REVIEW: Interspecies Transmission of Prions
E. G. Afanasieva, V. V. Kushnirov, and M. D. Ter-Avanesyan*
Bach Institute of Biochemistry, Russian Academy of Sciences, Leninsky pr. 33, 119071 Moscow, Russia; E-mail: mdter@inbi.ras.ru* To whom correspondence should be addressed.
Received February 21, 2011; Revision received March 21, 2011
Mammalian prions are infectious agents of proteinaceous nature that cause several incurable neurodegenerative diseases. Interspecies transmission of prions is usually impeded or impossible. Barriers in prion transmission are caused by small interspecies differences in the primary structure of prion proteins. The barriers can also depend on the strain (variant) of a transmitted prion. Interspecies barriers were also shown for yeast prions, which define some heritable phenotypes. Yeast prions reproduce all the main traits of prion transmission barriers observed for mammals. This allowed to show that the barrier in prion transmission can be observed even upon copolymerization of two prionogenic proteins. Available data allow elucidation of the mechanisms that impede prion transmission or make it impossible.
KEY WORDS: interspecies barrier, prions, PrP, [PSI+], yeast, Saccharomyces cerevisiaeDOI: 10.1134/S0006297911130013
Abbreviations: a.a., amino acid residue; PRNP, gene which encodes the PrP protein; PrP, mammalian prion protein; PrPC, normal cellular form of the prion protein; PrPSc, infectious form of the prion protein.
Prions were initially discovered as infectious agents of proteinaceous
nature. They cause transmissible spongiform encephalopathies in animals
and humans, such as Creutzfeldt–Jacob disease (CJD),
Gerstmann–Sträussler–Scheinker syndrome, and kuru in
humans, as well as sheep scrapie and bovine spongiform encephalopathy
(mad cow disease). These incurable diseases are accompanied by
morphological changes in brain tissue that involve accumulation of
amyloid-like structures. Despite the fact that some of these diseases
have been known for a long time (CJD – for around 100 years,
sheep scrapie – since the middle of 18th century), their
etiology became well understood only in the middle of the 20th
century.
Observation of the similar neuropathology of CJD, kuru, and scrapie has played a considerable role in the understanding of the etiology of these diseases. In 1959 W. Hadlow hypothesized that kuru, a disease that was widespread among the cannibalistic aboriginal population of New Guinea, was infectious. He proposed to verify this hypothesis by intracerebral inoculations of chimpanzees with brain homogenates obtained from deceased kuru patients [1]. This hypothesis was later confirmed by D. Gaidushek and his coauthors, who observed that chimpanzees inoculated with kuru-patient brain homogenates indeed developed a disease which was similar to scrapie in all of its pathological manifestations [2]. It was subsequently shown that scrapie was also a transmissible disease, since it could be transmitted to mice [3].
At about the same time it was observed that the causative agent of scrapie is unusually resistant to various treatments that inactivate most known viruses and bacteria, such as high temperature, formaldehyde fixation [4], and irradiation with UV light [5]. This led to the conclusion that the scrapie infectious agent can replicate in the absence of nucleic acids [6]. In 1967 D. Griffith proposed several hypotheses concerning the spread and transmission of these diseases, including what would later be known as the prion hypothesis: “…subunits can polymerize only in the presence of polymeric “condensation nuclei” [7]. Purification of the infectious material identified an agent with an approximate molecular mass of 27-30 kDa, whose major component was of a proteinaceous nature. The infectivity of this agent was partially inactivated by proteinase K, urea, and other agents that could disrupt protein structure. This agent was termed a prion (from proteinaceous infectious particle) and the protein was called PrP (Prion Protein) [8]. Identification of the PRNP gene, which encodes PrP [9], showed that a specific conformation of the protein, rather than the presence of the protein itself, was responsible for the disease. Later on, homologs of the PRNP gene were found not only in mammals, but also in birds [10] and fish [11]. Figure 1 shows an alignment of the amino acid sequences of PrP from several mammalian species.
The prion protein PrP is anchored on the external side of the cellular membrane via glycosylphosphatidylinositol and is expressed mostly in nervous and lymphoreticular tissue [12]. The function of this protein is still not clear, though recent work has demonstrated the role of neuronal PrPC in the formation of the myelin sheaths of nerve fibers [13].Fig. 1. Comparison of the primary structures of PrP proteins (without the signal sequence) from several mammalian species and of Sup35 from four species of Saccharomyces yeast. Amino acid sequences of the human PrP protein and the prion domain of the S. cerevisiae Sup35 protein are presented. Residues that differ in the proteins of other species are listed in the appropriate positions. Methionine-129 is highlighted in the human PrP protein.

The prion isoform of the PrP protein (PrPSc, where Sc stands for scrapie) differs from the normal, non-prion cellular form (PrPC, where C stands for cellular) by its secondary structure. PrPC is rich in α-helical regions and does not have a significant amount of β-structure, whereas the PrPSc form is mainly β-structured [14]. The infectious form of PrP, whether it arrived into an organism from the outside or was generated de novo, facilitates the transformation of normal PrPC into the pathogenic PrPSc, which is resistant to proteolysis and accumulates in brain tissues in the form of plaques consisting of rod-like or fibril-like aggregates of the protein.
One of the fundamental features of prions is their ability to exist in a range of different variants (strains), which differ in the conformation of the prion form of the PrP protein. Strains of PrPSc with different conformations show variation in the character of the prion disease: incubation periods, clinical manifestations, and brain damage patterns can all differ. These strain-specific variations are stably propagated in vivo. This means that if laboratory animals are infected with different strains of PrPSc, the same strain of prion that was used for infection will be maintained and propagated in these animals [15, 16].
