REVIEW: Multiple DNA Damage Recognition Factors Involved in Mammalian Nucleotide Excision Repair
K. Sugasawa
Biosignal Research Center, Organization of Advanced Science and Technology, Kobe University, 1-1 Rokkodai, Nada-ku, Kobe, Hyogo 657-8501, Japan; fax: (+81) 78-803-5970; E-mail: ksugasawa@garnet.kobe-u.ac.jp
Received September 14, 2010; Revision received October 5, 2010
The nucleotide excision repair (NER) subpathway operating throughout the mammalian genome is a versatile DNA repair system that can remove a wide variety of helix-distorting base lesions. This system contributes to prevention of blockage of DNA replication by the lesions, thereby suppressing mutagenesis and carcinogenesis. Therefore, it is of fundamental significance to understand how the huge genome can be surveyed for occurrence of a small number of lesions. Recent studies have revealed that this difficult task seems to be accomplished through sequential actions of multiple DNA damage recognition factors, including UV-DDB, XPC, and TFIIH. Notably, these factors adopt completely different strategies to recognize DNA damage. XPC detects disruption and/or destabilization of the base pairing, which ensures a broad spectrum of substrate specificity for global genome NER. In contrast, UV-DDB directly recognizes particular types of lesions, such as UV-induced photoproducts, thereby vitally recruiting XPC as well as further extending the substrate specificity. After DNA binding by XPC, moreover, the helicase activity associated with TFIIH scans a DNA strand to make a final search for the presence of aberrant chemical modifications of DNA. The combination of these different strategies makes a crucial contribution to simultaneously achieving efficiency, accuracy, and versatility of the entire repair system.
KEY WORDS: nucleotide excision repair, DNA damage recognition, xeroderma pigmentosum, XPC, UV-DDB, TFIIHDOI: 10.1134/S0006297911010044
Abbreviations: AAF, N-acetyl-2-aminofluorene; BHD, β-hairpin domain; CPDs, cyclobutane pyrimidine dimers; CS, Cockayne syndrome; NER, nucleotide excision repair; PCNA, proliferating cell nuclear antigen; 6-4PPs, pyrimidine-pyrimidone (6-4) photoproducts; RFC, replication factor C; RPA, replication protein A; TGD, transglutaminase-homology domain; TTD, trichothiodystrophy; UV-DDB, UV-damaged DNA-binding protein consisting of two subunits (DDB1 and DDB2); XP, xeroderma pigmentosum.
Genomic DNA is highly susceptible to damage caused by its intrinsic
instability, endogenously-produced factors like reactive oxygen
species, and environmental genotoxic agents such as radiation and
chemicals. DNA damage can interfere with various nuclear functions
including replication and transcription, thereby inducing mutations,
chromosomal aberrations, apoptosis of cells, etc. These deleterious
effects of DNA damage have been implicated in diverse human pathologies
such as cancer, neurological degeneration, and progeria. To cope with
these adverse effects, organisms have acquired multiple DNA repair
pathways during evolution (for review, see [1]).
Nucleotide excision repair (NER) is one of the most versatile DNA repair pathways, which eliminates a wide variety of DNA helix-distorting base lesions. Those substrates for NER include ultraviolet light (UV)-induced dipyrimidinic photolesions, i.e. cyclobutane pyrimidine dimers (CPDs) and pyrimidine-pyrimidone (6-4) photoproducts (6-4PPs), as well as intrastrand cross-links and bulky base adducts that can be induced by numerous chemical compounds [2]. Notably, these base lesions handled by NER do not share any common chemical structure. In humans, hereditary defects in NER are associated with several autosomal recessive disorders such as xeroderma pigmentosum (XP), Cockayne syndrome (CS), and trichothiodystrophy (TTD). So far 10 NER-deficient genetic complementation groups (XP-A through -G; CS-A and -B; TTD-A) have been identified, and all the responsible genes are already cloned [3]. It is now known that the protein products encoded by these genes are components of the NER molecular machinery.
In general, initial recognition of DNA damage is a key event regulating the whole repair process, although it has remained to be understood how cells survey the huge genome continuously to detect and remove a small number of lesions. This article overviews protein factors and molecular mechanisms underlying the lesion recognition for mammalian NER operating throughout the genome.
MOLECULAR MECHANISM OF MAMMALIAN NER
Two subpathways have been discerned for mammalian NER: global genome NER (GG-NER) is a general pathway that can handle lesions anywhere in the genome, whereas transcription-coupled NER (TC-NER) is specialized to remove lesions from the transcribed strand of transcriptionally-active genes (Scheme 1; see color insert). These NER subpathways seem to differ in their strategies for damage recognition. In TC-NER, elongating RNA polymerase II, which stumbles at a lesion on the template DNA strand, is supposed to trigger NER [4, 5]. Two CS gene products, CSA and CSB proteins, are specifically required for this process [6, 7]. On the other hand, lesion recognition for GG-NER depends on protein factors that possess specific binding capacity to DNA sites containing damage. In mammals, DNA binding by the XPC protein complex is necessary to initiate the GG-NER process [8-10]. In addition, for certain types of lesions the UV-damaged DNA-binding protein (UV-DDB) appears to promote the damage recognition by XPC [11-13].
A model for the molecular mechanism of mammalian NER
Scheme 1
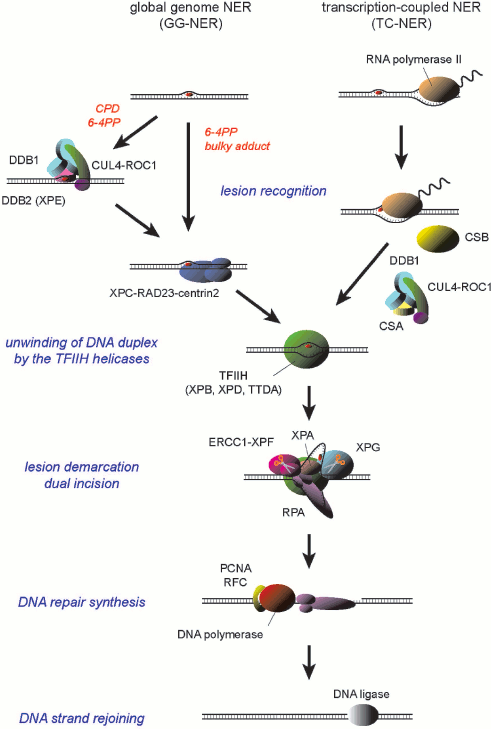
Despite the distinct molecular mechanisms underlying damage detection, the following steps of both subpathways depend on a common set of protein factors. One of the key factors is the transcription factor IIH (TFIIH), which is a multifunctional protein complex that is essential not only for NER but also for initiation of basal transcription. Among 10 subunits of TFIIH, XPB and XPD possess helicase activities [14, 15], which locally unwind DNA duplex around a lesion. The partially single-stranded state of DNA is stabilized through assembly of additional factors such as XPA, XPG, and replication protein A (RPA) [16-18], thereby demarcating the lesion for dual incision by two structure-specific NER endonucleases, ERCC1-XPF and XPG [19-21]. After removal of a 24-to-29-mer oligonucleotide containing the lesion, the resulting single-stranded gap is then filled by DNA repair synthesis. This process has been successfully reconstituted in vitro with DNA polymerase δ or ε (Pol δ/ε) in the presence of their elongation clamp, proliferating cell nuclear antigen (PCNA), and the clamp loader ATPase complex, replication factor C (RFC) [22, 23], whereas involvement of other DNA polymerases, such as Pol κ, has been also suggested [24, 25]. Final rejoining of the DNA strands may be accomplished by either DNA ligase I or XRCC1/DNA ligase III [23, 26, 27].
