REVIEW: The Phenoptosis Problem: What Is Causing the Death of an Organism? Lessons from Acute Kidney Injury
D. B. Zorov1,2*, E. Y. Plotnikov1,2, S. S. Jankauskas2,3, N. K. Isaev1,2, D. N. Silachev1,2, L. D. Zorova2,4, I. B. Pevzner2,3, N. V. Pulkova2,3, S. D. Zorov2,3, and M. A. Morosanova2,3
1Belozersky Institute of Physico-Chemical Biology, Lomonosov Moscow State University, 119991 Moscow, Russia; fax: (495) 939-0338; E-mail: zorov@genebee.msu.su2Institute of Mitoengineering, Lomonosov Moscow State University, 119991 Moscow, Russia
3Faculty of Bioengineering and Bioinformatics, Lomonosov Moscow State University, 119991 Moscow, Russia
4International Laser Center, Lomonosov Moscow State University, 119991 Moscow, Russia
* To whom correspondence should be addressed.
Received March 26, 2012; Revision received March 30, 2012
Programmed execution of various cells and intracellular structures is hypothesized to be not the only example of elimination of biological systems – the general mechanism can also involve programmed execution of organs and organisms. Modern rating of programmed cell death mechanisms includes 13 mechanistic types. As for some types, the mechanism of actuation and manifestation of cell execution has been basically elucidated, while the causes and intermediate steps of the process of fatal failure of organs and organisms remain unknown. The analysis of deaths resulting from a sudden heart arrest or multiple organ failure and other acute and chronic pathologies leads to the conclusion of a special role of mitochondria and oxidative stress activating the immune system. Possible mechanisms of mitochondria-mediated induction of the signaling cascades involved in organ failure and death of the organism are discussed. These mechanisms include generation of reactive oxygen species and damage-associated molecular patterns in mitochondria. Some examples of renal failure-induced deaths are presented with mechanisms and settings determined by some hypothetical super system rather than by the kidneys themselves. This system plays the key role in the process of physiological senescence and termination of an organism. The facts presented suggest that it is the immune system involved in mitochondrial signaling that can act as the system responsible for the organism’s death.
KEY WORDS: sudden death, multiple organ failure, apoptosis, kidney, mitochondria, antioxidantDOI: 10.1134/S0006297912070073
Abbreviations: AIF, apoptosis-inducing factor; DAMP, damage-associated molecular patterns; MODS, multiple organ dysfunction syndrome; mtDNA, mitochondrial DNA; PAMP, pathogen-associated molecular patterns; PARP, poly(ADP-ribose) polymerase; ROS, reactive oxygen species; SCA, sudden cardiac arrest; SkQ1, 10-(6′-plastoquinonyl) decyltriphenylphosphonium; SkQR1, 10-(6′-plastoquinonyl) decylrhodamine 19.
The current rapid development of molecular basics of biology and a major
redistribution of academic resources in this area have resulted in
lesser emphasis on old historically fundamental disciplines, in
particular, physiology of entire biological systems. As a result of
this, many data on intracellular and intermolecular interactions in
living cells are being accumulated, and in parallel with the progress
in these areas, there appear serious doubts whether there is a
relationship of these results to processes actually occurring in cells,
organs, and organisms.
Understanding of the mechanisms of the destruction of the whole system has proved to be one of the fundamental problems in biology and medicine. Large amounts of data and arguments about the mechanisms of cell death have been collected (although there is no consensus even in this area), while at the same time understanding of the causes of failure of functioning of an entire organ or an organism is so fragmentary and incomplete that it sometimes is just hard to comment. Even the search for information on this issue is extremely difficult, for it is virtually impossible to identify key words that could narrow down the search. For instance, when seeking a solution in PubMed and listing “death of an organism”, “death”, and “phenoptosis” as key words, the researcher will get data that will hardly be useful.
So, our first task is to formulate definitions. There are many problems in determining the moment of death of a biological organism. We leave aside these details, accepting to some extent the subjectivity of such concepts as the criteria for determining death. Such an approach, while being obviously not the best one, still seems to be acceptable within reasonable limits, for it is supported by most people. The second problem (which is the subject of discussion in this paper) is to identify the signals that trigger the process of eliminating of a biological system of a higher order, an entire organism. We need not only determine the nature of death signals, but also to identify the system responsible for the decision to eliminate the organism, that is, the receiver of these signals.
FROM SMALL TO LARGE
In 1999, V. P. Skulachev proposed a theory according to which the programs of destruction of biological structures of different levels follows a certain sequence: from mitoptosis through apoptosis and organoptosis to phenoptosis. At the same time, reactive oxygen species (ROS) were postulated to be the determining factor in termination of these biological structures [1, 2]. Some more details were added to this scheme later [3].
Mitoptosis, mitophagy, programmed destruction of mitochondria. Directed destruction of the entire mitochondria population in a reticulocyte at a certain stage of development of mammalian erythrocytes can serve as an example of mitoptosis, or the programmed degradation of mitochondria in nature [4].
The destruction of sperm mitochondria in the fertilized egg is typical for many species and apparently follows the same mitoptotic law [5, 6]. Autophagosomal degradation of mitochondria (mitophagy) is suggested to be the main mechanism of destruction of mitochondria [5-7]. Fragmentation of the mitochondrial reticulum [8-12], generation of a nonspecific conductance in the inner mitochondrial membrane (mitochondrial megachannel [13-15]), ROS-induced ROS release coupled to the appearance of this conductance [16, 17], and activation of degrading hydrolases [18, 19] are likely to precede mitophagy. However, the idea that ROS, especially of mitochondrial origin, play the major role in regulation of autophagosomal activity has been gaining more support recently due to growing amount of evidence [20].
Manifestations of mitoptosis are likely to be very diverse. Besides the illustrative examples of mitoptosis due to autophagosomal activation, there is also the case of mitoptosis with no apparent signs of autophagy, this process being coupled to release of mitochondria from a cell [21, 22].
Cell death. Programmed elimination of cells that can no longer be unambiguously interpreted as apoptosis, due to accumulation of new data, appears to be a very large area that requires substantially more factual material and discussion than can be provided in this article. The share of “non-programmed” cell death among the partners in the task of cell destruction decreases steadily, which may eventually result in the realization that there is no such “random” option of cell death, since detailed examination of these cascades reveals the existence of many elements of biological programs. The reason for this phenomenon lies in the fact that in all the described types of cell destruction, the process of proliferation of a death signal proves to be a multistage one, and as long as there are many stages, there appears a number of possibilities to regulate partial stages of the process, affecting the final result, and this fact per se proves the existence of a program. We would like to focus on only two things – the diversity of the ways to realize the programs of cell death and the role of mitochondria and ROS in a number of these manifestations. By the beginning of 2012, certain agreement has been reached on the existence of 13 regulated cell death programs [23]. Let us list them and provide their short description.
1. Caspase-dependent intrinsic apoptosis.
2. Caspase-independent intrinsic apoptosis.
Mitochondria play the key role in these two types of cell death: their outer membrane becomes permeabilized, mitochondrial transmembrane potential is reduced, and the proteins of the intermembrane space are released into the cytosol.
3. Extrinsic apoptosis by death receptors.
4. Extrinsic apoptosis by dependent receptors.
These two types of cell death are triggered by external stress signals that are captured and mediated by special transmembrane receptors; mitochondria do not participate in this process.
5. Necroptosis as a type of regulated necrosis (which was traditionally considered to be a random event) with specific characteristics different from apoptosis and autophagy. This type of cell death is triggered by ligands of tumor necrosis factor receptor (TNFR1); receptor-interacting protein kinases, RIP1, and RIP2 participate in this process.