INTERSPECIES TRANSMISSION OF PRION DISEASES
The ability of prions to be transmitted between different species was used to confirm the transmissibility of prion diseases and also allowed the creation of efficient experimental models in laboratory animals such as mice and hamsters. Experiments with these models showed that spongiform encephalopathies were most efficiently transmitted between animals of the same species or between closely related species. For instance, CJD can be transmitted between humans and it can also be transmitted from humans to chimpanzees; scrapie can be transmitted from sheep to goats, but cannot be transmitted to either chimpanzees or humans [8]. Bovine prion diseases can be transmitted to humans via meat consumption, though with low probability. Thus, though the interspecies transmission of prion diseases is possible, it is often limited by interspecies barriers. The term “interspecies barrier” may indicate the impossibility of prion transmission, a considerable increase in the incubation period of the disease after the infectious agent has been transmitted, or a decrease in the probability of disease transmission [17]. One of the first published works on interspecies prion transmission showed that the incubation period of the disease in hamsters injected intracerebrally with mink-derived infectious material was over 600 days, which is several times longer than the incubation period observed during infection with hamster-derived material [18].
Obviously, interspecies prion transmission barriers may be due to the differences in the amino acid sequences of PrP proteins. This hypothesis was verified experimentally. It was demonstrated that hamster prions efficiently infect transgenic mice with a hamster PRNP gene but do not infect wild-type mice [19]. Notably, the prion transmission barrier is not always symmetrical. There are cases in which prions cannot be transmitted from one species to another, while the barrier is non-existent or very weak in the reverse direction. For instance, infection of Syrian hamsters with mouse-derived prion material resulted in the emergence of pathological symptoms after 378 days [20, 21], while transmission of infectious material from Syrian hamsters to mice did not result in any pathological symptoms even after two years [22].
Barriers of interspecies prion transmission and their asymmetry can be observed in vitro. For instance, in a cell-free system hamster PrPC adopted a protease-resistant prion-like state only in the presence of PrPSc mouse protein. However, in the reverse experiment, where mouse PrPC was incubated in the presence of hamster PrPSc, the level of prion conversion was insignificant, which was in agreement with the data obtained in vivo [23].
It should be noted that the efficiency of interspecies transmission of the disease and duration of the incubation period can depend on the strain of the prion [24]. For instance, two different mink-derived isolates of the PrPSc prion were inoculated into Syrian hamsters and were observed to cause different incubation periods as well as characteristic brain damage patterns. In one case the disease was manifested 65 days after inoculation and was accompanied by hyperesthesia and cerebellar ataxia, while in the other case the disease was manifested only after 168 days with lethargic symptoms and no cerebellar ataxia. Probably the most illustrative example of the strain-dependence of prion transmission was observed during the use of laboratory animals for modeling a new human prion disease – nvCJD (a novel variant of CJD which appeared in humans after infection with mad cow disease). While conventional CJD is not easily transmitted to wild-type mice, it can efficiently be transmitted to transgenic mice producing human PrP. What is especially surprising, the nvCJD variant was more easily transmitted to wild-type mice as compared to mice homozygous for the human PRNP gene, despite the fact that the prion strains were maintained by the same protein, i.e. human PrP [25].
Thus, efficiency of interspecies prion transmission depends on the prion strain. However, sometimes interspecies transmission can alter the characteristics of the prion. For example, when sheep scrapie is transmitted to mice with an intermediary passage in white rats, the resulting scrapie symptoms are different compared to those observed after a direct transmission from sheep to mice [26]. These observations indicated that the infectious agent can experience changes in a new host, though the nature of these induced changes is not clear. As more and more cases of this scrapie “mutation” were observed [27], it became apparent that such changes of prion characteristics were not an exceptional occurrence [18, 27].
Variability of prions can facilitate the appearance of their heterogeneity that can be detected during interspecies transmission. The infectious agent extracted from a single mink with transmissible encephalopathy was introduced into seven Chinese hamsters, which manifested pathological symptoms 600 days after intracerebral inoculation. Subsequent inoculation of healthy hamsters with brain homogenates derived from hamsters from the first passage demonstrated two distinct strains of transmissible encephalopathy that differed in their incubation periods. Moreover, these two strains also showed different patterns of brain damage [28].
Prion heterogeneity was also observed during passages in cell cultures [29]. Two different cell lines of murine neuroblastoma N2a (PK1 and R33) were infected with brain homogenate from the same diseased mouse. After several generations, the characteristics of the original infectious agent changed in the PK1 cells: PK1-derived prions could no longer infect R33 cells, while the original prions extracted from the mouse brain could still do so. Cell cultures could be “cured” of the prion disease by swainsonine – an inhibitor of the Golgi apparatus α-mannosidase II, which is involved in the synthesis of N-linked glycans, though the sensitivity to this compound depends on the prion strain. It was observed that PK1 cells cultivated in the absence of swainsonine lost prions after the compound was added to the medium, while prions present in cells of the same line that were cultured in the presence of swainsonine were resistant to this treatment. This means that the original population of prions was initially heterogeneous and the cultivation conditions influenced the propagation of the different strains.
AMINO ACID SUBSTITUTIONS IN THE PrP PROTEIN THAT AFFECT PRION
TRANSMISSION
Experimental infection of two mustelid species, the black weasel and the mink, with mink-transmissible encephalopathy showed that the two species had different sensitivity to the infectious agent: the incubation periods for the weasels were 28-38 months and 4 months for the minks. The primary structures of the mink and black weasel PrP proteins differ by two amino acid residues: the mink PrP residues phenylalanine-179 and arginine-224 are substituted by lysine and glutamine respectively in weasels [30]. Notably, not only interspecies variations in amino acid sequences, but also allelic variants of PRNP can affect the efficiency of prion disease transmission. For instance, sheep homozygous for the PRNP allele that encodes PrP with a valine residue in position 136 are more susceptible to scrapie than sheep homozygous for the allele which encodes an alanine at the same position [31], while animals homozygous for the allele that encodes a protein with an arginine-171 residue are resistant to scrapie infection [31, 32].
