independently of p53 [354, 355], which enhances TSC2 phosphorylation and even more severe inhibition of TORC1.
CONTROL OF BALANCE BETWEEN PROLIFERATIVE SIGNALS AND AVAILABILITY
OF NUTRIENTS
Opposite effects resulting in TORC1 activation are mediated though TSC1–TSC2 activation by Akt (PKB) protein kinase in response to signals from growth factors and hormones. The signals emerge in response to availability of glucose (through the IGF-1/insulin pathway) or as result of stimulation with growth factors or cytokines. In the case of conflicting signals (TSC1/2 activation by AMPK, and inhibition by Akt) the effect of AMPK remains dominant. Thus, in the case of depleted energy the cell does not react to stimulating signals from growth factors.
Signaling from receptor of IGF-1 of insulin stimulates cell growth in response to glucose availability. p53 can intervene in this process even before the binding of IGF-1 to its receptor, as it activates at transcription level the IGF-BP3 gene whose product serves as the main carrier of IGF-1 in circulation that defines its bioavailability in tissues. The induction of IGF-BP3 results in attenuation of the signal through the insulin receptor not only for the given cell, but also in the surrounding. Thus, p53 interferes with the conductivity of the insulin pathway, which limits glucose consumption and thereby assists in tumor suppression.
The binding of insulin or IGF-1 to receptor activates phosphoinositidin-3-kinase (PI3K) that converts PIP2 to PIP3. The increased local concentration of PIP3 on the plasma membrane stimulates its binding to membrane proteins that contain plextrin-homologous domains (PHD), in particular the Akt proteins (or protein kinase B, PKB) and PDK1 (or PDPK1, phosphoinositide-dependent kinase-1). Due to close localization on the membrane, PDK1 phosphorylates Akt at Thr308, which stimulates the PK activity of Akt. However, complete activity of Akt kinase emerges only after additional phosphorylation at Ser473 catalyzed by TORC2 [324, 356, 357].
The activation of Akt is negatively regulated by the tumor suppressor product PTEN. PTEN is a lipid phosphatase that counteracts PI3K activity by conversion of PIP3 to PIP2, thereby lowering Akt activity. In cells that have lost PTEN activity this signal pathway is constitutively active, thus contributing to tumor development [358]. PTEN expression is controlled by p53 [359] due to which p53 inhibits Akt activity and provides additional antitumor activity. Protein phosphatase PP2A, dephosphorylating Akt at Thr308, also lowers Akt activity [360].
Akt PK modulates numerous processes by phosphorylation of various proteins [324, 361]. In particular, Akt stimulates cell proliferation by inhibition of p27Kipl and through activation of c-myc and cyclin D1. Akt can exhibit antiapoptotic effect through inhibition of Bad and transcription factors FoxO as well as through activation of NFkB. By its action against these and other targets, Akt stimulates glycolysis even under excess of oxygen, enhances the expression on plasma membrane of glucose transporters GLUT1 and GLUT4 [362, 363], and stimulates mitochondrion-bound hexokinase [364] that activates glycolysis and the pentose-phosphate pathway. Akt activates ATP-citrate lyase thereby increasing the de novo biosynthesis of fatty acids [365] required for building membranes in rapidly growing cells. Akt phosphorylates Mdm2 and increases its activity forcing it to the nucleus, which results in inhibition of p53 [366, 367]. Thus, Akt attenuates p53 and relieves its inhibitory effect on proliferation.
Inhibition of transcription factors FoxO significantly contributes to effects associated with Akt activation. Akt phosphorylates FoxO at three sites, one of which activates a site for its binding to proteins 14-3-3 [368, 369], which results in translocation of FoxO3a and FoxO1 from the nucleus into the cytoplasm. Inactivation of FOXO results in significant reorganization of metabolism due to inhibition of activity of genes regulated by these transcription factors [370]. At the same time, transcription factors FoxO are capable of directly blocking protein phosphatase PP2A [371] and thus stimulate Akt by counteracting its dephosphorylation at Thr308.
A significant number of biological effects of Akt are mediated by activation of TORC1 through the inhibition of TSC1–TSC2 complex. Akt is able to phosphorylate TSC2 at several sites [323], and specifically the phosphorylation at Ser939 and Ser981 create sites for binding to 14-3-3 proteins [330]. Although the detailed mechanism of TSC complex inactivation upon Akt phosphorylation is unclear [323], it is known that the complex can no longer act as GAP on small GTPase Rheb. This results in TORC1 activation, stimulation of protein synthesis, inhibition of autophagy, and cell mass increase. At the same time, TORC1 activation launches several feedbacks that lower activities of TORC1 and Akt. The activation of S6K results in not only phosphorylation of ribosomal protein S6 and stimulation of translation, but also phosphorylation of insulin receptor substrate (IRS1) protein, which attenuates signaling from insulin receptor to PI3K and lowers Akt activity. Besides, the TSC1–TSC2 complex reciprocally regulates TORC1 and TORC2, therefore simultaneously with TORC1 activation Akt inhibits TORC2, which correspondingly decreases activating phosphorylation of Akt itself at Ser473. In this respect there may be interesting implications related to the p53-dependent sestrins. Sestrins activate the TSC1–TSC2 complex, inhibit TORC1, and stimulate TORC2, thus activating Akt and enhancing expression of glucose transporters, which could lead to lowering glucose level in the blood, and thereby exhibit antidiabetic effect.
ROLE OF p53 IN CONTROL OF AUTOPHAGY
Prolonged restriction of external supply of nutrients launches a self-eating process or macroautophagy (autophagy), which plays an important role for cell survival. A cell sacrifices a part of its mass to get access to additional sources of energy and elements required for building new structures. Autophagy is associated with formation of autophagosomes, double membrane vesicles that engulf a part of the cytoplasm containing organelles, such as fragments of endoplasmic reticulum, endosomes, and mitochondria. Autophagosomes then fuse to a lysosome to form autolysosomes in which the captured structures are digested, thus supplying the cell with nutrients required for protein, carbohydrate, and lipid syntheses [348]. Besides, autophagy is important for removal from the cell worn-out or damaged structures and components, such as oxidized protein aggregates, defective organelles, in particular of damaged mitochondria whose presence is harmful for the cell and represents a danger for its survival. Defective mitochondria release certain diffusible compounds and ROS that serve as signals for initiation of autophagy [372], which results in elimination of undesirable sources of ROS that create mutagenic background and accelerate the aging process [373, 374]. Autophagy plays a particularly important role in long-lived non-dividing cells in which it allows repair of the cells during their extended life. However, as many components of the autophagy pathway functionally overlap with those required for apoptotic death, the two processes allow the cell to smartly balance between life and death for the sake of the organism’s survival [375].
Autophagy is a tightly regulated evolutionarily conserved process [376] that has recently become an object of active studies. Details of mechanisms and stages of autophagy can be obtained in a number of recent excellent reviews [377-379]. The most important negative regulator of autophagy is the mTOR pathway that stops autophagy by phosphorylation of at least two most important factors, protein homologs of the yeast Atg1 and Atg13 genes [331].
p53 can also control autophagy, but the outcome of the regulation is still subject to considerable controversy [380-383]. First, p53 can act in favor of stimulation of autophagy by inhibition of TORC1 activity [335] due to interaction of AMPK and p53-dependent sestrins with TSC1–TSC2 complex [351]. When energy level is low and ATP is depleted, AMPK phosphorylates and activates p53, which not only arrests cell divisions but induces SESN1 and SESN2 that force AMPK to phosphorylate and activate TSC2 [350]. p53 can also stimulate autophagy by inducing another p53-regulated gene DRAM, encoding a lysosomal protein [384]. The forced expression of DRAM (even in p53-deficient cells) stimulates autophagy, while autophagy can be inhibited by RNA interference against the DRAM gene. The DRAM function contributes to cell survival because ectopic upregulation of DRAM significantly increases clonogenic index [385, 386]. Simultaneously the DRAM gene product is involved in p53-dependent apoptosis [384], which confirms the interrelations of the processes. Another member of the p53 gene family, TA-p73, is also able to enhance autophagy but does not require the involvement of DRAM [380, 387]. p53 and p73 are related by their ability to induce a number of proapoptotic proteins of the Bcl family (Bax, Bad, Bnip3, Puma), which indirectly contribute to autophagy induction. These proapoptotic proteins are known to counteract the activity of antiapoptotic proteins Bcl2 and BclXL. However, along with the antiapoptotic function due to formation of protein complex with Bax, the Bcl2 and BclXL proteins can also inhibit initiation of autophagy through the formation of a protein complex with the most important autophagy regulator Beclin 1 [388].
Autophagy can be also induced by ARF protein that is a powerful inducer of p53 in response to oncogene activation [389]. However, ARF is also able to induce autophagy in the absence of p53 [389], especially its short isoform smARF, which is localized in mitochondria [390].
It is remarkable that p53 can also inhibit autophagy. p53 functions both in the nucleus and in the cytoplasm, where it is not only involved in induction of apoptosis by interaction with the Bcl family proteins, but also a powerful inhibitor of autophagy [382]. Cytoplasmic p53 is able to inhibit autophagy even in enucleated cells, and this property is remarkably preserved in p53 mutants [391]. Perhaps the compromised autophagy, characteristic of tumor cells, may be partially due to mutant p53 forms present in the cytoplasm. As nuclear p53 stimulates and cytoplasmic p53 inhibits autophagy, the balance between the two forms contributes to fine regulation of the process.
In cells that have deleted p53 gene, autophagy is substantially increased [380, 392], but this is not due to the absence of cytoplasmic p53. Enhanced autophagy is observed in p53-knockout mouse fibroblasts [382] as well as in the nematode C. elegans strain with deletion of the cep-1 gene, a homolog of p53 [392]. Cells fully devoid of p53 activity have an increased level of ROS, which by itself can enhance autophagy. TIGAR, one of the p53-induced genes, is able to inhibit autophagy due to its ability to decrease intracellular ROS [249]. In addition, p53-knockout cells have defects in some mitochondrial processes. p53 is required for biogenesis of mitochondria [215, 291], for repair of mitochondrial DNA [214], as well as for normal function of the mitochondrial ETC. The p53-regulated gene SCO2 is involved in the assembly of complex IV, and its absence impairs mitochondrial respiration [250, 251]. The p53 regulated AIF gene [253] encodes a mitochondrial protein with NADPH-oxidase activity, which participates in the assembly and function of complex I [255, 393]. As autophagy can be induced by signals from defective mitochondria, it is quite possible that partially defective mitochondria of p53-deficient cells also emit certain signals that stimulate autophagy.
Through its activity as a positive regulator of autophagy, p53 may contribute to cell survival under conditions of insufficient nutrient supply. Together with AMPK, p53 assists in the organism’s adaptation to starvation by mobilizing its internal resources. Certainly, this activity of p53 also aims better protection of the genome, as without the adaptation to starvation organisms would not be able to pass their hereditary information to progeny during famines.
p53 AND AGING OF THE ORGANISM
Functions of p53 that aim for the organism’s adaptation emerge even before birth: p53 regulates transcription of the LIF gene [38, 39], which is required for implantation of the embryo in the womb [394]. Later p53 is involved in embryonic development by preventing unscheduled reprogramming of stem cells. Only transient inactivation of p53 could allow reprogramming of induced pluripotent stem cells (iPS) by exogenous expression of certain transcription factors, an approach to regenerative medicine [395-397]. During the whole life, p53 protects genome integrity and contributes to repair and preservation of optimal homeostasis in each separate cell and in the whole organism.
The role of p53 in the aging process is complicated and controversial. Without functional p53 the lifetime is significantly decreased due to early development of malignancies. Mice with homozygous deletion of the p53 gene die mainly by the age of 9 months. The lifetime of patients with Li-Fraumeni syndrome (carrying hereditary defects in the p53 gene) is substantially reduced. However, accelerated senescence is observed in a mouse model expressing permanently activated p53, although they display enhanced antitumor protection [398]. These data could suggest that the organism pays with shorter life for better anticancer protection. However, lifetime is not reduced in mice with an additional copy of the normal p53 gene [399] as well as in mice with lower expression of Mdm2 [400], although the mice are more resistant to malignancy. On the other hand, two alleles containing either arginine or proline in position 72 of p53 differ in their ability to induce apoptosis. The Arg72 protein, corresponding to the evolutionarily younger allele, is more efficient in apoptosis induction; such protein is more actively transported into mitochondria where it contributes to the mitochondrial pore opening. Correspondingly, the Arg72 allele provides better protection against cancer [401, 402]. People with the more ancient Pro72 allele are diagnosed with cancer at a younger age, although they live longer after the diagnosis. They are more resistant to stresses and demonstrate somewhat decreased rate of organism aging [403, 404].
