Cerebral Ischemia–Reperfusion Induces GAPDH S-Nitrosylation and Nuclear Translocation
Chong Li1‡, Jun-Jun Feng1‡, Yong-Ping Wu2, and Guang-Yi Zhang1*
1Jiangsu Province Key Laboratory of Brain Disease Bioinformation and Research Center of Biochemistry and Molecular Biology, Xuzhou Medical College, 84 West Huai-hai Road, Xuzhou 221002, Jiangsu, China; fax: +86-516-8574-8486; E-mail: gyzhang@xzmc.edu.cn2Jiangsu Province Key Laboratory of Anesthesiology, Xuzhou Medical College, Xuzhou, Jiangsu, China
‡ These authors contributed equally to this work.
* To whom correspondence should be addressed.
Received December 2, 2011; Revision received March 10, 2012
Glyceraldehyde-3-phosphate dehydrogenase (GAPDH), a glycolytic enzyme, plays an important role in glycolysis. It was reported that GAPDH undergoes S-nitrosylation, which facilitated its binding to Siah1 and resulted in nuclear translocation and cell apoptosis. The results of this study show that GAPDH S-nitrosylation, Siah1 binding, translocation to nucleus, and concomitant neuron death occur during the early stages of reperfusion in the rat four-vessel occlusion ischemic model. N-Methyl-D-aspartate receptor antagonist MK801, neuronal nitric oxide synthase inhibitor 7-nitroindazole, or monoamine oxidase-B inhibitor (R)-(–)-deprenyl hydrochloride could inhibit GAPDH S-nitrosylation and translocation and exert neuroprotective effects.
KEY WORDS: GAPDH, S-nitrosylation, Siah1, deprenyl hydrochloride, cerebral ischemiaDOI: 10.1134/S0006297912060156
Abbreviations: BCIP, 5-bromo-4-chloro-3-indolyl-phosphate; DEP, (R)-(–)-deprenyl hydrochloride; DMSO, dimethylsulfoxide; GAPDH, glyceraldehyde-3-phosphate dehydrogenase; MAO-B, monoamine oxidase B; MCAo, middle cerebral artery occlusion; NBT, nitroblue tetrazolium; NMDA, N-methyl-D-aspartate; nNOS, neuronal nitric oxide synthase; NO, nitric oxide; NO-Cbl, nitrosylcobalamin; 7NI, 7-nitroindazole; 4-VO, four-vessel occlusion.
Stroke is one of the serious diseases that cause death. Calcium overload
and glutamic acid excitation toxicity has been thought to be the main
neuron death mechanism of brain stroke [1-3]. Recently, nitric oxide (NO) has been considered as
a mediator of excitation toxicity by activating a cGMP-independent
pathway in ischemia–reperfusion. On the other hand, NO can be
produced in nNOS (neuronal nitric oxide synthase)-catalyzed reactions
and cause protein S-nitrosylation, a modification that reversibly adds
NO to thiol moiety of reactive cysteine residues [3]. This direct chemical reaction with proteins
modified by NO occurs via a complex chemical mechanism without the
assistance of enzymes [4, 5].
NO biosynthesis by nNOS in brain is preferentially activated by calcium
influx through N-methyl-D-aspartate (NMDA)-type glutamate receptors [6-8]. As a general rule,
S-nitrosylation reactions result in specific physiological or
pathophysiological actions by modifying protein function [5, 9, 10].
It was reported that apoptosis-stimulated NO production could result in GAPDH S-nitrosylation at cysteine 150 [11]. S-Nitrosylated GAPDH triggered GAPDH binding to Siah1 (an E3 ubiquitin ligase), resulted in nuclear translocation of GAPDH, and ultimately led to apoptosis. GAPDH could stabilize Siah1 and promote the degradation of nuclear target proteins. From the endotoxin-induced macrophage and neuronal activation induced by glutamate, it was found that the NO–S-nitrosylation-GAPDH–Siah1 cascade might represent an important cytotoxic event [11].
The results of the present work suggest that during cerebral ischemia–reperfusion, NMDA receptor channel activation causes overloaded Ca2+ influx, activates nNOS and promotes GAPDH S-nitrosylation, further promotes the binding of GAPDH to Siah1, the nuclear translocation of GAPDH, and eventually leads to apoptosis. The effects of NMDA receptor antagonist MK801, nNOS inhibitor 7NI (7-nitroindazole), and monoamine oxidase inhibitor DEP ((R)-(–)-deprenyl hydrochloride) on GAPDH S-nitrosylation and nuclear translocation were also estimated, and a significant neuroprotective effect against cerebral ischemia–reperfusion was established.