Study of the PRNP gene allele frequencies in cannibalistic New Guinea aboriginal tribes shows that in humans the susceptibility to prion disease is considerably influenced by the valine/methionine polymorphism in position 129 of the PrP protein (Fig. 1): heterozygosity for these two alleles seems to increase resistance to kuru [33]. Apart from that, substitution of glycine for valine at position 127 increased kuru resistance in people homozygous for the methionine codon 129 [34]. Interestingly, this PrP polymorphism seems to affect interspecies prion transmission as well, since cases of nvCJD disease have only been observed in people homozygous for the methionine-129 PRNP allele [35]. Experiments using transgenic mice have demonstrated that the human valine-129 PrP protein cannot sustain the prion state characteristic of the mad cow disease strain, and infection of such mice is accompanied by alterations in the prion strain characteristics [36]. On the other hand, substitution of one amino acid residue in the PrP protein by another can lead to the formation of a protein that is not only incapable of taking on the PrPSc state, but also inhibits its propagation [37].
Thus, susceptibility to various forms of transmissible encephalopathy can depend not only on the interspecies variations in the amino acid sequence of the prion proteins, but also on the intraspecies polymorphism, with some amino acid substitutions increasing the susceptibility to infection and others lowering the probability of transmission.
PRIONS OF LOWER EUKARYOTES
Studies of the interspecies prion transmission barrier in animals are expensive as well as labor- and time-consuming. The use of mammalian cell cultures is more productive for these studies, but there are some difficulties with obtaining stable cell lines with PrP in the prion state. Finally, even though in vitro experiments provide information on the interactions between PrPSc and PrPC from various species, these data are not always in agreement with data obtained in vivo [38].
Apart from mammals, prions have also been discovered in lower eukaryotes, where they mediate the inheritance of phenotypic traits, thus acting as genetic determinants. Currently there are 10 known prion determinants in Saccharomyces cerevisiae yeast as well as one determinant in the filamentous fungus Podospora anserina. These genetic determinants are based on the prion properties of proteins with no common structure or functions and have different phenotypic manifestations. Overproduction of yeast prionogenic proteins facilitates the appearance of the corresponding prion determinant, and this appearance is usually dependent on the presence of the [PIN+] prion, which does not have a phenotypic manifestation of its own [39, 40].
Most of the prion determinants of lower eukaryotes are based on the ability of the respective proteins to form amyloid polymers that are resistant to strong detergents such as SDS or sarcosyl. Such polymers differ from other proteinaceous aggregates by their ability to catalyze their own growth, thus acting as a template or seed for protein polymerization. Prion inheritance is critically dependent on the activity of the Hsp104 chaperone and its co-chaperones that fragment prion fibrils and thus propagate them and increase the overall polymerization of the prion protein [41, 42]. In yeast, non-heritable detergent-resistant amyloids were also observed that are formed during the overproduction of Sup35 in cells bearing the [PIN+] prion. These amyloids are not fragmented by the Hsp104 chaperone, and thus they cannot be inherited and exist only due to efficient cross-seeding by [PIN+] prion polymers [43]. It can be supposed that recognition of yeast prion polymers by the Hsp104 chaperone and their subsequent fragmentation depend on the specific fold of the prion domains, which exposes certain hydrophobic amino acid residues [44].
Unlike mammalian prions, yeast prions do not cause cell death. Nevertheless, the issue of their biological role remains open. Some researchers presume that the prion conversion of a protein is an anomaly that disrupts the normal folding of a protein molecule, and thus prions are considered to be harmful due to the fact that the cell loses some functions due to protein aggregation and deactivation [45]. On the other hand, conversion of some proteins into a prion state may confer adaptive advantages to yeast cells [46-50].
As an object of study, yeast prions present a number of obvious advantages as compared to mammalian prions: experiments with yeast prions consume considerably less time and are not dangerous to researchers. Studies of yeast prions have provided conclusive evidence of the prion hypothesis and allow the modeling of most features of mammalian prions, such as strain variability and interspecies transmission barriers.
YEAST MODELS FOR THE STUDY OF INTERSPECIES TRANSMISSION
BARRIERS
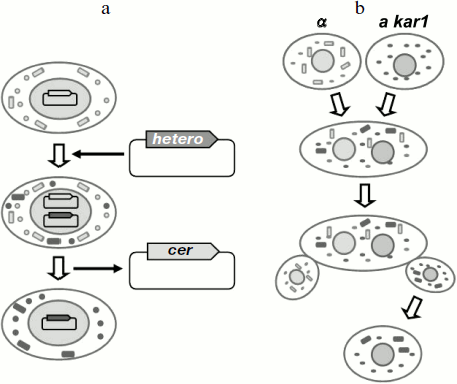
In yeast, interspecies prion transmission is usually studied using S. cerevisiae cells that harbor either the wild-type prion gene or its homolog from a different yeast species. Thus, in fact these experiments study the transmission of the prion state between proteins from different species in S. cerevisiae cells. Transmission of the prion state is accomplished by one of the following methods (Fig. 2). The first method is based on plasmid shuffling. Cells containing a prion determinant harbor a prion gene on a centromeric (single copy) plasmid, while the chromosomal copy of the gene is absent. Then the centromeric plasmid encoding the heterologous gene is introduced into the cells, and the original plasmid is lost. If the prion determinant persists after this procedure, then the protein encoded by the heterologous gene has received the prion state, i.e. prion transmission was successful. The second method is based on cytoduction – a method, in which tested yeast cells are mated with mutants defective for karyogamy. Then the cells fuse, but the nuclei remain unfused. During such fusion, the heterologous prion protein produced by the recipient cell comes into contact with the prion in the donor cell and can thus acquire the prion state. Subsequent mitotic division yields haploid cells bearing the nucleus of either one or the other mating partners. Transmission of the prion is monitored by the presence of the prion determinant in cells bearing the recipient nucleus.
Although at present many yeast prions are known, the interspecies transmission barriers have been demonstrated only for two of them, [PSI+] and [URE3] [51-57].Fig. 2. Methods for analyzing interspecies transmission of prions in S. cerevisiae yeast. a) A method based on plasmid shuffling; b) a method based on cytoduction; a and α, yeast mating types; kar1, a mutation which prevents karyogamy. Rectangles depict prion polymers, circles – the non-prion (monomeric) form of the prionogenic protein. Cer, a plasmid encoding the S. cerevisiae protein; hetero, a plasmid encoding the heterologous prionogenic protein.