Studying functions of p53 under physiologically normal conditions allows clearer understanding of its controversial role in aging. In the absence of strong stresses, p53 works in “background” or “maintenance” mode to ensure optimal balancing of metabolism, efficient repair of the genome and other structures, and better antioxidant defense. Therefore, the constitutive function of p53 assists in slowing the aging process.
Aging is manifested by exhausted regenerative potential of tissues, accumulation of molecular damage, and functional decline. ROS that actively damage DNA, proteins, and lipids play an important role in acceleration of aging. Aging cells display signs of senescence, they produce increased amounts of ROS, which results in further molecular lesions and exhaustion of replicative potential. The most important question is why do ROS begin to accumulate in an aging organism? Apparently, this is due to a certain weakness of the systems that are intended to timely remove ROS and provide repair of molecular damage in the cells and intracellular matrix. Autophagy is one of the systems intended to rejuvenate the organism.
Autophagosomes preferentially form around damaged mitochondria that release increased amounts of ROS. Autophagy is also induced by nutrient limitation, and the process removes the most damaged and worn-out portions of the cytoplasm. This is perhaps one of the mechanisms through which calorie restriction prolongs life [405, 406]. The “self-eating” process assists in rejuvenating of the cell and of the whole organism. However, the autophagy process attenuates with age [407-409], which results in accumulation of “molecular garbage” in cells, which is characteristic of aging. According to emerging concepts, the major mechanisms that drive aging lie in weakening of the rejuvenating cell repair mechanisms, rather than by ultimate accumulation of ROS and the products of molecular damage.
The main regulator of autophagy is the TORC1 complex that slows autophagy under sufficient supply of nutrients and ATP. The maintenance of high level of protein and lipid biosynthesis is necessary for the developing organism, but it becomes unneeded and harmful with age. However, TORC1 activity does not slow with age but instead increases [410, 411]. Many harmful conditions and habits, such as inappropriately high food consumption, obesity, development of insulin resistance due to continuous glucose overload, contribute to TORC1 acceleration with age. It is noteworthy that once, for any reason, the intracellular ROS level is increased, it further stimulates mechanisms that assist upregulation of TORC1, leading to weakened cell repair and accelerated aging. While in youth TORC1 promotes organism growth and development, after reaching puberty it starts working as an engine that drives the aging process [410]. Recent trials of TORC1 inhibitor rapamycin have demonstrated its significant life-extending effect in a genetically heterogeneous mouse population even if the application of rapamycin is started at an old age [412]. Notable extension of life was also demonstrated in trials in mice fed with antidiabetic drug metformin, which increases cytoplasmic AMP level and induces AMPK [413-416] thereby inhibiting TORC1, as well as in mice with knocked out gene for ribosomal protein S6 kinase, the TORC1 substrate [417]. However, clinical trials of different antioxidants did show any consistent life-extending effects, or lower incidents of old-age associated disease, and in some cases they even demonstrated somewhat increased morbidity and mortality [418]. Thus, TORC1 hyperactivity seems to be the most likely cause and driving force of the aging process and a promising target for therapy.
p53 inhibits TORC1 at several levels, which results in activation of autophagy. Thus, p53 contributes to the process of non-dividing cell repair, and therefore its “background” activity beyond severe stresses aims at inhibition of aging and extension of homeostasis both within individual cells and in the whole organism.
A different scenario emerges upon stresses when p53 function preserves the organism by restricting damaged cells. This process is inevitably associated with certain release of ROS, which produce not only direct damage to DNA and structural components of the cell, but also stimulates TORC1 [419, 420], thus contributing to accelerated aging of tissues and of the whole organism. The major practical conclusion from the controversial role of p53 in the aging process could be avoidance of any severe effects, stresses, and intoxications that are able to induce p53 stress response, which apparently contribute to premature aging.
THERAPEUTIC EFFECT ON p53-DEPENDENT MECHANISMS
In previous sections we have considered the main activities of the tumor suppressor p53. Clearly, there are two sides to p53, one is more gentle, which helps the cells to maintain stability of the genome under physiological conditions, and the other is more radical when elimination of severely damaged cells following different emergences is most appropriate to prevent genome alterations. Both sides of p53 aim for a common goal – to prevent origination and spreading of diseased cells. The constitutive activities of p53 are most important when considering measures for disease prevention, whereas the ability of p53 to kill abnormal cells can be used in cancer therapy.
At present there are several approaches under consideration that might use the suppressor properties of p53 in cancer therapy. Several recent reviews touch this subject [134, 135, 421-423]. The approaches include introduction into tumor cells of constructs expressing wild-type p53, the use of small compounds for pharmacological reactivation of broken p53-dependent mechanisms, the use of recombinant viruses that can replicate and induce death of exclusively tumor cells devoid of p53 activity, etc.
Hereditary deficiency of p53 activity results in increased probability of cancer. The Li-Fraumeni syndrome is associated with mutations in a single allele of the p53 gene and is characterized by independent development at relatively young age of several types of tumors in the same individual [28]. A polymorphism in p53 protein (Pro72 or Arg72) represents another very common clinically significant example. The Arg72 allele is associated with better ability to induce apoptosis and better protection against cancer [424, 425]. A significant number of people have weakened p53 function due to increased expression of Mdm2 protein, which originates from polymorphous replacement of a single nucleotide in the transcription control region of the MDM2 gene (SNP309). Individuals that have homozygous G allele of SNP309 on average develop malignancies earlier [63, 426]. There is also a connection between polymorphism in this region of the p73 gene and the risk of early cancer onset [426, 427].
There are still no real approaches to lowering the risk of cancer among individuals with the above-mentioned variations in the p53-dependent mechanisms. However, the fact that increased ROS level is registered in cells with compromised p53 function [35] forces consideration of application of antioxidants for prevention of cancer in the groups at risk. Increased Mdm2 activity could be pharmacologically managed by nutlin-type compounds that specifically destroy the complex of p53 with Mdm2 [428].
Despite its obvious role in prevention of malignancies, the activity of p53 could be compared with the double-edged sword. The stress-induced activation of p53 that occurs during cancer chemotherapy has a negative effect on normal cells, especially on hemopoietic, intestinal epithelium, and endothelial cells. Prolonged intoxications, pathologies that proceed with chronic inflammation, degenerative processes (such as Huntington’s disease [429, 430]) are accompanied by long-term p53-induced stress response in certain tissues, which creates a background that favors the development of pathology and accelerate aging processes in organs. Under some conditions compounds that transiently and selectively inhibit activity of p53 could be indicated [431]. The search for such inhibitors has identified two classes of small molecules [432, 433]. Although not being strictly specific towards p53, the compounds diminish some of the undesired side effects of p53. It was found that transient inhibition of p53 functions does not represent a hazard for genome stability, probably because as soon as the activity of p53 is restored the accumulated damage can be quickly and efficiently removed from the organism.
As p53 is a pleiotropic regulator, it simultaneously affects many metabolic processes. However, for therapeutic purposes effects of p53 on certain select targets can be preferred, as this would avoid some undesirable effects associated with activation of other p53 targets. Therefore, studying mechanisms that are associated with separate effectors of p53 may contribute to a more selective and less damaging approached for disease prevention and therapy.
During the last few years there have been substantial revisions in concepts on the role of p53 in the organism. Previously it was assumed that the tumor suppressor function of p53 is mediated exclusively in response to structural damage and alterations in cell physiology. Now it is found that p53 activities are extended to normal cells and that they efficiently contribute to genome stability even in the absence of stresses and lesions. The latter activities aim at maintenance of optimal intracellular homeostasis to support the conditions that prevent molecular damage. Both functions of p53 contribute to maintenance of high genetic stability of somatic cells, reduce probability of malignant transformation, and slow the aging process.
This work was supported by the Russian Foundation for Basic Research, grant for International Research Scholars from the Howard Hughes Medical Institute, a grant from the Program on Molecular and Cellular Biology, Russian Academy of Sciences, a grant from the “Dynasty” Foundation of Dmitry Zimin, and grants from the USA National Institutes of Health (R01 CA104903 and R01 AG025278).
REFERENCES
1.Oren, M., Maltzman, W., and Levine, A. J. (1981)
Mol Cell Biol., 1, 101-110.
2.Maltzman, W., and Czyzyk, L. (1984) Mol Cell
Biol., 4, 1689-1694.
3.Chance, B., Sies, H., and Boveris, A. (1979)
Physiol Rev., 59, 527-605.
4.Chang, C., Simmons, D. T., Martin, M. A., and Mora,
P. T. (1979) J. Virol., 31, 463-471.
5.DeLeo, A. B., Jay, G., Appella, E., Dubois, G. C.,
Law, L. W., and Old, L. J. (1979) Proc. Natl. Acad. Sci. USA,
76, 2420-2424.
6.Kress, M., May, E., Cassingena, R., and May, P.
(1979) J. Virol., 31, 472-483.
7.Lane, D. P., and Crawford, L. V. (1979)
Nature, 278, 261-263.
8.Linzer, D. I., and Levine, A. J. (1979)
Cell, 17, 43-52.
9.Chumakov, P. M., Jotseva, V. S., and Georgiev, G.
P. (1982) Doklady AN SSSR, 267, 1272-1275.
10.Oren, M., and Levine, A. J. (1983) Proc. Natl.
Acad. Sci. USA, 80, 56-59.
11.Jenkins, J. R., Rudge, K., and Currie, G. A.
(1984) Nature, 312, 651-654.
12.Parada, L. F., Land, H., Weinberg, R. A., Wolf,
D., and Rotter, V. (1984) Nature, 312, 649-651.
13.Jenkins, J. R., Rudge, K., Chumakov, P., and
Currie, G. A. (1985) Nature, 317, 816-818.
14.Wolf, D., Laver-Rudich, Z., and Rotter, V. (1985)
Mol. Cell Biol., 5, 1887-1893.
15.Bukhman, V. L., Ninkina, N. N., Chumakov, P. M.,
Khilenkova, M. A., and Samarina, O. P. (1987) Genetika,
23, 1547-1554.
16.Eliyahu, D., Goldfinger, N., Pinhasi-Kimhi, O.,
Shaulsky, G., Skurnik, Y., Arai, N., Rotter, V., and Oren, M. (1988)
Oncogene, 3, 313-321.
17.Finlay, C. A., Hinds, P. W., Tan, T. H., Eliyahu,
D., Oren, M., and Levine, A. J. (1988) Mol. Cell Biol.,
8, 531-539.
18.Halevy, O., Rodel, J., Peled, A., and Oren, M.
(1991) Oncogene, 6, 1593-1600.
19.Baker, S. J., Markowitz, S., Fearon, E. R.,
Willson, J. K., and Vogelstein, B. (1990) Science, 249,
912-915.
20.Eliyahu, D., Michalovitz, D., Eliyahu, S.,
Pinhasi-Kimhi, O., and Oren, M. (1989) Proc. Natl. Acad. Sci.
USA, 86, 8763-8767.
21.Finlay, C. A., Hinds, P. W., and Levine, A. J.
(1989) Cell, 57, 1083-1093.
22.Michalovitz, D., Halevy, O., and Oren, M. (1990)
Cell, 62, 671-680.
23.Diller, L., Kassel, J., Nelson, C. E., Gryka, M.
A., Litwak, G., Gebhardt, M., Bressac, B., Ozturk, M., Baker, S. J.,
Vogelstein, B., et al. (1990) Mol. Cell Biol., 10,
5772-5781.
24.Yonish-Rouach, E., Resnitzky, D., Lotem, J.,
Sachs, L., Kimchi, A., and Oren, M. (1991) Nature, 352,
345-347.
25.Wang, Y., Blandino, G., Oren, M., and Givol, D.
(1998) Oncogene, 17, 1923-1930.
26.Ventura, A., Kirsch, D. G., McLaughlin, M. E.,
Tuveson, D. A., Grimm, J., Lintault, L., Newman, J., Reczek, E. E.,
Weissleder, R., and Jacks, T. (2007) Nature, 445,
661-665.
27.Xue, W., Zender, L., Miething, C., Dickins, R.
A., Hernando, E., Krizhanovsky, V., Cordon-Cardo, C., and Lowe, S. W.
(2007) Nature, 445, 656-660.
28.Malkin, D. (1993) Cancer Genet.
