REVIEW: Transmembrane Electric Potential Difference in the Protein–Pigment Complex of Photosystem 2
M. D. Mamedov*, V. N. Kurashov, I. O. Petrova, and A. Yu. Semenov
Belozersky Institute of Physico-Chemical Biology, Lomonosov Moscow State University, 119991 Moscow, Russia; fax: (495) 939-3181; E-mail: mamedov@genebee.msu.ru* To whom correspondence should be addressed.
Received April 12, 2012
The protein–pigment complex of photosystem 2 (PS2) localized in the thylakoid membranes of higher plants, algae, and cyanobacteria is the main source of oxygen on Earth. The light-induced functioning of PS2 is directly linked to electron and proton transfer across the membrane, which results in the formation of transmembrane electric potential difference (ΔΨ). The major contribution to ΔΨ of the PS2 reaction center is due to charge separation between the primary chlorophyll donor P680 and the quinone acceptor QA, accompanied by re-reduction of P680+ by the redox-active tyrosine residue YZ. The processes associated with the uptake and release of protons on the acceptor and donor sides of the enzyme, respectively, are also coupled with ΔΨ generation. The objective of this work was to describe the mechanisms of ΔΨ generation associated with the S-state transitions of the water-oxidizing complex in intact PS2 complex and in PS2 preparation depleted of Mn4Ca cluster in the presence of artificial electron donors. The findings elucidate the mechanisms of electrogenic reactions on the PS2 donor side and may be a basis for development of an effective solar energy conversion system.
KEY WORDS: photosystem 2, reaction center, S-state transitions, proteoliposomes, electron donors, direct electrometric method, photoelectric response, electrogenicity, channelsDOI: 10.1134/S0006297912090015
Abbreviations: DAD, diaminodurene (2,3,5,6-tetramethyl-p-phenylenediamine); DCPIP, 2,6-dichlorophenolindophenol; DPC, 1,5-diphenylcarbazide; OEC, oxygen-evolving complex; PMS, phenazine methosulfate; PS, photosystem; RC, reaction center; TMPD, N,N,N′,N′-tetramethyl-p-phenylenediamine.
STRUCTURAL PECULIARITIES OF PHOTOSYSTEM 2
Life on planet Earth is to a large extent maintained by oxygenic photosynthesis because it is the main source of useful chemical energy in the biosphere and oxygen in the atmosphere of the Earth. Thylakoid membranes of oxygen-evolving organisms (cyanobacteria, algae, higher plants) contain three redox enzymes (photosystem 2 (PS2), cytochrome b6f-complex, photosystem 1 (PS1)) and ATP synthase. The main function of PS2 is to use solar energy for charge separation followed by two essential chemical reactions: water oxidation (with the release of protons and oxygen as side products) and plastoquinone reduction (with the uptake of protons). These reactions are spatially separated and occur on different sides of the PS2 membrane reaction center (RC).
The core complexes of PS2 are considered as a minimal unit capable of oxidizing water molecules. The structure of the PS2 core complexes isolated from two related thermophilic cyanobacteria, Thermosynechococcus elongatus and Th. vulcanus, was determined by X-ray structure analysis at a resolution of 3.8 to 1.9 Å [1-3]. Each monomer of the enzyme contains 20 protein subunits (total molecular weight of 350 kDa), with 17 of them being integral membrane proteins. In addition to protein subunits, one monomer comprises 35 chlorophyll a molecules, two pheophytin molecules, 11 β-carotene molecules, >20 lipid molecules, two plastoquinone molecules, two heme molecules, one nonheme iron ion, four manganese atoms, three or four calcium atoms, three chloride ions, one bicarbonate ion, and >15 detergent molecules. Each PS2 monomer also contains >1300 water molecules arranged in two layers at the surfaces of the stromal and lumenal sides of the enzyme, with the latter layer containing more water molecules than the former [3]. However, all organic and inorganic cofactors involved in the charge transfer reactions are located on subunits D1 and D2 of RC [4] (Fig. 1).
