Phosphatase Activity in Barley Proteins Tightly Bound to DNA and Its Development-Dependent Changes
K. Bielskiene1, D. Labeikyte2, N. Sjakste3,4, L. Bagdoniene2*, and B. Juodka2
1Laboratory of Molecular Oncology, Institute of Oncology, Vilnius University, P. Baublio 3b, Vilnius LT-08406, Lithuania2Department of Biochemistry and Biophysics, Vilnius University, M. K. Ciurlionio 21, Vilnius LT-03101, Lithuania, fax: 370-5-239-8231; E-mail: lida.bagdoniene@gf.vu.lt; lida.bagdoniene@gmail.com
3Faculty of Medicine, University of Latvia, Sarlotes 1a, Riga LV1001, Latvia
4Latvian Institute of Organic Synthesis, Aizkraukles 21, Riga LV 1006, Latvia
* To whom correspondence should be addressed.
Received February 29, 2012; Revision received March 11, 2012
The tightly bound proteins (TBPs), a protein group that remains attached to DNA either covalently or noncovalently after deproteinization, have been found in numerous eukaryotic species. Some TBPs isolated from mammalian and yeast cells possess phosphatase or kinase activity. The aim of this study was to characterize further TBPs in barley (Hordeum vulgare) cells. The spectra of TBPs varied in different organs of barley shoots (first leaves, coleoptile, and roots) and at different developmental stages of the plant. Some barley TBPs manifested phosphatase, probably Ser/Thr or dual Ser/Thr/Tyr activity. MALDI-TOF mass spectrometry of barley TBPs identified several proteins involved in chromatin rearrangement and regulation processes, including transcription factors, serpins, protein phosphatases and protein kinases, RNA helicases, and DNA topoisomerase II.
KEY WORDS: tightly bound proteins, nuclear matrix, phosphataseDOI: 10.1134/S0006297912060168
Abbreviations: IEF, isoelectrofocusing; MALDI TOF-TOF, matrix-assisted laser desorption/ionization tandem time-of-flight; MS, mass spectrometry; TBPs, proteins tightly bound to DNA.
Eukaryotic genes are supposed to be organized into higher-order
structures formed by discrete and topologically independent domains
organized in loops attached to the nuclear matrix. The loop
organization of chromatin could be important not only for compaction of
the fiber, but also for regulation of gene expression and replication.
Polypeptides involved in specific structural organization and modeling
of the chromatin fibers and chromosome territories are mostly insoluble
nuclear matrix proteins with strong affinity to DNA [1-5]. Functions and existence of
the nuclear matrix is a still-debated question, but it is evident that
proteins tightly bound to DNA (TBPs) are involved in chromatin folding
and reorganization during the cell cycle. TBPs are operationally
defined as polypeptides that are able to form permanent or transient
tight complexes with DNA, which may be stabilized by covalent bonds,
and cannot be detached from DNA by standard deproteinization procedures
or by treatment with strong dissociating agents such as sarcosyl, urea,
guanidine hydrochloride, etc. [6]. Serpins Spi-1,
Spi-2, and Spi-3 [7], 16 kDa protein C1D [8], some polypeptides with phosphatase and kinase
activities, and other unidentified polypeptides [9,
10] belong to the TBP group. Very similar sets of
TBPs possessing phosphatase and kinase activities were found in yeast
and murine myeloid erythroleukemia (MEL) cells [11, 12]. It was hypothesized
more than fifty years ago that interactions between DNA and proteins
could be covalent. This hypothesis was recently proved when it was
shown that 0.1-0.3% of TBPs are bound to DNA via a phosphotriester bond
[13-15]. It is supposed that
such proteins are important for differentiation and regulation of gene
activity.
Despite a great deal of research, the functional significance of TBPs is not yet clear. Unfortunately, most of the previous studies on TBPs were performed on animal tissues, and very little is known about TBPs of higher plants. Characterization of plant TBPs appears to be a challenging problem, as plant genomes are organized in a specific way and chromatin organization also possesses some peculiarities compared to animal genomes. To fill this gap, we set the following goals in this work: 1) to characterize the polypeptide spectrum of TBPs in different barley shoot organs; 2) to identify a set of TBPs by means of mass-spectrometry and protein database analysis; 3) to determine the affinity of barley TBPs to DNA; 4) to characterize phosphatase activity of barley TBPs in various shoot organs and during different plant development stages. Barley shoots were chosen as an object as a useful model for plant development studies. Some preliminary work on barley TBPs was already performed by our group [16, 17]. It was shown that the spectrum of TBPs appeared to be organ and developmental-stage specific and thus promising for further investigations.
MATERIALS AND METHODS
Plant material. Seeds of the barley cultivar Auksiniai 3 were obtained from the Botanical Garden of Vilnius University (Kairenai, Lithuania). Etiolated shoots were grown for 3-5 days at a 24°C in darkness. Coleoptiles, first leaves, and roots were dissected from shoots of Zadoks 07 (coleoptile emerged stage) and 10 (first leaf through coleoptile) developmental stages [18]. Dissected coleoptiles, roots, and first leaf tissue were combined into one sample for each tissue at both developmental stages and used for bulk DNA extraction.