MATERIALS AND METHODS
Antibodies and reagents. Rabbit polyclonal anti-GAPDH, anti-Siah1 (used for i.p.) and anti-actin (sc-10731) antibodies were obtained from Santa Cruz Biotechnology (USA). Anti-H2B (#2722) antibodies were obtained from Cell Signaling Biotechnology (USA). The secondary anti-rabbit IgG antibodies (T6778) were purchased from Sigma (USA). Nitrocellulose filters were from Amersham (USA). BCIP/NBT color development substrate (5-bromo-4-chloro-3-indolyl-phosphate/nitroblue tetrazolium) was obtained from Promega (USA). DEP was from Tocris (USA). MK801, 7-NI, and other chemicals were all from Sigma.
Induction of ischemia. Adult male Sprague–Dawley rats weighing 200-250 g were used (Shanghai Experimental Animal Center, Chinese Academy of Sciences, Shanghai, China). Transient cerebral ischemia was induced by a four-vessel occluded (4-VO) method as described previously [12]. Rats were anesthetized with chloral hydrate (300 mg/kg, intraperitoneally) before both vertebral arteries were occluded permanently by electrocautery and both carotid arteries were isolated. Then the rats were allowed to recover for 24 h, and both carotid arteries were occluded with aneurysm clips to induce cerebral ischemia without the administration of chloral hydrate. After occlusion for 15 min, the aneurysm clips were removed for reperfusion. Rats that lost their righting reflex within 30 sec and those whose pupils were dilated and unresponsive to light were selected for the experiments. Sham control rats were treated using the same surgical procedures except that the carotid arteries were not occluded.
Model of transient focal ischemia. Adult male Sprague–Dawley rats weighing 200-250 g were used. Transient focal ischemia was induced by intraluminal occlusion of the left middle cerebral artery (MCAo) as described before [13] with slight modification. Rats were anaesthetized with chloral hydrate (300 mg/kg, intraperitoneally). Mean arterial blood pressure was monitored during MCAo through a tail cuff, and arterial blood gas was analyzed at 30 min before or after the onset of ischemia. The animals underwent left MCA occlusion for 90 min and then reperfusion for 24 h. After recovering from anesthesia, the animals were maintained in an air-conditioned room at 37°C.
Administration of drugs. Rats were injected intraperitoneally with DEP (20 mg/kg) in 0.9% NaCl solution 90 min before ischemia or 3 h after ischemia. 7-NI (25 mg/kg) dissolved in 1% dimethylsulfoxide (DMSO) was intraperitoneally injected 20 min before ischemia. MK801 in 0.9% NaCl solution was intraperitoneally injected 60 min before ischemia. Control rats were intraperitoneally given corresponding solvent, 0.9% NaCl or 1% DMSO.
Sample preparation. Rats were decapitated immediately at different time-points of reperfusion, and then the hippocampal CA1 were isolated and quickly frozen in liquid nitrogen. Tissues were homogenized in ice-cold homogenization buffer containing 50 mM MOPS-NaOH (pH 7.4), 100 mM KCl, 320 mM sucrose, 50 mM NaF, 0.5 mM MgCl2, 0.2 mM dithiothreitol (omitted when S-nitrosylation was tested), 1 mM EDTA, 1 mM EGTA, 1 mM Na3VO4, 20 mM sodium pyrophosphate, 20 mM β-phosphoglycerol, 1 mM p-nitrophenyl phosphate, 1 mM benzamidine, 1 mM phenylmethylsulfonyl fluoride (PMSF), 5 µg/ml leupeptin, 5 µg/ml aprotinin, and 5 µg/ml pepstatin A. The homogenates were centrifuged at 800g for 10 min at 4°C. Protein concentration was determined by the method of Lowry et al. [14]. Samples were stored at –80°C and were thawed only once before being used.