PS2 can be functionally subdivided into three structural domains: central photochemical, plastoquinone-reducing, and water-oxidizing. Four chlorophyll molecules and two pheophytin molecules are arranged into two branches associated with proteins D1 and D2 and located in the central part of the enzyme. The quinone acceptor complex of PS2 consists of two plastoquinone molecules with significantly different properties. The primary (QA) and secondary (QB) quinone acceptors function as one- and two-electron carriers, respectively [5]. The water-oxidizing site in thylakoids is located on the lumenal side of PS2. Based on the latest X-ray data with resolution of 1.9 Å, all components of the oxygen-evolving complex (OEC) described by the formula Mn4CaO5 have been detected [3]. In addition, all amino acid ligands of the OEC have been identified, and the bound water molecules have been found for the first time (in particular, the molecules close to the OEC and tyrosine YZ).Fig. 1. Redox cofactors of PS2 core complex. The image of the OEC is adapted from article [4].
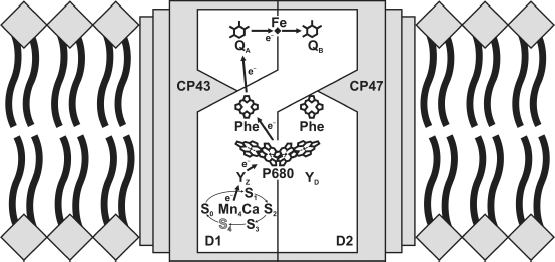
The PS2 core complex, due to its 3D structure and availability of site-specific mutants, is a convenient object for the study of structure–function interactions.
FUNCTIONING OF PS2
Light energy absorbed by the pigments of integral antenna proteins CP43 and CP47 is transferred during sub-microsecond times to the primary electron donor P680, which is a chlorophyll a dimer. The primary charge separation in RC, including a series of highly optimized electron transfer processes (see Fig. 1 and [6]), has been studied by various methods such as absorption and fluorescent spectroscopy (including femtosecond time-resolved spectroscopy), electrochromic shift of optical absorption bands and experiments with hole burning, etc. The data obtained by differential optical absorption spectroscopies at 20-fsec resolution under physiological conditions (278 K) in the PS2 core complexes from spinach show that the primary electron transfer reaction occurs at a time constant of ~0.9 psec [6] and is determined by charge separation within P680. The subsequent electron transfer from chlorophyll, which occurs during 13-14 psec, corresponds to formation of the secondary radical pair P680+PheD1– [6] and is stabilized as a result of rapid electron transfer to the tightly bound quinone QA on the stromal/cytoplasmic side of the membrane in ~200 psec. Thus, excitation energy (P680)* is used for electron transfer to a distance of ~23 Å. It should be noted that only the D1 branch is functionally active in PS2 [6, 7].
The hole on P680 is filled by electron transfer from redox-active tyrosine-161 (YZ) of the PS2 subunit D1. This electron transfer is coupled with the proton escape from YZ to the closest histidine (His190), which results in formation of the neutral tyrosine radical (YZ·) [8, 9]. The kinetics of P680+ reduction in PS2 complexes with intact OEC is characterized by time constants of 25-70 nsec (“fast” nanosecond kinetics), 300-600 nsec (“slow” nanosecond kinetics), and 15-50 µsec (microsecond kinetics) [10].
The electron from QA is further transferred to a distance of ~17 Å to the secondary quinone acceptor QB during 0.1-0.3 msec without detected oxidation–reduction involvement of an iron ion [5]. The subsequent turnovers of the enzyme include the same reactions but with different kinetics at some stages due to charge accumulation on the manganese cluster and on QB. The transfer of the second electron to QB− causes the uptake of two protons from the water phase, the release of formed plastoquinol QBH2 from the binding site in the protein, and its substitution by a molecule of oxidized plastoquinone from the membrane pool [5].
During each catalytic cycle, two water molecules are transformed into an oxygen molecule and four protons in the cycle of five intermediates called S-states (S0→S4) (Fig. 1). Since the S1 state (basic state) is most stable in the dark, the first four flashes cause transitions S1→S2, S2→S3, S3→S0, and S0→S1. Molecular oxygen is released during the S3→S0 transition via intermediate state S4 [11-13]. The kinetics of S1 to S3 transitions vary from 50 to 300 µsec, while the terminal reaction YZ·S3→YZS0 + O2 + 2 H+ occurs on the millisecond time scale [11]. The overall catalytic cycle can be presented as a scheme including eight stages of alternating reactions of electron transfer and proton release followed by the final stage – release of an oxygen molecule [12, 13].
Thus, single-electron processes (P680→QA, YZ→P680+) in PS2 are coupled with four-electron oxidation of water (2 H2O → 4 e– + 4 H+ + O2) and two-electron plastoquinone reduction (PQ + 2 e– + 2 H+ → PQH2), which makes this enzyme one of the most complex energy converters.