DNA isolation. Plant tissues were frozen in liquid nitrogen and ground in a mortar up to a fine powder. DNA was extracted from the plant material according to the previously described protocol of chloroform–isoamyl alcohol extraction [19] with some modifications. Cells (10 g) were suspended in extraction buffer (100 mM Tris-HCl, pH 8.0, 500 mM NaCl, 50 mM EDTA, 1.25% SDS) at a cell to buffer ratio of 1 : 1.6 (v/v) and incubated at 65°C for 30 min. Then extraction was performed with one volume of chloroform–isoamyl alcohol mixture (24 : 1 v/v). The final mixture was centrifuged at 4°C for 15 min at 2800g, cold ethanol (1 : 2 v/v) was added for DNA precipitation, centrifuged for 30 min at 2800g, rinsed with 70% ethanol, and air-dried. Dry DNA was dissolved in 3 ml of TE buffer (10 mM Tris-HCl, pH 8.0, 1 mM EDTA). Digestion with RNase A (Serva, Germany) (10 μg/ml) was performed for 3 h at room temperature. DNA was extracted with 3 ml of chloroform–isoamyl alcohol mixture as before (24 : 1) and centrifuged at 2800g (4°C). DNA was precipitated with two volumes of ethanol and 0.1 volume of 3 M sodium acetate, collected by centrifugation at 4°C for 30 min at 9000g, rinsed with 70% ethanol, and air-dried. Dried DNA was dissolved in 1 ml of the TE buffer and stored at 4°C.
TBP sample preparation. Isolated DNA (5 mg/ml) was diluted with 50 mM Tris-HCl buffer, pH 8.0, containing 1 mM MgCl2 and benzonase (Merck, Germany) was added (0.1 U/µg DNA). The reaction was continued while the sample was dialyzed for 16 h at room temperature against 1-1.5 liters of the same buffer and, subsequently, for 16-18 h at 4°C against 1-1.5 liters of the TE buffer. The resulting protein solutions were used for phosphatase assays, PAGE, and two-dimensional IEF/PAGE analysis.
DNA fragment preparation. To obtain DNA fragments, the digest was incubated with proteinase K (0.5 mg/ml) and SDS (0.5%) overnight at 37ºC and deproteinized with chloroform. Remnants of DNA were precipitated with ethanol [20].
SDS-PAGE. TBPs (70-100 µg) were mixed with the sample buffer, heated to 100oC for 5 min, and cooled [21]. SDS-PAGE gels (10-12%) were run at 120 V constant voltages for 4 h and stained with Coomassie brilliant blue dye.
Western blotting. Protein samples were fractionated by SDS-PAGE, and the gels and 0.45 μm pore size nitrocellulose filter (Bio-Rad, USA) were preincubated for 15 min in transfer buffer (19 mM glycine, 25 mM Tris, 0.1% SDS, 20% methanol). Standard electroblotting was performed for 3 h by applying a constant 200 mA current.
Determination of TBP phosphatase activity using MUP. 4-Methylumbelliferyl phosphate (MUP) (Boehringer Mannheim, Germany) was used as substrate at a concentration of 2 mM. Hydrolysis of MUP was followed fluorimetrically (excitation and emission wavelengths were 340 and 424 nm, respectively). The assay was carried out using 10 µg TBPs in 300 µl of 50 mM Tris-HCl (pH 7.0-9.5) or 50 mM Mes-NaOH (pH 5.5-7.0) buffer for 30 min.
Detection of TBP phosphatase activity in situ. TBPs were fractionated by SDS-PAGE and electroblotted onto nitrocellulose filters. Protein blots were incubated overnight at 4ºC in renaturation buffer (100 mM Hepes-NaOH (Roth), pH 7.4, containing 0.2 CHAPS (Roth), 10 mM MgCl2, 50 mM KCl, and 1 BSA (Roth). Phosphatase activity was visualized in situ by incubation of protein blots in a solution containing 50 mM Tris-HCl, pH 7.0, 0.5 agarose, 50 µg/ml 5-bromo-4-chloro-3-indolyl phosphate (BCIP) (Sigma, USA) and 1 mg/ml nitro blue tetrazolium (NBT) (Sigma) for 2-3 h. Protein bands with phosphatase activity were colored blue.
Two-dimensional gel separation (IEF/SDS-PAGE). IEF/SDS-PAGE was performed with a Multiphor II device (Amersham Biosciences, UK). Samples (20-50 µg TBPs) were purified with 2-D Clean-Up Kit (Amersham) according to manufacturer’s recommendation and dissolved in sample buffer (12 g urea, 50 mg DTT, 0.13 ml Pharmalit 3-10, 0.13 ml Triton X-100 and water to 25 ml). For the first dimension, 13 cm Immobiline Dry Strips pH 4-7 (Amersham Biosciences) were used. Dry strips were rehydrated, reduced, and alkylated according to manufacturer’s recommendations. For the second dimension, 8-18% Excell gels (Amersham Biosciences) were used. SDS-PAGE buffers: anode (0.45 M Tris-acetate, pH 6.6, 4 g/liter SDS, 0.05 g/liter Orange G); cathode (0.08 M Tris, 0.8 M Tricine and 6 g/liter SDS, pH 7.1). Gels were visualized with Coomassie brilliant blue dye and silver [22].
Tryptic in-gel digestion. The areas of the gel were cut out and subjected to in-gel tryptic digestion overnight [22], and the gel slices were dehydrated with 50% acetonitrile and then dried completely using a centrifugal evaporator (DNA Mini; Eppendorf, Germany). Protein spots were rehydrated in 20 μl of 25 mM ammonium bicarbonate (pH 8.3) containing 20 μg/ml of modified trypsin (Promega, USA). Once this solution had been fully absorbed by the gel, a trypsin-free buffer was added just enough to cover the slice, and the samples were incubated overnight at 37°C. The tryptic peptides were subsequently extracted from the gel slices as follows. Any extraneous solution remaining after the digestion was removed and placed in a fresh tube. The gel slices were first subjected to aqueous extraction and then to organic extraction with 5% trifluoroacetic acid in 50% acetonitrile, shaking occasionally. The digestion and extract solutions were then combined and evaporated to dryness.