THE XPC COMPLEX: A VERSATILE DAMAGE DETECTOR FOR GG-NER
The human XPC gene encodes a 106-kDa basic protein (theoretical pI ~ 9.03) consisting of 940 amino acids. The XPC protein exists in vivo as a stable heterotrimeric complex containing one of the two human homologs of Saccharomyces cerevisiae Rad23p (RAD23A or RAD23B) and centrin-2, which has been also known as a crucial component of the centrosome [28-30]. The interaction with RAD23 markedly stabilizes the XPC protein both in vivo and in vitro, which is thus important to maintain cellular GG-NER activity [31-34]. In all human cell lines so far tested, RAD23B is always expressed at much higher levels than RAD23A, whereas these proteins in total are present in a large excess (~20-fold) over XPC [33, 34]. The two RAD23 proteins similarly bind to XPC in vitro, so that a majority of XPC is bound in vivo to RAD23B rather than RAD23A, presumably reflecting the difference in their protein levels [35, 36]. Centrin-2 is a small calcium-binding protein that belongs to the calmodulin superfamily and contains four conserved EF-hand motifs. An α-helix in the C-terminal domain of XPC has been identified as the site that binds to centrin-2 through hydrophobic interactions [37-39]. Although centrin-2 is dispensable for reconstitution of the cell-free NER reaction, it significantly enhances DNA binding and damage recognition functions of the XPC complex.
The purified XPC complex appears to have the ability to specifically bind to a variety of defined DNA lesions: for example, the UV-induced 6-4PP, a guanine modification with N-acetyl-2-aminofluorene (AAF), and an artificial cholesteryl moiety [9, 40, 41], which are completely different from each other in their chemical structure but, nevertheless, are excised by NER in vitro. The specific binding was demonstrated with conventional electrophoretic mobility shift and DNase I footprinting assays, as well as by direct observation using atomic force microscopy. Further extensive biochemical analyses eventually revealed that the XPC complex does not directly recognize the damaged DNA structures per se. Instead, it prefers some specific DNA secondary structures containing a junction between double-stranded DNA and a single-stranded 3′-overhang [42].
The proposed model for the XPC–DNA interaction, which was deduced from biochemical data, has been recently corroborated by the X-ray crystal structure of the complex involving the S. cerevisiae XPC homolog Rad4p and DNA duplex with a site-specific lesion (a UV-induced CPD within a 3-base bubble structure) [43]. Rad4p and XPC share a considerable (but not extensive) amino acid sequence homology, which is particularly pronounced within their C-terminal domains that are responsible for several important functions of these proteins, such as DNA binding [44]. From the structural point of view, the C-terminal domain can be further divided into four subdomains: transglutaminase-homology domain (TGD) and three consecutive β-hairpin domains (designated as BHD1, BHD2, and BHD3, respectively). The solved crystal structure indicates that TGD and BHD1 together interact with the undamaged part of the DNA duplex that is located 3′ to the damaged bases, while double-stranded DNA on the other side makes no contact with the proteins. On the other hand, BHD2 and BHD3 bind to a short DNA segment very close to the damaged site, mainly interacting with the undamaged strand. As a result, two undamaged bases opposite the lesion are flipped out from DNA, being captured by BHD2 and BHD3. In addition, insertion of the BHD3 β-hairpin into the major groove induces a ~42° bend of the DNA, so that the damaged bases seem to be also flipped out and structurally disordered without any interaction with the protein. Therefore, these findings perfectly coincide with the previous notion that XPC binds to the end of double-stranded DNA with a 3′-overhang.
Taken together with the biochemical and structural studies, the XPC complex should be considered as a structure-specific DNA binding factor rather than a damaged DNA binding factor. Its specific binding depends solely on the presence of some bases that are prevented from the canonical Watson–Crick base pairing, but not on any particular chemical structure of the damaged DNA. Thus it has been plausibly approved that such biochemical properties of XPC provide a molecular basis for the extremely broad substrate specificity exhibited by GG-NER.
FUNCTIONS OF UV-DDB IN GG-NER DAMAGE RECOGNITION
Since XPC recognizes the presence of unpaired bases, its binding affinity for a certain lesion appears to vary depending on the degree of DNA helical distortion induced by the lesion. Notably, a major UV-induced photolesion, CPD, is associated with only a subtle structural distortion: in fact, previous NMR studies revealed that if a CPD is present in DNA duplex, both affected pyrimidine residues still retain hydrogen bonding with the opposite purine bases [45]. As a result, XPC by itself poorly recognizes the CPD [41, 46], and this problem is overcome, at least partially, by the aid of UV-DDB, another damage recognition factor involved in GG-NER.
UV-DDB was initially identified as a protein factor that strongly binds to UV-irradiated DNA [47]. Later the protein factor responsible for this binding activity was purified, which revealed that UV-DDB is a complex that consists of two subunits, designated as DDB1 and DDB2, respectively [48]. DNA binding assays with substrates containing a defined lesion indicate that purified UV-DDB recognizes UV-induced 6-4PP with particularly high specificity and, unlike XPC, it also exhibits moderate but significant affinity for CPD [49-53]. Moreover, UV-DDB is also capable of binding to abasic sites as well as bubble-like DNA structures of relatively small size, although the functional relevance of such binding still remains to be elucidated.