Cytogenet., 66, 83-92.
29.Donehower, L. A., Harvey, M., Slagle, B. L.,
McArthur, M. J., Montgomery, C. A., Jr., Butel, J. S., and Bradley, A.
(1992) Nature, 356, 215-221.
30.Baker, S. J., Fearon, E. R., Nigro, J. M.,
Hamilton, S. R., Preisinger, A. C., Jessup, J. M., van Tuinen, P.,
Ledbetter, D. H., Barker, D. F., Nakamura, Y., White, R., and
Vogelstein, B. (1989) Science, 244, 217-221.
31.Brosh, R., and Rotter, V. (2009) Nat. Rev.
Cancer, 9, 701-713.
32.Chumakov, P. M. (2007) Biochemistry
(Moscow), 72, 1399-1421.
33.Lane, D. P. (1992) Nature, 358,
15-16.
34.Vousden, K. H., and Prives, C. (2009)
Cell, 137, 413-431.
35.Sablina, A. A., Budanov, A. V., Ilyinskaya, G.
V., Agapova, L. S., Kravchenko, J. E., and Chumakov, P. M. (2005)
Nat. Med., 11, 1306-1313.
36.Bensaad, K., and Vousden, K. H. (2005) Nat.
Med., 11, 1278-1279.
37.Hirota, Y., Daikoku, T., Tranguch, S., Xie, H.,
Bradshaw, H. B., and Dey, S. K. (2010) J. Clin. Invest.,
120, 803-815.
38.Hu, W., Feng, Z., Teresky, A. K., and Levine, A.
J. (2007) Nature, 450, 721-724.
39.Kang, H. J., Feng, Z., Sun, Y., Atwal, G.,
Murphy, M. E., Rebbeck, T. R., Rosenwaks, Z., Levine, A. J., and Hu, W.
(2009) Proc. Natl. Acad. Sci. USA, 106, 9761-9766.
40.Zhao, T., and Xu, Y. (2010) Trends Cell
Biol., 20, 170-175.
41.Olovnikov, I. A., Kravchenko, J. E., and
Chumakov, P. M. (2009) Semin. Cancer Biol., 19,
32-41.
42.Hollstein, M., and Hainaut, P. (2010) J.
Pathol., 220, 164-173.
43.Farnebo, M. (2009) Cell Cycle, 8,
2343-2346.
44.Mahmoudi, S., Henriksson, S., Corcoran, M.,
Mendez-Vidal, C., Wiman, K. G., and Farnebo, M. (2009) Mol.
Cell, 33, 462-471.
45.Galban, S., Martindale, J. L., Mazan-Mamczarz,
K., Lopez de Silanes, I., Fan, J., Wang, W., Decker, J., and Gorospe,
M. (2003) Mol. Cell Biol., 23, 7083-7095.
46.Mazan-Mamczarz, K., Galban, S., Lopez de Silanes,
I., Martindale, J. L., Atasoy, U., Keene, J. D., and Gorospe, M. (2003)
Proc. Natl. Acad. Sci. USA, 100, 8354-8359.
47.Grover, R., Ray, P. S., and Das, S. (2008)
Cell Cycle, 7, 2189-2198.
48.Grover, R., Candeias, M. M., Fahraeus, R., and
Das, S. (2009) Oncogene, 28, 2766-2772.
49.Bourdon, J. C., Fernandes, K., Murray-Zmijewski,
F., Liu, G., Diot, A., Xirodimas, D. P., Saville, M. K., and Lane, D.
P. (2005) Genes Dev., 19, 2122-2137.
50.Ghosh, A., Stewart, D., and Matlashewski, G.
(2004) Mol. Cell Biol., 24, 7987-7997.
51.Janicke, R. U., Graupner, V., Budach, W., and
Essmann, F. (2009) Biol. Chem., 390, 951-963.
52.Asher, G., Reuven, N., and Shaul, Y. (2006)
Bioessays, 28, 844-849.
53.Buryanovsky, L., Fu, Y., Boyd, M., Ma, Y., Hsieh,
T. C., Wu, J. M., and Zhang, Z. (2004) Biochemistry, 43,
11417-11426.
54.Vella, F., Ferry, G., Delagrange, P., and Boutin,
J. A. (2005) Biochem. Pharmacol., 71, 1-12.
55.Pearson, K. J., Baur, J. A., Lewis, K. N.,
Peshkin, L., Price, N. L., Labinskyy, N., Swindell, W. R., Kamara, D.,
Minor, R. K., Perez, E., Jamieson, H. A., Zhang, Y., Dunn, S. R.,
Sharma, K., Pleshko, N., Woollett, L. A., Csiszar, A., Ikeno, Y., le
Couteur, D., Elliott, P. J., Becker, K. G., Navas, P., Ingram, D. K.,
Wolf, N. S., Ungvari, Z., Sinclair, D. A., and de Cabo, R. (2008)
Cell Metab., 8, 157-168.
56.Barger, J. L., Kayo, T., Vann, J. M., Arias, E.
B., Wang, J., Hacker, T. A., Wang, Y., Raederstorff, D., Morrow, J. D.,
Leeuwenburgh, C., Allison, D. B., Saupe, K. W., Cartee, G. D.,
Weindruch, R., and Prolla, T. A. (2008) PLoS One, 3,
e2264.
57.Haupt, Y., Maya, R., Kazaz, A., and Oren, M.
(1997) Nature, 387, 296-299.
58.Honda, R., Tanaka, H., and Yasuda, H. (1997)
FEBS Lett., 420, 25-27.
59.Kubbutat, M. H., Jones, S. N., and Vousden, K. H.
(1997) Nature, 387, 299-303.
60.Wu, X., Bayle, J. H., Olson, D., and Levine, A.
J. (1993) Genes Dev., 7, 1126-1132.
61.Shvarts, A., Steegenga, W. T., Riteco, N., van
Laar, T., Dekker, P., Bazuine, M., van Ham, R. C., van der Houven, van
Oordt, W., Hateboer, G., van der Eb, A. J., and Jochemsen, A. G. (1996)
Embo J., 15, 5349-5357.
62.Matijasevic, Z., Krzywicka-Racka, A., Sluder, G.,
and Jones, S. N. (2008) Cell Cycle, 7, 2967-2973.
63.Bond, G. L., Hirshfield, K. M., Kirchhoff, T.,
Alexe, G., Bond, E. E., Robins, H., Bartel, F., Taubert, H., Wuerl, P.,
Hait, W., Toppmeyer, D., Offit, K., and Levine, A. J. (2006) Cancer
Res., 66, 5104-5110.
64.Jin, Y., Lee, H., Zeng, S. X., Dai, M. S., and
Lu, H. (2003) Embo J., 22, 6365-6377.
65.Leng, R. P., Lin, Y., Ma, W., Wu, H., Lemmers,
B., Chung, S., Parant, J. M., Lozano, G., Hakem, R., and Benchimol, S.
(2003) Cell, 112, 779-791.
66.Dornan, D., Wertz, I., Shimizu, H., Arnott, D.,
Frantz, G. D., Dowd, P., O’Rourke, K., Koeppen, H., and Dixit, V.
M. (2004) Nature, 429, 86-92.
67.Sdek, P., Ying, H., Chang, D. L., Qiu, W., Zheng,
H., Touitou, R., Allday, M. J., and Xiao, Z. X. (2005) Mol.
Cell, 20, 699-708.
68.Shimada, M., Kitagawa, K., Dobashi, Y., Isobe,
T., Hattori, T., Uchida, C., Abe, K., Kotake, Y., Oda, T., Suzuki, H.,
Hashimoto, K., and Kitagawa, M. (2009) Cancer Sci., 100,
866-872.
69.Yang, W., Rozan, L. M., McDonald, E. R., 3rd,
Navaraj, A., Liu, J. J., Matthew, E. M., Wang, W., Dicker, D. T., and
El-Deiry, W. S. (2007) J. Biol. Chem., 282,
3273-3281.
70.Rajendra, R., Malegaonkar, D., Pungaliya, P.,
Marshall, H., Rasheed, Z., Brownell, J., Liu, L. F., Lutzker, S.,
Saleem, A., and Rubin, E. H. (2004) J. Biol. Chem., 279,
36440-36444.
71.Yamasaki, S., Yagishita, N., Sasaki, T.,
Nakazawa, M., Kato, Y., Yamadera, T., Bae, E., Toriyama, S., Ikeda, R.,
Zhang, L., Fujitani, K., Yoo, E., Tsuchimochi, K., Ohta, T., Araya, N.,
Fujita, H., Aratani, S., Eguchi, K., Komiya, S., Maruyama, I., Higashi,
N., Sato, M., Senoo, H., Ochi, T., Yokoyama, S., Amano, T., Kim, J.,
Gay, S., Fukamizu, A., Nishioka, K., Tanaka, K., and Nakajima, T.
(2007) Embo J., 26, 113-122.
72.Allton, K., Jain, A. K., Herz, H. M., Tsai, W.
W., Jung, S. Y., Qin, J., Bergmann, A., Johnson, R. L., and Barton, M.
C. (2009) Proc. Natl. Acad. Sci. USA, 106,
11612-11616.
73.Tai, E., and Benchimol, S. (2009) Proc. Natl.
Acad. Sci. USA, 106, 11431-11432.
74.Lowe, S. W., and Sherr, C. J. (2003) Curr.
Opin. Genet. Dev., 13, 77-83.
75.Sherr, C. J. (2006) Nat. Rev. Cancer,
6, 663-673.
76.Chen, D., Kon, N., Li, M., Zhang, W., Qin, J.,
and Gu, W. (2005) Cell, 121, 1071-1083.
77.Dai, M. S., Zeng, S. X., Jin, Y., Sun, X. X.,
David, L., and Lu, H. (2004) Mol. Cell Biol., 24,
7654-7668.
78.Lohrum, M. A., Ludwig, R. L., Kubbutat, M. H.,
Hanlon, M., and Vousden, K. H. (2003) Cancer Cell, 3,
577-587.
79.Zhang, Y., Wolf, G. W., Bhat, K., Jin, A., Allio,
T., Burkhart, W. A., and Xiong, Y. (2003) Mol. Cell Biol.,
23, 8902-8912.
80.Zhang, Y., and Lu, H. (2009) Cancer Cell,
16, 369-377.
81.Li, M., Chen, D., Shiloh, A., Luo, J., Nikolaev,
A. Y., Qin, J., and Gu, W. (2002) Nature, 416,
648-653.
82.Li, M., Brooks, C. L., Kon, N., and Gu, W. (2004)
Mol. Cell, 13, 879-886.
83.Cummins, J. M., and Vogelstein, B. (2004) Cell
Cycle, 3, 689-692.
84.Tang, J., Qu, L. K., Zhang, J., Wang, W.,
Michaelson, J. S., Degenhardt, Y. Y., El-Deiry, W. S., and Yang, X.
(2006) Nat. Cell Biol., 8, 855-862.
85.Song, M. S., Song, S. J., Kim, S. Y., Oh, H. J.,
and Lim, D. S. (2008) Embo J., 27, 1863-1874.
86.Kruse, J. P., and Gu, W. (2008) Cell,
133, 930-931.
87.Kruse, J. P., and Gu, W. (2009) Cell,
137, 609-622.
88.Appella, E., and Anderson, C. W. (2001) Eur.
J. Biochem., 268, 2764-2772.
89.Shieh, S. Y., Ikeda, M., Taya, Y., and Prives, C.
(1997) Cell, 91, 325-334.
90.Shieh, S. Y., Ahn, J., Tamai, K., Taya, Y., and
Prives, C. (2000) Genes Dev., 14, 289-300.
91.Ashcroft, M., Kubbutat, M. H., and Vousden, K. H.
(1999) Mol. Cell Biol., 19, 1751-1758.
92.Blattner, C., Tobiasch, E., Litfen, M.,
Rahmsdorf, H. J., and Herrlich, P. (1999) Oncogene, 18,
1723-1732.
93.Marchenko, N. D., Zaika, A., and Moll, U. M.
(2000) J. Biol. Chem., 275, 16202-16212.
94.Marchenko, N. D., Wolff, S., Erster, S., Becker,
K., and Moll, U. M. (2007) Embo J., 26, 923-934.
95.Chipuk, J. E., Kuwana, T., Bouchier-Hayes, L.,
Droin, N. M., Newmeyer, D. D., Schuler, M., and Green, D. R. (2004)
Science, 303, 1010-1014.
96.Chipuk, J. E., and Green, D. R. (2006) Cell
Death Differ., 13, 994-1002.