Mass-spectrometry (MALDI TOF-TOF MS). For the MALDI TOF-TOF analysis, the peptides were redissolved in 3 μl of 50% acetonitrile and 0.1% trifluoroacetic acid solution and then placed with a matrix (α-cyano-4-hydroxycinnamic acid) on the target plate. The analysis was performed on a 4800 Plus MALDI TOF-TOF analyzer (Applied Biosystems, Canada) (in the Lithuanian Proteomic Center) and externally calibrated using synthetic peptides with known masses (4700 Cal Mix 1; Applied Biosystems). The MS spectra were obtained in the positive ionization mode at 3.080 kV, and the MS/MS spectra were obtained in the positive ionization mode at 3.780 kV. The mass information generated from the composite spectrum was submitted to a search performed within the MSDB and UniProtKB-SwissProt databases, using the GPS Explorer software (Applied Biosystems) based on the Mascot search engine.
DNA-binding protein blot assay was performed as described [23]. Nuclear proteins were separated by one dimension 12% SDS-PAGE and electroblotted to 0.45 µm nitrocellulose filters in 19 mM glycine, 25 mM Tris, 0.1% SDS, and 20% methanol. Protein blots were blocked overnight at room temperature in blocking buffer (10 mM Tris-HCl, pH 7.5, 50 mM NaCl, 2 mM EDTA, 1% BSA), incubated for 1 h in binding buffer (10 mM Tris-HCl, pH 7.4, 50 mM NaCl, 2 mM EDTA, 0.05% BSA), and transferred into a fresh portion of the binding buffer supplemented with 10 ng/ml of radioactively labeled DNA probe (10,000-70,000 cpm/ml) and 100 ng/ml of competitor DNA (Eco R1 digested plasmid pUC19). TBP-bound DNA fragments obtained according to exhaustive DNase digestion procedure was used as probes. Binding was carried out overnight at 37°C in hybridization oven under gentle agitation. The membranes were washed three times for 15 min in 100 ml of binding buffer and autoradiographed.
DNA probe labeling. Different DNA fractions were labeled with [α32P]dATP (Hartmann Analytic, Germany) using a DecaLabel DNA Labeling Kit (UAB Fermentas, Lithuania). Unincorporated nucleotides were removed by selective precipitation of DNA with ethanol in the presence of ammonium acetate.
Western blotting with anti Topo II antibody. The proteins separated on SDS-PAGE were electroblotted onto 0.45 µm nitrocellulose filters in Tris-glycine buffer (25 mM Tris, 19 mM glycine, 0.1 SDS containing 20 methanol) at 200 mA for 3 h at room temperature. The protein blots were preincubated in TST buffer (50 mM Tris-HCl, pH 7.5, 150 mM NaCl, 0.05 Tween 20 with 5 BSA) for 20 h at 4°C. Anti Topo II antibodies (Abcam Ab 4517) diluted 1 : 5000 in TS buffer with 5 BSA were added, and incubation was performed for 12 h at 4°C. After extensive washing with the TST buffer, the membranes were incubated with 1 : 5000 dilution of anti-rabbit antibodies conjugated with horseradish peroxidase and the ECL detection system.
RESULTS
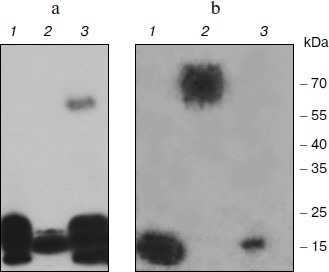
Affinity of the TBP-bound DNA to nuclear proteins. To test the ability of TBP complexes to be restored in vitro, we performed DNA-binding protein blot assay with electrophoretically fractionated TBPs extracted from different barley organs and TBP-bound DNA. Results are shown in Fig. 1. DNA fragments isolated from the tight DNA–protein complexes of coleoptiles manifested high affinity to low molecular weight TBPs proteins extracted from all organs. It also formed complexes with a 60 kDa protein from the coleoptile nuclei (Fig. 1a). DNA fragments isolated from complexes with root TBPs also had high affinity to low molecular weight nuclear proteins from roots and coleoptiles, and among the nuclear proteins extracted from leaves these DNA fragments recognized a 70 kDa peptide (Fig. 1b). Thus TBP complexes can form in vitro, both DNA and protein components possessing high affinity to each other.
Spectra of TBPs in barley shoot organs. Separation of barley TBPs obtained after benzonase digestion of DNA in 2D gel electrophoresis is presented in Fig. 2. Piloting experiments indicated that barley TBPs were mostly acidic, thus isoelectrofocusing was performed in the pH range from 4.0 to 6.8. In coleoptiles (Fig. 2, a and b), numerous well-resolved spots with molecular masses ranging from 15 to 70 kDa were observed. Transition from Zadoks 07 to Zadoks 10 stage was followed by an increase in the number of the spots, predominantly due to polypeptides with pI of 4.5-5.5 and molecular masses of 35-70 kDa. In leaves (Fig. 2, c and d), TBPs with pI higher than 6.0 were not observed. The number of the most acidic polypeptides decreased in the course of development. In roots (Fig. 2, e and f), transition to Zadoks 10 stage was followed by an increase in spots with molecular masses of about 20 and 35 kDa. Thus the TBP spectrum in barley shoot organs appears to be tissue- and developmental stage-specific.Fig. 1. DNA-binding protein blot assay with electrophoretically fractionated TBPs isolated from the roots (1), leaves (2), and coleoptiles (3) and incubated with TBP-associated DNA from coleoptile (a) or roots (b).