Nuclear extraction. The homogenates were centrifuged at 800g for 10 min at 4°C. Supernatants (cytosolic fraction) were collected, and protein concentrations were determined. The nuclear pellets were extracted with 20 mM Hepes-NaOH (pH 7.9), 20% glycerol, 420 mM NaCl, 0.5 mM MgCl2, 1 mM EDTA, 1 mM EGTA, 1 mM dithiothreitol, and enzyme inhibitors for 30 min at 4°C with constant agitation. After centrifugation at 12,000g for 15 min at 4°C, supernatants (nuclear fraction) were collected, and protein concentrations were determined.
Determination of protein S-nitrosylation. S-Nitrosylated GAPDH was assayed by biotin switch assay as described by Jaffrey et al. [9] using low-light conditions and opaque tubes. The hippocampi were homogenized in HEN buffer (250 mM Hepes-NaOH, pH 7.7, 1 mM EDTA, 0.1 mM neocuproine). Free thiols were blocked by methylation with methyl methanethiosulfonate. Unreacted methyl methanethiosulfonate was removed by protein precipitation with 10 volumes of acetone (–20°C). Cysteine residues that had been S-nitrosylated by nitric oxide donor NO-Cbl (nitrosylcobalamin) were converted to free thiols with sodium ascorbate (1 mM final concentration), which does not react with methylated thiols. The free thiols were then biotinylated with biotin-hexyl pyridyldithiopropionamide (HPDP) at 25°C for 1 h. Proteins were precipitated by chilled acetone, and the pellet was resuspended in HENS buffer (250 mM Hepes-NaOH, pH 7.7, 1 mM EDTA, 0.1 mM neocuproine, 1% SDS). Biotinylated proteins were precipitated with streptavidin-agarose and eluted from the beads with a solution containing 20 mM Hepes-NaOH (pH 7.7), 100 mM NaCl, 1 mM EDTA, and 100 mM 2-mercaptoethanol.
Immunoprecipitation and immunoblotting. Tissue homogenates (400 μg of protein) were diluted fourfold with immunoprecipitation buffer (50 mM Hepes-NaOH buffer (pH 7.4) containing 10% glycerol, 150 mM NaCl, 1% Triton X-100, 0.5% Nonidet P-40, 1 mM EDTA, 1 mM EGTA, 1 mM PMSF, and 1 mM Na3VO4). Samples were preincubated for 1 h with 20 µl of protein A-Sepharose CL-4B (Amersham Biosciences, Sweden) at 4°C and centrifuged to remove proteins that had adhered nonspecifically to protein A. The supernatants were incubated with 1-2 µg of primary antibodies overnight at 4°C. Protein A was added to the tube for another 2 h incubation. The samples were centrifuged at 10,000g for 2 min at 4°C, and the pellets were washed three times with the immunoprecipitation buffer. Bound proteins were eluted by boiling at 100°C for 5 min in SDS-PAGE loading buffer and then isolated by centrifugation. The supernatants were separated on polyacrylamide gels and then electrotransferred to nitrocellulose membrane [15]. After blocking for 3 h in Tris-buffered saline with 0.1% Tween 20 (TBST) and 3% bovine serum albumin, the membranes were incubated overnight at 4°C with primary antibodies in TBST containing 3% bovine serum albumin. The membranes were then washed and incubated with alkaline phosphatase-conjugated secondary antibodies in TBST for 2 h and developed using NBT/BCIP color substrate (Promega). The density of the bands on the membrane was scanned and analyzed with LabWorks image analysis software (UVP, Inc.).
Histological analysis and immunohistochemistry. Rats were perfusion-fixed with 4% paraformaldehyde in 0.1 M sodium phosphate buffer (pH 7.4) under anesthesia. Brains were removed quickly and further fixed with the same fixation solution overnight at 4°C. Post-fixed brains were embedded in paraffin, followed by preparation of coronal sections (5 µm thick) using a microtome. The paraffin-embedded brain sections were deparaffinized with xylene and rehydrated with ethanol at graded concentrations of 100-70% (v/v), followed by washing with water. The sections were stained with 0.1% (w/v) cresyl violet and examined by light microscopy, and the number of surviving hippocampal CA1 pyramidal cells/mm of length was counted as the neuronal density. Immunoreactivity was determined by the avidin/biotin/peroxidase method. Sections were deparaffinized with xylene and rehydrated with ethanol at graded concentrations and distilled water. High temperature antigen retrieval was performed in 1 mM Na-citrate buffer. To block endogenous peroxidase activity, sections were incubated for 30 min in 1% H2O2. After being blocked with 5% (v/v) normal goat serum in phosphate buffered saline for 1 h at 37°C, the sections were incubated with rabbit polyclonal anti-GAPDH antibodies (1 : 100 dilution) at 4°C for 2 days. These sections were then incubated overnight with biotinylated goat anti-rabbit secondary antibody and subsequently with avidin-conjugated horseradish peroxidase for 1 h at 37°C. Finally, the sections were incubated with the peroxidase substrate diaminobenzidine until the desired stain intensity developed.