6. Autophagic cell death is characterized by massive vacuolization of the cytoplasm. Normally, autophagosomes (bilayer membrane vesicles containing degenerating cytoplasmic organelles and the cytosol) fuse with lysosomes, this process resulting in the internal autophagosome components being digested by acidic lysosome hydrolases. When this fusion is blocked, accumulation of autophagosomes causes cell death.
7. Mitotic catastrophe takes place when something goes wrong in the course of mitosis; such cells die during mitosis or a little later. Extensive changes in the nucleus (micronucleation or multinucleation) are often a marker of mitotic catastrophe. Micronucleation is often the result of chromosomes or their fragments being unevenly distributed between daughter cells. Multinucleation is characterized by the presence of two or more nuclei of the same or different sizes, this resulting from improper cytokinesis.
8. Anoikis is the type of cell death determined by the interaction of cells with extracellular matrix via integrins and the receptors of growth factors. Violation of this interaction triggers anoikis, which later follows the stages of controlled apoptosis.
9. Cornification, or keratinization, is characteristic of the cells of the outer layer of epidermis, where stratum corneum is formed as a result of death of keratinocytes. This structure contains not only dead cells, but also different proteins and lipids, which provide the skin with structural stability, elasticity, mechanical resistance, and hydrophobicity.
10. Entosis, or cell cannibalism, is expressed phenotypically (cell in a cell), in particular, in samples of tumors. It is triggered by loss of extracellular matrix and is not accompanied by activation of caspases. Internalized cells appear to be perfectly normal at first, but then they disappear, presumably due to digestion.
11. Netosis is the type of cell death accompanied by the release of DNA from neutrophils and eosinophils; this DNA together with associated proteins forms NET (neutrophil extracellular traps) for protection against bacterial. Chromatin condensation and destruction of nuclear and other intracellular membranes are observed in netotic cells before DNA release. Netosis is suppressed by inhibitors of NADPH-oxidase, which produces superoxide anion necessary for triggering autophagy.
12. Parthanatos represents the case of regulated necrosis. This process is named after the Greek god of death Thanatos (the word meaning death in Greek; Thanatos was the son of the god of eternal darkness, Tartarus, and the goddess of night, Nyx). PolyADP-ribose (PAR) polymerases (PARPs) participate in triggering of this type of cell death, in particular, PARP1, which is responsible for reparation of moderately damaged DNA. NAD and ATP are used in the course of this process; as a result, their reserve is depleted and toxic PAR is accumulated in mitochondria. The latter substance causes the reduction of mitochondrial membrane potential; AIF (apoptosis-inducing factor) is released from mitochondria and becomes bound to PAR, thus triggering parthanatos. This type of caspase-independent cell death is observed in cases of brain stroke, neurodegeneration, diabetes, and inflammation.
13. Pyroptosis is characterized by activation of caspase 1 and the participation of the inflammasome. As a result, pyrogenic interleukins IL1β and IL18 are released. These compounds often cause activation of other caspases, perforation of the cell membrane, swelling, and osmotic lysis of the cell. In particular, this type of cell death is observed in macrophages during bacterial infection, but it may also have a viral nature or take place in brain cells in case of stroke. It does not depend on PARP activation or DNA fragmentation.
Simple logic tells us that regulation of the majority of these programs is both energy- and redox-dependent, and hence it will be determined by mitochondria, including not only ATP production, but also other mitochondrial functions, in particular maintenance of cell redox homeostasis [24]. In a number of cases, e.g. when pathways 1, 2, 4, 6, 11 become activated, mitochondrial restructuring, such as permeabilization of outer and inner membranes, occurs. This phenomenon probably determines the release of certain components that trigger terminal and irreversible stages of apoptotic cell degradation (cytochrome c, AIF, SMAC, DIABLO, endonuclease G, serine protease Omi/HTRA2) or the innate immune response (in the case of mitochondrial DNA being released from mitochondria [25-30]). The role of ROS in each of the modalities is not fully elucidated, but in the case of the above-described pathways connected to the restructuring of mitochondria, their involvement is obligatory, even though in the case of netosis it is ROS produced by NADPH-oxidase (NOX2), rather than mitochondrial ROS, that play the key role [31].
Death/failure of organs. It is not easy to document the acute death of an organ with the exception of sudden cardiac arrest (SCA) (which is, by the way, responsible for almost 15% of the mortality rate in the USA [32]). In the majority of other cases, a rapid (acute) or slow (chronic) progressive weakening of the functioning of an organ (e.g. thymus involution, liver failure caused by cirrhosis, development of acute or chronic renal failure, virus-induced decline of immunity, muscle atrophy, loss of structures responsible for cognitive brain activity, development of blindness, deafness, etc.) occurs.
Chronic renal failure [33] and even the very procedure of hemodialysis (which is done to artificially replace the activity of damaged kidneys but provokes oxidative stress and inflammation [34, 35]) are factors increasing the risk of SCA. These factors seem to cause SCA or, more precisely, they correlate with SCA frequency [33]. In addition, SCA often develops in patients with ischemic heart disease, left ventricular hypertrophy and myocardial fibrosis, calcification of blood vessels, excessive sympathetic activity, systematic inflammation, in diabetics, in the elderly, etc. By the way, about two thirds of deaths caused by sudden cardiac arrest have no apparent reason (accepted risk markers are absent) and appear as the first clinical event [32], although about one third of all the cases are connected to long-term concomitant pathologies of various types that place individuals into risk categories. Taking into account these data on the contribution of this pathology in the overall picture of mortality, nearly 10% of all deaths are caused by a sudden and unexplainable cardiac arrest.
The clearly programmed syndrome of multiple simultaneous failure of a number of organs (multiple organ failure (MOF) syndrome) is obviously a particular example and a source of phenoptotic death of a system. The mechanism of rapid or acute phenoptosis may be based on this phenomenon, and that is why it requires special consideration. The following phenomena are considered to be the main causes of this syndrome: those coupled to ischemia/reperfusion; accompanied by excessive ROS generation; activation of neutrophils and their attachment to endothelium followed by infiltration into tissues; impaired intestinal barrier function leading to penetration of bacteria and/or endotoxins; the state of hyperinflammation alternating with immunosuppression facilitating bacterial infection, etc. [36-40].
In 2011 an article was published with the rather provocative title “Mitochondrial Dysfunction and Biogenesis: Do ICU Patients Die from Mitochondrial Failure?” [41]. The authors believe the body’s response to initial stimuli, such as trauma, ischemia, burns, infection, or stress are composed of and regulated by mediators of different humoral systems, such as complement, coagulation system, etc., or are the result of the action of such cells as monocytes/macrophages, i.e. associated with the release of cytokines, proteases, ROS, and reactive nitrogen species. In principle, this response is originally supposed to rescue the organism, but hypermobilization, having crossed a certain threshold, can lead to the complete imbalance of mediator system and can be transformed into a damaging stimulus culminating in MOF. In the next section we will consider the possible role of mitochondria in this imbalance.
Death of an organism. Any pathology accompanied by acute renal failure immediately increases mortality by 15-60%, a fact indicating the special role of kidneys in mortality [42]. In the case of intensive care unit patients, deterioration of oxygen supply and hyperactive inflammation caused by ischemia are critical.