INTERSPECIES TRANSMISSION OF [URE3]
The [URE3] determinant represents the prion state of the Ure2 protein, which is a transcriptional regulator of nitrogen catabolite repression. The prionogenic domain of Ure2 is an amino-terminal sequence (a.a. 1-94) that is rich in asparagine and glutamine residues [58], while the carboxyl-terminal domain (a.a. 95-354) is responsible for catabolite repression [59]. Transition of the N-terminal domain of Ure2 into the prion state, known as [URE3], inactivates the protein, which in turn activates the transcription of genes involved in the transport of “non-preferable” sources of nitrogen, which is repressed in the presence of “preferable” nitrogen sources, such as ammonium sulfate or glutamine. Thus, [URE3] cells can be phenotypically detected by their ability to utilize ureidosuccinate from medium rich in ammonium salts. Currently there are also alternative methods for the phenotypic detection of [URE3].
Ure2 proteins from closely related yeast species of the Saccharomyces genus have been shown to recreate most of the traits of interspecies transmission barriers observed for PrP proteins [57]. Notably, the level of identity between the Ure2 proteins was similar to that of the PrP proteins from different mammalian species. Specifically, it was demonstrated that the efficiency of [URE3] transmission could vary from 0 to almost 100% depending on the difference between the amino acid sequences of the interacting proteins and the variant (in yeast prion strains are usually termed variants) of the prion to be transmitted. Interestingly, the conformation of the transmitted prion can be maintained by the heterologous recipient protein. This was observed during the transmission of the prion state from the S. cerevisiae Ure2 protein to the S. mikatae Ure2 protein and then back onto S. cerevisiae Ure2. Thus, the S. mikatae Ure2 could seemingly maintain and propagate the prion conformation that it received from the S. cerevisiae protein.
The results of these studies of the prion transmission barrier on the Ure2 protein confirm the utility of using a yeast model for elucidating the fundamental characteristics of interspecies transmission barriers in mammals. However, the most comprehensive studies of the interspecies transmission of prions were conducted using the [PSI+] prion.
[PSI+] PRION
The [PSI+] determinant is probably the best-studied yeast prion. [PSI+] has a nonsense-suppressor phenotype, which is due to the aggregation and partial inactivation of the Sup35 (eRF3) translation termination factor [41, 60, 61]. The Sup35 protein consists of three domains [62]. Its amino-terminal N domain (a.a. 1-123) is responsible for the prion properties of the protein. This domain is unusually rich in asparagine and glutamine residues (almost 50% combined). This domain allows the protein to polymerize in [PSI+] cells [63, 64]. The Sup35 prion domain can be divided into two structurally distinct areas. The beginning of the prion domain (a.a. 1-40) is especially rich in asparagine and glutamine, while the remaining area (a.a. 41-97) contains five and a half imperfect oligopeptide repeats. The carboxyl-terminal C domain (a.a. 254-685) is responsible for the translation termination activity of the protein. The middle M domain (a.a. 124-253) is rich in charged residues: 42% of lysine and glutamate combined. The role of this domain remains unclear; it does not have a definite structure and is probably a spacing element between the N and C domains [65]. The N and M domains are much less conserved than the C domain. Similarly to mammalian prions and the yeast [URE3] and [PIN+] determinants, [PSI+] can exist in distinct variants with different properties [66, 67]. These differences reflect the distinct conformations of Sup35 in prion polymers [68, 69].
The modular structure of Sup35 is very convenient for using [PSI+] as an instrument for the analysis of the prionogenic potential of other proteins. For this purpose, the prion domain of Sup35 is exchanged for a different polypeptide sequence, and this chimeric protein is then tested for its ability to form a prion with a suppressor phenotype similar to [PSI+] [70]. Exchange of the C-terminal domain of Sup35 for GFP (green fluorescent protein) or any other fluorescent protein allows researchers to microscopically monitor the aggregation state of the protein. Fluorescent tagging of two prionogenic proteins allows monitoring of colocalization of the aggregates formed by these two proteins, which indicates their co-aggregation. This approach was successfully used for demonstration of the prion nature of Sup35 proteins from various yeast species and for the detection of interspecies transmission barriers in yeast.
TRANSMISSION OF THE PRION STATE BETWEEN Sup35 PROTEINS FROM
DIFFERENT YEAST GENERA
The first experiments concerning the transmission of the prion state between different Sup35 proteins involved studies of the prion properties of Sup35 proteins from evolutionarily distant species of yeast. The prion domains of these proteins have considerably diverging amino acid sequences, but similar amino acid content, being rich in glutamine and asparagine residues. It was demonstrated that Sup35 proteins from Pichia methanolica, Kluyveromyces lactis, Debaryomyces hansenii, Candida maltosa, and C. albicans cannot receive the prion state from the S. cerevisiae Sup35 protein [51-55, 71]. The impossibility of transmission in this case seems to be related to the impossibility of interaction between these proteins and the prion form of S. cerevisiae Sup35, and this was directly demonstrated for P. methanolica Sup35 [52].
The inability of divergent Sup35 proteins to interact was also observed in vitro using a peptide microarray [72]. This method identified short peptide regions within the S. cerevisiae Sup35 prion domain that could initiate polymerization upon incubation with full-length protein. Notably, none of these peptides could initiate the polymerization of C. albicans Sup35, and vice versa, peptides from the prion domain of C. albicans could not stimulate the polymerization of S. cerevisiae Sup35. Stimulation of polymerization between proteins could be observed if the prion domains were introduced with short identical amino acid sequences. Notably, this work did not assess whether the forming amyloid polymers were of prion nature.