Data analysis and statistics. Values are expressed as the means ± S.D. and were obtained from at least four rats. Statistical analysis of the results was carried out by one-way analysis of variance (ANOVA), followed by Duncan’s new multiple range method or the Newman–Keuls test. P values <0.05 were considered significant.
RESULTS
GAPDH S-nitrosylation was mediated by endogenous NO and promoted the binding of GAPDH with Siah1. As shown in Fig. 1A, GAPDH was S-nitrosylated, the level of which significantly elevated at the 6-h time point of reperfusion on the rat 4-VO cerebral ischemia model. To elucidate whether S-nitrosylation of GAPDH was induced by endogenous NO, the nNOS inhibitor 7-NI was used. We further used NMDA receptor antagonist MK801 and monoamine oxidase-B (MAO-B) inhibitor DEP. As shown in Fig. 1B, administration of 7NI, MK801, or DEP markedly suppressed the S-nitrosylation of GAPDH. As shown in Fig. 1C, GAPDH S-nitrosylation facilitated its combination with Siah1. Administration of 7NI, MK801, or DEP markedly suppressed this combination. These results suggest that GAPDH S-nitrosylation was mediated by endogenous NO and promoted the combination of GAPDH with Siah1.
GAPDH S-nitrosylation promoted nuclear translocation of GAPDH and Siah1. To investigate whether GAPDH S-nitrosylation could promote the nuclear translocation of GADPH and Siah1, we examined the time course of GAPDH in nuclei. As shown in Fig. 2A, nuclear GAPDH reached its peak level at the 6-h time point after reperfusion, which was coincident to the peak level of GAPDH S-nitrosylation. Administration of 7-NI, MK801, or DEP also markedly suppressed the nuclear translocation of GADPH at the 6-h time point, as shown in Figs. 2B and 2C. The combination of GADPH with Siah1 at the 6-h time point was also examined. As shown in Fig. 2D, administration of 7-NI, MK801, or DEP markedly suppressed the quantity of binding GAPDH with Siah1. The subcellular location of GADPH was also examined by immunohistochemistry, and similar results were observed (Fig. 3). These results suggest that GAPDH S-nitrosylation not only promoted the binding but also the nuclear translocation of GAPDH and Siah1.Fig. 1. GAPDH S-nitrosylation and GAPDH-Siah1 association was inhibited by MK801, 7NI, and DEP in rat cerebral ischemia and reperfusion. A) Time course of S-nitrosylated GAPDH in hippocampal CA1 derived from sham-treated rats or rats with 15 min ischemia at various time points of reperfusion. B) NMDA receptor antagonist MK801, nNOS inhibitor 7NI, and MAO-B inhibitor DEP inhibited GAPDH S-nitrosylation at the 6-h time point of reperfusion. C) Co-immunoprecipitation analysis of GAPDH with Siah1 in cytosol. MK801, 7-NI, and DEP inhibited GAPDH and Siah1 association at 6 h of reperfusion. S-Nitrosylation of GAPDH was determined by the biotin-switch assay. Immunoblotting directly with anti-actin antibody was also performed. Bands were scanned, and the intensities were represented as fold versus sham treatment. Data are means ± S.D. from four independent experiments. * P < 0.05 versus sham group; ** P < 0.05 versus vehicle group.