Generally speaking, cell death of any type – either caused by one of the non-programmed mechanisms that have not been described in this paper or following one of the 13 described programmed mechanisms (although one may expect a combination of different death types in one organ and organism) – can be a reason for organ(s) failure. At the same time, massive cell death is not necessarily a part of hyperinflammatory response, even though the total disruption of organ functioning is a frequent event. For example, post-mortem histological examination of animals or humans, including those who have died from MOF, often shows no signs of necrotic or apoptotic cell death in a failed tissue [43-46]. However, signs of mitochondrial dysfunction in biopsy samples from experimental animals, sepsis patients, or patients with transplanted organ failure have been repeatedly described [47-54].
Briefly summarizing the information on the genesis of MOF, which is the root cause of posttraumatic death, one can notice that it occurs as a result of impaired inflammatory response and can have bimodal character. Having experienced serious damaging effects of infectious and noninfectious nature (trauma, burns, acute pancreatitis), an individual enters the early stage of hyperinflammation (i.e. systemic inflammatory response syndrome (SIRS) develops). When the primary damaging factor is strong enough, it may cause early MOF (one-hit model). If the system manages to withstand the blow, MOF may still develop, but it will happen later as a secondary inflammatory injury response (two-hit model) [55-57].
In the late 70s-early 80s, uncontrolled infection was considered to be the main cause of MOF [58-60]. In this case, sepsis with possible circulation of bacteria and their components (e.g. bacterial DNA) in blood (the fact leading to immune response) was seen as the intermediate stage between the initial damage and MOF. However, blood proves to be sterile in the case of the syndrome of systemic inflammatory response observed in trauma or hemorrhagic shock. In such a situation, both systemic inflammatory syndrome and MOF can be observed in approximately 30% of patients. The clinical picture of these phenomena is not distinguishable from that of bacterial sepsis, and mortality is very high in both cases [56, 57]. It turns out that extensive injuries often lead to a sepsis-like state and systemic inflammation can be described as one of pathogenic components of MOF.
It should be noted that sepsis causes massive cell death (of necrotic and apoptotic types) in the lymphoid organs (spleen, thymus) and lymphoid cells of non-lymphoid organs (e.g. intestines). Cell death in non-lymphoid cells [46] is rarely observed, this fact indicating the extreme lymphatic focus of sepsis-induced lesions. In the course of sepsis, lymphocytes undergo mitochondria-mediated accelerated apoptosis, while neutrophils experience delayed apoptosis. Given the fact that a pronounced neutrophil apoptosis was shown in sepsis patients, the delayed neutrophil apoptosis was suggested to contribute significantly to organ damage and mortality of patients [61, 62].
Sepsis is known to be accompanied by coagulopathy. It is not really clear whether coagulopathy bears pathogenic significance in sepsis, for anticoagulants have both positive (or neutral) and negative effect on the mortality of sepsis patients in different studies (this mortality reaches almost 40% on the 28th day of the disease [63]). However, microvascular obstruction of an organ can cause changes characteristic of ischemia/reperfusion of tissue with mitochondria playing the key role in pathogenesis [64, 65], and the inflammatory component being a mandatory part of the pathogenesis [66, 67].
It should be noted that mitochondria are increasingly becoming the object of research on this problem. Researchers see mitochondria as one of the elements clearly involved in the solution of the dilemma posed by nature – to live or not to live, whether it be a cell, an organ, or an organism [41, 68, 69]. Moreover, the following phenomenon seems to be of no less significance: fairly obvious mitochondria-produced elements, such as ROS [1-3, 26, 41, 70, 71] or ATP [72-74] are the most frequent pathogenic factors that are considered to be decisive for the solution of this dilemma. These elements participate in numerous physiological reactions determining the possible modulation of innate and acquired immunity.
Other recently discovered products of mitochondria seem to require special attention, for they have been found to directly determine the activation of innate immunity. They are part of a large group called DAMPs (damage-associated molecular patterns) [30, 75]. This name comes from the analogy to PAMPs (bacterial pathogen-associated molecular patterns) [76]. PAMPs are elements of bacterial cells, such as lipopolysaccharide, peptidoglycan, fibronectin, hyaluronan, cardiolipin, bacterial DNA, formylmethionine, etc. PAMPs activate specific receptors, PRRs (pattern-recognition receptors), which recognize molecules of evolutionarily distant strangers. Eleven known Toll receptors are examples of classical PRRs [75]. Activation of the immune system by DAMPs occurs in a sterile environment, that is, it is not accompanied by bacterial infection, although all its symptoms appear like a septic response. Mitochondrial DAMPs include the following compounds: N-formylmethionine proteins (similar to prokaryotes, which again confirms the prokaryotic origin of mitochondria), cardiolipin (similar to bacteria), mitochondrial DNA (mtDNA, which, as in bacteria, has cyclic structure), ATP, ROS (which are also produced in bacteria), and cytochrome c (also present in bacteria, though somewhat different [77]). This comparison shows that, given the similarity in the nature of the antigens, the basic response to mitochondrial DAMPs may be similar to the response to bacterial PAMPs as in both cases it activates innate immunity.
The previously mentioned fatal systemic inflammatory response syndrome caused by trauma has been shown to be mediated by DAMPs [78-80]. Cell damage is accompanied by the release of DAMPs into the bloodstream. These DAMPs activate neutrophils, and their excessive activation can cause organ damage [81]. The concentration of mtDNA in the blood of patients who have experienced severe trauma is thousands of times higher than normal [82]. The mtDNA can cause neutrophil activation due to interaction with various receptors, including Toll receptors, and it can also be produced by eosinophils due to mtDNA release from cells [28]. It seems to be quite interesting that mtDNA, along with other DNA released from cells, can constitute a primitive antibacterial defense by building a special above-mentioned extracellular trap (NET) made of DNA and antibacterial proteins. This trap, besides having the useful antibacterial function, can cause thrombosis, which can be effectively treated with anticoagulants based on DNase and heparin [83, 84]. Together these data indicate the complex and fatal relationships between the products of mitochondrial decay and immune system elements, which include the possibility of amplifying the signal of cell death and later, the death of the entire organ.
The natural conclusion is that this modulation of mitochondrial activity, and above all, in a manner that will preserve the integral properties of mitochondria regardless of the strength of the damage, can form a strategic direction of therapy of mito-, apo-, organo-, and, possibly, phenoptosis.
We have already mentioned that one (but not the only one) approach to the maintenance of mitochondrial homeostasis is based on the controlled regulation of ROS level, which obeys the simple law multet nocem (from Latin too much is harmful) [24], but too little is also bad [85]. A partial solution to this problem may include the use of antioxidants in the compulsory rationing of ROS production, while avoiding “overdosing” of these antioxidants, which might block a number of intrinsic physiological reactions that require normal ROS levels. Mitochondria-targeted antioxidants [86-96] prove to be effective antioxidants of a new generation. Later we will discuss their role in the treatment of renal pathologies and in studying the mechanism of the organism’s death.
LESSONS FROM RENAL FAILURE
Kidney pathologies are among the most severe and difficult to treat diseases. As a result, acute or chronic renal failure develops leading to a terminal stage, which finishes with death when no medical intervention is provided. When kidneys cannot properly fulfill their excretory function, it becomes necessary to exogenously purify blood from toxic products (through so-called hemodialysis), apply cell therapy, or transplant a donor organ. Since the kidney is a paired organ, the deficiency of one kidney is often offset by heavier load on the other kidney, and thus there is no significant effect on the state of the whole organism. However, when deprived of such a safety option, that is, in the case of only one kidney functioning, any remaining organ pathologies are almost always fatal.