The presence of identical short amino acid sequences can also mediate interactions between the prion and normal forms of heterotypical Sup35 in vivo. The prion state of prionogenic yeast proteins is usually obtained by transient overproduction of the appropriate protein [73]. Overproduction of C. albicans Sup35 in S. cerevisiae cells did not facilitate the formation of prions by the endogenous S. cerevisiae Sup35. However, overproduction of a chimeric variant of C. albicans Sup35 in which a short (a.a. 8-26) stretch was replaced with an appropriate stretch of S. cerevisiae Sup35 did induce [PSI+], i.e. it facilitated the transition of S. cerevisiae Sup35 into a prion state [51]. However, it is unclear whether this case is indeed interspecies transmission of the prion state, or rather that overproduction of the chimeric protein stimulated de novo appearance of [PSI+]. The latter possibility is in agreement with the results obtained during study of [PSI+] S. cerevisiae cells producing a chimeric Sup35 protein with a P. methanolica prion domain. Despite the fact that this combination of proteins was shown to exhibit a prion transmission barrier [52, 71], N. Vishveshwara and S. Liebman discovered that the chimeric protein can acquire the prion state, albeit with low probability. Notably, different prion variants of the chimeric protein were formed during this process. The authors stress than the formation of various [PSI+] variants is characteristic of de novo formation, rather than interspecies transmission [74].
However, further studies showed that interspecies transmission of the prion state between structurally dissimilar Sup35 proteins is possible. It is known that the S. cerevisiae Sup35 protein can form structurally distinct fibrils at different temperatures (4 and 37°C), and that infection of yeast with these fibrils results in the formation of different [PSI+] variants [55]. Fibrils formed at 4°C, but not at 37°C, stimulated the in vitro polymerization of the NM fragment of C. albicans Sup35. Moreover, infection with fibrils formed at 4°C could result, albeit with low frequency, in the appearance of the [PSI+] phenotype in the cells synthesizing C. albicans Sup35. Prion aggregates extracted from such cells could infect cells producing either C. albicans or S. cerevisiae Sup35.
TRANSMISSION OF THE PRION STATE BETWEEN Sup35 PROTEINS FROM
CLOSELY RELATED YEAST SPECIES
Sup35 proteins with low sequence identity are not a sufficiently adequate model of the mammalian prion transmission barrier, since in most cases such proteins cannot interact. The sequence identity of PrP proteins in mammals is considerably higher, which means that there is at least a possibility of interaction between these proteins. For instance, hamster PrP expressed in a culture of murine neuroblastoma cells disrupted the propagation of a prion state maintained on murine PrP [37], which indicates that these two proteins interacted. For this reason, modeling mammalian prion transmission barriers in yeast requires the use of prion proteins derived from closely related species, which have a level of sequence identity comparable to that of mammalian PrP proteins (Fig. 1).
A model of interspecies transmission of [PSI+] based on closely related species of yeast was used by B. Chen et al. [56]. In that study the researchers observed that [PSI+] is not transmitted from S. cerevisiae Sup35 to either S. bayanus or S. paradoxus Sup35. Despite this observation, the heterologous Sup35 proteins could efficiently co-aggregate with the prion form of S. cerevisiae Sup35. This allowed the authors to conclude that the prion state could not be transmitted, despite efficient co-aggregation of the prionogenic proteins. Unfortunately, the next publication by these authors [75] showed that the data on which this important conclusion was based were unconvincing. It turned out that the prion state of S. cerevisiae Sup35 could be transmitted onto the heterologous proteins with an efficiency varying from 12 to 93%, and that the efficiency of the co-aggregation was not as high as was reported previously and the measurements of this parameter were not sufficiently accurate.
To provide a convincing proof of the existence of a barrier for the transmission of the prion state between interacting proteins it was necessary to find cases in which the prion state is not transmitted between homologs of Sup35, but the heterologous protein is able to co-aggregate with the protein in prion conformation. Such results were obtained by E. Afanasieva et al. in the study of prion transmission between the S. cerevisiae Sup35 protein and hybrid Sup35 proteins. These proteins included previously studied Sup35 prion domains from S. bayanus and S. paradoxus as well as those from S. mikatae and S. kudriavzevii, all of which were fused to the MC domain of S. cerevisiae [76]. Since prion transmission is known to depend on the prion variant, the study analyzed the transmission of four different [PSI+] variants.
Study of the ability of these proteins to form detergent-resistant polymers in [PSI+] cells showed that the lack of prion transmission could be observed despite copolymerization between the prion form of S. cerevisiae Sup35 and the heterologous protein. In some cases the heterologous proteins could almost completely convert into the polymeric form, while in other cases they were barely detectable in the polymer fraction. Notably, the appearance of Sup35 with a heterologous prion domain in the detergent-resistant polymeric fraction could lead to efficient (up to 25%) loss of the [PSI+] determinant, irrespective of the amount of heterologous protein in this fraction. This indicated that the hybrid Sup35 molecules interacted with the prion polymers of S. cerevisiae Sup35. In some cases the observed transmission barrier was due to the lack of interaction between the heterologous protein and the prion form of S. cerevisiae Sup35. All the observed effects, i.e. [PSI+] transmission barrier, efficiency of prion loss, and the relative amount of aggregated hybrid Sup35 depended on the origin of the Sup35 prion domain and the [PSI+] variant.
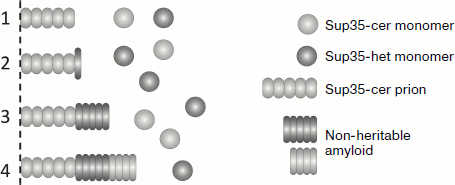
The data allowed the authors to conclude that the barrier in [PSI+] transmission between Sup35 proteins from closely related species of yeast could be caused by several reasons (Fig. 3). Despite the structural similarity between the prion domains, such proteins may be unable to interact. However, the barrier can exist even when the heterologous proteins interact. In this case a single or a few molecules of heterologous protein bind onto the amyloid polymer, but they cannot continue polymerization. Alternatively, molecules of the heterologous protein can polymerize, but do not take on a prion fold, and thus they form non-heritable polymers. Thus, even if the heterologous protein can efficiently copolymerize with the prion template, its heritability can be lost, thus causing a prion transmission barrier. Amyloids of Sup35 with similar characteristics were previously observed in [PIN+] cells overproducing Sup35. It was demonstrated that their non-heritable nature was due to their inability to be fragmented by the Hsp104 chaperone, and thus these amyloids could not be propagated [43]. It seems that formation of non-heritable amyloids is a general rule for the cases in which a prion initiates the polymerization of another protein, since this was observed in several other similar cases [77].