97.Vaseva, A. V., and Moll, U. M. (2009) Biochim.
Biophys. Acta, 1787, 414-420.
98.Nakano, K., and Vousden, K. H. (2001) Mol.
Cell, 7, 683-694.
99.Yu, J., Zhang, L., Hwang, P. M., Kinzler, K. W.,
and Vogelstein, B. (2001) Mol. Cell, 7, 673-682.
100.Oda, E., Ohki, R., Murasawa, H., Nemoto, J.,
Shibue, T., Yamashita, T., Tokino, T., Taniguchi, T., and Tanaka, N.
(2000) Science, 288, 1053-1058.
101.Miyashita, T., and Reed, J. C. (1995)
Cell, 80, 293-299.
102.Yao, H., Li, P., Venters, B. J., Zheng, S.,
Thompson, P. R., Pugh, B. F., and Wang, Y. (2008) J. Biol.
Chem., 283, 20060-20068.
103.Shen, Y., and Shenk, T. (1994) Proc. Natl.
Acad. Sci. USA, 91, 8940-8944.
104.Li, Y. Z., Lu, D. Y., Tan, W. Q., Wang, J. X.,
and Li, P. F. (2008) Mol. Cell Biol., 28, 564-574.
105.Robles, A. I., Bemmels, N. A., Foraker, A. B.,
and Harris, C. C. (2001) Cancer Res., 61, 6660-6664.
106.Moroni, M. C., Hickman, E. S., Lazzerini
Denchi, E., Caprara, G., Colli, E., Cecconi, F., Muller, H., and Helin,
K. (2001) Nat. Cell Biol., 3, 552-558.
107.Fortin, A., Cregan, S. P., MacLaurin, J. G.,
Kushwaha, N., Hickman, E. S., Thompson, C. S., Hakim, A., Albert, P.
R., Cecconi, F., Helin, K., Park, D. S., and Slack, R. S. (2001) J.
Cell Biol., 155, 207-216.
108.Alberts, B. (2008) in Molecular Biology of
the Cell, 5th Edn., Garland Science, New York.
109.Owen-Schaub, L. B., Zhang, W., Cusack, J. C.,
Angelo, L. S., Santee, S. M., Fujiwara, T., Roth, J. A., Deisseroth, A.
B., Zhang, W. W., Kruzel, E., et al. (1995) Mol. Cell Biol.,
15, 3032-3040.
110.Wu, G. S., Burns, T. F., McDonald, E. R., 3rd,
Jiang, W., Meng, R., Krantz, I. D., Kao, G., Gan, D. D., Zhou, J. Y.,
Muschel, R., Hamilton, S. R., Spinner, N. B., Markowitz, S., Wu, G.,
and El-Deiry, W. S. (1997) Nat. Genet., 17, 141-143.
111.Yoshida, K., and Miki, Y. (2010) Cancer
Sci., 101, 831-835.
112.Kuribayashi, K., Krigsfeld, G., Wang, W., Xu,
J., Mayes, P. A., Dicker, D. T., Wu, G. S., and El-Deiry, W. S. (2008)
Cancer Biol. Ther., 7, 2034-2038.
113.Maecker, H. L., Koumenis, C., and Giaccia, A.
J. (2000) Cancer Res., 60, 4638-4644.
114.Rikhof, B., Corn, P. G., and El-Deiry, W. S.
(2003) Cancer Biol. Ther., 2, 707-712.
115.Attardi, L. D., Reczek, E. E., Cosmas, C.,
Demicco, E. G., McCurrach, M. E., Lowe, S. W., and Jacks, T. (2000)
Genes Dev., 14, 704-718.
116.Lin, Y., Ma, W., and Benchimol, S. (2000)
Nat. Genet., 26, 122-127.
117.Fiscella, M., Zhang, H., Fan, S., Sakaguchi,
K., Shen, S., Mercer, W. E., Vande Woude, G. F., O’Connor, P. M.,
and Appella, E. (1997) Proc. Natl. Acad. Sci. USA, 94,
6048-6053.
118.Bourdon, J. C., Renzing, J., Robertson, P. L.,
Fernandes, K. N., and Lane, D. P. (2002) J. Cell Biol.,
158, 235-246.
119.Oda, K., Arakawa, H., Tanaka, T., Matsuda, K.,
Tanikawa, C., Mori, T., Nishimori, H., Tamai, K., Tokino, T., Nakamura,
Y., and Taya, Y. (2000) Cell, 102, 849-862.
120.Nakamura, Y. (2004) Cancer Sci.,
95, 7-11.
121.Polyak, K., Xia, Y., Zweier, J. L., Kinzler, K.
W., and Vogelstein, B. (1997) Nature, 389, 300-305.
122.Hwang, P. M., Bunz, F., Yu, J., Rago, C., Chan,
T. A., Murphy, M. P., Kelso, G. F., Smith, R. A., Kinzler, K. W., and
Vogelstein, B. (2001) Nat. Med., 7, 1111-1117.
123.Chang, T. C., Wentzel, E. A., Kent, O. A.,
Ramachandran, K., Mullendore, M., Lee, K. H., Feldmann, G., Yamakuchi,
M., Ferlito, M., Lowenstein, C. J., Arking, D. E., Beer, M. A., Maitra,
A., and Mendell, J. T. (2007) Mol. Cell, 26, 745-752.
124.He, L., He, X., Lim, L. P., de Stanchina, E.,
Xuan, Z., Liang, Y., Xue, W., Zender, L., Magnus, J., Ridzon, D.,
Jackson, A. L., Linsley, P. S., Chen, C., Lowe, S. W., Cleary, M. A.,
and Hannon, G. J. (2007) Nature, 447, 1130-1134.
125.Hermeking, H. (2007) Cancer Cell,
12, 414-418.
126.Raver-Shapira, N., Marciano, E., Meiri, E.,
Spector, Y., Rosenfeld, N., Moskovits, N., Bentwich, Z., and Oren, M.
(2007) Mol. Cell, 26, 731-743.
127.Tarasov, V., Jung, P., Verdoodt, B., Lodygin,
D., Epanchintsev, A., Menssen, A., Meister, G., and Hermeking, H.
(2007) Cell Cycle, 6, 1586-1593.
128.El-Deiry, W. S., Tokino, T., Velculescu, V. E.,
Levy, D. B., Parsons, R., Trent, J. M., Lin, D., Mercer, W. E.,
Kinzler, K. W., and Vogelstein, B. (1993) Cell, 75,
817-825.
129.El-Deiry, W. S. (1998) Semin. Cancer
Biol., 8, 345-357.
130.Zhu, J., and Chen, X. (2000) Mol. Cell
Biol., 20, 5602-5618.
131.Hermeking, H., and Benzinger, A. (2006)
Semin. Cancer Biol., 16, 183-192.
132.Helton, E. S., and Chen, X. (2007) J. Cell
Biochem., 100, 883-896.
133.Harms, K., Nozell, S., and Chen, X. (2004)
Cell Mol. Life Sci., 61, 822-842.
134.Almazov, V. P., Kochetkov, D. A., and Chumakov,
P. M. (2007) Mol. Biol. (Moscow), 41, 947-463.
135.Selivanova, G. (2010) Semin. Cancer
Biol., 20, 46-56.
136.Macip, S., Igarashi, M., Berggren, P., Yu, J.,
Lee, S. W., and Aaronson, S. A. (2003) Mol. Cell Biol.,
23, 8576-8585.
137.Zhang, H. (2007) J. Cell Physiol.,
210, 567-574.
138.Wynford-Thomas, D. (1996) Oncol. Res.,
8, 387-398.
139.Artandi, S. E., and DePinho, R. A. (2010)
Carcinogenesis, 31, 9-18.
140.Kanaya, T., Kyo, S., Hamada, K., Takakura, M.,
Kitagawa, Y., Harada, H., and Inoue, M. (2000) Clin. Cancer
Res., 6, 1239-1247.
141.Xu, D., Wang, Q., Gruber, A., Bjorkholm, M.,
Chen, Z., Zaid, A., Selivanova, G., Peterson, C., Wiman, K. G., and
Pisa, P. (2000) Oncogene, 19, 5123-5133.
142.Shats, I., Milyavsky, M., Tang, X., Stambolsky,
P., Erez, N., Brosh, R., Kogan, I., Braunstein, I., Tzukerman, M.,
Ginsberg, D., and Rotter, V. (2004) J. Biol. Chem., 279,
50976-50985.
143.Won, J., Chang, S., Oh, S., and Kim, T. K.
(2004) Proc. Natl. Acad. Sci. USA, 101, 11328-11333.
144.Stewart, S. A., and Weinberg, R. A. (2006)
Annu. Rev. Cell Dev. Biol., 22, 531-557.
145.Macip, S., Igarashi, M., Fang, L., Chen, A.,
Pan, Z. Q., Lee, S. W., and Aaronson, S. A. (2002) Embo J.,
21, 2180-2188.
146.Kortlever, R. M., Higgins, P. J., and Bernards,
R. (2006) Nat. Cell Biol., 8, 877-884.
147.Leal, J. F., Fominaya, J., Cascon, A.,
Guijarro, M. V., Blanco-Aparicio, C., Lleonart, M., Castro, M. E.,
Ramon, Y. C. S., Robledo, M., Beach, D. H., and Carnero, A. (2008)
Oncogene, 27, 1961-1970.
148.Cosme-Blanco, W., Shen, M. F., Lazar, A. J.,
Pathak, S., Lozano, G., Multani, A. S., and Chang, S. (2007) EMBO
Rep., 8, 497-503.
149.Van Nguyen, T., Puebla-Osorio, N., Pang, H.,
Dujka, M. E., and Zhu, C. (2007) J. Exp. Med., 204,
1453-1461.
150.Barboza, J. A., Liu, G., Ju, Z., El-Naggar, A.
K., and Lozano, G. (2006) Proc. Natl. Acad. Sci. USA,
103, 19842-19847.
151.Choudhury, A. R., Ju, Z., Djojosubroto, M. W.,
Schienke, A., Lechel, A., Schaetzlein, S., Jiang, H., Stepczynska, A.,
Wang, C., Buer, J., Lee, H. W., von Zglinicki, T., Ganser, A.,
Schirmacher, P., Nakauchi, H., and Rudolph, K. L. (2007) Nat.
Genet., 39, 99-105.
152.Toledo, F., Krummel, K. A., Lee, C. J., Liu, C.
W., Rodewald, L. W., Tang, M., and Wahl, G. M. (2006) Cancer
Cell, 9, 273-285.
153.Yu, J., and Zhang, L. (2003) Cancer
Cell, 4, 248-249.
154.Michalak, E. M., Villunger, A., Adams, J. M.,
and Strasser, A. (2008) Cell Death Differ., 15,
1019-1029.
155.Liu, G., Parant, J. M., Lang, G., Chau, P.,
Chavez-Reyes, A., El-Naggar, A. K., Multani, A., Chang, S., and Lozano,
G. (2004) Nat. Genet., 36, 63-68.
156.Kunz, C., Pebler, S., Otte, J., and von der
Ahe, D. (1995) Nucleic Acids Res., 23, 3710-3717.
157.Bian, J., and Sun, Y. (1997) Mol. Cell
Biol., 17, 6330-6338.
158.Zou, Z., Gao, C., Nagaich, A. K., Connell, T.,
Saito, S., Moul, J. W., Seth, P., Appella, E., and Srivastava, S.
(2000) J. Biol. Chem., 275, 6051-6054.
159.Sager, R., Sheng, S., Pemberton, P., and
Hendrix, M. J. (1997) Adv. Exp. Med. Biol., 425,
77-88.
160.Mashimo, T., Watabe, M., Hirota, S., Hosobe,
S., Miura, K., Tegtmeyer, P. J., Rinker-Shaeffer, C. W., and Watabe, K.
(1998) Proc. Natl. Acad. Sci. USA, 95, 11307-11311.
161.Malik, F. A., Sanders, A. J., and Jiang, W. G.
(2009) Histol. Histopathol., 24, 519-530.
162.Komarova, E. A., Diatchenko, L., Rokhlin, O.
W., Hill, J. E., Wang, Z. J., Krivokrysenko, V. I., Feinstein, E., and
Gudkov, A. V. (1998) Oncogene, 17, 1089-1096.
163.Buckbinder, L., Talbott, R., Velasco-Miguel,
S., Takenaka, I., Faha, B., Seizinger, B. R., and Kley, N. (1995)
Nature, 377, 646-649.