The recently solved X-ray crystal structure of UV-DDB has provided novel insights to the damage recognition mechanism by UV-DDB [54]. The DDB1 subunit is composed of three consecutive β-propeller domains (designated as BPA, BPB, and BPC, respectively), while DDB2 also contains a β-propeller that is exclusively responsible for interaction with DNA. DDB2 has an N-terminal extension with a helix-loop-helix structure, which is involved in interaction with DDB1. Upon binding to DNA containing a 6-4PP, a conserved hairpin on one end of the DDB2 β-propeller is inserted into the minor groove, inducing a ~40° bend of the DNA duplex. As a result, the two pyrimidine residues comprising a 6-4PP are flipped out into a binding pocket of DDB2. Notably, this binding pocket appears to be sized to accommodate two nucleotides, strongly suggesting that UV-DDB has evolved especially to cope with UV-induced dipyrimidinic photolesions. Therefore, in a striking contrast to XPC, UV-DDB seems to be a specialized damage detector that directly interacts with the damaged bases themselves.
It has been shown that the genetic complementation group E of XP is caused by mutations in the DDB2 gene, so that cells from XP-E patients lack the specific DNA binding activity [55-57]. On the contrary, no mutation has been identified in the DDB1 gene from patients with XP or other NER-deficient disorders. Despite the obvious biochemical activity, cellular and clinical phenotypes associated with the lack of UV-DDB are complicated and confusing in some sense. Among the XP complementation groups, XP-E patients show the mildest symptoms and, in line with this, fibroblast cells from the patients only show mild sensitivity to killing by UV and relatively high levels of UV-induced unscheduled DNA synthesis (~50% of normal cells) [58, 59]. The latter phenotype is probably due to competence of XP-E cells to remove 6-4PPs at nearly normal levels, whereas repair of CPD is compromised more profoundly: this appears paradoxical if one considers the binding nature of UV-DDB to these UV photolesions. It should be noted that UV-DDB is not an essential factor to reconstitute the in vitro NER reaction with purified proteins, although it may have some stimulatory effects under certain conditions [22, 60]. On the other hand, no GG-NER occurs in XP-C cells that normally express UV-DDB, indicating that UV-DDB never functionally substitutes for the essential factor XPC, but rather plays some auxiliary roles in damage recognition for GG-NER.
The technique of UV irradiation through micropore membrane filters has been used to induce local DNA damage within the nucleus and visualize recruitment of NER factors to the damaged sites. Notably, UV-DDB accumulates into the damaged subnuclear domain even in the absence of XPC, indicating that it functions at very early stages of GG-NER [61]. More detailed analyses using a similar technique revealed that UV-DDB indeed facilitates recruitment of XPC as well as other NER proteins to the sites containing UV-induced DNA lesions [11-13]. In line with this, it has been shown that UV-DDB directly interacts with XPC [52]. Although interaction between UV-DDB and XPA has been also reported [62], XPA does not accumulate to the local UV damage in the absence of functional XPC, even if UV-DDB is normally expressed. Nevertheless, the UV-DDB-mediated recruitment of XPC is obviously relevant to repair of CPD, because this lesion easily escapes detection by XPC as discussed above.
In contrast to CPD, quite some number of previous works on in vivo NER kinetics failed to show considerable effects of UV-DDB on removal of 6-4PP. However, several recent studies have indicated that UV-DDB can indeed facilitate recruitment of XPC to 6-4PP as well, which becomes discernible especially when the number of generated lesions is low enough [12, 63]. Taken together, it can be concluded that UV-DDB facilitates recognition for GG-NER of both major UV photolesions, CPD and 6-4PP, through vital recruitment of XPC: this mechanism is particularly relevant when cells are exposed to physiologically low doses of UV.
ROLES FOR CUL4 UBIQUITIN LIGASE IN GG-NER
As mentioned above, UV-DDB was first purified as the DDB1-DDB2 heterodimer but, more recently it has been shown to interact in vivo with several additional factors. One of such factors is the E3 ubiquitin ligase complex, which is composed of a member of the cullin family (CUL4A), a RING domain protein (RBX/ROC1), and the COP9 signalosome complex [64]. Notably, the CSA protein, which is involved in the TC-NER pathway of NER, also turned out to bind DDB1 as well as the CUL4A E3 complex. Although DDB1 was originally identified as part of the DNA damage binding factor, it is currently considered as an adaptor mediating between CUL4 ubiquitin ligase and a member of the DCAF (DDB1-CUL4 associating factor) family, which interacts with specific targets for ubiquitylation [65]. Besides DDB2 and CSA, a number of DCAFs have been so far identified.
After cells are treated with UV, UV-DDB becomes tightly bound to chromatin and the associating ubiquitin ligase seems to be activated as judged from conjugation of CUL4A to NEDD8 and dissociation of the COP9 signalosome [64]. The recombinant DDB1–DDB2–CUL4A–ROC1 complex was expressed in and purified from insect cells, which was used for reconstitution of in vitro ubiquitylation reactions involving the ubiquitin activating enzyme (E1) and the ubiquitin conjugating enzyme (E2) [52]. In this system XPC, DDB2, and CUL4A are subject to polyubiquitylation. In fact, it has been demonstrated that XPC undergoes transient ubiquitylation upon UV irradiation of cells, which however seems to be reversible, not inducing degradation of the protein. In a striking contrast, DDB2 is clearly degraded by the 26S proteasome in UV-irradiated cells [66-68]. When DDB2 is polyubiquitylated in vitro, UV-DDB appears to be prevented from interaction with the UV photolesions (both CPD and 6-4PP), whereas ubiquitylated XPC still exhibits DNA binding capacity. Based on these findings, it has been proposed that one of the roles for the DDB2 ubiquitylation may be promoting transfer of the UV photolesion from UV-DDB (a strong binder) to XPC (a weak binder) (Scheme 2; see color insert). Nevertheless, the biological significance of XPC ubiquitylation as well as DDB2 degradation remains to be understood.