Photochemical processes. Light flash-induced excitation of chlorophyll P680 in the PS2 RC causes electron transfer across the dielectric layer of the thylakoid membrane to a distance of ~35 Å. As a result, the protons in the donor and acceptor regions of the enzyme are transferred in opposite directions. Since these vectorial processes have components perpendicular to the plane of the membrane, they are coupled with generation of transmembrane electric potential difference (ΔΨ) (Fig. 2).
Only under certain conditions (the pathway of the transfer reaction must have a component perpendicular to the plane of the membrane; charge transfer must occur in a region of the protein with dielectric permittivity of less than 40 [14]; and the transported charge must not be locally compensated (as it is, e.g. when electrons and protons are transported together on plastoquinone)) the electron and proton transfers are accompanied by ΔΨ generation.Fig. 2. Electrogenic stages of charge transfer in PS2. The scheme does not show electrogenesis determined by protonation of doubly reduced QB.
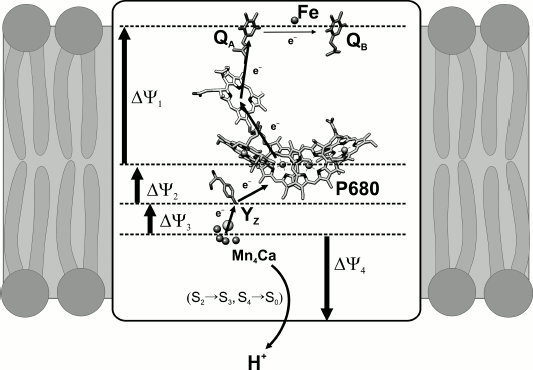
The generation of ΔΨ coupled to the vectorial reactions of charge transfer in PS2 has been registered in different photosynthesizing preparations (chloroplasts, thylakoids, PS2 membrane fragments, PS2 core complexes) by different methods (microelectrodes, modified patch clamp, electroluminescence, electrochemical shift of pigment absorption bands in light-harvesting complexes, and electrochemical methods) [14].
The kinetics of ΔΨ generation determined by the early stages of electron transfer in RC was measured by the light gradient method (based on ΔΨ measurement by silver chloride electrodes located in the lower and upper parts of the measuring cell) on chloroplasts and PS2 membrane fragments [15, 16]. The authors arrived at a conclusion that the primary charge separation in PS2 includes two electrogenic stages with approximately the same contributions to the total amplitudes of transmembrane electric potential – electron transfer between P680 and pheophytin and further electron transfer to QA [15] (Fig. 2).
Study of the catalytic cycle of water oxidation. The typical rate of water oxidation by the PS2 complex under stationary illumination is 100-200 turnovers of the enzyme per second. During this process, each turnover of the enzyme is characterized by a set of distinguishable intermediates formed during time periods from microseconds to several milliseconds from the starting moment of the reaction. Registration of the kinetics of transfers between the intermediates in a single turnover of the enzyme allows the study of molecular mechanisms of separate charge transfer reactions, while measurement under equilibrium conditions gives only the overall view of the light-dependent processes in PS2.
Significant advantages for studying the mechanism of ΔΨ generation associated with charge transfer within the enzyme are provided by isolated PS2 core complexes incorporated into phospholipid liposomes studied by the direct electrometric method [14, 17]. The principle of direct electrometry developed in our laboratory consists in the fusion of closed vesicles or liposomes containing protein complexes with a lipid-coated thin collodion film and ΔΨ measurement with macroelectrodes immersed in electrolyte buffer solution on the two sides of the artificial membrane. This method is exceptionally sensitive and allows registration of intraprotein charge transfer to a distance of >0.5 Å in the direction perpendicular to the plane of the membrane. Note that the kinetics of electron transfer can be recorded by different methods of spectroscopy, while the kinetics of vectorial proton transfer can be measured with high time resolution only by electrometry.
The results obtained by this method have shown that the relative contribution to the total ΔΨ (~17%) attributed to the electron transfer from tyrosine YZ to photooxidized P680 in the PS2 core complexes incorporated into liposomes [18, 19] is close to the ΔΨ value measured previously by the light gradient method in PS2 membrane fragments (~16%) oriented in a microcoaxial cell [16]. In the present work we have reviewed and summarized the results of recording the kinetics of separate stages of ΔΨ generation determined by charge transfer on the donor side of PS2 core complexes incorporated into proteoliposomes under conditions of single actuation of the enzyme.