Mass spectral analysis indicated (see table in Supplement at the site of Biochemistry (Moscow) (http://protein.bio.msu.ru/biokhimiya)) that barley TBPs are homologous to several transcription factors, such as Pur-alpha 1, Pur b, auxin-responsive protein IAA16 (spots 2 and 6 in coleoptile TBPs). In leaves, TBP transcription factors were numerous, such as GATA transcriptional activator (spots 15 and 17), homeobox-leucine zipper protein ROC1, ABA-inducible bHLH (spot 15), transcription factor GTE (spot 20), and transcriptional adapters ADA2a and ADA2b (spots 16 and 22). Mediator complex (mediator of RNA polymerase II transcription activator), a protein providing scaffolding for RNA polymerase and the general transcription factors [23-25], was identified in spot 6 among coleoptile TBPs.Fig. 2. Barley TBPs obtained after digestion of 0.5 mg DNA with benzonase. The gels were stained with Coomassie brilliant blue R-250. a) Coleoptile, Zadoks stage 07; b) coleoptile, Zadoks stage 10; c) first leaves, Zadoks stage 07; d) first leaves, Zadoks stage 10; e) roots, Zadoks stage 07; f) roots, Zadoks stage 10. Positions of the molecular mass markers (kDa) are indicated on the left, and pI values are indicated below the figures. The marked polypeptides were digested with trypsin and analyzed with mass spectrometry.

Peptides involved in biotic and abiotic stress response were also found among TBPs. We have identified the two-component response regulator ARR15 (spot 5), which acts as negative regulator of cytokinin signaling [26]; probable disease resistance proteins [27] (spots 7-9, 11, 13); zinc finger CCCH domain-containing proteins (spots 1, 2, and 20). The CCCH domain-containing proteins repress jasmonic acid (JA) signaling in promoting leaf senescence and regulate panicle development and pollination/fertilization processes [28, 29]. Heat shock protein beta-11 (spot 7) is also considered as a stress protein [30, 31].
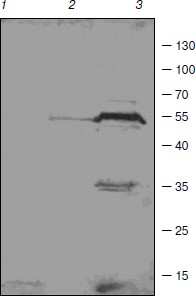
Some enzymes of DNA replication and repair, RNA processing, and chromatin remodeling were also identified among barley TBPs. RNA maturase was identified in spot 31. Several identified polypeptides are homologous to RNA helicases (spots 10, 22, 24, 29). These enzymes interact with double-stranded DNA and topoisomerase II [32]. Topoisomerase II was identified in spots 26 and 30. Due to great interest in this enzyme and contradictions concerning its presence in TBP fractions, the presence of topoisomerase II in our TBP preparations was verified by means of Western blotting with anti-topo II antibody (Fig. 3). Western blotting revealed binding of several peptides to anti-topo II antibody in TBP preparations obtained from coleoptile by exhaustive DNase digestion, TBPs isolated from leaves gave much weaker signal, and the enzyme was not detected by means of the Western blotting in the root TBP preparations (Fig. 3).
Two DNA repair proteins were identified among barley TBPs. Cell cycle checkpoint protein RAD17 involved in regulation of DNA double strand break repair [33] was identified in spot 11. DNA-damage-repair/toleration protein DRT100 was identified in spot 1; the function of this protein in plants has not been revealed yet [34].Fig. 3. Western blots of TBPs isolated from roots (1), leaves (2), and coleoptiles (3) and assayed with anti-topoisomerase II antibody.
Several identified polypeptides participate in chromatin remodeling processes. FACT complex sub SPT16 (spot 22) is a component of the FACT complex, a main chromatin factor that performs rearrangement of the nucleosomes [35]. High-mobility group protein HMG1/2-like protein identified in spot 16 is involved in transcription and recombination processes [36, 37]. Nuclear actin was also found among TBPs, in spot 12. Transposon-related proteins like transposon CACTA protein (spot 26) and AC9 transposase (spot 19) were also identified. Other proteins involved in nuclear regulation pathways, like nuclear hormone receptor family member nhr-28 (spot 11), Ran-binding protein 1 (spot 3), and cyclin-B1-4 (spot 10) also appeared to be tightly bound to DNA.
Using MALDI TOF-TOF analysis we have also identified some proteins involved in the ubiquitination pathway. RING finger protein 150 was identified in spot 11; this protein mediates protein–protein interactions that are relevant to a variety of cellular processes in plants [38]. F-box protein (At2g44130), a critical protein for the controlled degradation of cellular regulatory proteins via the ubiquitin pathway, was identified in spots 13 and 14. Small ubiquitin-related modifier 1 (spot 5) is an ubiquitin-like protein that can be covalently attached to target lysines as a monomer [39]. U-box domain-containing protein 57 (spot 19) functions as an E3 ubiquitin ligase. This enzyme was also identified in spot 26.
Finally, we have revealed the well-characterized TBPs such as serpins [7, 17], protein kinases, and phosphatases [11, 12, 40]. Serpin-Z5 (spot 21) belongs to the serine protease (serpin) family, which controls cell proteolysis and is important for plant growth, development, responses to stress, and defense against insects and pathogens. Serpins irreversibly inhibit endogenous and exogenous target proteinases [41]. We have also revealed numerous phosphatases — these were serine/threonine-protein phosphatase BSL (spot 23), serine/threonine phosphatase PP2C (spots 7 and 10), PP1 (spots 3 and 4), and various serine/threonine protein kinases (spots 2, 4, 10, 13, 14, 18, and 28). This confirms that the phosphatase and kinase activities are main features of TBPs [11, 13, 40]. This finding motivated us to perform a set of experiments aimed on characterization of enzymatic activities in barley TBPs.