Possible roles of UV-DDB-CUL4 ligase-mediated ubiquitylation in damage recognition for GG-NER. When UV-DDB recognizes and binds to a lesion within a nucleosome core, the associating CUL4 ubiquitin ligase may ubiquitylate histones. This may lead to dissociation of the histone octamer, thereby allowing XPC to get access to the lesion. Then the ubiquitin ligase may further ubiquitylate XPC and DDB2, presumably promoting transfer of the lesion from UV-DDB to XPC
Scheme 2
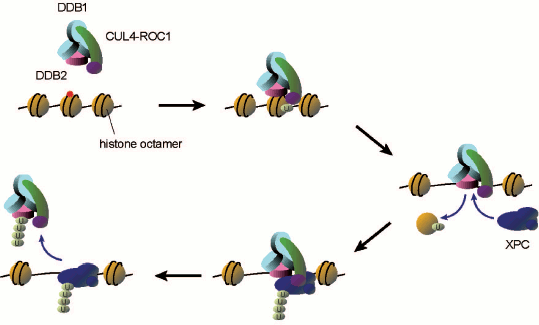
The CUL4A ubiquitin ligase associated with UV-DDB has been implicated also in UV-induced ubiquitylation of histones, H2A [69] and/or H3/H4 [70]. It has been reported that the monoubiquitylation of H3 and H4 in vitro results in dissociation of histone octamers from DNA (Scheme 2). Notably, if DNA damage is located within a nucleosome core, accessibility to XPC as well as other NER proteins is severely impaired [71-73], suggesting that roles for UV-DDB and the associating ubiquitin ligase may involve remodeling of chromatin structures around the lesion site. In addition, histone acetyltransferases CBP/p300 associate in vivo with UV-DDB [68, 74], which may thus coordinate multiple post-translational modifications of histones (and/or other proteins) to prepare the local chromatin environment for efficient repair reactions.
More recently, Cul4a-deficient mice have been generated, which provide novel insights into the roles of this E3 component in regulation of NER and other cellular UV responses [75]. In addition to the GG-NER proteins, the DDB1-CUL4A ligases are also involved in ubiquitylation of various proteins such as a DNA replication licensing factor CDT1 [76, 77] as well as a DNA damage checkpoint regulator p21 [78-80]. Despite these crucial targets for ubiquitylation, animals with Cul4a deletion turned out apparently healthy. This is due, at least partly, to functional redundancy between Cul4a and Cul4b, another CUL4 ortholog present in mammals, so that additional suppression of Cul4b expression resulted in growth arrest of Cul4a–/– fibroblasts. Surprisingly, on the other hand, disruption of the Cul4a gene alone results in marked elevation of the steady-state protein levels of DDB2, XPC, and p21. Consequently, cellular GG-NER and DNA damage checkpoint activities are simultaneously enhanced, while mice with skin-specific Cul4a abrogation exhibit extraordinary hyper-resistance to UVB-induced skin tumors. Thus CUL4A seems to limit both GG-NER and damage checkpoint activities by mediating constitutive degradation of the key factors, although the biological relevance of these findings still remains to be understood. Since the UV-induced XPC ubiquitylation does not seem to induce its degradation, the reason for the increased XPC protein levels in the Cul4a-deficient cells should also be clarified.
BIPARTITE SUBSTRATE DISCRIMINATION FOR GG-NER
Although UV-DDB intervenes in recognition of UV-induced photolesions, direct binding by XPC is a more general pathway for initiation of GG-NER as a versatile repair system, which can handle numerous chemically-induced bulky adducts, for instance. As discussed above, XPC only samples the presence of disrupted or destabilized base pairs within DNA duplex, but not any specific chemical structures associated with lesions. On the other hand, a series of biochemical studies have proposed the “bipartite” substrate discrimination model for NER, telling that efficient substrates for in vitro NER must possess at least two structural elements: one is disruption of the canonical Watson–Crick base pairing (this is exactly the determinant of the XPC binding specificity), and the other is some aberrant chemical modification of DNA (this is generically referred to as “damage”).
This notion originated from in vitro studies using the C4′ pivaloyl adduct to a deoxyribose residue in the DNA backbone [81]. Although this structure itself, which hardly interferes with base pairing nearby, is poorly excised in the cell-free NER system, the same lesion becomes a good substrate if placed in the close vicinity of a small bubble structure. Later we showed that XPC efficiently recognizes and binds to some perturbed DNA structures with a single-stranded segment, such as bubbles and loops, regardless of the presence or absence of damage [41]. However, in vitro NER with the same set of DNA substrates demonstrated that dual incision absolutely depends on the presence of DNA damage. These findings all together plausibly point that, to induce the productive NER process, DNA binding by XPC is not sufficient and the presence of damage needs to be somehow verified thereafter.
The second damage verification step is important as a “safety lock” of the NER system, which prevents adverse incision at undamaged DNA sites that can even challenge the genome integrity. However, the precise molecular mechanism underlying the damage verification has been elusive. One of the factors that have been implicated in this process is TFIIH, which contains two helicase subunits, XPB and XPD, and is probably recruited by XPC through direct interaction [82-84]. In general, helicases are supposed to bind a DNA strand and translocate along it by using the energy of ATP hydrolysis, thereby displacing the complementary strand. Based on such properties, the helicase activities associated with TFIIH seem to be suitable for scanning a DNA strand and detecting subtle alteration of the chemical structure: just like DNA/RNA polymerases that are blocked by a lesion on the template DNA strand. Indeed, it was reported that the strand displacement as well as ATPase activities of the S. cerevisiae XPD homolog, Rad3p, were inhibited when the DNA substrates were damaged [85], suggesting that its translocation along a DNA strand is blocked by the presence of lesions. This blockage of the TFIIH helicase translocation may underlie the damage verification for NER.
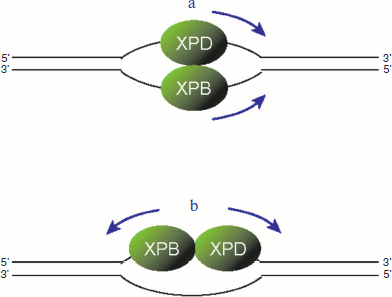
It should be noted that the two helicases associated with TFIIH have different polarities: XPB moves on a DNA strand from 3′ to 5′ direction, whereas XPD exhibits the opposite 5′ to 3′ directionality [86, 87]. These biochemical natures raise the possibility that, after TFIIH is recruited to the site of damage, the two helicases may associate with different DNA strands and move towards the same direction (figure, panel (a); see color insert). In this model, the damaged and undamaged strands can be discriminated by which helicase is blocked [88, 89]. Another possibility is that both helicases may bind to the same strand and move apart from each other (figure, panel (b); see color insert). In this case, the damaged strand needs to be discriminated before the TFIIH helicases are loaded.