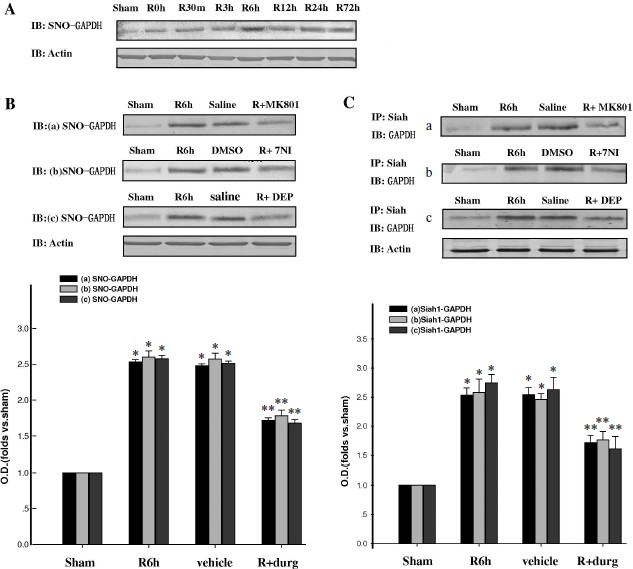
Fig. 2. GAPDH nuclear translocation was inhibited by MK801, 7NI, and DEP in rat cerebral ischemia and reperfusion. A) Time course of GAPDH level in the nucleus during cerebral ischemia and reperfusion in hippocampal CA1. B, C) Nuclear translocation of GAPDH was inhibited by MK801, 7-NI, and DEP at 6 h of reperfusion. D) MK801, 7-NI, and DEP inhibited the GAPDH and Siah1 association in nucleus at 6 h of reperfusion. Immunoblotting was directly performed with anti-GAPDH or anti-actin antibody. Bands were scanned, and the intensities were represented as fold versus sham treatment. Data are means ± S.D. from four independent experiments (n = 4). * P < 0.05 versus sham group; ** P < 0.05 versus saline group.
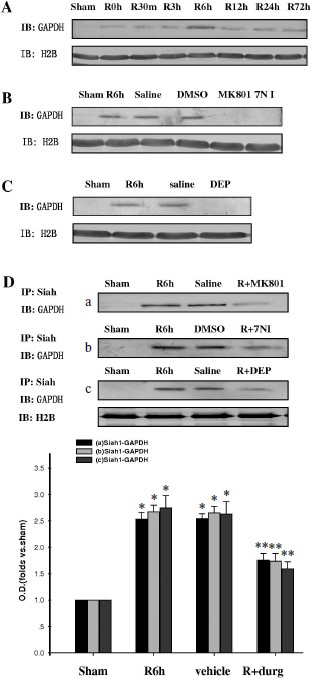
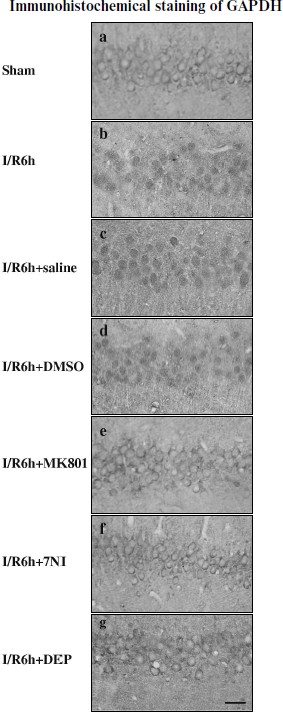
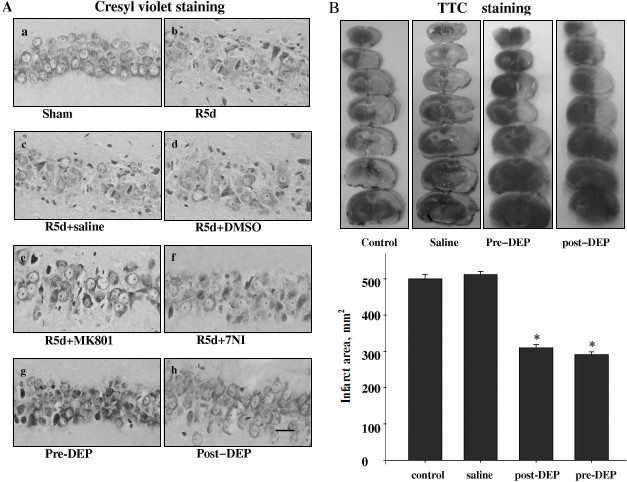
Inhibition of GAPDH S-nitrosylation exerted a neuroprotective effect against cerebral ischemia–reperfusion injury. The specific nuclear translocation of GAPDH was involved in apoptosis [16]. The neuroprotection of GAPDH S-nitrosylation inhibition by the drugs 7-NI, MK801, or DEP was studied. We used cresyl violet staining to test the survival of hippocampal CA1 pyramidal cells after ischemia–reperfusion. Those cells with round and pale stained nuclei were counted as normal cells, whereas shrunken cells with pyknotic nuclei were regarded as dead cells. As shown in Fig. 4, 15 min of ischemia followed by 5 days of reperfusion led to severe cell death (R5d group; Fig. 4Ab). Administration of 7-NI or MK801 showed significant neuroprotection against apoptosis (Figs. 4Ae and 4Af). DEP administrated before (pre-DEP group) ischemia or 3 h after ischemia (post-DEP group) could dramatically decrease the neuronal degeneration (Figs. 4Ag and 4Ah). At the same time, vehicle controls did not show any protection (Figs. 4Ac and 4Ad). The numbers of surviving pyramidal cells in the CA1 region of sham operation group, ischemia insult group, saline treatment group, DMSO treatment group, MK801 treatment group, 7-NI treatment group, pre-DEP treatment group, and post-DEP treatment group were 207 ± 8, 15 ± 5, 15 ± 5, 15 ± 7, 153 ± 5, 161 ± 9, 169 ± 5 and 176 ± 6.Fig. 3. Immunohistochemical staining of GAPDH in coronal sections of hippocampus. Expression and subcellular localization of GAPDH after different treatment is shown. Example of immunohistochemical staining sections of hippocampus of sham operated rats (a), rats subjected to 15 min of ischemia followed by 6 h of reperfusion (b), and rats subjected to 15 min of ischemia followed by 6 h of reperfusion with administration of saline (c), DMSO (d), MK801 (e), 7-NI (f), or DEP (g). The boxed areas in the left column are shown at higher magnification in the right columns. Magnification is ×400 in panels (a)-(g); scale bar 10 µm.