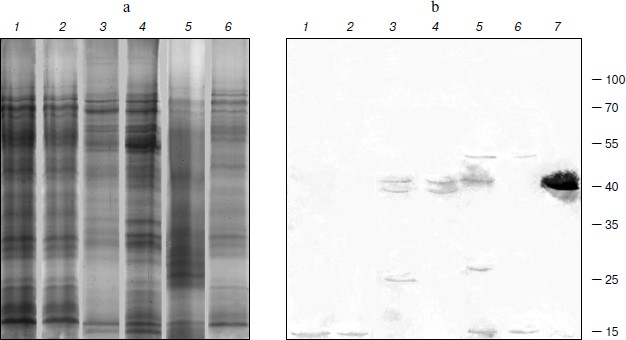
TBPs with phosphatase activity. The nature and the role of phosphorylation/dephosphorylation of TBPs are not fully understood at present. We have tried thus to examine whether barley TBPs possess this activity and whether it differs in plant organs or is modified during different developmental stages of the plant. Phosphatase activity was really detected in barley TBPs, but only in preparations obtained after hydrolysis of TBP–DNA complexes with benzonase (0.1 U/µg DNA). No activity was found when other protocols of TBP isolation were applied. Control experiments showed that benzonase used in our experiments was not contaminated with phosphatase. TBPs with phosphatase activity were identified in preparations obtained from plants both of Zadoks 07 and 10 development stages and in all organs: coleoptile, first leaves, and roots. To establish the polypeptides manifesting phosphatase activity, we employed the SDS-PAGE denaturation–renaturation scheme [42]. The SDS-PAGE-separated TBPs (Fig. 4a) were blotted on a nitrocellulose membrane, renatured overnight, and exposed in situ to NBT and BCIP. As seen in Fig. 4b, different polypeptides showed phosphatase activity in different organs and their development stages. In barley roots of Zadoks stages 07 and 10, the phosphatase activity was identified in ~10 kDa TBPs (Fig. 4b, lanes 1 and 2), in first leaves in ~38 and ~40 kDa TBPs (lanes 3 and 4), and in coleoptile ~70 and 10 kDa polypeptides (lanes 5 and 6) possessed phosphatase activity. In coleoptile of Zadoks stage 10 (Fig. 4b, lane 5) and first leaves of this stage, TBPs of 40 and 25 kDa manifested phosphatase activity. In first leaves of Zadoks stage 10, an additional 20 kDa polypeptide with phosphatase activity, absent from other organs, was identified. Thus, the number of TBPs that exhibit phosphatase activity in different barley organs increases during development (transition from Zadoks stage 07 to Zadoks stage 10). However, this effect was not observed with root TBPs.
To characterize the phosphatase activity of TBPs, we measured the pH dependence of this activity. As seen in Fig. 5, the appearance of pH profiles of TBP phosphatase activity depends on organs and development stages of barley. Phosphatase activity of root TBPs of both Zadoks 07 and 10 stages was maximal at pH 8.5. Optimal pH for phosphatases from leave and coleoptile TBPs was 7.8. In Zadoks stage 10 coleoptile, the TBP phosphatases manifested an additional peak at pH 8.5. Probably, these differences were due to the presence of different phosphatases in TBPs.Fig. 4. Detection of barley TBP phosphatase activity. a) Barley TBPs obtained after digestion of 0.5 mg DNA with benzonase were separated by 12% SDS-PAGE and stained with Coomassie blue G-colloidal dye. b) In situ staining for phosphatase activity with NBT and BCIP. 1) Roots, Zadoks stage 10; 2) roots, Zadoks stage 07; 3) first leaves, Zadoks stage 10; 4) first leaves, Zadoks stage 07; 5) coleoptiles, Zadoks stage 10; 6) coleoptiles, Zadoks stage 07; 7) alkaline phosphatase control. Positions of molecular mass markers (kDa) are indicated on the right.
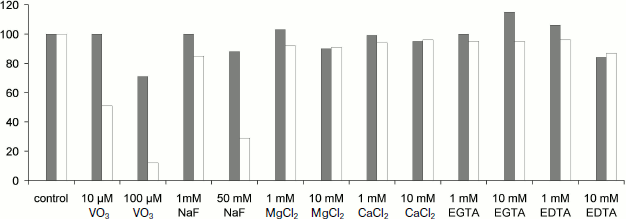
To reveal the type of TBP phosphatases, inhibitor analysis was performed. Phosphatase activity in TBPs of different barley organs and development stages was assayed using a standard phosphatase assay (substrate MUP) in the presence of various additions. The results are shown in Fig. 6.Fig. 5. Effect of pH on phosphatase activity. a) Roots, Zadoks stage 07; b) roots, Zadoks stage 10; c) first leaves, Zadoks stage 07; d) first leaves, Zadoks stage 10; e) coleoptiles, Zadoks stage 07; f) coleoptiles, Zadoks stage 10. Phosphatase activity was measured as MUP hydrolysis rate expressed as fluorescence change per 5 min at room temperature. The assay was carried out using 10 µg TBPs in 300 µl 50 mM Tris-HCl (pH 7.0-9.5) or 50 mM Mes-NaOH (pH 5.5-7.0).