164.Feng, Z. (2010) Cold Spring Harb. Perspect.
Biol., 2, a001057.
165.Levine, A. J., Feng, Z., Mak, T. W., You, H.,
and Jin, S. (2006) Genes Dev., 20, 267-275.
166.Neuberg, M., Buckbinder, L., Seizinger, B., and
Kley, N. (1997) Endocrine, 7, 107-109.
167.Dameron, K. M., Volpert, O. V., Tainsky, M. A.,
and Bouck, N. (1994) Science, 265, 1582-1584.
168.Schultz, G. S., and Wysocki, A. (2009) Wound
Repair Regen., 17, 153-162.
169.Nishimori, H., Shiratsuchi, T., Urano, T.,
Kimura, Y., Kiyono, K., Tatsumi, K., Yoshida, S., Ono, M., Kuwano, M.,
Nakamura, Y., and Tokino, T. (1997) Oncogene, 15,
2145-2150.
170.Van Meir, E. G., Polverini, P. J., Chazin, V.
R., Su Huang, H. J., de Tribolet, N., and Cavenee, W. K. (1994) Nat.
Genet., 8, 171-176.
171.Yu, X., Harris, S. L., and Levine, A. J. (2006)
Cancer Res., 66, 4795-4801.
172.Lespagnol, A., Duflaut, D., Beekman, C., Blanc,
L., Fiucci, G., Marine, J. C., Vidal, M., Amson, R., and Telerman, A.
(2008) Cell Death Differ., 15, 1723-1733.
173.Yu, X., Riley, T., and Levine, A. J. (2009)
FEBS J., 276, 2201-2212.
174.Sadowski, L., Pilecka, I., and Miaczynska, M.
(2009) Exp. Cell Res., 315, 1601-1609.
175.Schorey, J. S., and Bhatnagar, S. (2008)
Traffic, 9, 871-881.
176.Amzallag, N., Passer, B. J., Allanic, D.,
Segura, E., Thery, C., Goud, B., Amson, R., and Telerman, A. (2004)
J. Biol. Chem., 279, 46104-46112.
177.Di Guglielmo, G. M., le Roy, C., Goodfellow, A.
F., and Wrana, J. L. (2003) Nat. Cell Biol., 5,
410-421.
178.Sigismund, S., Woelk, T., Puri, C., Maspero,
E., Tacchetti, C., Transidico, P., Di Fiore, P. P., and Polo, S. (2005)
Proc. Natl. Acad. Sci. USA, 102, 2760-2765.
179.Saksena, S., Sun, J., Chu, T., and Emr, S. D.
(2007) Trends Biochem. Sci., 32, 561-573.
180.Levine, A. J., Hu, W., and Feng, Z. (2006)
Cell Death Differ., 13, 1027-1036.
181.Kim, E., and Deppert, W. (2006) Cell Death
Differ., 13, 885-889.
182.Bakalkin, G., Selivanova, G., Yakovleva, T.,
Kiseleva, E., Kashuba, E., Magnusson, K. P., Szekely, L., Klein, G.,
Terenius, L., and Wiman, K. G. (1995) Nucleic Acids Res.,
23, 362-369.
183.Lee, S., Cavallo, L., and Griffith, J. (1997)
J. Biol. Chem., 272, 7532-7539.
184.Wetzel, C. C., and Berberich, S. J. (2001)
Biochim. Biophys. Acta, 1517, 392-397.
185.Zotchev, S. B., Protopopova, M., and
Selivanova, G. (2000) Nucleic Acids Res., 28,
4005-4012.
186.Bakhanashvili, M., Hizi, A., and Rahav, G.
(2010) Cell Cycle, 9, 1380-1389.
187.Mummenbrauer, T., Janus, F., Muller, B.,
Wiesmuller, L., Deppert, W., and Grosse, F. (1996) Cell,
85, 1089-1099.
188.Walter, K., Warnecke, G., Bowater, R., Deppert,
W., and Kim, E. (2005) J. Biol. Chem., 280,
42497-42507.
189.Huang, P. (1998) Oncogene, 17,
261-270.
190.Melle, C., and Nasheuer, H. P. (2002)
Nucleic Acids Res., 30, 1493-1499.
191.Bakhanashvili, M. (2001) Eur. J.
Biochem., 268, 2047-2054.
192.Bakhanashvili, M. (2001) Oncogene,
20, 7635-7644.
193.Lilling, G., Elena, N., Sidi, Y., and
Bakhanashvili, M. (2003) Oncogene, 22, 233-245.
194.Bakhanashvili, M., Novitsky, E., Lilling, G.,
and Rahav, G. (2004) Oncogene, 23, 6890-6899.
195.Janus, F., Albrechtsen, N., Dornreiter, I.,
Wiesmuller, L., Grosse, F., and Deppert, W. (1999) Cell Mol. Life
Sci., 55, 12-27.
196.Blander, G., Kipnis, J., Leal, J. F., Yu, C.
E., Schellenberg, G. D., and Oren, M. (1999) J. Biol. Chem.,
274, 29463-29469.
197.Dutta, A., Ruppert, J. M., Aster, J. C., and
Winchester, E. (1993) Nature, 365, 79-82.
198.Garkavtsev, I. V., Kley, N., Grigorian, I. A.,
and Gudkov, A. V. (2001) Oncogene, 20, 8276-8280.
199.Hanson, S., Kim, E., and Deppert, W. (2005)
Oncogene, 24, 1641-1647.
200.Herring, C. J., West, C. M., Wilks, D. P.,
Davidson, S. E., Hunter, R. D., Berry, P., Forster, G., MacKinnon, J.,
Rafferty, J. A., Elder, R. H., Hendry, J. H., and Margison, G. P.
(1998) Br. J. Cancer, 78, 1128-1133.
201.Jayaraman, L., Murthy, K. G., Zhu, C., Curran,
T., Xanthoudakis, S., and Prives, C. (1997) Genes Dev.,
11, 558-570.
202.Sturzbecher, H. W., Donzelmann, B., Henning,
W., Knippschild, U., and Buchhop, S. (1996) Embo J., 15,
1992-2002.
203.Wang, X. W., Vermeulen, W., Coursen, J. D.,
Gibson, M., Lupold, S. E., Forrester, K., Xu, G., Elmore, L., Yeh, H.,
Hoeijmakers, J. H., and Harris, C. C. (1996) Genes Dev.,
10, 1219-1232.
204.Xanthoudakis, S., Miao, G., Wang, F., Pan, Y.
C., and Curran, T. (1992) Embo J., 11, 3323-3335.
205.Dregoesc, D., Rybak, A. P., and Rainbow, A. J.
(2007) DNA Repair (Amst.), 6, 588-601.
206.Hwang, B. J., Ford, J. M., Hanawalt, P. C., and
Chu, G. (1999) Proc. Natl. Acad. Sci. USA, 96,
424-428.
207.Liu, G., and Chen, X. (2006) Mol. Cell
Biol., 26, 1398-1413.
208.Chen, J., and Sadowski, I. (2005) Proc.
Natl. Acad. Sci. USA, 102, 4813-4818.
209.Scherer, S. J., Maier, S. M., Seifert, M.,
Hanselmann, R. G., Zang, K. D., Muller-Hermelink, H. K., Angel, P.,
Welter, C., and Schartl, M. (2000) J. Biol. Chem., 275,
37469-37473.
210.Shimodaira, H., Yoshioka-Yamashita, A.,
Kolodner, R. D., and Wang, J. Y. (2003) Proc. Natl. Acad. Sci.
USA, 100, 2420-2425.
211.Xu, J., and Morris, G. F. (1999) Mol. Cell
Biol., 19, 12-20.
212.Kimura, T., Takeda, S., Sagiya, Y., Gotoh, M.,
Nakamura, Y., and Arakawa, H. (2003) Nat. Genet., 34,
440-445.
213.Kolberg, M., Strand, K. R., Graff, P., and
Andersson, K. K. (2004) Biochim. Biophys. Acta, 1699,
1-34.
214.Wang, J., Lohman, G. J., and Stubbe, J. (2009)
Biochemistry, 48, 11612-11621.
215.Lebedeva, M. A., Eaton, J. S., and Shadel, G.
S. (2009) Biochim. Biophys. Acta, 1787, 328-334.
216.Jackson, A. L., and Loeb, L. A. (2001)
Mutat. Res., 477, 7-21.
217.Beckman, K. B., and Ames, B. N. (1997) J.
Biol. Chem., 272, 19633-19636.
218.Miller, D. M., Buettner, G. R., and Aust, S. D.
(1990) Free Radic. Biol. Med., 8, 95-108.
219.Valko, M., Leibfritz, D., Moncol, J., Cronin,
M. T., Mazur, M., and Telser, J. (2007) Int. J. Biochem. Cell
Biol., 39, 44-84.
220.Muller, F. L., Liu, Y., and van Remmen, H.
(2004) J. Biol. Chem., 279, 49064-49073.
221.Lambeth, J. D. (2004) Nat. Rev.
Immunol., 4, 181-189.
222.Muller, F. L., Lustgarten, M. S., Jang, Y.,
Richardson, A., and van Remmen, H. (2007) Free Radic. Biol.
Med., 43, 477-503.
223.Miwa, S., Muller, F. L., and Beckman, K. B.
(2008) in Aging Medicine: Oxidative Stress in Aging: From Model
Systems to Human Diseases (Miwa, K. B. B. S., and Muller, F. L.,
eds.) Humana Press, Totowa, N. J., pp. 11-35.
224.Ho, Y. S., Xiong, Y., Ma, W., Spector, A., and
Ho, D. S. (2004) J. Biol. Chem., 279, 32804-32812.
225.Andziak, B., O’Connor, T. P., and
Buffenstein, R. (2005) Mech. Ageing Dev., 126,
1206-1212.
226.Rhee, S. G., Chae, H. Z., and Kim, K. (2005)
Free Radic. Biol. Med., 38, 1543-1552.
227.Martindale, J. L., and Holbrook, N. J. (2002)
J. Cell Physiol., 192, 1-15.
228.Bensaad, K., and Vousden, K. H. (2007)
Trends Cell Biol., 17, 286-291.
229.Desaint, S., Luriau, S., Aude, J. C.,
Rousselet, G., and Toledano, M. B. (2004) J. Biol. Chem.,
279, 31157-31163.
230.Liu, G., and Chen, X. (2002) Oncogene,
21, 7195-7204.
231.Rivera, A., and Maxwell, S. A. (2005) J.
Biol. Chem., 280, 29346-29354.
232.Yoon, K. A., Nakamura, Y., and Arakawa, H.
(2004) J. Hum. Genet., 49, 134-140.
233.Hussain, S. P., Amstad, P., He, P., Robles, A.,
Lupold, S., Kaneko, I., Ichimiya, M., Sengupta, S., Mechanic, L.,
Okamura, S., Hofseth, L. J., Moake, M., Nagashima, M., Forrester, K.
S., and Harris, C. C. (2004) Cancer Res., 64,
2350-2356.
234.O’Connor, J. C., Wallace, D. M.,
O’Brien, C., and Cotter, T. G. (2008) Invest. Ophthalmol. Vis.
Sci., 49, 4237-4244.
235.Velasco-Miguel, S., Buckbinder, L., Jean, P.,
Gelbert, L., Talbott, R., Laidlaw, J., Seizinger, B., and Kley, N.
(1999) Oncogene, 18, 127-137.
236.Budanov, A. V., Shoshani, T., Faerman, A.,
Zelin, E., Kamer, I., Kalinski, H., Gorodin, S., Fishman, A., Chajut,
A., Einat, P., Skaliter, R., Gudkov, A. V., Chumakov, P. M., and
Feinstein, E. (2002) Oncogene, 21, 6017-6031.
237.Bensaad, K., Tsuruta, A., Selak, M. A., Vidal,
M. N., Nakano, K., Bartrons, R., Gottlieb, E., and Vousden, K. H.
(2006) Cell, 126, 107-120.
238.Hu, W., Zhang, C., Wu, R., Sun, Y., Levine, A.,
and Feng, Z. (2010) Proc. Natl. Acad. Sci. USA, 107,
7455-7460.
239.Cano, C. E., Gommeaux, J., Pietri, S., Culcasi,
M., Garcia, S., Seux, M., Barelier, S., Vasseur, S., Spoto, R. P.,
Pebusque, M. J., Dusetti, N. J., Iovanna, J. L., and Carrier, A. (2009)
Cancer Res., 69, 219-226.