Since nearly all RCs in dark-adapted PS2 preparations with active OEC are in state S1 until the first light flash, the kinetics of photoresponse shows an additional electrogenic stage with a characteristic time (τ) of 30-65 µsec (pH 6.5) and an amplitude of 2.5-3.5% of the YZ•QA− phase as a response to the first laser flash, in addition to rapid ΔΨ generation due to charge separation between P680 and quinone QA and re-reduction of P680+ via electron transfer from YZ [18, 19]. Such characteristic times are close to the rate of manganese ion oxidation during the S1→S2 transition measured by X-ray absorption spectroscopy with high time resolution [12]. Comparison of the kinetics of photoelectric responses of PS2 core complexes depleted of the Mn4Ca cluster and the same preparations but treated with iron (II) ions (in the latter case, the high-affinity manganese-binding site is inhibited) suggested that manganese oxidation at the low-affinity site was non-electrogenic [20]. This is evidence that the electrogenic reduction of tyrosine YZ• occurs by way of vectorial electron transfer from manganese in the high-affinity site.
One of the methods for distinguishing protolytic reactions from other processes involved in potential photogeneration is to study the dependence of the charge transfer reaction on pH or comparison of the reaction rate constants for H2O and D2O. The absence of the influence of heavy water on the kinetics of the electrogenic phase during the S1→S2 transition of the OEC demonstrates that proton release does not take place on the donor side of the enzyme (data not shown).
In response to the second laser flash (transition S2→S3), the kinetics of the photoelectric response shows an additional electrogenic phase with a characteristic time of ~240-300 µsec (pH 6.5) and an amplitude of ~5-7% of the fast-phase amplitude YZ•QA− [18, 19]. The authors of work [13] suggested deprotonation at CP43 Arg357 structurally linked to the manganese cluster in the course of S2→S3 transition, after the formation of radical YZ• and until oxidation of manganese [13, 21, 22]. This process is a limiting stage during the S2→S3 transition.
The latest electrometric experiments show that the reaction rate constant of the S2→S3 transition decreases in the presence of D2O, which favors proton transfer to the water phase (data not shown). The dependence of the kinetics of the S2→S3 transition on D2O has been shown previously for PS2 membrane fragments by pulse absorption spectroscopy [23].
The photoelectric response induced by the third laser flash (transition S3→S4→S0) contains an additional electrogenic component with τ ~ 4.5-6 msec and relative amplitude 4-6% of the amplitude of the YZ•QA− phase [18, 19]. It is assumed that in the intact PS2 complexes two protons are cleaved from water molecules during the terminal transition, and these data are in agreement with the role of amino acid groups close to the OEC in proton release [12, 13, 21, 22]. However, the formation of state S4 does not include electron transfer from the manganese complex to radical YZ•, and it seems that the central event is deprotonation of the manganese complex (or its environment) with a characteristic time of ~200 µsec [12, 13]. The latter was shown for PS2 membrane fragments and thylakoids by absorption spectroscopy [12].
The absence of an electrogenic phase with the characteristic time of ~200 µsec in the kinetics of the photoelectric response induced by the third laser flash demonstrates that the proton transfer during the S3→S4 transition is not vectorial, but electrically neutral. Also, the reduction of manganese through electron transfer from the water molecule during the S4→S0 transition is most likely not electrogenic either. Therefore, the observed electrogenesis in response to the third light flash is probably determined by proton transfer from the manganese complex or amino acids close to the latter into the water phase during the S4→S0 transition.
The similar amplitudes of electrogenic reactions during OEC transitions S2→S3 and S4→S0 give grounds to believe that protons cover equal distances. In contrast, the kinetics of the S2→S3 transition shows a more significant (nearly twofold) isotopic effect compared to the S4→S0 transition.
It should be noted that the contributions of the S2→S3 and S4→S0 transitions to the total electrogenesis in thylakoids measured by the electrochromic shift of the absorption bands of antenna carotenoids is approximately twofold higher than those revealed by direct electrometry in the core complexes [18]. In addition, as distinct from the intact PS2 core complexes with the terminal transition S4→S0 characterized by the time of ~4.6-6 msec, the value of this parameter in thylakoids and PS2 membrane fragments is ~1-1.2 msec [18]. This difference may be due to the increase in dielectric permittivity around the manganese cluster as a result of removal of peripheral proteins. Figure 3 schematically shows the properties of individual S-states and electrogenic reactions during the S-transitions of the OEC.