Inhibition of GAPDH S-nitrosylation exerted a neuroprotective effect against focal ischemia–reperfusion injury. We also applied DEP on the MACo model to examine the protective effect against focal ischemia–reperfusion injury. DEP was administrated before or 3 h after ischemia on the MACo model. TTC staining results show that the infract area were decreased clearly in both pre-DEP treatment group and post-DEP treatment group (Fig. 4B).Fig. 4. Neuroprotection of MK801, 7NI, and DEP on hippocampal CA1 pyramidal neuron against ischemia reperfusion. A) Cresyl violet staining was performed in sections from the hippocampus in sham operation (a), rats subjected to 5 days of reperfusion after global ischemia (b), administration of saline (c), DMSO (d), MK801 (e), 7-NI (f), and DEP (pre-DEP) (g) 20 min before ischemia, or administration of DEP 3 h after global ischemia (post-DEP) (h). Cresyl violet staining data were obtained from six animals and a typical experiment is presented. B) Representative appearance of TTC 4% stained of coronal section showing infarct area consistently identified in the cortex and striatum of the left cerebral hemisphere as distinct pale-stained area in the rats subjected to 90 min ischemia and 24 h reperfusion (control saline) or attenuation of infract area treatment with DEP. Data were obtained from ten animals, and the results of typical experiment are presented. The amount of injury was expressed as a ratio of the total infarct volume to the total volume of the damaged hemisphere. * P < 0.05 versus saline groups.
DISCUSSION
In the animal model of cerebral ischemia, morphological changes of neurons, for example cell volume shrinkage, nuclear condensation or DNA breakage [17, 18], biochemical evidence such as expression of Bcl-2 family [19], cytochrome c release [20], and activation of caspase family members are termed as parameters or mechanisms that caused apoptosis [21, 22]. GAPDH has recently been found, in addition to the well-known major role in the glycolytic pathway, to combine with the cell membrane and be involved in endocytosis [23, 24], tRNA nuclear transport [25], transcription activity stimulation [26], DNA replication and repair regulation [17, 27], and cell apoptosis. In some stimulus conditions, GAPDH overexpression and nuclear accumulation can cause cell apoptosis, and GAPDH antisense oligonucleotide can inhibit the expression of GADPH and apoptosis [28]. It was reported that S-nitrosylated GAPDH could bind with Siah1 and migrate to the nucleus in staurosporine-induced HEK293 cell apoptosis [11]. In the present work, we found that during cerebral ischemia–reperfusion, NO production was stimulated and resulted in GAPDH S-nitrosylation, which triggered the GAPDH binding to Siah1, nuclear translocation, and ultimately led to cell death.