It is possible that S. cerevisiae Sup35 can join a polymer after a heterologous protein molecule and continue polymerizing, but these molecules will also have a non-prion (non-heritable) fold. Notably, in all the studied cases, binding of the heterologous protein to the S. cerevisiae prion polymers occurred with low probability, since frequent binding would have resulted in a complete or nearly complete loss of [PSI+].Fig. 3. Possible mechanisms of copolymerization between Sup35 proteins with a heterologous prion domain (Sup35-het) and the prion form of the S. cerevisiae Sup35 protein (Sup35-cer). For simplicity, only one fiber end is shown. Hybrid Sup35: 1) does not interact with Sup35-cer; 2) blocks further polymerization; 3, 4) polymerizes in a non-heritable fold. In (4) further polymerization of Sup35-cer is possible, albeit in a non-heritable fold.
MUTATIONS IN THE SUP35 GENE WHICH INFLUENCE
[PSI+] TRANSMISSION
Study of the interspecies transmission barrier in mammals showed that even single amino acid substitutions in the PrP protein can have a strong effect on an organism’s susceptibility to prion infection. Similar results were obtained for S. cerevisiae yeast prions: [PSI+] transmission barriers can be the result of mutations in the SUP35 gene, in other words, single amino acid substitutions can make a protein unable to receive the prion state. Such an effect was observed for the PNM2 mutation of the SUP35 gene (G58D substitution in the Sup35 prion domain). Moreover, production of this mutant protein in [PSI+] cells interfered with the propagation of the prion determinant, even though the mutation itself did not prevent the protein from acquiring the prion state upon overproduction [78, 79]. The prion transmission barrier depends on the [PSI+] variant – some variants sustained by wild-type Sup35 could be transmitted to the protein encoded by the PNM2 mutation of the SUP35 gene (E. Afanasieva, unpublished data). Interestingly, not all sequence differences in prionogenic molecules result in prion transmission barriers, since not only the quantity, but also the “quality” of these differences is important. Thus, whereas even single amino acid substitutions could prevent Sup35 from receiving the prion state from a wild-type protein [78, 80, 81], Sup35 with a prion domain which differed from the S. cerevisiae Sup35 by 13 amino acid residues could easily accept the prion state (E. Afanasieva, unpublished data).
Despite the fact that single amino acid substitutions can prevent a protein from receiving prion conformations, Sup35 with a truncated prion domain can maintain [PSI+] [82]. Sequential deletions of the Sup35 prion domain’s oligopeptide repeats from the C-terminus showed that even though the [PSI+] maintained by these proteins changed its phenotypic manifestation, it still reverted to the original phenotype when the prion state was transmitted back onto the full-length protein. One of these deletion variants could only maintain certain [PSI+] variants.
Identification and study of mammalian proteins would have been impossible without their interspecies transmission, since it was this feature that allowed the creation of efficient experimental systems based on the use of laboratory animals. However, the interspecies prion transmission barriers attracted special attention only after the discovery that prion diseases could be contracted by humans through food. It was inexplicable why cows could contract sheep scrapie, while scrapie, unlike mad cow disease, could not be transmitted to humans. Studies conducted during recent years have shown that the interspecies transmission barrier depends not only on the primary structure of prion proteins, but also on the prion strain. Since the range of prion conformations that a protein can acquire is limited by its primary structure, it is reasonable to assume that higher identity of interacting proteins provides a higher number of similar prion conformations in which these proteins can exist, thus making it easier for these proteins to transmit the prion state to each other.
Discovery of prions in lower eukaryotes, especially in yeast, which is one of the most convenient eukaryotic organisms for molecular-biological research, resulted in considerable progress in the study of interspecies prion transmission barriers. During the last decade, this model has not only recreated all the major features of prion transmission in mammals, but has also yielded data that elucidate the possible causes of impaired or non-existent prion transmission. In fact, the prion transmission barriers exist despite the physical interaction between heterologous proteins. The heterologous protein can interact with the end of the prion fibril and thus block further growth. More surprising was the observation that a heterologous protein could copolymerize with the prion form of the resident protein, but in doing so formed non-heritable (non-prion) polymers, which could not be recognized by chaperones and were not fragmented. The interspecies prion transmission barrier involving the formation of non-prion amyloids can hardly be recreated in in vitro systems, since these experiments can yield information on a heterologous protein’s ability to join prion polymers, but do not give insight into the nature of these new polymers (such as their infective potential or heritability).
Notably, the strain-dependence of the prion transmission barrier makes the term “interspecies barrier” somewhat erroneous, since a barrier between two species may or may not exist depending on the prion strain. On the other hand, single amino acid substitutions can make prionogenic proteins unable to receive a certain prion state. Thus, a more appropriate term for inefficient or impossible transmission would be a “transmission barrier”.
This work was supported by the Russian Foundation for Basic Research grants 08-04-00062-a and 11-04-00442-a as well as a Wellcome Trust grant No. 081991/Z/07/Z.
REFERENCES
1.Hadlow, W. J. (1959) Lancet, 274,
289-290.
2.Gajdusek, D. C., Gibbs, C. J., and Alpers, M.
(1966) Nature, 209, 794-796.
3.Chandler, R. L. (1961) Lancet, 277,
1378-1379.
4.Pattison, I. H. (1965) J. Comp. Pathol.,
75, 159-164.
5.Alper, T., Haig, D. A., and Clarke, M. C. (1966)
Biochem. Biophys. Res. Commun., 22, 278-284.
6.Alper, T., Cramp, W. A., Haig, D. A., and Clarke,
M. C. (1967) Nature, 214, 764-766.
7.Griffith, J. S. (1967) Nature, 215,
1043-1044.
8.Prusiner, S. B. (1982) Science, 216,
136-144.
9.Oesch, B., Westaway, D., Waelchli, M., McKinley, M.
P., Kent, S. B., Aebersold, R., Barry, R. A., Tempst, P., Teplow, D.
B., and Hood, L. E. (1985) Cell, 40, 735-746.
10.Gabriel, J. M., Oesch, B., Kretzschmar, H.,
Scott, M., and Prusiner, S. B. (1992) Proc. Natl. Acad. Sci.
USA, 89, 9097-9101.