During the catalytic cycle of oxygen formation from water in PS2, protons must be released into the water phase from the manganese complex immersed deep in the protein matrix. Therefore, there must be special pathways for proton transfer in the hydrophobic part of the enzyme [24, 25]. Such a pathway for proton release must be effective enough not to retard the S-cycle. In addition to effectiveness, there is also a question of direction. It should be noted that the pathways of proton transfer in PS2 have been studied much less than the pathways of electron transfer. The protein environment cannot provide the supply of protons to the water phase [26]. In addition to the identification of amino acid residues around the manganese cluster, similar but not identical predictions for proton channels have been obtained in three works on intraprotein channel modeling [27-29]. Recent studies of molecular dynamics of the sequence of amino acid residues in this region has revealed a series of residues and water molecules linked by hydrogen bonds that leads from the manganese cluster to the lumen [30]. Three redox-active manganese atoms, an oxygen atom O(5) (probably OH–), two water molecules bound to Mn(4), and the side chains of D61−/R357+ are included in the proton release pathway [9, 31].Fig. 3. Scheme of transitions between the S-states of PS2 OEC. Percentages denote the relative contribution of electrogenic reactions. In dark-adapted PS2 samples, OEC are mainly in state S1.
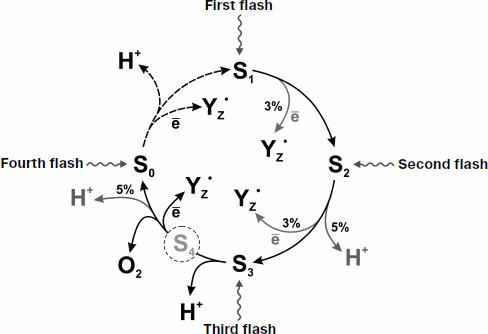
Although the authors of work [32] supposed that YZ was a part of the pathway of proton release to the lumen and that there was a potential pathway of proton release from YZ in the latter 3D structure of PS2 core complexes [3], it should be noted that the experimental data clearly support the “fluctuating” model, according to which the YZ proton never leaves the YZ-His190 site [8, 9].
From our point of view, investigation of the effect of zinc ions on the kinetics of proton transfer reactions during S-transitions to PS2 core complexes incorporated into liposomes is of certain interest. Since the phospholipid membrane is actually impermeable for bivalent cations, their binding site in the protein must be located on the outer side of the membrane. Therefore, zinc added from outside could selectively inhibit the phase of ΔΨ generation associated with proton transfer from the manganese complex or its immediate environment to the water phase (during transitions S2→S3 and S4→S0). It is known that zinc ions inhibit the reduction of the secondary quinone in bacterial RCs, suppressing the proton transfer in intraprotein proton channels [33]. Zinc ions added from outside to cytochrome oxidase-containing proteoliposomes specifically inhibit the slow electrogenic phase of proton transfer coupled with the transition of cytochrome oxidase from the oxo ferrous into the oxidized state [34]. The results obtained by direct electrometry [35] and X-ray absorption spectroscopy [36] suggest that zinc in the bc1 complex binds at the site of ubiquinol oxidase (QO) and prevents the release of protons to the water phase. The possible influence of Zn2+ on the kinetics of proton transfer reactions during OEC transitions S2→S3 and S4→S0 in PS2 complexes has not yet been studied.
Reduction of tyrosine cation radical YZ. Photooxidized P680 is reduced by electron transfer from tyrosine YZ that, in turn, accepts an electron from the manganese complex. YZ is usually considered as a part of the single-electron “wiring” of the OEC but not as a part of the pentametal Mn4Ca cluster [13, 32]. The important question is how YZ couples the single-electron photochemical reaction and the four-electron catalytic oxidation of water, effectively controlling the water oxidation process. In this context, it is important to know in detail the protein environment of YZ and its interaction with water molecules and the manganese cluster in PS2. It is supposed that YZ is located in a hydrophobic environment and does not directly interact with the substrate water in PS2 [37].