The levels of products of nitrogen metabolism (such as creatinine or urea) in the peripheral blood determine the proper functioning of kidneys. Their increase above certain standards indicates the development of renal failure.
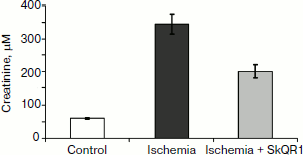
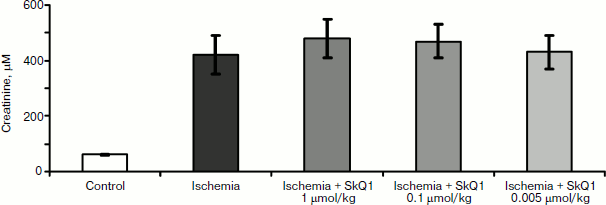
In experiments with rats, if one of the two kidneys is removed, and the remaining kidney is exposed to 90-min-long ischemia followed by reperfusion, after some time acute renal failure develops, leading to a significant increase in the level of creatinine and urea in the blood. But if the rats are treated with mitochondria-targeted antioxidant SkQR1 one day prior to kidney ischemia, the severity of renal failure is significantly reduced (see Fig. 1, the third day after ischemia). The same kind of manipulation with another mitochondrial antioxidant, SkQ1, did not lead to restoration of renal function (Fig. 2).
Fig. 1. Effect of SkQR1 (1 μmol/kg, intraperitoneal injection one day prior to 90-min ischemia) on the creatinine concentration in blood. Ischemia/reperfusion leads to a marked increase in creatinine concentration, this fact indicating the development of renal failure; SkQR1 prevents renal dysfunction. From [89], reprinted with permission.
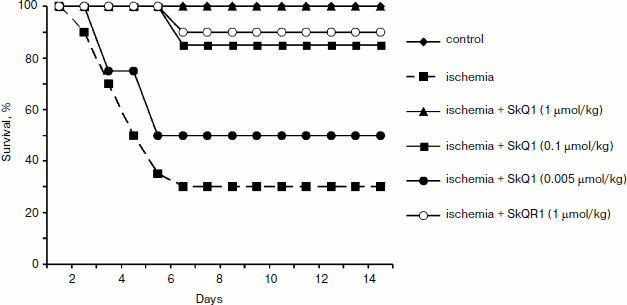
Different results were obtained when studying the survival of animals. Both antioxidants almost completely abolished animal death, which was as high as 70% in the absence of the antioxidants (Fig. 3).Fig. 2. Effect of different SkQ1 concentrations (intraperitoneal injection one day prior to 90-min ischemia) on the development of renal failure. SkQ1 did not prevent the increase in creatinine concentration in blood after ischemia/reperfusion. From [89], reprinted with permission.
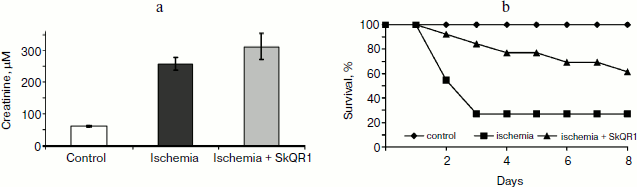
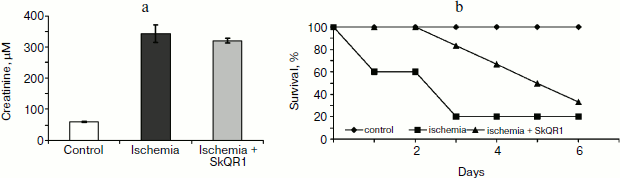
In another case, when the nephroprotective antioxidant SkQR1 was used in the treatment protocol, i.e. this compound was introduced immediately after ischemia, no nephroprotective properties could be observed, even though survival was shown to improve (though not as much as in case of prophylactic treatment). In the control case, higher values of nitrogen metabolism products in the bloodstream were indicative of severe renal failure (Fig. 4).Fig. 3. Survival of rats with one kidney subjected to 90-min ischemia followed by reperfusion. SkQ1 or SkQR1 were injected intraperitoneally one day prior to ischemia. From [89], reprinted with permission.
Thus, we can conclude that contrary to popular belief, it was not the renal failure that was the cause of animal death, but some control system, and it was this system that became the target of our antioxidants.Fig. 4. Effect of SkQR1 on renal function (a) and survival (b) of animals exposed to 40-min kidney ischemia. The antioxidant was injected after ischemia at a dose of 100 nmol/kg every 12 h for 2 days. For methods, see [94].
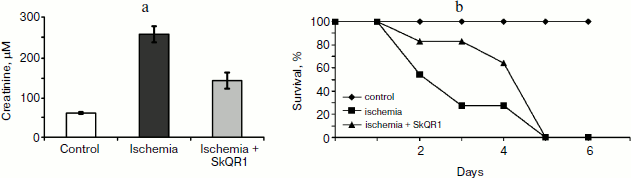
Let us support this conclusion with one more piece of experimental evidence. The next experiment was carried out while maintaining the body temperature at 34°C (warm ischemia), whereas in the previous examples the temperature of the anesthetized animal was naturally reduced because of anesthesia and was not artificially maintained at a normal level (cold ischemia). In the case of warm ischemia, we also observed the effect of divergence between the renal failure and survival of the animal. Figure 5 shows an example of an attempt of 2-day treatment of renal failure caused by warm ischemia. SkQR1 treatment was stopped after 2 days. It is evident that renal failure could be observed in both groups of animals (SkQR1-treated and controls), and as for survival, both groups showed an obvious hysteresis response, and the fatal outcome occurred simultaneously (Fig. 5). The latter observation is probably indicative of the fact that the abolition of antioxidant treatment led to the abolition of its positive effect on the survival mediated by the aforementioned unidentified system.
When prophylaxis (SkQR1-treatment introduced 3 h prior to ischemia) was combined with treatment (100 nmol/kg every 12 h after ischemia for 2 days followed by the abolition of the drug), the surviving animals demonstrated normalization of renal activity (Fig. 6), and the survival rate was slightly higher, but by day 5 it became the same as in the control group. This indicated (i) the short longevity of the signal canceling the order of the animal’s death coming from a system of non-renal nature when antioxidant treatment was stopped, and (ii) that under these conditions later normalization of renal function could no longer save the animal from death that was triggered by renal failure.Fig. 5. Effect of SkQR1 on renal function (a) and survival (b) of animals exposed to 90-min warm kidney ischemia. The antioxidant was injected after ischemia at a dose of 100 nmol/kg every 12 h for 2 days. For methods, see [89, 94].
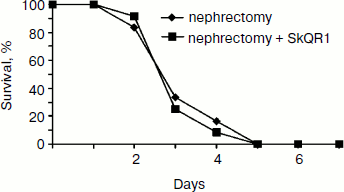
As one would expect, the removal of both kidneys caused rapid death of the animals regardless of SkQR1 treatment (Fig. 7).Fig. 6. Effect of SkQR1 on renal function (a) and survival (b) of animals exposed to 90-min warm kidney ischemia. The antioxidant was injected prior to ischemia and after ischemia at a dose of 100 nmol/kg every 12 h for 2 days. For methods, see [89, 94].