240.Ding, B., Chi, S. G., Kim, S. H., Kang, S.,
Cho, J. H., Kim, D. S., and Cho, N. H. (2007) J. Cell Sci.,
120, 2284-2294.
241.Budanov, A. V., Sablina, A. A., Feinstein, E.,
Koonin, E. V., and Chumakov, P. M. (2004) Science, 304,
596-600.
242.Chen, L., Xie, Q. W., and Nathan, C. (1998)
Mol. Cell, 1, 795-805.
243.Woo, H. A., Kang, S. W., Kim, H. K., Yang, K.
S., Chae, H. Z., and Rhee, S. G. (2003) J. Biol. Chem.,
278, 47361-47364.
244.Wood, Z. A., Poole, L. B., and Karplus, P. A.
(2003) Science, 300, 650-653.
245.Wood, Z. A., Schroder, E., Robin Harris, J.,
and Poole, L. B. (2003) Trends Biochem. Sci., 28,
32-40.
246.Yang, K. S., Kang, S. W., Woo, H. A., Hwang, S.
C., Chae, H. Z., Kim, K., and Rhee, S. G. (2002) J. Biol. Chem.,
277, 38029-38036.
247.Biteau, B., Labarre, J., and Toledano, M. B.
(2003) Nature, 425, 980-984.
248.Okar, D. A., Manzano, A., Navarro-Sabate, A.,
Riera, L., Bartrons, R., and Lange, A. J. (2001) Trends Biochem.
Sci., 26, 30-35.
249.Bensaad, K., Cheung, E. C., and Vousden, K. H.
(2009) Embo J., 28, 3015-3026.
250.Matoba, S., Kang, J. G., Patino, W. D., Wragg,
A., Boehm, M., Gavrilova, O., Hurley, P. J., Bunz, F., and Hwang, P. M.
(2006) Science, 312, 1650-1653.
251.Ma, W., Sung, H. J., Park, J. Y., Matoba, S.,
and Hwang, P. M. (2007) J. Bioenerg. Biomembr., 39,
243-246.
252.Liu, B., Chen, Y., and St Clair, D. K. (2008)
Free Radic. Biol. Med., 44, 1529-1535.
253.Stambolsky, P., Weisz, L., Shats, I., Klein,
Y., Goldfinger, N., Oren, M., and Rotter, V. (2006) Cell Death
Differ., 13, 2140-2149.
254.Punj, V., and Chakrabarty, A. M. (2003) Cell
Microbiol., 5, 225-231.
255.Vahsen, N., Cande, C., Briere, J. J., Benit,
P., Joza, N., Larochette, N., Mastroberardino, P. G., Pequignot, M. O.,
Casares, N., Lazar, V., Feraud, O., Debili, N., Wissing, S.,
Engelhardt, S., Madeo, F., Piacentini, M., Penninger, J. M., Schagger,
H., Rustin, P., and Kroemer, G. (2004) Embo J., 23,
4679-4689.
256.Klein, J. A., Longo-Guess, C. M., Rossmann, M.
P., Seburn, K. L., Hurd, R. E., Frankel, W. N., Bronson, R. T., and
Ackerman, S. L. (2002) Nature, 419, 367-374.
257.Joza, N., Susin, S. A., Daugas, E., Stanford,
W. L., Cho, S. K., Li, C. Y., Sasaki, T., Elia, A. J., Cheng, H. Y.,
Ravagnan, L., Ferri, K. F., Zamzami, N., Wakeham, A., Hakem, R.,
Yoshida, H., Kong, Y. Y., Mak, T. W., Zuniga-Pflucker, J. C., Kroemer,
G., and Penninger, J. M. (2001) Nature, 410, 549-554.
258.Cande, C., Vahsen, N., Garrido, C., and
Kroemer, G. (2004) Cell Death Differ., 11, 591-595.
259.Yu, S. W., Wang, H., Poitras, M. F., Coombs,
C., Bowers, W. J., Federoff, H. J., Poirier, G. G., Dawson, T. M., and
Dawson, V. L. (2002) Science, 297, 259-263.
260.Andrabi, S. A., Kim, N. S., Yu, S. W., Wang,
H., Koh, D. W., Sasaki, M., Klaus, J. A., Otsuka, T., Zhang, Z.,
Koehler, R. C., Hurn, P. D., Poirier, G. G., Dawson, V. L., and Dawson,
T. M. (2006) Proc. Natl. Acad. Sci. USA, 103,
18308-18313.
261.Yu, S. W., Andrabi, S. A., Wang, H., Kim, N.
S., Poirier, G. G., Dawson, T. M., and Dawson, V. L. (2006) Proc.
Natl. Acad. Sci. USA, 103, 18314-18319.
262.Ye, H., Cande, C., Stephanou, N. C., Jiang, S.,
Gurbuxani, S., Larochette, N., Daugas, E., Garrido, C., Kroemer, G.,
and Wu, H. (2002) Nat. Struct. Biol., 9, 680-684.
263.Susin, S. A., Lorenzo, H. K., Zamzami, N.,
Marzo, I., Brenner, C., Larochette, N., Prevost, M. C., Alzari, P. M.,
and Kroemer, G. (1999) J. Exp. Med., 189, 381-394.
264.Miramar, M. D., Costantini, P., Ravagnan, L.,
Saraiva, L. M., Haouzi, D., Brothers, G., Penninger, J. M., Peleato, M.
L., Kroemer, G., and Susin, S. A. (2001) J. Biol. Chem.,
276, 16391-16398.
265.Urbano, A., Lakshmanan, U., Choo, P. H., Kwan,
J. C., Ng, P. Y., Guo, K., Dhakshinamoorthy, S., and Porter, A. (2005)
Embo J., 24, 2815-2826.
266.Kondoh, H., Lleonart, M. E., Gil, J., Wang, J.,
Degan, P., Peters, G., Martinez, D., Carnero, A., and Beach, D. (2005)
Cancer Res., 65, 177-185.
267.Ruiz-Lozano, P., Hixon, M. L., Wagner, M. W.,
Flores, A. I., Ikawa, S., Baldwin, A. S., Jr., Chien, K. R., and
Gualberto, A. (1999) Cell Growth Differ., 10,
295-306.
268.Kondoh, H., Lleonart, M. E., Bernard, D., and
Gil, J. (2007) Histol. Histopathol., 22, 85-90.
269.Essmann, F., Pohlmann, S., Gillissen, B.,
Daniel, P. T., Schulze-Osthoff, K., and Janicke, R. U. (2005) J.
Biol. Chem., 280, 37169-37177.
270.Achanta, G., Sasaki, R., Feng, L., Carew, J.
S., Lu, W., Pelicano, H., Keating, M. J., and Huang, P. (2005) Embo
J., 24, 3482-3492.
271.Cho, Y., Gorina, S., Jeffrey, P. D., and
Pavletich, N. P. (1994) Science, 265, 346-355.
272.Velu, C. S., Niture, S. K., Doneanu, C. E.,
Pattabiraman, N., and Srivenugopal, K. S. (2007) Biochemistry,
46, 7765-7780.
273.Sun, X. Z., Vinci, C., Makmura, L., Han, S.,
Tran, D., Nguyen, J., Hamann, M., Grazziani, S., Sheppard, S., Gutova,
M., Zhou, F., Thomas, J., and Momand, J. (2003) Antiox. Redox.
Signal., 5, 655-665.
274.Buzek, J., Latonen, L., Kurki, S., Peltonen,
K., and Laiho, M. (2002) Nucleic Acids Res., 30,
2340-2348.
275.Ueno, M., Masutani, H., Arai, R. J., Yamauchi,
A., Hirota, K., Sakai, T., Inamoto, T., Yamaoka, Y., Yodoi, J., and
Nikaido, T. (1999) J. Biol. Chem., 274, 35809-35815.
276.Seo, Y. R., Kelley, M. R., and Smith, M. L.
(2002) Proc. Natl. Acad. Sci. USA, 99, 14548-14553.
277.Seemann, S., and Hainaut, P. (2005)
Oncogene, 24, 3853-3863.
278.Tomasini, R., Samir, A. A., Carrier, A.,
Isnardon, D., Cecchinelli, B., Soddu, S., Malissen, B., Dagorn, J. C.,
Iovanna, J. L., and Dusetti, N. J. (2003) J. Biol. Chem.,
278, 37722-37729.
279.Yoshida, K., Liu, H., and Miki, Y. (2006) J.
Biol. Chem., 281, 5734-5740.
280.Di Stefano, V., Blandino, G., Sacchi, A.,
Soddu, S., and D’Orazi, G. (2004) Oncogene, 23,
5185-5192.
281.Okamura, S., Arakawa, H., Tanaka, T.,
Nakanishi, H., Ng, C. C., Taya, Y., Monden, M., and Nakamura, Y. (2001)
Mol. Cell, 8, 85-94.
282.Gommeaux, J., Cano, C., Garcia, S., Gironella,
M., Pietri, S., Culcasi, M., Pebusque, M. J., Malissen, B., Dusetti,
N., Iovanna, J., and Carrier, A. (2007) Mol. Cell Biol.,
27, 2215-2228.
283.Tomasini, R., Seux, M., Nowak, J., Bontemps,
C., Carrier, A., Dagorn, J. C., Pebusque, M. J., Iovanna, J. L., and
Dusetti, N. J. (2005) Oncogene, 24, 8093-8104.
284.Nowak, J., and Iovanna, J. L. (2009)
Autophagy, 5, 383-384.
285.Nowak, J., Archange, C., Tardivel-Lacombe, J.,
Pontarotti, P., Pebusque, M. J., Vaccaro, M. I., Velasco, G., Dagorn,
J. C., and Iovanna, J. L. (2009) Mol. Biol. Cell, 20,
870-881.
286.Hanahan, D., and Weinberg, R. A. (2000)
Cell, 100, 57-70.
287.DeBerardinis, R. J., Lum, J. J.,
Hatzivassiliou, G., and Thompson, C. B. (2008) Cell Metab.,
7, 11-20.
288.Narkar, V. A., Downes, M., Yu, R. T., Embler,
E., Wang, Y. X., Banayo, E., Mihaylova, M. M., Nelson, M. C., Zou, Y.,
Juguilon, H., Kang, H., Shaw, R. J., and Evans, R. M. (2008)
Cell, 134, 405-415.
289.Bauer, D. E., Harris, M. H., Plas, D. R., Lum,
J. J., Hammerman, P. S., Rathmell, J. C., Riley, J. L., and Thompson,
C. B. (2004) FASEB J., 18, 1303-1305.
290.Yeung, S. J., Pan, J., and Lee, M. H. (2008)
Cell Mol. Life Sci., 65, 3981-3999.
291.Ibrahim, M. M., Razmara, M., Nguyen, D.,
Donahue, R. J., Wubah, J. A., and Knudsen, T. B. (1998) Biochim.
Biophys. Acta, 1403, 254-264.
292.Ramanathan, A., Wang, C., and Schreiber, S. L.
(2005) Proc. Natl. Acad. Sci. USA, 102, 5992-5997.
293.Zhou, S., Kachhap, S., and Singh, K. K. (2003)
Mutagenesis, 18, 287-292.
294.Vousden, K. H. (2009) Biochem. Soc.
Trans., 37, 511-517.
295.Vousden, K. H., and Ryan, K. M. (2009) Nat.
Rev. Cancer, 9, 691-700.
296.Schwartzenberg-Bar-Yoseph, F., Armoni, M., and
Karnieli, E. (2004) Cancer Res., 64, 2627-2633.
297.Mathupala, S. P., Heese, C., and Pedersen, P.
L. (1997) J. Biol. Chem., 272, 22776-22780.
298.Smith, T. A. (2000) Br. J. Biomed. Sci.,
57, 170-178.
299.Corcoran, C. A., Huang, Y., and Sheikh, M. S.
(2006) Cancer Biol. Ther., 5, 1610-1613.
300.Lyakhov, I. G., Krishnamachari, A., and
Schneider, T. D. (2008) Nucleic Acids Res., 36,
3828-3833.
301.Tian, W. N., Braunstein, L. D., Apse, K., Pang,
J., Rose, M., Tian, X., and Stanton, R. C. (1999) Am. J.
Physiol., 276, C1121-1131.
302.Weintraub, H., Hauschka, S., and Tapscott, S.
J. (1991) Proc. Natl. Acad. Sci. USA, 88, 4570-4571.
303.Araki, N., Morimasa, T., Sakai, T., Tokuoh, H.,
Yunoue, S., Kamo, M., Miyazaki, K., Abe, K., Saya, H., and Tsugita, A.