Two possible models for translocation by TFIIH helicases. a) The XPB and XPD helicases may bind to different strands and move toward the same direction. b) Alternatively, the two helicases may bind to the same strand and move to the opposite directions
THE NER MACHINERY CAN SEARCH FOR A LESION
As discussed in the previous section, the NER machinery first samples disrupted base pairs and then verifies if any abnormal chemical structure is present. Unexpectedly, we recently found that these two events are spatially separable [90]. This finding was based on a series of in vitro NER reactions using defined DNA substrates that contained both a UV-induced photolesion and a small bubble or loop structure with a certain distance in between. Since CPD induced only a marginal distortion of the DNA helical structure, it was poorly recognized and excised by in vitro NER. However, the same lesion became excised very efficiently when a 3-base bubble structure was inserted about 60 bases 5′ to it. Footprinting analyses revealed that XPC was targeted to the bubble site, rather than to the CPD site, indicating that the NER machinery has a potential to find a lesion at a rather distal position from the site where XPC initially binds. Surprisingly, this stimulatory effect on excision of CPD was completely abrogated when a bubble structure was present 3′ to the lesion. The observed position specificity could be plausibly explained if one assumes that a certain NER factor may search for a lesion by moving along a specific DNA strand in 5′ to 3′ direction from the XPC-bound site. It should be noted that this directionality of the damage search coincides with the polarity of the XPD helicase, as discussed above, supporting the role of this helicase in damage verification. Furthermore, these findings strongly argue against the model that the XPB and XPD helicases may scan individual DNA strands, thereby equally contributing to discrimination between the damaged and undamaged strands.
Given that only one DNA strand is subjected to scanning for damage, it is particularly important to understand how the correct (damaged) strand can be selected. This depends, at least partly, on the binding polarity of XPC. As suggested from our biochemical data [42] as well as from the crystal structure of Rad4p [43], a bubble structure allows XPC to bind in two distinct orientations, whereas the binding is constrained in a defined polarity if a loop structure is used, in which only one strand has unpaired bases. When a 3-base loop, instead of a bubble, was inserted 5′ to a CPD, stimulation of in vitro NER incision at the CPD site was observed only when the “undamaged” strand was looped out. In striking contrast, excision of CPD was totally abrogated if the lesion-containing strand had a loop. We propose that, after XPC interacts with a single-stranded segment, it may guide the XPD helicase in TFIIH to be loaded onto the opposite strand and start scanning. Notably, although this model is based on biochemical studies using somewhat artificial substrates, it can be plausibly extrapolated to the normal NER reaction, where XPC needs to recognize and interact with widowed bases opposite a lesion. In addition, a series of biochemical studies revealed that precise incision sites by NER varied depending on the type of lesions (15-24 nucleotides 5′ and 2-8 nucleotides 3′ to a lesion, respectively), while the total length of the excised oligonucleotides was largely constant (24-29 nucleotides) [91-93]. Given that a sliding complex, presumably driven by the XPD helicase, unwinds DNA duplex and collides with a lesion from the 5′ side, the above model also explains the asymmetric dual incision pattern observed with eukaryotic NER.
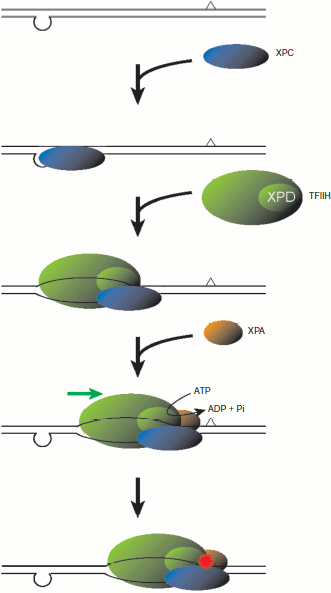
Assembly of NER factors at the CPD site, which was stimulated by a distal loop structure, could be observed with purified proteins and the defined DNA substrate immobilized on paramagnetic beads [90]. In these experiments, not only TFIIH but also XPC and XPA seemed to be present at the CPD site in a mutually dependent manner, suggesting that these three factors may translocate along a DNA strand as a ternary complex (Scheme 3; see color insert). This model implies that, even though XPC initially binds to a suspicious site in a “wrong” orientation, the site is cleared upon recruitment of TFIIH and XPA, thereby providing XPC with a chance to retry. Exactly as expected, the ATPase activity of XPD was required for translocation by the NER factors from the loop site to CPD, while ATP hydrolysis by XPB also appeared to be important. The intrinsic helicase activity associated with XPB appeared much weaker than that of XPD [86], and mutational analyses of XPB in the recombinant TFIIH complex have recently revealed that its ATPase, but not helicase activity is necessary for in vitro NER [94]. These results suggest that energy produced by the XPB ATPase may be consumed mainly in opening of DNA duplex, rather than in translocation on a DNA strand. This is quite reminiscent of its role in the promoter opening for transcriptional initiation, whereas the ATPase activity of XPD is known to be dispensable for the transcriptional function of TFIIH [95]. There are at least two possibilities for the role of the XPB ATPase in NER. First, upon recruitment of TFIIH by XPC, XPB may unwind DNA duplex to some extent, which may then allows loading of XPD onto a specific strand. Alternatively, after the XPD helicase encounters and stumbles at a lesion, XPB may further open the duplex, thereby promoting assembly and/or stability of the pre-incision complex.
A model for the molecular mechanism underlying damage search and verification. First, XPC recognizes the presence of unpaired bases (in the bottom strand, in this figure) and binds 5′ to the distorted site. Then TFIIH is loaded and, presumably together with XPC and XPA, scans the opposite (top) strand in 5′ to 3′ direction, which is mainly driven by the XPD helicase. When the translocation by XPD is blocked by some aberrant modification of the DNA chemical structure, the presence of a lesion is verified, thereby leading to the subsequent lesion demarcation and dual incision.
Scheme 3
As discussed above, damage recognition for NER in the mammalian global genome is accomplished through concerted actions of multiple protein factors including XPC, UV-DDB, and TFIIH. It should be noted that these factors rely upon completely different criteria from each other to detect abnormalities of DNA structure. XPC only recognizes the presence of unpaired bases, regardless whether DNA is indeed damaged or not, thereby conferring the broad substrate specificity on the whole system. In contrast, UV-DDB directly interacts with specific types of modified bases, especially dinucleotide photolesions induced by UV irradiation, and promotes recruitment of XPC. Since XPC alone poorly recognizes CPD, UV-DDB helps further extending the substrate specificity of GG-NER. Finally, the XPD helicase in TFIIH scans a DNA strand in 5′ to 3′ direction to make a fine inspection for unusual chemical modification. Once the movement of XPD is blocked, presence of damage is verified, leading to the subsequent demarcation and excision of the lesion. The combination of these different strategies must make a crucial contribution to simultaneously achieving efficiency, accuracy, and versatility of the repair system.