Together the above-described data suggest renal failure to be a necessary, but not sufficient condition for the organism’s death, which is also determined by some other factors which, while being rather obscure, are surely of mitochondrial nature, given the mitochondria-targeted nature of the antioxidants used in this work. One needs to remember that antioxidants, while circulating through the whole organism, could interact with all mitochondria in all organs that have no permeability barriers. It seems that in the case illustrated by Fig. 3, one antioxidant (SkQR1) both reduced renal failure and “did not allow” the system controlling the animal death to eliminate the organism, while another antioxidant (SkQ1) worked only at the level of the second system. Basically, we can also make certain conclusions about the nature of the interaction of these systems. In the first case, it is the death-triggering signal that is eliminated, and in the second case the signal transmission to the next level is blocked while the signal itself is preserved.Fig. 7. Effect of SkQR1 on the survival of animals exposed to bilateral nephrectomy. The antioxidant was injected at a dose of 100 nmol/kg every 12 h during the entire period until the death of the animals. For methods, see [94].
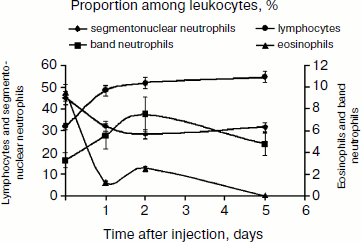
A single dose of SkQR1 changes the blood formula (see Fig. 8), significantly reducing the number of neutrophils (segmentonuclear granulocytes) and increasing the number of lymphocytes, a fact indicating direct or indirect impact of the antioxidant on other organs. It is worth noting that SkQ1 showed similar effect on leukocyte counts (data not shown). Neutrophils are considered to be a potent source of free radicals; kidney tissue infiltration by neutrophils and ROS generation in the process of phagocytosis of damaged cells play an important role in the pathogenesis of ischemic kidney damage, and that is why reduction in the number of neutrophils in blood may contribute to the protective effect of mitochondrial antioxidants. On the other hand, absolute and relative increases of lymphocytes in blood are observed, and it is lymphocytes that are involved in regeneration processes. This may increase the regenerative potential of the damaged organ.
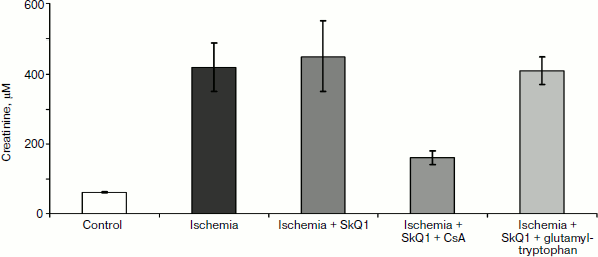
In any case, mitochondria-targeted antioxidants obviously affect the immune system. This impact is aimed at suppression of the innate (nonspecific) immunity, i.e. neutrophils, and stimulation of the specific component (lymphocytes), which includes many regulatory cells (T-helper cells of several types, T-suppressors, T-regulators) as well as antibody-producing cells. In this regard, the results of certain experiments seem to be quite interesting: the use of SkQ1 prior to 90-min-long kidney ischemia did not prevent renal failure (although it abolished death, see above), but when combined with the immunosuppressant cyclosporin, SkQ1 showed a pronounced nephroprotective effect (see Fig. 9). However, the use of SkQ1 together with an immunostimulator (glutamyl-tryptophan dipeptide, thymogen) had no effect on renal function, which remained impaired after ischemia (Fig. 9).Fig. 8. Change in blood leukocyte counts after a single injection of 1 μmol/kg SkQR1 to intact animals. For methods, see [89].
Concluding this review, we would like to avoid the temptation to present an optimistic picture of the complete or even partial understanding of the processes leading to individual death. We are still in the process of accumulating data and trying to comprehend the causes of death. A huge number of deaths have no visible reasons – for instance, we have mentioned the syndrome of sudden cardiac arrest [32] or multiple organ failure syndrome [46, 55]. Psychoemotional reasons for cardiac pathologies also fit into this category, e.g. Takotsubo cardiomyopathy or the “broken heart syndrome” [97-99], which often develops as a result of severe stress, sadness, or grief (this pathology constitutes 2% of all cardiac infarctions) [100]. Sudden death in epilepsy is another case of unexplainable death [101].Fig. 9. Effect of 1 μmol/kg SkQ1 injected together with immunosuppressant (cyclosporin A, CsA) and immunostimulant glutamyl-tryptophan on renal function (creatinine concentration). SkQ1 was administered one day prior to 90-min kidney ischemia, and immunomodulators were administered 2 days prior to ischemia. For methods, see [89].
It may seem strange, but the cause of death of most cancer patients is often unclear; these deaths prove to be suspiciously variable and multi-faceted [102, 103], even though we can assume that about 50% of cancer patients die from complications [104] that may be mediated, for instance, by thromboembolism, with all the above-described effects of the induction of oxidative stress [105].
The very multi-stage process of dying (http://dying.about.com/od/thedyingprocess/a/process.htm) resembles the process of poisoning. It is for this reason that we overviewed the impact of the decay products of cells and mitochondria on living systems, e.g. some of the physiologically active toxic substances whose actions is largely mediated by the immune system, which sometimes reports the impending death [106]. The important role of the immune system as a powerful tool used for the organism’s death is quite obvious. Transformation of mitochondria into cell killers is no longer a hypothesis; it has become a well-known fact, even though there continue unceasing debates on the scrupulous details of the mechanism of this transformation. Are mitochondria to some extent the murderers of organs and organisms? This question seems to be of a different order, the hypothesis being partially supported by logical and factual evidence presented in this article. Confirmation or rejection of this hypothesis requires a complete understanding of the mechanism of individual death; it is also necessary to understand completely and correctly the causes of death, which would require the joined efforts of scientists, physicians, statisticians, or simply attentive and logically thinking people.
This work was supported by grants of the Russian Foundation for Basic Research 11-04-01307 (DBZ), 11-04-00771 (EYP), 12-04-00025 (NKI) and a grant from the President of the Russian Federation MK-729.2012.4 (DNS).
REFERENCES
1.Skulachev, V. P. (1999) Biochemistry
(Moscow), 64, 1418-1426.
2.Skulachev, V. P. (1999) Mol. Aspects Med.,
20, 139-184.
3.Skulachev, V. P. (2002) Ann. N.Y. Acad.
Sci., 959, 214-237.
4.Gasko, O., and Danon, D. (1972) Exp. Cell.
Res., 75, 159-169.
5.Sutovsky, P., Moreno, R. D., Ramalho-Santos, J.,
Dominko, T., Simerly, C., and Schatten, G. (1999) Nature,
402, 371-372.
6.Al Rawi, S., Louvet-Vallee, S., Djeddi, A., Sachse,
M., Culetto, E., Hajjar, C., Boyd, L., Legouis, R., and Galy, V. (2011)
Science, 334, 1144-1147.
7.Ciechanover, A. (1994) Cell, 79,
13-21.
8.Vorobjev, I. A., and Zorov, D. B. (1983) FEBS
Lett., 163, 311-314.
9.Skulachev, V. P., Bakeeva, L. E., Chernyak, B. V.,
Domnina, L. V., Minin, A. A., Pletjushkina, O. Y., Saprunova, V. B.,
Skulachev, I. V., Tsyplenkova, V. G., Vasiliev, J. M., Yaguzhinsky, L.
S., and Zorov, D. B. (2004) Mol. Cell. Biochem., 256/257,
341-358.
10.Plotnikov, E. Y., Vasileva, A. K.,
Arkhangelskaya, A. A., Pevzner, I. B., Skulachev, V. P., and Zorov, D.
B. (2008) FEBS Lett., 582, 3117-3124.
11.Twig, G., Hyde, B., and Shirihai, O. S. (2008)
Biochim. Biophys. Acta, 1777, 1092-1097.