(2000) Electrophoresis, 21, 1880-1889.
304.Jaksch, M., Paret, C., Stucka, R., Horn, N.,
Muller-Hocker, J., Horvath, R., Trepesch, N., Stecker, G., Freisinger,
P., Thirion, C., Muller, J., Lunkwitz, R., Rodel, G., Shoubridge, E.
A., and Lochmuller, H. (2001) Hum. Mol. Genet., 10,
3025-3035.
305.Okorokov, A. L., and Milner, J. (1999) Mol.
Cell Biol., 19, 7501-7510.
306.McLure, K. G., Takagi, M., and Kastan, M. B.
(2004) Mol. Cell Biol., 24, 9958-9967.
307.Warburg, O. (1956) Science, 123,
309-314.
308.Warburg, O., Posener, K., and Negelein, E.
(1924) Biochem. Zeitschrift., 152, 319-344.
309.Warburg, O. H. (1947) in Ideen zur
Fermentchemie der Tumoren, Abh. Deutschen Akad. Wissenschaften,
Berlin.
310.Bonnet, S., Archer, S. L., Allalunis-Turner,
J., Haromy, A., Beaulieu, C., Thompson, R., Lee, C. T., Lopaschuk, G.
D., Puttagunta, L., Harry, G., Hashimoto, K., Porter, C. J., Andrade,
M. A., Thebaud, B., and Michelakis, E. D. (2007) Cancer Cell,
11, 37-51.
311.Coghlan, A. (2007) New Scientist,
193, 13-13.
312.Dhar, S., and Lippard, S. J. (2009) Proc.
Natl. Acad. Sci. USA, 106, 22199-22204.
313.Wang, T., Marquardt, C., and Foker, J. (1976)
Nature, 261, 702-705.
314.Pfeiffer, T., Schuster, S., and Bonhoeffer, S.
(2001) Science, 292, 504-507.
315.Gillies, R. J., and Gatenby, R. A. (2007) J.
Bioenerg. Biomembr., 39, 251-257.
316.Ren, B., Yee, K. O., Lawler, J., and
Khosravi-Far, R. (2006) Biochim. Biophys. Acta, 1765,
178-188.
317.Kandt, R. S., Haines, J. L., Smith, M.,
Northrup, H., Gardner, R. J., Short, M. P., Dumars, K., Roach, E. S.,
Steingold, S., Wall, S., et al. (1992) Nat. Genet., 2,
37-41.
318.Van Slegtenhorst, M., de Hoogt, R., Hermans,
C., Nellist, M., Janssen, B., Verhoef, S., Lindhout, D., van den
Ouweland, A., Halley, D., Young, J., Burley, M., Jeremiah, S.,
Woodward, K., Nahmias, J., Fox, M., Ekong, R., Osborne, J., Wolfe, J.,
Povey, S., Snell, R. G., Cheadle, J. P., Jones, A. C., Tachataki, M.,
Ravine, D., Sampson, J. R., Reeve, M. P., Richardson, P., Wilmer, F.,
Munro, C., Hawkins, T. L., Sepp, T., Ali, J. B., Ward, S., Green, A.
J., Yates, J. R., Kwiatkowska, J., Henske, E. P., Short, M. P., Haines,
J. H., Jozwiak, S., and Kwiatkowski, D. J. (1997) Science,
277, 805-808.
319.Crino, P. B., Nathanson, K. L., and Henske, E.
P. (2006) N. Engl. J. Med., 355, 1345-1356.
320.Benvenuto, G., Li, S., Brown, S. J., Braverman,
R., Vass, W. C., Cheadle, J. P., Halley, D. J., Sampson, J. R.,
Wienecke, R., and DeClue, J. E. (2000) Oncogene, 19,
6306-6316.
321.Chong-Kopera, H., Inoki, K., Li, Y., Zhu, T.,
Garcia-Gonzalo, F. R., Rosa, J. L., and Guan, K. L. (2006) J. Biol.
Chem., 281, 8313-8316.
322.Gao, X., and Pan, D. (2001) Genes Dev.,
15, 1383-1392.
323.Huang, J., and Manning, B. D. (2008)
Biochem. J., 412, 179-190.
324.Guertin, D. A., and Sabatini, D. M. (2007)
Cancer Cell, 12, 9-22.
325.Sarbassov, D. D., Ali, S. M., and Sabatini, D.
M. (2005) Curr. Opin. Cell Biol., 17, 596-603.
326.Wullschleger, S., Loewith, R., and Hall, M. N.
(2006) Cell, 124, 471-484.
327.Sarbassov, D. D., Ali, S. M., Kim, D. H.,
Guertin, D. A., Latek, R. R., Erdjument-Bromage, H., Tempst, P., and
Sabatini, D. M. (2004) Curr. Biol., 14, 1296-1302.
328.Cybulski, N., and Hall, M. N. (2009) Trends
Biochem. Sci., 34, 620-627.
329.Yang, Q., and Guan, K. L. (2007) Cell
Res., 17, 666-681.
330.Cai, S. L., Tee, A. R., Short, J. D., Bergeron,
J. M., Kim, J., Shen, J., Guo, R., Johnson, C. L., Kiguchi, K., and
Walker, C. L. (2006) J. Cell Biol., 173, 279-289.
331.Jung, C. H., Ro, S. H., Cao, J., Otto, N. M.,
and Kim, D. H. (2010) FEBS Lett., 584, 1287-1295.
332.Corradetti, M. N., and Guan, K. L. (2006)
Oncogene, 25, 6347-6360.
333.Huang, J., Dibble, C. C., Matsuzaki, M., and
Manning, B. D. (2008) Mol. Cell Biol., 28, 4104-4115.
334.Feng, Z., Hu, W., de Stanchina, E., Teresky, A.
K., Jin, S., Lowe, S., and Levine, A. J. (2007) Cancer Res.,
67, 3043-3053.
335.Feng, Z., Zhang, H., Levine, A. J., and Jin, S.
(2005) Proc. Natl. Acad. Sci. USA, 102, 8204-8209.
336.Carling, D. (2004) Trends Biochem. Sci.,
29, 18-24.
337.Shaw, R. J. (2006) Curr. Opin. Cell
Biol., 18, 598-608.
338.Carling, D., Sanders, M. J., and Woods, A.
(2008) Int. J. Obes. (Lond.), 32, Suppl. 4, S55-59.
339.Suter, M., Riek, U., Tuerk, R., Schlattner, U.,
Wallimann, T., and Neumann, D. (2006) J. Biol. Chem.,
281, 32207-32216.
340.Yoo, L. I., Chung, D. C., and Yuan, J. (2002)
Nat. Rev. Cancer, 2, 529-535.
341.Jorgensen, S. B., and Rose, A. J. (2008)
Front. Biosci., 13, 5589-5604.
342.Inoki, K., Ouyang, H., Zhu, T., Lindvall, C.,
Wang, Y., Zhang, X., Yang, Q., Bennett, C., Harada, Y., Stankunas, K.,
Wang, C. Y., He, X., MacDougald, O. A., You, M., Williams, B. O., and
Guan, K. L. (2006) Cell, 126, 955-968.
343.Warden, S. M., Richardson, C., O’Donnell,
J., Jr., Stapleton, D., Kemp, B. E., and Witters, L. A. (2001)
Biochem. J., 354, 275-283.
344.Zeng, P. Y., and Berger, S. L. (2006) Cancer
Res., 66, 10701-10708.
345.Okoshi, R., Ozaki, T., Yamamoto, H., Ando, K.,
Koida, N., Ono, S., Koda, T., Kamijo, T., Nakagawara, A., and Kizaki,
H. (2008) J. Biol. Chem., 283, 3979-3987.
346.Jones, R. G., Plas, D. R., Kubek, S., Buzzai,
M., Mu, J., Xu, Y., Birnbaum, M. J., and Thompson, C. B. (2005) Mol.
Cell, 18, 283-293.
347.Buzzai, M., Jones, R. G., Amaravadi, R. K.,
Lum, J. J., DeBerardinis, R. J., Zhao, F., Viollet, B., and Thompson,
C. B. (2007) Cancer Res., 67, 6745-6752.
348.Lum, J. J., Bauer, D. E., Kong, M., Harris, M.
H., Li, C., Lindsten, T., and Thompson, C. B. (2005) Cell,
120, 237-248.
349.Brugarolas, J., Lei, K., Hurley, R. L.,
Manning, B. D., Reiling, J. H., Hafen, E., Witters, L. A., Ellisen, L.
W., and Kaelin, W. G., Jr. (2004) Genes Dev., 18,
2893-2904.
350.Budanov, A. V., and Karin, M. (2008)
Cell, 134, 451-460.
351.Maiuri, M. C., Malik, S. A., Morselli, E.,
Kepp, O., Criollo, A., Mouchel, P. L., Carnuccio, R., and Kroemer, G.
(2009) Cell Cycle, 8, 1571-1576.
352.Greer, E. L., Oskoui, P. R., Banko, M. R.,
Maniar, J. M., Gygi, M. P., Gygi, S. P., and Brunet, A. (2007) J.
Biol. Chem., 282, 30107-30119.
353.Greer, E. L., Dowlatshahi, D., Banko, M. R.,
Villen, J., Hoang, K., Blanchard, D., Gygi, S. P., and Brunet, A.
(2007) Curr. Biol., 17, 1646-1656.
354.Lee, J. H., Budanov, A. V., Park, E. J., Birse,
R., Kim, T. E., Perkins, G. A., Ocorr, K., Ellisman, M. H., Bodmer, R.,
Bier, E., and Karin, M. (2010) Science, 327,
1223-1228.
355.Nogueira, V., Park, Y., Chen, C. C., Xu, P. Z.,
Chen, M. L., Tonic, I., Unterman, T., and Hay, N. (2008) Cancer
Cell, 14, 458-470.
356.Sarbassov, D. D., Guertin, D. A., Ali, S. M.,
and Sabatini, D. M. (2005) Science, 307, 1098-1101.
357.Alessi, D. R., James, S. R., Downes, C. P.,
Holmes, A. B., Gaffney, P. R., Reese, C. B., and Cohen, P. (1997)
Curr. Biol., 7, 261-269.
358.Vogelstein, B., and Kinzler, K. W. (2004)
Nat. Med., 10, 789-799.
359.Stambolic, V., MacPherson, D., Sas, D., Lin,
Y., Snow, B., Jang, Y., Benchimol, S., and Mak, T. W. (2001) Mol.
Cell, 8, 317-325.
360.Padmanabhan, S., Mukhopadhyay, A., Narasimhan,
S. D., Tesz, G., Czech, M. P., and Tissenbaum, H. A. (2009)
Cell, 136, 939-951.
361.Carracedo, A., and Pandolfi, P. P. (2008)
Oncogene, 27, 5527-5541.
362.Hill, M. M., Clark, S. F., Tucker, D. F.,
Birnbaum, M. J., James, D. E., and Macaulay, S. L. (1999) Mol. Cell
Biol., 19, 7771-7781.
363.Young, C. D., and Anderson, S. M. (2008)
Breast Cancer Res., 10, 202.
364.Robey, R. B., and Hay, N. (2006)
Oncogene, 25, 4683-4696.
365.Berwick, D. C., Hers, I., Heesom, K. J., Moule,
S. K., and Tavare, J. M. (2002) J. Biol. Chem., 277,
33895-33900.
366.Mayo, L. D., and Donner, D. B. (2001) Proc.
Natl. Acad. Sci. USA, 98, 11598-11603.
367.Zhou, B. P., Liao, Y., Xia, W., Zou, Y., Spohn,
B., and Hung, M. C. (2001) Nat. Cell Biol., 3,
973-982.
368.Brunet, A., Bonni, A., Zigmond, M. J., Lin, M.
Z., Juo, P., Hu, L. S., Anderson, M. J., Arden, K. C., Blenis, J., and
Greenberg, M. E. (1999) Cell, 96, 857-868.
369.Brunet, A., Kanai, F., Stehn, J., Xu, J.,
Sarbassova, D., Frangioni, J. V., Dalal, S. N., DeCaprio, J. A.,
Greenberg, M. E., and Yaffe, M. B. (2002) J. Cell Biol.,
156, 817-828.
370.Gross, D. N., van den Heuvel, A. P., and
Birnbaum, M. J. (2008) Oncogene, 27, 2320-2336.
371.Ni, Y. G., Wang, N., Cao, D. J., Sachan, N.,
Morris, D. J., Gerard, R. D., Kuro, O. M., Rothermel, B. A., and Hill,
J. A. (2007) Proc. Natl. Acad. Sci. USA, 104,
20517-20522.