nNOS was activated by over-influx of calcium from activated NMDA receptors in the early stage of ischemia–reperfusion. nNOS-derived NO promoted GAPDH S-nitrosylation and its combination with Siah1. Siah1 contains specific nuclear localization signal and thus facilitates the nuclear translocation of GAPDH, finally leading to apoptosis. When given NMDA receptor antagonist MK801 or nNOS inhibitor 7NI, GAPDH S-nitrosylation and nuclear translocation was inhibited, and neuron apoptosis was also suppressed. In our previous studies, we reported that nNOS activity was significantly increased in the early stage of cerebral ischemia–reperfusion at the time point of 6 h [7], which may increase NO production and enable increased S-nitrosylation of GAPDH.
It was reported that GAPDH thiols were oxidized under oxidative stress during ischemia–reperfusion [29]. Oxidation of SH-groups of the active site of GAPDH could enhance its binding with DNA and the accumulation of GAPDH in the nucleus [30]. GAPDH S-nitrosylation can also promote its nuclear translocation [11]. Our results showed that during cerebral ischemia–reperfusion the nuclear GAPDH was significantly increased at the 6-h time point. GAPDH has no nuclear localization signal, but from the present results we can see that S-nitrosylated GAPDH could bind with Siah1 and be transferred into the nucleus. Siah1 is unstable and will undergo degradation in the nucleus, but its stability is increased upon GAPDH binding [11].
When given monoamine oxidase inhibitor DEP, GAPDH S-nitrosylation and nuclear translocation was inhibited and neuron apoptosis was also suppressed. DEP could act on the polyamine site of NMDA receptor to prevent calcium influx [31]. Furthermore, DEP was found to interact with GADPH and exert a neuroprotective effect, which may contribute to the improvements in Parkinson’s and Huntington’s diseases [32]. A recent study also showed that a cytosolic protein, GOSPEL, could bind to S-nitrosylated GADPH and exert neuroprotective effects [33]. From these reports and the results of the present study, we conclude that DEP can also inhibit GADPH S-nitrosylation and reduce neuron apoptosis in brain ischemia–reperfusion injury.
This work was supported by grants from the National Natural Science Foundation of China (Nos. 30870543, 31000360), the Education Departmental Nature Science Funds of Jiangsu Province (09KJB310015, 10KJA310053), the Science and Technology Bureau of Xuzhou, Jiangsu Province (XF10C077), and the Priority Academic Program Development of Jiangsu Higher Education Institutions (PAPD). Dr. Chong Li was supported by “New Century Excellent Talents in University (NCET)” program of the Chinese Ministry of Education and “Six Talent Peaks Program” of Jiangsu Province of China.
REFERENCES
1.Leist, M., and Nicotera, P. (1998) Rev. Physiol.
Biochem. Pharmacol., 132, 79-125.
2.Lee, J. M., Zipfel, G. J., and Choi, D. W. (1999)
Nature, 399, A7-14.
3.Hess, D. T., Matsumoto, A., Kim, S. O., Marshall,
H. E., and Stamler, J. S. (2005) Nat. Rev. Mol. Cell Biol.,
6, 150-166.
4.Stamler, J. S., Simon, D. I., Osborne, J. A.,
Mullins, M. E., Jaraki, O., Michel, T., Singel, D. J., and Loscalzo, J.
(1992) Proc. Natl. Acad. Sci. USA, 89, 444-448.
5.Ahern, G. P., Klyachko, V. A., and Jackson, M. B.
(2002) Trends Neurosci., 25, 510-517.
6.Sattler, R., Xiong, Z., Lu, W. Y., Hafner, M.,
MacDonald, J. F., and Tymianski, M. (1999) Science, 284,
1845-1848.
7.Yu, H. M., Xu, J., Li, C., Zhou, C., Zhang, F.,
Han, D., and Zhang, G. Y. (2008) Neuroscience, 155,
1120-1132.
8.Radi, R. (2004) Proc. Natl. Acad. Sci. USA,
101, 4003-4008.
9.Jaffrey, S. R., Erdjument-Bromage, H., Ferris, C.
D., Tempst, P., and Snyder, S. H. (2001) Nat. Cell Biol.,
3, 193-197.
10.Christopherson, K. S., Hillier, B. J., Lim, W.