11.Rivera-Milla, E., Stuermer, C. A. O., and
Malaga-Trillo, E. (2003) Trends Genet., 19, 72-75.
12.Simons, K., and Ehehalt, R. J. (2002) Clin.
Invest., 110, 597-603.
13.Bremer, J., Baumann, F., Tiberi, C., Wessig, C.,
Fischer, H., Schwarz, P., Steele, A. D., Toyka, K. V., Nave, K.-A.,
Weis, J., and Aguzzi, A. (2010) Nat. Neurosci., 13,
310-318.
14.Pan, K. M., Baldwin, M., Nguyen, J., Gasset, M.,
Serban, A., Groth, D., Mehlhorn, I., Huang, Z., Fletterick, R. J., and
Cohen, F. E. (1993) Proc. Natl. Acad. Sci. USA, 90,
10962-10966.
15.Bessen, R. A., Kocisko, D. A., Raymond, G. J.,
Nandan, S., Lansbury, P. N., Jr., and Caughey, B. (1995) Nature,
375, 698-700.
16.Telling, G. C., Parchi, P., DeArmond, S. J.,
Cortell, P., Montagna, P., Gabizon, R., Mastrianni, J., Lugaresi, E.,
Gambetti, P., and Prusiner, S. B. (1996) Science, 274,
2079-2082.
17.Fraser, H., and Dickinson, A. G. (1973) J.
Comp. Pathol., 83, 29-40.
18.Kimberlin, R. H., and Walker, C. A. (1986) J.
Gen. Virol., 67, 255-263.
19.Scott, M., Foster, D., Mirenda, C., Serban, D.,
Coufal, F., Waelchli, M., Torchia, M., Groth, D., Carlson, G.,
DeArmond, S. J., Westaway, D., and Prusiner, S. B. (1989) Cell,
59, 847-857.
20.Kimberlin, R. H., and Walker, C. A. (1978) J.
Gen. Virol., 39, 487-496.
21.Kimberlin, R. H., Cole, S., and Walker, C. A.
(1987) J. Gen. Virol., 68, 1875-1881.
22.Kimberlin, R. H., Walker, C. A., and Fraser, H.
(1989) J. Gen. Virol., 70, 2017-2025.
23.Kocisko, D. A., Priola, S. A., Raymond, G. J.,
Chesebro, B., Lansbury, P. T., and Caughey, B. (1995) Proc. Natl.
Acad. Sci. USA, 92, 3923-3927.
24.Bessen, R. A., and Marsh, R. F. (1992) J.
Virol., 66, 2096-2101.
25.Hill, A. F., Desbruslais, M., Joiner, S., Sidle,
K. C., Gowland, I., Collinge, J., Doey, L. J., and Lantos, P. (1997)
Nature, 389, 448-450.
26.Pattison, I. H., and Jones, K. M. (1968) Res.
Vet. Sci., 9, 408-410.
27.Bruce, M. E., and Dickinson, A. G. (1987) J.
Gen. Virol., 68, 79-89.
28.Kimberlin, R. H., Cole, S., and Walker, C. A.
(1986) Neuropathol. Appl. Neurobiol., 12, 197-206.
29.Li, J., Browning, S., Mahal, S. P., Oelschlegel,
A. M., and Weissmann, C. (2010) Science, 327,
869-872.
30.Bartz, J. C., McKenzie, D. I., Bessen, R. A.,
Marsh, R. F., and Aiken, J. M. (1994) J. Gen. Virol., 75,
2947-2953.
31.Bossers, A., Schreuder, B. E., Muileman, I. H.,
Belt, P. B., and Smits, M. A. (1996) J. Gen. Virol., 77,
2669-2673.
32.Westaway, D., Cooper, C., Turner, S., Da
Costa, M., Carlson, G. A., and Prusiner, S. B. (1994) Proc.
Natl. Acad. Sci. USA, 91, 6418-6422.
33.Mead, S., Stumpf, M. P., Whitfield, J., Beck, J.
A., Poulter, M., Campbell, T., Uphill, J. B., Goldstein, D., Alpers,
M., Fisher, E. M., and Collinge, J. (2003) Science, 300,
640-643.
34.Mead, S., Whitfield, J., Poulter, M., Shah, P.,
Uphill, J., Campbell, T., Al-Dujaily, H., Hummerich, H., Beck, J.,
Mein, C. A., Verzilli, C., Whittaker, J., Alpers, M. P., and Collinge,
J. (2009) N. Engl. J. Med., 361, 2056-2065.
35.Collinge, J., and Clark, A. R. (2007)
Science, 318, 930-936.
36.Wadsworth, J. D. F., Asante, E. A., Desbruslais,
M., Linehan, J. M., Joiner, S., Gowland, I., Welch, J., Stone, L.,
Lloid, S. E., Hill, A. F., Brander, S., and Collinge, J. (2004)
Science, 306, 1793-1796.
37.Priola, S. A., and Chesebro, B. (1995) J.
Virol., 69, 7754-7758.
38.Vanik, D. L., Surewicz, K. A., and Surewicz, W.
K. (2004) Mol. Cell, 14, 139-145.
39.Derkatch, I. L., Bradley, M. E., Zhou, P.,
Chernoff, Y. O., and Liebman, S. W. (1997) Genetics, 147,
507-519.
40.Bradley, M. E., and Liebman, S. W. (2004) Mol.
Microbiol., 51, 1649-1659.
41.Paushkin, S. V., Kushnirov, V. V., Smirnov, V.
N., and Ter-Avanesyan, M. D. (1996) EMBO J., 15,
3127-3134.
42.Kushnirov, V. V., and Ter-Avanesyan, M. D. (1998)
Cell, 94, 13-16.
43.Salnikova, A. B., Kryndushkin, D. S., Smirnov, V.
N., Kushnirov, V. V., and Ter-Avanesyan, M. D. (2005) J. Biol.
Chem., 280, 8808-8812.
44.Alexandrov, I. M., Vishnevskaya, A. B.,
Ter-Avanesyan, M. D., and Kushnirov, V. V. (2008) J. Biol.
Chem., 283, 15185-15192.