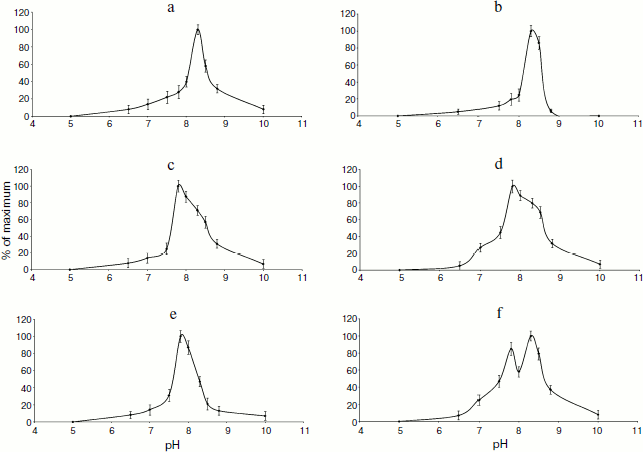
Fig. 6. Effects of additions on TBPs phosphatase activity of barley Zadoks stage 10 first leaves (white columns) and coleoptiles (gray columns). Phosphatase activity was measured as MUP hydrolysis rate expressed as fluorescence change per 5 min at room temperature. The assay was carried out using 10 µg TBPs in 300 µl 50 mM Tris-HCl, pH 8.0. All determinations were made in triplicate, and the values were normalized to untreated control.
DISCUSSION
Our attempt to characterize barley TBPs performed in this work revealed great diversity of this protein group. Some conclusions can be drawn. Most of the transcription factors identified among TBPs participate in cell growth, biotic or abiotic stress response, differentiation, and development processes. These transcription factors interact with DNA via purine-rich double-stranded telomeric repeated sequence (Pur-alpha 1), GATA motif (GATA transcriptional activator), noncanonical helix–loop–helix motif (ABA-INDUCIBLE bHLH), and leucine zipper (homeobox-leucine zipper protein ROC1). Thus, our results and results of the previous work [43] show that many transcription factors containing different DNA binding domains are resistant to a deproteinization procedure. The interactions of DNA with corresponding domains of transcription factors appeared to be resistant to salts, detergents, and organic solvents.
In the present work, we proved unequivocally the presence of topoisomerase II in TBP preparations. Although topoisomerases are considered to be classical examples of proteins covalently bound to DNA [44], several authors failed to find topoisomerases among proteins remaining attached to DNA after deproteinization [45]. Finding of topo II motifs among the DNA sequences tightly bound to TBPs [46, 47] and mass spectral analysis indicate the presence of topo II in tightly bound DNA–protein complexes.
The presence of RNA maturases among TBPs seems to be particularly interesting. These enzymes interact with both RNA and DNA. The DNA-binding domains of the enzyme protrude inside the DNA double helix, and DNA forms a curvature in the site of interaction with the enzyme [48]. According to our previous results, DNA fraction bound to TBPs is enriched in bent sequences and Z DNA [46, 47, 49]. Finding of DNA repair proteins among TBPs also confirmed the results of our previous studies, in which several DNA repair proteins (RAD 7, RHC31) were identified among yeast TBPs [50].
Chromosomal high-mobility group protein HMG1/2-like protein identified as a TBP participates in many DNA transactions such as transcription and recombination. The assembly of the nucleoprotein complexes often requires the DNA to be specifically folded by DNA-bending proteins. In plants, various chromatin-associated high mobility group proteins of the HMGI and HMGI/Y families have been identified that have the potential to act as structural factors that modulate DNA structure [36, 37]. Another newly identified TBP, nuclear actin, attracts interest of many researchers because of its possible role in genome organization. Long-range directed interphase chromatin movement seems to require actin polymerization, as the expression of mutant actin unable to polymerize prevents chromatin relocation [51]. Some data strongly indicate that nuclear actin is involved in gene transcription by all the three polymerases [52].
The presence of the members of the ubiquitin–proteasome pathway among TBPs appears to be a well-established fact [43, 50]. This was confirmed in the present study. It is known that many proteins containing a RING finger play a key role in the ubiquitination pathway. RING finger protein 150 has been shown to mediate protein–protein interactions that are relevant to a variety of cellular processes in plants [38]. F-box proteins have been shown to be critical for the controlled degradation of cellular regulatory proteins via the ubiquitin pathway. F-box proteins specifically recruit the targets to be ubiquitinated, mainly through protein–protein interaction modules such as WD-40 domains or leucine-rich repeats (LRRs) [53]. Small ubiquitin-related modifier 1 is an ubiquitin-like protein, which can be covalently attached to target lysines as a monomer. This protein functions as an antagonist of ubiquitin in the degradation process [39]. U-box domain-containing protein 57 functions as an E3 ubiquitin ligase in the protein ubiquitination pathway. Our data support the hypothesis on the involvement of proteasomes in transcription regulation [54]. Serpins, a protein class commonly found among TBPs [7, 17], were represented in barley cells by Serpin-Z5, which controls cell proteolysis and is important for plant growth, development, responses to stress, and defense against insects and pathogens. Serpins irreversibly inhibit endogenous and exogenous target proteinases [46].