Even though XPC happens to bind a DNA site without damage, the NER machinery seems to have the potential to search around for damage, raising the possibility that this could function as a damage surveillance system in cells that are not particularly treated with DNA damaging agents. For instance, some AT-rich regions in the genome, including replication origins and transcriptional promoters, may open spontaneously under topological stresses caused by altered chromatin structures, thereby possibly serving as “docking sites” for XPC. Moreover, XPC is able to bind to the abasic site [96] and probably to the single strand break, both of which are the most frequent types of spontaneous DNA damage. UV-DDB also exhibits a considerable affinity for the abasic site [49, 53, 54], which could be relevant in recruiting XPC and initiating a damage search. Together with prerequisite remodeling of chromatin structure, further studies would be necessary to understand in vivo regulation of NER.
REFERENCES
1.Friedberg, E. C., Walker, G. C., Siede, W., Wood,
R. D., Schultz, R. A., and Ellenberger, T. (2006) DNA Repair and
Mutagenesis, Second Edition, ASM Press, Washington, DC.
2.Gillet, L. C., and Scharer, O. D. (2006) Chem.
Rev., 106, 253-276.
3.Bootsma, D., Kraemer, K. H., Cleaver, J. E., and
Hoeijmakers, J. H. J. (2001) in The Metabolic and Molecular Basis of
Inherited Disease, Vol. 1 (Scriver, C. R., Beaudet, A. L., Sly, W.
S., and Valle, D., eds.) McGraw-Hill Book Co., New York, NY, pp.
677-703.
4.Svejstrup, J. Q. (2002) Nat. Rev. Mol. Cell.
Biol., 3, 21-29.
5.Tornaletti, S., and Hanawalt, P. C. (1999)
Biochimie, 81, 139-146.
6.Van Hoffen, A., Natarajan, A. T., Mayne, L. V., van
Zeeland, A. A., Mullenders, L. H. F., and Venema, J. (1993) Nucleic
Acids Res., 21, 5890-5895.
7.Venema, J., Mullenders, L. H. F., Natarajan, A. T.,
van Zeeland, A. A., and Mayne, L. V. (1990) Proc. Natl. Acad. Sci.
USA, 87, 4707-4711.
8.Riedl, T., Hanaoka, F., and Egly, J.-M. (2003)
EMBO J., 22, 5293-5303.
9.Sugasawa, K., Ng, J. M. Y., Masutani, C., Iwai, S.,
van der Spek, P. J., Eker, A. P. M., Hanaoka, F., Bootsma, D., and
Hoeijmakers, J. H. J. (1998) Mol. Cell, 2, 223-232.
10.Volker, M., Mone, M. J., Karmakar, P., van
Hoffen, A., Schul, W., Vermeulen, W., Hoeijmakers, J. H. J., van Driel,
R., van Zeeland, A. A., and Mullenders, L. H. F. (2001) Mol.
Cell, 8, 213-224.
11.Fitch, M. E., Nakajima, S., Yasui, A., and Ford,
J. M. (2003) J. Biol. Chem., 278, 46906-46910.
12.Moser, J., Volker, M., Kool, H., Alekseev, S.,
Vrieling, H., Yasui, A., van Zeeland, A. A., and Mullenders, L. H. F.
(2005) DNA Repair (Amst.), 4, 571-582.
13.Wang, Q. E., Zhu, Q., Wani, G., Chen, J., and
Wani, A. A. (2004) Carcinogenesis, 25, 1033-1043.
14.Schaeffer, L., Moncollin, V., Roy, R., Staub, A.,
Mezzina, M., Sarasin, A., Weeda, G., Hoeijmakers, J. H. J., and Egly,
J.-M. (1994) EMBO J., 13, 2388-2392.
15.Schaeffer, L., Roy, R., Humbert, S., Moncollin,
V., Vermeulen, W., Hoeijmakers, J. H. J., Chambon, P., and Egly, J.-M.
(1993) Science, 260, 58-63.
16.Evans, E., Fellows, J., Coffer, A., and Wood, R.
D. (1997) EMBO J., 16, 625-638.
17.Evans, E., Moggs, J. G., Hwang, J. R., Egly,
J.-M., and Wood, R. D. (1997) EMBO J., 16, 6559-6573.
18.Mu, D., Wakasugi, M., Hsu, D. S., and Sancar, A.
(1997) J. Biol. Chem., 272, 28971-28979.
19.Cloud, K. G., Shen, B., Strniste, G. F., and
Park, M. S. (1995) Mutat. Res., 347, 55-60.
20.O’Donovan, A., Davies, A. A., Moggs, J. G.,
West, S. C., and Wood, R. D. (1994) Nature, 371,
432-435.
21.Sijbers, A. M., de Laat, W. L., Ariza, R. R.,
Biggerstaff, M., Wei, Y.-F., Moggs, J. G., Carter, K. C., Shell, B. K.,
Evans, E., de Jong, M. C., Rademakers, S., de Rooij, J., Jaspers, N. G.
J., Hoeijmakers, J. H. J., and Wood, R. D. (1996) Cell,
86, 811-822.
22.Aboussekhra, A., Biggerstaff, M., Shivji, M. K.
K., Vilpo, J. A., Moncollin, V., Podust, V. N., Protic, M., Hubscher,
U., Egly, J.-M., and Wood, R. D. (1995) Cell, 80,
859-868.
23.Araujo, S. J., Tirode, F., Coin, F., Pospiech,
H., Syvaoja, J. E., Stucki, M., Hubscher, U., Egly, J.-M., and Wood, R.
D. (2000) Genes Dev., 14, 349-359.
24.Ogi, T., and Lehmann, A. R. (2006) Nat. Cell
Biol., 8, 640-642.
25.Ogi, T., Limsirichaikul, S., Overmeer, R. M.,
Volker, M., Takenaka, K., Cloney, R., Nakazawa, Y., Niimi, A., Miki,
Y., Jaspers, N. G. J., Mullenders, L. H. F., Yamashita, S., Fousteri,
M. I., and Lehmann, A. R. (2010) Mol. Cell, 37,
714-727.
26.Moser, J., Kool, H., Giakzidis, I., Caldecott,
K., Mullenders, L. H. F., and Fousteri, M. I. (2007) Mol. Cell,
27, 311-323.
27.Shivji, M. K. K., Podust, V. N., Hubscher, U.,
and Wood, R. D. (1995) Biochemistry, 34, 5011-5017.
28.Araki, M., Masutani, C., Takemura, M., Uchida,
A., Sugasawa, K., Kondoh, J., Ohkuma, Y., and Hanaoka, F. (2001) J.