372.Scherz-Shouval, R., and Elazar, Z. (2007)
Trends Cell Biol., 17, 422-427.
373.Droge, W., and Kinscherf, R. (2008) Antiox.
Redox. Signal., 10, 661-678.
374.Shintani, T., and Klionsky, D. J. (2004)
Science, 306, 990-995.
375.Gozuacik, D., and Kimchi, A. (2007) Curr.
Top. Dev. Biol., 78, 217-245.
376.Mizushima, N., Levine, B., Cuervo, A. M., and
Klionsky, D. J. (2008) Nature, 451, 1069-1075.
377.Glick, D., Barth, S., and Macleod, K. F. (2010)
J. Pathol., 221, 3-12.
378.Fimia, G. M., and Piacentini, M. (2010) Cell
Mol. Life Sci., 67, 1581-1588.
379.Wang, R. C., and Levine, B. (2010) FEBS
Lett., 584, 1417-1426.
380.Maiuri, M. C., Galluzzi, L., Morselli, E.,
Kepp, O., Malik, S. A., and Kroemer, G. (2010) Curr. Opin. Cell
Biol., 22, 181-185.
381.Tasdemir, E., Chiara Maiuri, M., Morselli, E.,
Criollo, A., D’Amelio, M., Djavaheri-Mergny, M., Cecconi, F.,
Tavernarakis, N., and Kroemer, G. (2008) Autophagy, 4,
810-814.
382.Tasdemir, E., Maiuri, M. C., Galluzzi, L.,
Vitale, I., Djavaheri-Mergny, M., D’Amelio, M., Criollo, A.,
Morselli, E., Zhu, C., Harper, F., Nannmark, U., Samara, C., Pinton,
P., Vicencio, J. M., Carnuccio, R., Moll, U. M., Madeo, F.,
Paterlini-Brechot, P., Rizzuto, R., Szabadkai, G., Pierron, G.,
Blomgren, K., Tavernarakis, N., Codogno, P., Cecconi, F., and Kroemer,
G. (2008) Nat. Cell Biol., 10, 676-687.
383.Galluzzi, L., Morselli, E., Kepp, O., Maiuri,
M. C., and Kroemer, G. (2010) Cell Cycle, 9, 250-255.
384.Crighton, D., Wilkinson, S., and Ryan, K. M.
(2007) Autophagy, 3, 72-74.
385.Kerley-Hamilton, J. S., Pike, A. M.,
Hutchinson, J. A., Freemantle, S. J., and Spinella, M. J. (2007)
Biochim. Biophys. Acta, 1769, 209-219.
386.Kondo, Y., Kanzawa, T., Sawaya, R., and Kondo,
S. (2005) Nat. Rev. Cancer, 5, 726-734.
387.Crighton, D., O’Prey, J., Bell, H. S.,
and Ryan, K. M. (2007) Cell Death Differ., 14,
1071-1079.
388.Lomonosova, E., and Chinnadurai, G. (2008)
Oncogene, 27, Suppl. 1, S2-19.
389.Abida, W. M., and Gu, W. (2008) Cancer
Res., 68, 352-357.
390.Reef, S., Zalckvar, E., Shifman, O., Bialik,
S., Sabanay, H., Oren, M., and Kimchi, A. (2006) Mol. Cell,
22, 463-475.
391.Morselli, E., Tasdemir, E., Maiuri, M. C.,
Galluzzi, L., Kepp, O., Criollo, A., Vicencio, J. M., Soussi, T., and
Kroemer, G. (2008) Cell Cycle, 7, 3056-3061.
392.Tavernarakis, N., Pasparaki, A., Tasdemir, E.,
Maiuri, M. C., and Kroemer, G. (2008) Autophagy, 4,
870-873.
393.Hangen, E., Blomgren, K., Benit, P., Kroemer,
G., and Modjtahedi, N. (2010) Trends Biochem. Sci., 35,
278-287.
394.Stewart, C. L., Kaspar, P., Brunet, L. J.,
Bhatt, H., Gadi, I., Kontgen, F., and Abbondanzo, S. J. (1992)
Nature, 359, 76-79.
395.Hong, H., Takahashi, K., Ichisaka, T., Aoi, T.,
Kanagawa, O., Nakagawa, M., Okita, K., and Yamanaka, S. (2009)
Nature, 460, 1132-1135.
396.Utikal, J., Polo, J. M., Stadtfeld, M.,
Maherali, N., Kulalert, W., Walsh, R. M., Khalil, A., Rheinwald, J. G.,
and Hochedlinger, K. (2009) Nature, 460, 1145-1148.
397.Marion, R. M., Strati, K., Li, H., Murga, M.,
Blanco, R., Ortega, S., Fernandez-Capetillo, O., Serrano, M., and
Blasco, M. A. (2009) Nature, 460, 1149-1153.
398.Tyner, S. D., Venkatachalam, S., Choi, J.,
Jones, S., Ghebranious, N., Igelmann, H., Lu, X., Soron, G., Cooper,
B., Brayton, C., Hee Park, S., Thompson, T., Karsenty, G., Bradley, A.,
and Donehower, L. A. (2002) Nature, 415, 45-53.
399.Garcia-Cao, I., Garcia-Cao, M.,
Martin-Caballero, J., Criado, L. M., Klatt, P., Flores, J. M., Weill,
J. C., Blasco, M. A., and Serrano, M. (2002) Embo J., 21,
6225-6235.
400.Mendrysa, S. M., O’Leary, K. A., McElwee,
M. K., Michalowski, J., Eisenman, R. N., Powell, D. A., and Perry, M.
E. (2006) Genes Dev., 20, 16-21.
401.Van Heemst, D., Mooijaart, S. P., Beekman, M.,
Schreuder, J., de Craen, A. J., Brandt, B. W., Slagboom, P. E., and
Westendorp, R. G. (2005) Exp. Gerontol., 40, 11-15.
402.Bonafe, M., Salvioli, S., Barbi, C., Trapassi,
C., Tocco, F., Storci, G., Invidia, L., Vannini, I., Rossi, M., Marzi,
E., Mishto, M., Capri, M., Olivieri, F., Antonicelli, R., Memo, M.,
Uberti, D., Nacmias, B., Sorbi, S., Monti, D., and Franceschi, C.
(2004) Cell Death Differ., 11, 962-973.
403.Orsted, D. D., Bojesen, S. E., Tybjaerg-Hansen,
A., and Nordestgaard, B. G. (2007) J. Exp. Med., 204,
1295-1301.
404.Rodier, F., Campisi, J., and Bhaumik, D. (2007)
Nucleic Acids Res., 35, 7475-7484.
405.Holloszy, J. O., and Fontana, L. (2007) Exp.
Gerontol., 42, 709-712.
406.Blagosklonny, M. V. (2010) Cell Cycle,
9, 683-688.
407.Levine, B., and Kroemer, G. (2009) Cell
Death Differ., 16, 1-2.
408.Salminen, A., and Kaarniranta, K. (2009)
Trends Mol. Med., 15, 217-224.
409.Yen, W. L., and Klionsky, D. J. (2008)
Physiology (Bethesda), 23, 248-262.
410.Blagosklonny, M. V. (2008) Cell Cycle,
7, 3344-3354.
411.Blagosklonny, M. V. (2009) Cell Cycle,
8, 4055-4059.
412.Harrison, D. E., Strong, R., Sharp, Z. D.,
Nelson, J. F., Astle, C. M., Flurkey, K., Nadon, N. L., Wilkinson, J.
E., Frenkel, K., Carter, C. S., Pahor, M., Javors, M. A., Fernandez,
E., and Miller, R. A. (2009) Nature, 460, 392-395.
413.Anisimov, V. N., Berstein, L. M., Egormin, P.
A., Piskunova, T. S., Popovich, I. G., Zabezhinski, M. A., Kovalenko,
I. G., Poroshina, T. E., Semenchenko, A. V., Provinciali, M., Re, F.,
and Franceschi, C. (2005) Exp. Gerontol., 40,
685-693.
414.Anisimov, V. N., Berstein, L. M., Egormin, P.
A., Piskunova, T. S., Popovich, I. G., Zabezhinski, M. A., Tyndyk, M.
L., Yurova, M. V., Kovalenko, I. G., Poroshina, T. E., and Semenchenko,
A. V. (2008) Cell Cycle, 7, 2769-2773.
415.Anisimov, V. N., Egormin, P. A., Bershtein, L.
M., Zabezhinskii, M. A., Piskunova, T. S., Popovich, I. G., and
Semenchenko, A. V. (2005) Bull. Exp. Biol. Med., 139,
721-723.
416.Anisimov, V. N., Egormin, P. A., Piskunova, T.
S., Popovich, I. G., Tyndyk, M. L., Yurova, M. N., Zabezhinski, M. A.,
Anikin, I. V., Karkach, A. S., and Romanyukha, A. A. (2010) Cell
Cycle, 9, 188-197.
417.Selman, C., Tullet, J. M., Wieser, D., Irvine,
E., Lingard, S. J., Choudhury, A. I., Claret, M., Al-Qassab, H.,
Carmignac, D., Ramadani, F., Woods, A., Robinson, I. C., Schuster, E.,
Batterham, R. L., Kozma, S. C., Thomas, G., Carling, D., Okkenhaug, K.,
Thornton, J. M., Partridge, L., Gems, D., and Withers, D. J. (2009)
Science, 326, 140-144.
418.Howes, R. M. (2006) Ann. N. Y. Acad.
Sci., 1067, 22-26.
419.Radisavljevic, Z. M., and Gonzalez-Flecha, B.
(2004) J. Cell Biochem., 91, 1293-1300.
420.Bae, G. U., Seo, D. W., Kwon, H. K., Lee, H.
Y., Hong, S., Lee, Z. W., Ha, K. S., Lee, H. W., and Han, J. W. (1999)
J. Biol. Chem., 274, 32596-32602.
421.Lane, D. P., and Lain, S. (2002) Trends Mol.
Med., 8, S38-42.
422.Staples, O. D., Steele, R. J., and Lain, S.
(2008) Surgeon, 6, 240-243.
423.Selivanova, G. (2001) Curr. Opin. Invest.
Drugs, 2, 1136-1141.
424.Dumont, P., Leu, J. I., Della Pietra, A. C.,
3rd, George, D. L., and Murphy, M. (2003) Nat. Genet.,
33, 357-365.
425.Pietsch, E. C., Humbey, O., and Murphy, M. E.
(2006) Oncogene, 25, 1602-1611.
426.Whibley, C., Pharoah, P. D., and Hollstein, M.
(2009) Nat. Rev. Cancer, 9, 95-107.
427.Jun, H. J., Park, S. H., Lee, W. K., Choi, J.
E., Jang, J. S., Kim, E. J., Cha, S. I., Kim, D. S., Kam, S., Kim, C.
H., Kang, Y. M., Jung, T. H., and Park, J. Y. (2007) Mol.
Carcinog., 46, 100-105.
428.Vassilev, L. T., Vu, B. T., Graves, B.,
Carvajal, D., Podlaski, F., Filipovic, Z., Kong, N., Kammlott, U.,
Lukacs, C., Klein, C., Fotouhi, N., and Liu, E. A. (2004)
Science, 303, 844-848.
429.Feng, Z., Jin, S., Zupnick, A., Hoh, J., de
Stanchina, E., Lowe, S., Prives, C., and Levine, A. J. (2006)
Oncogene, 25, 1-7.
430.Bae, B. I., Xu, H., Igarashi, S., Fujimuro, M.,
Agrawal, N., Taya, Y., Hayward, S. D., Moran, T. H., Montell, C., Ross,
C. A., Snyder, S. H., and Sawa, A. (2005) Neuron, 47,
29-41.
431.Gudkov, A. V., and Komarova, E. A. (2005)
Biochem. Biophys. Res. Commun., 331, 726-736.
432.Komarov, P. G., Komarova, E. A., Kondratov, R.
V., Christov-Tselkov, K., Coon, J. S., Chernov, M. V., and Gudkov, A.
V. (1999) Science, 285, 1733-1737.
433.Strom, E., Sathe, S., Komarov, P. G., Chernova,
O. B., Pavlovska, I., Shyshynova, I., Bosykh, D. A., Burdelya, L. G.,
Macklis, R. M., Skaliter, R., Komarova, E. A., and Gudkov, A. V. (2006)
Nat. Chem. Biol., 2, 474-479.