A., and Bredt, D. S. (1999) J. Biol. Chem., 274,
27467-27473.
11.Hara, M. R., Agrawal, N., Kim, S. F., Cascio, M.
B., Fujimuro, M., Ozeki, Y., Takahashi, M., Cheah, J. H., Tankou, S.
K., Hester, L. D., Ferris, C. D., Hayward, S. D., Snyder, S. H., and
Sawa, A. (2005) Nat. Cell Biol., 7, 665-674.
12.Pulsinelli, W. A., and Brierley, J. B. (1979)
Stroke, 10, 267-272.
13.Nagasawa, H., and Kogure, K. (1989)
Stroke, 20, 1037-1043.
14.Lowry, O. H., Rosebrough, N. J., Farr, A. L., and
Randall, R. J. (1951) J. Biol. Chem., 193, 265-275.
15.Kozutsumi, Y., Normington, K., Press, E.,
Slaughter, C., Sambrook, J., and Gething, M. J. (1989) J. Cell Sci.
Suppl., 11, 115-137.
16.Ishitani, R., Tanaka, M., Sunaga, K., Katsube,
N., and Chuang, D. M. (1998) Mol. Pharmacol., 53,
701-707.
17.MacManus, J. P., Hill, I. E., Huang, Z. G.,
Rasquinha, I., Xue, D., and Buchan, A. M. (1994) Neuroreport,
5, 493-496.
18.Li, Y., Chopp, M., Jiang, N., Yao, F., and
Zaloga, C. (1995) J. Cereb. Blood Flow Metab., 15,
389-397.
19.Gillardon, F., Lenz, C., Waschke, K. F.,
Krajewski, S., Reed, J. C., Zimmermann, M., and Kuschinsky, W. (1996)
Brain Res. Mol. Brain Res., 40, 254-260.
20.Fujimura, M., Morita-Fujimura, Y., Murakami, K.,
Kawase, M., and Chan, P. H. (1998) J. Cereb. Blood Flow Metab.,
18, 1239-1247.
21.Namura, S., Zhu, J., Fink, K., Endres, M.,
Srinivasan, A., Tomaselli, K. J., Yuan, J., and Moskowitz, M. A. (1998)
J. Neurosci., 18, 3659-3668.
22.Velier, J. J., Ellison, J. A., Kikly, K. K.,
Spera, P. A., Barone, F. C., and Feuerstein, G. Z. (1999) J.
Neurosci., 19, 5932-5941.
23.Caswell, A. H., and Corbett, A. M. (1985) J.
Biol. Chem., 260, 6892-6898.
24.Robbins, A. R., Ward, R. D., and Oliver, C.
(1995) J. Cell Biol., 130, 1093-1104.
25.Singh, R., and Green, M. R. (1993)
Science, 259, 365-368.
26.Morgenegg, G., Winkler, G. C., Hubscher, U.,
Heizmann, C. W., Mous, J., and Kuenzle, C. C. (1986) J.
Neurochem., 47, 54-62.
27.Baxi, M. D., and Vishwanatha, J. K. (1995)
Biochemistry, 34, 9700-9707.
28.Ishitani, R., Sunaga, K., Hirano, A., Saunders,
P., Katsube, N., and Chuang, D. M. (1996) J. Neurochem.,
66, 928-935.
29.Hwang, I. K., Yoo, K. Y., Kim, D. W., Choi, J.
H., Lee, I. S., and Won, M. H. (2007) Neurochem. Res.,
32, 1530-1538.
30.Arutyunova, E. I., Danshina, P. V., Domnina, L.
V., Pleten, A. P., and Muronetz, V. I. (2003) Biochem. Biophys. Res.
Commun., 307, 547-552.
31.Lange, K. W., Riederer, P., and Youdim, M. B.
(1994) Clin. Pharmacol. Ther., 56, 734-741.
32.Carlile, G. W., Chalmers-Redman, R. M., Tatton,
N. A., Pong, A., Borden, K. E., and Tatton, W. G. (2000) Mol.
Pharmacol., 57, 2-12.
33.Sen, N., Hara, M. R., Ahmad, A. S., Cascio, M.
B., Kamiya, A., Ehmsen, J. T., Agrawal, N., Hester, L., Dore, S.,
Snyder, S. H., and Sawa, A. (2009) Neuron, 63, 81-91.