Biol. Chem., 276, 18665-18672.
29.Masutani, C., Sugasawa, K., Yanagisawa, J.,
Sonoyama, T., Ui, M., Enomoto, T., Takio, K., Tanaka, K., van der Spek,
P. J., Bootsma, D., Hoeijmakers, J. H. J., and Hanaoka, F. (1994)
EMBO J., 13, 1831-1843.
30.Shivji, M. K. K., Eker, A. P. M., and Wood, R. D.
(1994) J. Biol. Chem., 269, 22749-22757.
31.Batty, D., Rapic’-Otrin, V., Levine, A. S.,
and Wood, R. D. (2000) J. Mol. Biol., 300, 275-290.
32.Ng, J. M. Y., Vermeulen, W., van der Horst, G. T.
J., Bergink, S., Sugasawa, K., Vrieling, H., and Hoeijmakers, J. H. J.
(2003) Genes Dev., 17, 1630-1645.
33.Okuda, Y., Nishi, R., Ng, J. M. Y., Vermeulen,
W., van der Horst, G. T. J., Mori, T., Hoeijmakers, J. H. J., Hanaoka,
F., and Sugasawa, K. (2004) DNA Repair (Amst.), 3,
1285-1295.
34.Sugasawa, K., Masutani, C., Uchida, A., Maekawa,
T., van der Spek, P. J., Bootsma, D., Hoeijmakers, J. H. J., and
Hanaoka, F. (1996) Mol. Cell. Biol., 16, 4852-4861.
35.Masutani, C., Araki, M., Sugasawa, K., van der
Spek, P. J., Yamada, A., Uchida, A., Maekawa, T., Bootsma, D.,
Hoeijmakers, J. H. J., and Hanaoka, F. (1997) Mol. Cell. Biol.,
17, 6915-6923.
36.Sugasawa, K., Ng, J. M. Y., Masutani, C.,
Maekawa, T., Uchida, A., van der Spek, P. J., Eker, A. P. M.,
Rademakers, S., Visser, C., Aboussekhra, A., Wood, R. D., Hanaoka, F.,
Bootsma, D., and Hoeijmakers, J. H. J. (1997) Mol. Cell. Biol.,
17, 6924-6931.
37.Bunick, C. G., Miller, M. R., Fuller, B. E.,
Fanning, E., and Chazin, W. J. (2006) Biochemistry, 45,
14965-14979.
38.Nishi, R., Okuda, Y., Watanabe, E., Mori, T.,
Iwai, S., Masutani, C., Sugasawa, K., and Hanaoka, F. (2005) Mol.
Cell. Biol., 25, 5664-5674.
39.Popescu, A., Miron, S., Blouquit, Y., Duchambon,
P., Christova, P., and Craescu, C. T. (2003) J. Biol. Chem.,
278, 40252-40261.
40.Janicijevic, A., Sugasawa, K., Shimizu, Y.,
Hanaoka, F., Wijgers, N., Djurica, M., Hoeijmakers, J. H. J., and
Wyman, C. (2003) DNA Repair (Amst.), 2, 325-336.
41.Sugasawa, K., Okamoto, T., Shimizu, Y., Masutani,
C., Iwai, S., and Hanaoka, F. (2001) Genes Dev., 15,
507-521.
42.Sugasawa, K., Shimizu, Y., Iwai, S., and Hanaoka,
F. (2002) DNA Repair (Amst.), 1, 95-107.
43.Min, J.-H., and Pavletich, N. P. (2007)
Nature, 449, 570-575.
44.Legerski, R., and Peterson, C. (1992)
Nature, 359, 70-73.
45.McAteer, K., Jing, Y., Kao, J., Taylor, J.-S.,
and Kennedy, M. A. (1998) J. Mol. Biol., 282,
1013-1032.
46.Kusumoto, R., Masutani, C., Sugasawa, K., Iwai,
S., Araki, M., Uchida, A., Mizukoshi, T., and Hanaoka, F. (2001)
Mutat. Res., 485, 219-227.
47.Chu, G., and Chang, E. (1988) Science,
242, 564-567.
48.Keeney, S., Chang, G. J., and Linn, S. (1993)
J. Biol. Chem., 268, 21293-21300.
49.Fujiwara, Y., Masutani, C., Mizukoshi, T., Kondo,
J., Hanaoka, F., and Iwai, S. (1999) J. Biol. Chem., 274,
20027-20033.
50.Payne, A., and Chu, G. (1994) Mutat. Res.,
310, 89-102.
51.Reardon, J. T., Nichols, A. F., Keeney, S.,
Smith, C. A., Taylor, J. S., Linn, S., and Sancar, A. (1993) J.
Biol. Chem., 268, 21301-21308.
52.Sugasawa, K., Okuda, Y., Saijo, M., Nishi, R.,
Matsuda, N., Chu, G., Mori, T., Iwai, S., Tanaka, K., Tanaka, K., and
Hanaoka, F. (2005) Cell, 121, 387-400.
53.Wittschieben, B. O., Iwai, S., and Wood, R. D.
(2005) J. Biol. Chem., 280, 39982-39989.
54.Scrima, A., Konickova, R., Czyzewski, B. K.,
Kawasaki, Y., Jeffrey, P. D., Groisman, R., Nakatani, Y., Iwai, S.,
Pavletich, N. P., and Thoma, N. H. (2008) Cell, 135,
1213-1223.
55.Itoh, T., Linn, S., Ono, T., and Yamaizumi, M.
(2000) J. Invest. Dermatol., 114, 1022-1029.
56.Nichols, A. F., Ong, P., and Linn, S. (1996)
J. Biol. Chem., 271, 24317-24320.
57.Rapic-Otrin, V., Navazza, V., Nardo, T., Botta,
E., McLenigan, M., Bisi, D. C., Levine, A. S., and Stefanini, M. (2003)
Hum. Mol. Genet., 12, 1507-1522.
58.Keeney, S., Eker, A. P. M., Brody, T., Vermeulen,
W., Bootsma, D., Hoeijmakers, J. H. J., and Linn, S. (1994) Proc.
Natl. Acad. Sci. USA, 91, 4053-4056.
59.Rapic Otrin, V., Kuraoka, I., Nardo, T.,
McLenigan, M., Eker, A. P., Stefanini, M., Levine, A. S., and Wood, R.
D. (1998) Mol. Cell. Biol., 18, 3182-3190.
60.Wakasugi, M., Shimizu, M., Morioka, H., Linn, S.,
Nikaido, O., and Matsunaga, T. (2001) J. Biol. Chem.,
276, 15434-15440.