45.Nakayashiki, T., Kurtzman, C. P., Edskes, H. K.,
and Wickner, R. B. (2005) Proc. Natl. Acad. Sci. USA,
102, 10575-10580.
46.True, H. L., Berlin, I., and Lindquist, S. L.
(2004) Nature, 431, 184-187.
47.Mironova, L. N., Goginashvili, A. I., and
Ter-Avanesyan, M. D. (2008) Mol. Biol. (Moscow), 42,
798-808.
48.Tyedmers, J., Madariaga, M. L., and Lindquist, S.
(2008) PLoS Biol., 6, 2605-2613.
49.Halfmann, R., and Lindquist, S. (2010)
Science, 330, 629-632.
50.Tuite, M. F., and Serio, T. R. (2010) Nat.
Rev. Mol. Cell. Biol., 11, 823-833.
51.Santoso, A., Chien, P., Osherovich, L. Z., and
Weissman, J. S. (2000) Cell, 100, 277-288.
52.Kushnirov, V. V., Kochneva-Pervukhova, N. V.,
Chechenova, M. B., Frolova, N. S., and Ter-Avanesyan, M. D. (2000)
EMBO J., 19, 324-331.
53.Chien, P., and Weissman, J. S. (2001)
Nature, 410, 223-227.
54.Hara, H., Nakayashiki, T., Crist, C. G., and
Nakamura, Y. (2003) Genes Cells, 8, 925-939.
55.Tanaka, M., Chien, P., Yonekura, K., and
Weissman, J. S. (2005) Cell, 121, 49-62.
56.Chen, B., Newnam, G. P., and Chernoff, Y. O.
(2005) Proc. Natl. Acad. Sci. USA, 104, 2791-2796.
57.Edskes, H. K., McCann, L. M., Hebert, A. M., and
Wickner, R. B. (2009) Genetics, 181, 1159-1167.
58.Komar, A. A., Lesnik, T., Cullin, C., Merrick, W.
C., Trachsel, H., and Altmann, M. (2003) EMBO J., 22,
1199-1209.
59.Coschigano, P. W., and Magasanik, B. (1991)
Mol. Cell. Biol., 11, 822-832.
60.Patino, M. M., Liu, J. J., Glover, J. R., and
Lindquist, S. (1996) Science, 273, 622-626.
61.Kryndushkin, D. S., Alexandrov, I. M.,
Ter-Avanesyan, M. D., and Kushnirov, V. V. (2003) J. Biol.
Chem., 278, 49636-49643.
62.Kushnirov, V. V., Ter-Avanesyan, M. D., Telckov,
M. V., Surguchov, A. P., Smirnov, V. N., and Inge-Vechtomov, S. G.
(1988) Gene, 66, 45-54.
63.Ter-Avanesyan, M. D., Dagkesamanskaya, A. R.,
Kushnirov, V. V., and Smirnov, V. N. (1994) Genetics,
137, 671-676.
64.Paushkin, S. V., Kushnirov, V. V., Smirnov, V.
N., and Ter-Avanesyan, M. D. (1997) Mol. Cell. Biol., 17,
2798-2805.
65.Baxa, U., Keller, P. W., Cheng, N., Wall, J. S.,
and Steven, A. C. (2011) Mol. Microbiol., 79,
523-532.
66.Derkatch, I. L., Chernoff, Y. O., Kushnirov, V.
V., Inge-Vechtomov, S. G., and Liebman, S. W. (1996) Genetics,
144, 1375-1386.
67.Kochneva-Pervukhova, N. V., Chechenova, M. B.,
Valouev, I. A., Kushnirov, V. V., Smirnov, V. N., and Ter-Avanesyan, M.
D. (2001) Yeast, 18, 489-497.
68.Tanaka, M., Chien, P., Naber, N., Cooke, R., and
Weissman, J. S. (2004) Nature, 428, 323-328.
69.King, C. Y., and Diaz-Avalos, R. (2004)
Nature, 428, 319-323.
70.Kushnirov, V. V., Vishnevskaya, A. B.,
Alexandrov, I. M., and Ter-Avanesyan, M. D. (2007) Prion,
1, 179-184.
71.Chernoff, Y. O., Galkin, A. P., Lewitin, E.,
Chernova, T. A., Newnam, G. P., and Belenkiy, S. M. (2000) Mol.
Microbiol., 35, 865-876.
72.Tessier, P. M., and Lindquist, S. (2007)
Nature, 447, 556-561.
73.Wickner, R. B. (1994) Science, 264,
566-569.
74.Vishveshwara, N., and Liebman, S. W. (2009)
BMC Biol., 7, 26.
75.Chen, B., Bruce, K. L., Newnam, G. P., Gyoneva,
S., Romanyuk, A. V., and Chernoff, Y. O. (2010) Mol. Microbiol.,
76, 1483-1499.
76.Afanasieva, E. G., Kushnirov, V. V., Tuite, M.
F., and Ter-Avanesyan, M. D. (2011) J. Biol. Chem., 286,
15773-15780.
77.Urakov, V. N., Vishnevskaya, A. B., Alexandrov,
I. M., Kushnirov, V. V., Smirnov, V. N., and Ter-Avanesyan, M. D.
(2010) Prion, 4, 45-52.
78.Doel, M. S., McCready, S. J., Nierras, C. R., and
Cox, B. S. (1994) Genetics, 137, 659-670.
79.Kochneva-Pervukhova, N. V., Paushkin, S. V.,
Kushnirov, V. V., Cox, B. S., Tuite, M. F., and Ter-Avanesyan, M. D.
(1998) EMBO J., 17, 5805-5810.
80.DePace, A. H., Santoso, A., Hillner, P., and
Weissman, J. S. (1998) Cell, 93, 1241-1252.
81.Chang, H-Y., Lin, J-Y., Lee, H-C., Wang, H. L.,
and King, C. Y. (2008) Proc. Natl. Acad. Sci. USA, 105,
13345-13350.
82.Shkundina, I. S., Kushnirov, V. V., Tuite, M. F.,
and Ter-Avanesyan, M. D. (2006) Genetics, 172,
827-835.