12.Gomes, L. C., and Scorrano, L. (2008) Biochim.
Biophys. Acta, 1777, 860-866.
13.Zorov, D. B., Kinnally, K. W., and Tedeschi, H.
(1992) J. Bioenerg. Biomembr., 24, 119-124.
14.Zoratti, M., and Szabo, I. (1995) Biochim.
Biophys. Acta, 1241, 139-176.
15.Elmore, S. P., Qian, T., Grissom, S. F., and
Lemasters, J. J. (2001) FASEB J., 15, 2286-2287.
16.Zorov, D. B., Filburn, C. R., Klotz, L. O.,
Zweier, J. L., and Sollott, S. J. (2000) J. Exp. Med.,
192, 1001-1014.
17.Zorov, D. B., Juhaszova, M., and Sollott, S. J.
(2006) Biochim. Biophys. Acta, 1757, 509-517.
18.Zorov, D. B., Kobrinsky, E., Juhaszova, M., and
Sollott, S. J. (2004) Circ. Res., 95, 239-252.
19.Lee, J., Giordano, S., and Zhang, J. (2012)
Biochem. J., 441, 523-540.
20.Scherz-Shouval, R., and Elazar, Z. (2007)
Trends Cell Biol., 17, 422-427.
21.Lyamzaev, K. G., Nepryakhina, O. K., Saprunova,
V. B., Bakeeva, L. E., Pletjushkina, O. Y., Chernyak, B. V., and
Skulachev, V. P. (2008) Biochim. Biophys. Acta, 1777,
817-825.
22.Bisharyan, Y., and Clark, T. G. (2011)
Mitochondrion, 11, 909-918.
23.Galluzzi, L., Vitale, I., Abrams, J. M., Alnemri,
E. S., Baehrecke, E. H., Blagosklonny, M. V., Dawson, T. M., Dawson, V.
L., El-Deiry, W. S., Fulda, S., Gottlieb, E., Green, D. R., Hengartner,
M. O., Kepp, O., Knight, R. A., Kumar, S., Lipton, S. A., Lu, X.,
Madeo, F., Malorni, W., Mehlen, P., Nunez, G., Peter, M. E.,
Piacentini, M., Rubinsztein, D. C., Shi, Y., Simon, H. U.,
Vandenabeele, P., White, E., Yuan, J., Zhivotovsky, B., Melino, G., and
Kroemer, G. (2012) Cell Death Differ., 19, 107-120.
24.Zorov, D. B., Krasnikov, B. F., Kuzminova, A. E.,
Vysokikh, M., and Zorova, L. D. (1997) Biosci. Rep., 17,
507-520.
25.Zorov, D. B. (1996) Biochemistry (Moscow),
61, 939-946.
26.Zorov, D. B. (1996) Biochim. Biophys.
Acta, 1275, 10-15.
27.Brinkmann, V., Reichard, U., Goosmann, C.,
Fauler, B., Uhlemann, Y., Weiss, D. S., Weinrauch, Y., and Zychlinsky,
A. (2004) Science, 303, 1532-1535.
28.Yousefi, S., Gold, J. A., Andina, N., Lee, J. J.,
Kelly, A. M., Kozlowski, E., Schmid, I., Straumann, A., Reichenbach,
J., Gleich, G. J., and Simon, H. U. (2008) Nat. Med., 14,
949-953.
29.Remijsen, Q., Kuijpers, T. W., Wirawan, E.,
Lippens, S., Vandenabeele, P., and Vanden Berghe, T. (2011) Cell
Death Differ., 18, 581-588.
30.Krysko, D. V., Agostinis, P., Krysko, O., Garg,
A. D., Bachert, C., Lambrecht, B. N., and Vandenabeele, P. (2011)
Trends Immunol., 32, 157-164.
31.Steinberg, B. E., and Grinstein, S. (2007)
Sci. STKE, 2007, pe11.
32.Myerburg, R. J. (2002) J. Cardiovasc.
Electrophysiol., 13, 709-723.
33.De Bie, M. K., Buiten, M. S., Rabelink, T. J.,
and Jukema, J. W. (2012) Heart, 98, 335-341.
34.Pupim, L. B., Himmelfarb, J., McMonagle, E.,
Shyr, Y., and Ikizler, T. A. (2004) Kidney Int., 65,
2371-2379.
35.Zanetti, M., Barazzoni, R., Gortan Cappellari,
G., Burekovic, I., Bosutti, A., Stocca, A., Bianco, F., Ianche, M.,
Panzetta, G., and Guarnieri, G. (2011) J. Ren. Nutr., 21,
401-409.
36.Welbourn, C. R., Goldman, G., Paterson, I. S.,
Valeri, C. R., Shepro, D., and Hechtman, H. B. (1991) Br. J.
Surg., 78, 651-655.
37.Koziol, J. M., Rush, B. F., Jr., Smith, S. M.,
and Machiedo, G. W. (1988) J. Trauma, 28, 10-16.
38.Rush, B. F., Jr., Sori, A. J., Murphy, T. F.,
Smith, S., Flanagan, J. J., Jr., and Machiedo, G. W. (1988) Ann.
Surg., 207, 549-554.
39.Marshall, J. C., Christou, N. V., and Meakins, J.
L. (1993) Ann. Surg., 218, 111-119.
40.Partrick, D. A., Moore, F. A., Moore, E. E.,
Barnett, C. C., Jr., and Silliman, C. C. (1996) New Horiz.,
4, 194-210.
41.Kozlov, A. V., Bahrami, S., Calzia, E., Dungel,
P., Gille, L., Kuznetsov, A. V., and Troppmair, J. (2011) Ann.
Intens. Care, 1, 41.
42.Srisawat, N., Hoste, E. E., and Kellum, J. A.
(2010) Blood Purif., 29, 300-307.
43.Singer, M., de Santis, V., Vitale, D., and
Jeffcoate, W. (2004) Lancet, 364, 545-548.
44.Watanabe, E., Muenzer, J. T., Hawkins, W. G.,
Davis, C. G., Dixon, D. J., McDunn, J. E., Brackett, D. J., Lerner, M.
R., Swanson, P. E., and Hotchkiss, R. S. (2009) Lab. Invest.,
89, 549-561.
45.Azevedo, L. C. (2010) Endocr. Metab. Immune
Disord. Drug Targets, 10, 214-223.
46.Hotchkiss, R. S., Swanson, P. E., Freeman, B. D.,
Tinsley, K. W., Cobb, J. P., Matuschak, G. M., Buchman, T. G., and
Karl, I. E. (1999) Crit. Care Med., 27, 1230-1251.
47.Fredriksson, K., Tjader, I., Keller, P.,
Petrovic, N., Ahlman, B., Scheele, C., Wernerman, J., Timmons, J. A.,
and Rooyackers, O. (2008) PLoS One, 3, e3686.
48.Brealey, D., Brand, M., Hargreaves, I., Heales,
S., Land, J., Smolenski, R., Davies, N. A., Cooper, C. E., and Singer,
M. (2002) Lancet, 360, 219-223.
49.Vanhorebeek, I., de Vos, R., Mesotten, D.,
Wouters, P. J., de Wolf-Peeters, C., and van den Berghe, G. (2005)
Lancet, 365, 53-59.
50.Duclos-Vallee, J. C., Vittecoq, D., Teicher, E.,
Feray, C., Roque-Afonso, A. M., Lombes, A., Jardel, C., Gigou, M.,
Dussaix, E., Sebagh, M., Guettier, C., Azoulay, D., Adam, R., Ichai,
P., Saliba, F., Roche, B., Castaing, D., Bismuth, H., and Samuel, D.