Our study has also revealed a phosphatase activity of barley TBPs intrinsic for numerous peptides of the fraction. The spectrum of TBPs with this activity varied in different barley organs and was dependent on development stage, and the pH-dependence of the enzymes varied in different organs and was bimodal in one case. This suggests the existence of numerous proteins with phosphatase activity among TBPs. Our attempt to classify the TBPs phosphatase activity by means of inhibitory analysis also supports the above conclusion. Most cell phosphatases can be identified as Ser/Thr or Tyr phosphatases [55]. Ser/Thr phosphatases are divided into protein phosphatase P (PPP) and protein phosphatase M (PPM). The PPM family mainly consists of PP2C phosphatases. The PPP family contains protein phosphatases 1 (PP1), 2A (PP2A, 2B (PP2B), PP5, PP7. Tyr phosphatases (PTPases) are divided into two families – A and B [56, 57]. Protein phosphatases can be distinguished according the sensitivity to specific protein phosphatase inhibitors. A type Tyr phosphatases (PTPases) are less sensitive to NaVO3 than B type PTPases. IC50 for A type PTPases is 40 μM, but IC50 for B type PTPases is 2-6 μM. Ser/Thr phosphatases are insensitive to micromolar NaVO3 concentrations (10-100 μM). Barley leaf TBP phosphatases were sensitive to both NaVO3, even when at low concentration, and NaF. This indicates that both protein phosphatases (PPases) and Tyr phosphatases are found among barley TBPs. TBP phosphatases are unlikely to belong to PP-1 or PP-2A classes, as divalent metal ions do not affect the activity of these phosphatases [58]. It is known that some Ser/Thr phosphatases are activated by divalent metal ions: PP-2B is activated by Ca2+, PP-7 by Ca2+ and Mg2+, and PP-2C by Mg2+ [59]. Barley Zadoks stage 10 coleoptile TBP phosphatases were activated by millimolar concentrations of EGTA. This indicates that these TBP phosphatases are PP-2C, keeping in mind that part of them could belong to Tyr phosphatase type. Finding of a protein homologous to serine/threonine-protein phosphatase BSL, serine/threonine phosphatase PP2C, and various other serine/threonine protein kinases confirms the results of enzymatic activity measurements and indicates that the phosphatase activity is one of the main features of TBPs [11-13, 40].
Taken together, our data indicate that TBPs are represented in barley plants by a large set of proteins including DNA binding proteins of different classes and several groups of enzymes. The number and identity of TBPs depend on the plant organ and its developmental stage. Barley TBPs can form complexes with DNA in vitro. Phosphatase activity is a characteristic feature of barley TBPs.
We would like to thank Mindaugas Valius and Marija Ger for help in mass spectral analysis.
This work was supported by Lithuanian State Science and Studies Foundation grant No. T-07176.
REFERENCES
1.Berezney, R. (2002) Advan. Enzyme Regul.,
42, 39-52.
2.Cockell, M., and Gasser, S. M. (1999) Curr. Net.
Develop., 9, 199-205.
3.Foster, H. A., and Bridger, J. M. (2005)
Chromosoma, 114, 212-229.
4.Trinkle-Mulcahy, L., and Lamond, A. I. (2008)
FEBS Lett., 582, 1960-1970.
5.Takata, H., Nishijima, H., Ogura, S., Sakaguchi,
T., Bubulya, P. A., Mochizuki, T., and Shibahara, K. (2009) Genes to
Cells, 14, 975-990.
6.Neuer-Nitsche, B., Plagens, U., and Werner, D. J.
(1983) Mol. Biol., 164, 213-235.
7.Rothbarth, K., Kempf, T., Juodka, B., Glaser, T.,
Stammer, H., and Werner, D. (2001) Eur. J. Cell. Biol.,
80, 341-348.
8.Rothbarth, K., Spiess, E., Juodka, B., Yavuzer, U.,
Nehls, P., Stammer, H., and Werner, D. (1999) J. Cell. Sci.,
112, 2223-2232.
9.Avramova, Z., Michailov, I., and Tsanev, R. (1989)
Biochim. Biophys. Acta, 1007, 109-111.
10.Juodka, B., Spiess, E., Angiolillo, A., Joswig,
G., Rothbarth, K., and Werner, D. (1995) Nucleic Acids Res.,
23, 1359-1366.
11.Borutinskaite, V., Bagdoniene, L., Labeikyte, D.,
and Juodka, B. (2004) Biologija, 4, 11-14.
12.Labeikyte, D., Bagdoniene, L., and Juodka, B.
(1999) Biologija, 4, 22-27.
13.Juodka, B., Pfutz, M., and Werner, D. (1991)
Nucleic Acids Res., 19, 6391-6398.
14.Labeikyte, D., Bagdoniene, L., Sasnauskiene, S.,
Sabaliauskiene, V., and Juodka, B. (2002) Biologija, 2,
50-55.
15.Tsanev, R., and Avramova, Z. (1994)
Chromosoma, 103, 293-301.
16.Sjakste, T., Bielskiene, K., Roder, M., Sugoka,
O., Labeikyte, D., Bagdoniene, L., Juodka, B., Vassetzky, Y., and
Sjakste, N. (2009) BMC Plant Biol., 9, 56.
17.Bielskiene, K., Bagdoniene, L., Labeikyte, D.,
Juodka, B., and Sjakste, N. (2009) Biologija, 55,
7-13.
18.Anderson, P. M., Oelke, E. A., and Simons, S. R.
(1995)
[http://www.extension.umn.edu/distribution/cropsystems/DC2547.html].
19.Plaschke, J., Ganal, M. W., and Roder, M. S.
(1995) Theor. Appl. Genet., 91, 1001-1007.
20.Avramova, Z., Georgiev, O., and Tsanev, R. (1994)
DNA Cell Biol., 13, 539-548.
21.Laemmli, U. K. (1970) Nature, 227,
680-685.
22.Shevcenko, A., Wilm, M., Vorm, O., and Mann, M.