61.Wakasugi, M., Kawashima, A., Morioka, H., Linn,
S., Sancar, A., Mori, T., Nikaido, O., and Matsunaga, T. (2002) J.
Biol. Chem., 277, 1637-1640.
62.Wakasugi, M., Kasashima, H., Fukase, Y., Imura,
M., Imai, R., Yamada, S., Cleaver, J. E., and Matsunaga, T. (2009)
Nucleic Acids Res., 37, 516-525.
63.Nishi, R., Alekseev, S., Dinant, C., Hoogstraten,
D., Houtsmuller, A. B., Hoeijmakers, J. H. J., Vermeulen, W., Hanaoka,
F., and Sugasawa, K. (2009) DNA Repair (Amst.), 8,
767-776.
64.Groisman, R., Polanowska, J., Kuraoka, I.,
Sawada, J., Saijo, M., Drapkin, R., Kisselev, A. F., Tanaka, K., and
Nakatani, Y. (2003) Cell, 113, 357-367.
65.Lee, J., and Zhou, P. (2007) Mol. Cell,
26, 775-780.
66.Chen, X., Zhang, J., Lee, J., Lin, P. S., Ford,
J. M., Zheng, N., and Zhou, P. (2006) Mol. Cell, 22,
489-499.
67.Fitch, M. E., Cross, I. V., Turner, S. J.,
Adimoolam, S., Lin, C. X., Williams, K. G., and Ford, J. M. (2003)
DNA Repair (Amst.), 2, 819-826.
68.Rapic’-Otrin, V., McLenigan, M. P., Bisi,
D. C., Gonzalez, M., and Levine, A. S. (2002) Nucleic Acids
Res., 30, 2588-2598.
69.Kapetanaki, M. G., Guerrero-Santoro, J., Bisi, D.
C., Hsieh, C. L., Rapic-Otrin, V., and Levine, A. S. (2006) Proc.
Natl. Acad. Sci. USA, 103, 2588-2593.
70.Wang, H., Zhai, L., Xu, J., Joo, H. Y., Jackson,
S., Erdjument-Bromage, H., Tempst, P., Xiong, Y., and Zhang, Y. (2006)
Mol. Cell, 22, 383-394.
71.Hara, R., Mo, J., and Sancar, A. (2000) Mol.
Cell. Biol., 20, 9173-9181.
72.Hara, R., and Sancar, A. (2002) Mol. Cell.
Biol., 22, 6779-6787.
73.Yasuda, T., Sugasawa, K., Shimizu, Y., Iwai, S.,
Shiomi, T., and Hanaoka, F. (2005) DNA Repair (Amst.), 4,
389-395.
74.Datta, A., Bagchi, S., Nag, A., Shiyanov, P.,
Adami, G. R., Yoon, T., and Raychaudhuri, P. (2001) Mutat. Res.,
486, 89-97.
75.Liu, L., Lee, S., Zhang, J., Peters, S. B.,
Hannah, J., Zhang, Y., Yin, Y., Koff, A., Ma, L., and Zhou, P. (2009)
Mol. Cell, 34, 451-460.
76.Higa, L. A., Mihaylov, I. S., Banks, D. P.,
Zheng, J., and Zhang, H. (2003) Nat. Cell Biol., 5,
1008-1015.
77.Hu, J., McCall, C. M., Ohta, T., and Xiong, Y.
(2004) Nat. Cell Biol., 6, 1003-1009.
78.Abbas, T., Sivaprasad, U., Terai, K., Amador, V.,
Pagano, M., and Dutta, A. (2008) Genes Dev., 22,
2496-2506.
79.Kim, Y., Starostina, N. G., and Kipreos, E. T.
(2008) Genes Dev., 22, 2507-2519.
80.Nishitani, H., Shiomi, Y., Iida, H., Michishita,
M., Takami, T., and Tsurimoto, T. (2008) J. Biol. Chem.,
283, 29045-29052.
81.Hess, M. T., Schwitter, U., Petretta, M., Giese,
B., and Naegeli, H. (1997) Proc. Natl. Acad. Sci. USA,
94, 6664-6669.
82.Araujo, S. J., Nigg, E. A., and Wood, R. D.
(2001) Mol. Cell. Biol., 21, 2281-2291.
83.Li, R.-Y., Calsou, P., Jones, C. J., and Salles,
B. (1998) J. Mol. Biol., 281, 211-218.
84.Yokoi, M., Masutani, C., Maekawa, T., Sugasawa,
K., Ohkuma, Y., and Hanaoka, F. (2000) J. Biol. Chem.,
275, 9870-9875.
85.Naegeli, H., Bardwell, L., and Friedberg, E. C.
(1992) J. Biol. Chem., 267, 392-398.
86.Coin, F., Marinoni, J. C., Rodolfo, C., Fribourg,
S., Pedrini, A. M., and Egly, J.-M. (1998) Nat. Genet.,
20, 184-188.
87.Sung, P., Bailly, V., Weber, C., Thompson, L. H.,
Prakash, L., and Prakash, S. (1993) Nature, 365,
852-855.
88.Dip, R., Camenisch, U., and Naegeli, H. (2004)
DNA Repair (Amst.), 3, 1409-1423.
89.Wood, R. D. (1999) Biochimie, 81,
39-44.
90.Sugasawa, K., Akagi, J., Nishi, R., Iwai, S., and
Hanaoka, F. (2009) Mol. Cell, 36, 642-653.
91.Matsunaga, T., Mu, D., Park, C. H., Reardon, J.
T., and Sancar, A. (1995) J. Biol. Chem., 270,
20862-20869.
92.Moggs, J. G., Yarema, K. J., Essigmann, J. M.,
and Wood, R. D. (1996) J. Biol. Chem., 271,
7177-7186.
93.Svoboda, D. L., Taylor, J. S., Hearst, J. E., and
Sancar, A. (1993) J. Biol. Chem., 268, 1931-1936.
94.Coin, F., Oksenych, V., and Egly, J.-M. (2007)
Mol. Cell, 26, 245-256.
95.Winkler, G. S., Araujo, S. J., Fiedler, U.,
Vermeulen, W., Coin, F., Egly, J.-M., Hoeijmakers, J. H. J., Wood, R.
D., Timmers, H. T., and Weeda, G. (2000) J. Biol. Chem.,
275, 4258-4266.
96.Shimizu, Y., Iwai, S., Hanaoka, F., and Sugasawa,
K. (2003) EMBO J., 22, 164-173.