(2005) J. Hepatol., 42, 341-349.
51.Gellerich, F. N., Trumbeckaite, S., Hertel, K.,
Zierz, S., Muller-Werdan, U., Werdan, K., Redl, H., and Schlag, G.
(1999) Shock, 11, 336-341.
52.Mima, A., Shiota, F., Matsubara, T., Iehara, N.,
Akagi, T., Abe, H., Nagai, K., Matsuura, M., Murakami, T., Kishi, S.,
Araoka, T., Kishi, F., Kondo, N., Shigeta, R., Yoshikawa, K., Kita, T.,
Doi, T., and Fukatsu, A. (2011) Ren. Fail., 33,
622-625.
53.Fragaki, K., Cano, A., Benoist, J. F., Rigal, O.,
Chaussenot, A., Rouzier, C., Bannwarth, S., Caruba, C., Chabrol, B.,
and Paquis-Flucklinger, V. (2011) Mitochondrion, 11,
533-536.
54.Von Dessauer, B., Bongain, J., Molina, V.,
Quilodran, J., Castillo, R., and Rodrigo, R. (2011) J. Crit.
Care, 26, 103 e1-7.
55.Moore, F. A., Sauaia, A., Moore, E. E., Haenel,
J. B., Burch, J. M., and Lezotte, D. C. (1996) J. Trauma,
40, 501-510; discussion 510-512.
56.Ni Choileain, N., and Redmond, H. P. (2006)
Surgeon, 4, 23-31.
57.Lenz, A., Franklin, G. A., and Cheadle, W. G.
(2007) Injury, 38, 1336-1345.
58.Polk, H. C., Jr., and Shields, C. L. (1977)
Surgery, 81, 310-313.
59.Fry, D. E., Pearlstein, L., Fulton, R. L., and
Polk, H. C., Jr. (1980) Arch. Surg., 115, 136-140.
60.Bell, R. C., Coalson, J. J., Smith, J. D., and
Johanson, W. G., Jr. (1983) Ann. Intern. Med., 99,
293-298.
61.Jimenez, M. F., Watson, R. W., Parodo, J., Evans,
D., Foster, D., Steinberg, M., Rotstein, O. D., and Marshall, J. C.
(1997) Arch. Surg., 132, 1263-1269; discussion
1269-1270.
62.Taneja, R., Parodo, J., Jia, S. H., Kapus, A.,
Rotstein, O. D., and Marshall, J. C. (2004) Crit. Care Med.,
32, 1460-1469.
63.Stearns-Kurosawa, D. J., Osuchowski, M. F.,
Valentine, C., Kurosawa, S., and Remick, D. G. (2011) Annu. Rev.
Pathol., 6, 19-48.
64.Plotnikov, E. Y., Kazachenko, A. V., Vyssokikh,
M. Y., Vasileva, A. K., Tcvirkun, D. V., Isaev, N. K., Kirpatovsky, V.
I., and Zorov, D. B. (2007) Kidney Int., 72,
1493-1502.
65.Plotnikov, E. Y., Chupyrkina, A. A., Jankauskas,
S. S., Pevzner, I. B., Silachev, D. N., Skulachev, V. P., and Zorov, D.
B. (2011) Biochim. Biophys. Acta, 1812, 77-86.
66.Bonventre, J. V., and Weinberg, J. M. (2003)
J. Am. Soc. Nephrol., 14, 2199-2210.
67.Lien, Y. H., Lai, L. W., and Silva, A. L. (2003)
Life Sci., 74, 543-552.
68.Singer, M. (2007) Crit. Care Med.,
35, S441-448.
69.Carre, J. E., Orban, J. C., Re, L., Felsmann, K.,
Iffert, W., Bauer, M., Suliman, H. B., Piantadosi, C. A., Mayhew, T.
M., Breen, P., Stotz, M., and Singer, M. (2010) Am. J. Respir. Crit.
Care Med., 182, 745-751.
70.Zorov, D. B., Bannikova, S. Y., Belousov, V. V.,
Vyssokikh, M. Y., Zorova, L. D., Isaev, N. K., Krasnikov, B. F., and
Plotnikov, E. Y. (2005) Biochemistry (Moscow), 70,
215-221.
71.Bulua, A. C., Simon, A., Maddipati, R.,
Pelletier, M., Park, H., Kim, K. Y., Sack, M. N., Kastner, D. L., and
Siegel, R. M. (2011) J. Exp. Med., 208, 519-533.
72.Leist, M., Single, B., Castoldi, A. F., Kuhnle,
S., and Nicotera, P. (1997) J. Exp. Med., 185,
1481-1486.
73.Eguchi, Y., Shimizu, S., and Tsujimoto, Y. (1997)
Cancer Res., 57, 1835-1840.
74.Izyumov, D. S., Avetisyan, A. V., Pletjushkina,
O. Y., Sakharov, D. V., Wirtz, K. W., Chernyak, B. V., and Skulachev,
V. P. (2004) Biochim. Biophys. Acta, 1658, 141-147.
75.Seong, S. Y., and Matzinger, P. (2004) Nat.
Rev. Immunol., 4, 469-478.
76.Janeway, C. A., Jr. (1989) Cold Spring Harb.
Symp. Quant. Biol., 54, Pt. 1, 1-13.
77.Stevens, J. M. (2011) Metallomics,
3, 319-322.
78.Kleindienst, A., Hesse, F., Bullock, M. R., and
Buchfelder, M. (2007) Prog. Brain Res., 161, 317-325.
79.Cohen, M. J., Brohi, K., Calfee, C. S., Rahn, P.,
Chesebro, B. B., Christiaans, S. C., Carles, M., Howard, M., and
Pittet, J. F. (2009) Crit. Care, 13, R174.
80.Xiang, M., and Fan, J. (2011) Mol. Med.,
16, 69-82.
81.Zhang, Q., Raoof, M., Chen, Y., Sumi, Y., Sursal,
T., Junger, W., Brohi, K., Itagaki, K., and Hauser, C. J. (2010)
Nature, 464, 104-107.
82.Zhang, Q., Itagaki, K., and Hauser, C. J. (2010)
Shock, 34, 55-59.
83.Fuchs, T. A., Brill, A., Duerschmied, D.,
Schatzberg, D., Monestier, M., Myers, D. D., Jr., Wrobleski, S. K.,
Wakefield, T. W., Hartwig, J. H., and Wagner, D. D. (2010) Proc.
Natl. Acad. Sci. USA, 107, 15880-15885.
84.Brill, A., Fuchs, T. A., Savchenko, A., Thomas,
G. M., Martinod, K., De Meyer, S. F., Bhandari, A. A., and Wagner, D.
D. (2012) J. Thromb. Haemost., 10, 136-144.
85.Droge, W. (2002) Physiol. Rev., 82,
47-95.
86.Skulachev, V. P. (2007) Biochemistry
(Moscow), 72, 1385-1396.
87.Murphy, M. P., and Smith, R. A. (2007) Annu.
Rev. Pharmacol. Toxicol., 47, 629-656.
88.Antonenko, Y. N., Avetisyan, A. V., Bakeeva, L.
E., Chernyak, B. V., Chertkov, V. A., Domnina, L. V., Ivanova, O. Y.,
Izyumov, D. S., Khailova, L. S., Klishin, S. S., Korshunova, G. A.,
Lyamzaev, K. G., Muntyan, M. S., Nepryakhina, O. K., Pashkovskaya, A.