(1996) Annal. Chem., 68, 850-858.
23.Luderus, M. E., de Graaf, A., Mattia, E., den
Blaauwen, J. L., Grande, M. A., de Jong, L., and van Driel, R. (1992)
Cell, 70, 949-959.
24.Kwon, J. Y., Park, J. M., Gim, B. S., Han, S. J.,
Lee, J., and Kim, Y. J. (1999) Proc. Natl. Acad. Sci. USA,
96, 14990-14995.
25.Backstrom, S., Elfving, N., Nilsson, R., Wingsle,
G., and Bjorklund, S. (2007) Mol. Cell, 26, 717-729.
26.D’Agostino, I. B., Deruere, J., and Kieber,
J. J. (2000) Plant Physiol., 124, 1706-1717.
27.Martin, G. B., Bogdanove, A. J., and Sessa, G.
(2003) Annu. Rev. Plant Biol., 54, 23-61.
28.Kong, Z., Li, M., Yang, W., Xu, W., and Xue, Y.
(2006) Plant Physiol., 141, 1376-1388.
29.Wang, D., Guo, Y., Wu, C., Yang, G., Li, Y., and
Zheng, C. (2008) BMC Genom., 9, 44-44.
30.Marvin, M., O’Rourke, D., Kurihara, T.,
Juliano, C. E., Harrison, K. L., and Hutson, L. D. (2008) Dev.
Dyn., 237, 454-463.
31.Efeoglu, B. G. U. (2009) J. Sci.,
22, 67-75.
32.Fuller-Pace, F. V. (2006) FEBS Lett.,
413, 50-54.
33.Heitzeberg, F., Chen, I. P., Hartung, F., Orel,
N., Angelis, K. J., and Puchta, H. (2004) Plant J., 38,
954-968.
34.Pang, Q., Hays, J. B., and Rajagopal, I. (1992)
Proc. Natl. Acad. Sci. USA, 89, 8073-8077.
35.Duroux, M., Houben, A., Ruzicka, K., Friml, J.,
and Grasser, K. D. (2004) Plant J., 40, 660-671.
36.Grasser, K. D., Wohlfarth, T., Baeumlein, H., and
Feix, G. (1993) Plant Mol. Biol., 23, 619-625.
37.Grasser, K. D., Krohn, N. M., Lichota, J., and
Stemmer, Ch. (2000) Physiol. Plant., 110, 427-435.
38.Lim, S. D., Yim, W. C., Moon, J. C., Kim, D. S.,
Lee, B. M., and Jang, C. S. (2010) Plant Mol. Biol., 72,
369-380.
39.Kurepa, J., Walker, J. M., Smalle, J., Gosink, M.
M., Davis, S. J., Durham, T. L., Sung, D. Y., and Vierstra, R. D.
(2003) J. Biol. Chem., 278, 6862-6872.
40.Loeffler, H., Spiess, E., Juodka, B., Stammer,
H., and Werner, D. (1996) Eur. J. Biochem., 240,
600-608.
41.Roberts, T. H., and Hejgaard, J. (2008) Funct.
Integr. Genom., 8, 1-27.
42.Meikrantz, W., Smith, D., and Sladicka, M. (1991)
J. Cell. Sci., 98, 303-307.
43.Sjakste, N., Bagdoniene, L., Gutcaits, A.,
Labeikyte, D., Bielskiene, K., Trapina, I., Muiznieks, I., Vassetzky,
Y., and Sjakste, T. (2010) Biochemistry (Moscow), 75,
1240-1251.
44.Drygin, Y. F. (1998) Nucleic Acids Res.,
26, 4791-4796.
45.Avramova, Z., and Tsanev, R. J. (1987) Mol.
Biol., 196, 437-440.
46.Bagdoniene, L., Bonikataite, K., Borutinskaite,
V., Labeikyte, D., and Juodka, B. (2005) Biologija, 4,
1-8.
47.Labeikyte, D., Bagdoniene, L., Jonusiene, V.,
Sasnauskiene, S., and Juodka, B. (2005) Biologija, 1,
1-5.
48.Belfort, M. (2003) Genes Dev., 17,
2860-2863.
49.Sjakste, N. (1997) Proc. Latv. Acad. Sci.,
51, 61-63.
50.Bagdoniene, L., Borutinskaite, V., Navakauskiene,
R., and Juodka, B. (2008) Biologija, 54, 227-232.
51.Dundr, M., Ospina, J. K., Sung, M. H., John, S.,
Upender, M., Ried, T., Hager, G. L., and Matera, A. G. (2007) J.
Cell. Biol., 179, 1095-1103.
52.Percipalle, P. (2009) Cell Mol. Life Sci.,
66, 2151-2165.
53.Andrade, M. A., Gonzalez-Guzman, M., Serrano, R.,
and Rodriguez, P. L. (2001) Plant Mol. Biol., 46,
603-614.
54.Collins, G., and Tansey, W. (2006) Curr. Opin.
Genet. Dev., 16, 197-202.
55.Gordon, J. A. (1991) Meth. Enzymol.,
201, 477-482.
56.Dombradi, V. (2002) Eur. J. Biochem.,
269, 1049-1049.
57.Moorhead, G., Wever, V., Templeton, G., and Kerk,
D. (2009) Biochem. J., 417, 401-409.
58.Bollen, M., and Beullens, M. (2002) Trends
Biochem. Sci., 12, 138-146.
59.Hardie, D. G., et al. (1993) Protein
Phosphorylation: A Practical Approach, Vol. 62, IRL Press, New
York.
Supplementary TABLE (PDF)