A., Pletjushkina, O. Y., Pustovidko, A. V., Roginsky, V. A.,
Rokitskaya, T. I., Ruuge, E. K., Saprunova, V. B., Severina, I. I.,
Simonyan, R. A., Skulachev, I. V., Skulachev, M. V., Sumbatyan, N. V.,
Sviryaeva, I. V., Tashlitsky, V. N., Vassiliev, J. M., Vyssokikh, M.
Y., Yaguzhinsky, L. S., Zamyatnin, A. A., Jr., and Skulachev, V. P.
(2008) Biochemistry (Moscow), 73, 1273-1287.
89.Bakeeva, L. E., Barskov, I. V., Egorov, M. V.,
Isaev, N. K., Kapelko, V. I., Kazachenko, A. V., Kirpatovsky, V. I.,
Kozlovsky, S. V., Lakomkin, V. L., Levina, S. B., Pisarenko, O. I.,
Plotnikov, E. Y., Saprunova, V. B., Serebryakova, L. I., Skulachev, M.
V., Stelmashook, E. V., Studneva, I. M., Tskitishvili, O. V.,
Vasilyeva, A. K., Victorov, I. V., Zorov, D. B., and Skulachev, V. P.
(2008) Biochemistry (Moscow), 73, 1288-1299.
90.Agapova, L. S., Chernyak, B. V., Domnina, L. V.,
Dugina, V. B., Efimenko, A. Y., Fetisova, E. K., Ivanova, O. Y.,
Kalinina, N. I., Khromova, N. V., Kopnin, B. P., Kopnin, P. B.,
Korotetskaya, M. V., Lichinitser, M. R., Lukashev, A. L., Pletjushkina,
O. Y., Popova, E. N., Skulachev, M. V., Shagieva, G. S., Stepanova, E.
V., Titova, E. V., Tkachuk, V. A., Vasiliev, J. M., and Skulachev, V.
P. (2008) Biochemistry (Moscow), 73, 1300-1316.
91.Neroev, V. V., Archipova, M. M., Bakeeva, L. E.,
Fursova, A., Grigorian, E. N., Grishanova, A. Y., Iomdina, E. N.,
Ivashchenko, Zh. N., Katargina, L. A., Khoroshilova-Maslova, I. P.,
Kilina, O. V., Kolosova, N. G., Kopenkin, E. P., Korshunov, S. S.,
Kovaleva, N. A., Novikova, Y. P., Philippov, P. P., Pilipenko, D. I.,
Robustova, O. V., Saprunova, V. B., Senin, I. I., Skulachev, M. V.,
Sotnikova, L. F., Stefanova, N. A., Tikhomirova, N. K., Tsapenko, I.
V., Shchipanova, A. I., Zinovkin, R. A., and Skulachev, V. P. (2008)
Biochemistry (Moscow), 73, 1317-1328.
92.Anisimov, V. N., Bakeeva, L. E., Egormin, P. A.,
Filenko, O. F., Isakova, E. F., Manskikh, V. N., Mikhelson, V. M.,
Panteleeva, A. A., Pasyukova, E. G., Pilipenko, D. I., Piskunova, T.
S., Popovich, I. G., Roshchina, N. V., Rybina, O. Y., Saprunova, V. B.,
Samoylova, T. A., Semenchenko, A. V., Skulachev, M. V., Spivak, I. M.,
Tsybul’ko, E. A., Tyndyk, M. L., Vyssokikh, M. Y., Yurova, M. N.,
Zabezhinsky, M. A., and Skulachev, V. P. (2008) Biochemistry
(Moscow), 73, 1329-1342.
93.Skulachev, V. P., Anisimov, V. N., Antonenko, Y.
N., Bakeeva, L. E., Chernyak, B. V., Erichev, V. P., Filenko, O. F.,
Kalinina, N. I., Kapelko, V. I., Kolosova, N. G., Kopnin, B. P.,
Korshunova, G. A., Lichinitser, M. R., Obukhova, L. A., Pasyukova, E.
G., Pisarenko, O. I., Roginsky, V. A., Ruuge, E. K., Senin, I. I.,
Severina, I. I., Skulachev, M. V., Spivak, I. M., Tashlitsky, V. N.,
Tkachuk, V. A., Vyssokikh, M. Y., Yaguzhinsky, L. S., and Zorov, D. B.
(2009) Biochim. Biophys. Acta, 1787, 437-461.
94.Plotnikov, E. Y., Silachev, D. N., Chupyrkina, A.
A., Danshina, M. I., Jankauskas, S. S., Morosanova, M. A., Stelmashook,
E. V., Vasileva, A. K., Goryacheva, E. S., Pirogov, Y. A., Isaev, N.
K., and Zorov, D. B. (2010) Biochemistry (Moscow), 75,
145-150.
95.Kapay, N. A., Isaev, N. K., Stelmashook, E. V.,
Popova, O. V., Zorov, D. B., Skrebitsky, V. G., and Skulachev, V. P.
(2011) Biochemistry (Moscow), 76, 1367-1370.
96.Skulachev, M. V., Antonenko, Y. N., Anisimov, V.
N., Chernyak, B. V., Cherepanov, D. A., Chistyakov, V. A., Egorov, M.
V., Kolosova, N. G., Korshunova, G. A., Lyamzaev, K. G., Plotnikov, E.
Y., Roginsky, V. A., Savchenko, A. Y., Severina, I. I., Severin, F. F.,
Shkurat, T. P., Tashlitsky, V. N., Shidlovsky, K. M., Vyssokikh, M. Y.,
Zamyatnin, A. A., Jr., Zorov, D. B., and Skulachev, V. P. (2012)
Curr. Drug Targets, 12, 800-826.
97.Wittstein, I. S., Thiemann, D. R., Lima, J. A.,
Baughman, K. L., Schulman, S. P., Gerstenblith, G., Wu, K. C., Rade, J.
J., Bivalacqua, T. J., and Champion, H. C. (2005) N. Engl. J.
Med., 352, 539-548.
98.Lyon, A. R., Rees, P. S., Prasad, S.,
Poole-Wilson, P. A., and Harding, S. E. (2008) Nat. Clin. Pract.
Cardiovasc. Med., 5, 22-29.
99.Nykamp, D., and Titak, J. A. (2010) Ann.
Pharmacother., 44, 590-593.
100.Kurowski, V., Kaiser, A., von Hof, K.,
Killermann, D. P., Mayer, B., Hartmann, F., Schunkert, H., and Radke,
P. W. (2007) Chest, 132, 809-816.
101.Surges, R., and Sander, J. W. (2012) Curr.
Opin. Neurol., 25, 201-207.
102.Houten, L., and Reilley, A. A. (1980) J.
Surg. Oncol., 13, 111-116.
103.Kefford, R. F., Cooney, N. J., Woods, R. L.,
Fox, R. M., and Tattersall, M. H. (1981) Eur. J. Cancer Clin.
Oncol., 17, 1117-1124.
104.Ambrus, J. L., Ambrus, C. M., Mink, I. B., and
Pickren, J. W. (1975) J. Med., 6, 61-64.
105.Rahr, H. B., and Sorensen, J. V. (1992)
Blood Coagul. Fibrinolysis, 3, 451-460.
106.Simmons, E. M., Himmelfarb, J., Sezer, M. T.,
Chertow, G. M., Mehta, R. L., Paganini, E. P., Soroko, S., Freedman,
S., Becker, K., Spratt, D., Shyr, Y., and Ikizler, T. A. (2004)
Kidney Int., 65, 1357-1365.