It is also known that the properties of YZ in PS2 lacking the manganese cluster may be crucially different from those in the intact PS2. It has been supposed that in such preparations YZ is located in a hydrophilic environment and contacts the water phase [38-44]. Some substances, viz. manganese, ascorbate, N,N,N′,N′-tetramethyl-p-phenylenediamine (TMPD), 2,3,5,6-tetramethyl-p-phenylenediamine (DAD), 2,6-dichlorophenolindophenol (DCPIP), phenazine methosulfate (PMS), 1,5-diphenylcarbazide (DPC), benzidine, hydroxylamine, and hydrazine are able to act as electron donors in the absence of the manganese cluster [40-44]. Ascorbate seems to be the only alternative electron donor capable of supplying electrons to the PS2 RC in sufficient amounts in the absence of the manganese cluster in vivo [45].
Lipophilic electron donors. The extremely asymmetric orientation of PS2 core complexes in liposomes (the donor side outside the membrane) makes it possible to use the direct electrometric method for studying the mechanism of interaction between the PS2 RC depleted of Mn4Ca cluster and artificial electron donors. Increase in relative contributions of the slow components of membrane potential decrease associated with recombination of charges between QA and YZ• (~20-200 msec) in the presence of both Mn2+ (4 Mn per P680) and the reduced forms of lipophilic redox mediators (TMPD, DCPIP, DAD, PMS) and DPC is evidence of their ability to effectively interact with YZ•. At certain concentrations of these substances, an additional slow electrogenic phase appears in the kinetics of the photoelectric response to a light flash, contributing 15 to 25% to the total electrogenesis [44].
Experimental results lead to the conclusion that artificial electron donors are arranged in the following order in accordance with the degree of efficiency in reducing the radical YZ•: PMS > TMPD > DAD > DPC > DCPIP. Under anaerobic conditions, PMS proved to be even more effective. However, it is still unclear whether the reaction rate is saturated at increased PMS concentration. Saturation is observed in the experiments, e.g. with TMPD, where the maximum Kv value of 400-500 sec−1 is reached at 4 mM of the mediator.
Synthetic Mn-containing complexes. In recent years, various Mn-containing complexes have been synthesized as models of the OEC manganese cluster. The direct electrometric method applied for proteoliposomes containing PS2 core complexes depleted of manganese ions showed that the addition of a synthetic trinuclear manganese complex to the measuring medium resulted in extra generation of photoelectric response with an amplitude of ~25% of the fast (YZ•QA−) phase and was characterized by τ ~160 msec [46]. This phase was attributed to electron transfer from the donor binding site on the protein–water interface to the oxidized cofactor immersed deep in the protein. Previously, it was demonstrated that the rate of oxygen release in PS2 membrane fragments depleted of manganese ions in the presence of synthetic complex is higher than in the presence of MnCl2 [47].
Hydrophilic electron donors. There are substantial differences in the mechanism of reduction of tyrosine YZ• between small hydrophilic NH2OH/NH2NH2 and large lipophilic compounds DPC/TMPD/DCPIP/DAD. The increase in relative contributions of the slow components of membrane potential decay in the presence of NH2OH/NH2NH2 implies the prevention of charge recombination between QA− and YZ• by way of effective electron transfer to tyrosine YZ·. However, the absence of additional electrogenesis in the kinetics of photoelectric response is evidence of the non-electrogenic character of this reaction.
Mechanism of electron transfer. Photosynthetic RCs are generally an ideal object for studying long-distance electron transfer [48, 49]. Before discussing the mechanism of electron transfer in PS2 RC in the presence of artificial electron donors, it is important to note that the direct electrometric method used in our previous experiments showed the presence of an extra electrogenic phase in the millisecond time domain in the kinetics of photoelectric response during the reduction of the primary electron donors P870 in bacterial RC [50, 51] and P700 in cyanobacterial complexes of PS1 [52], oxidized under the influence of a laser flash, by artificial redox-active compounds such as TMPD, DCPIP, and PMS. Since the contribution of this phase (~20%) to the total photoelectric response was approximately equal to the contribution of the phase observed in the presence of cytochrome c2 in the case of bacterial RC [51] and cytochrome c6 [53] or plastocyanin [54] in PS1, it was concluded that the electrogenic reduction of P870+/P700+ by redox-active compounds results from the vectorial transfer of electrons from the protein–water interface to P870/P700 immersed in the protein.
We believe that additional electrogenesis observed in the presence of TMPD, DAD, DCPIP, DPC, and synthetic trinuclear manganese complex is also determined by the vectorial electron transfer from the protein–water interface (Fig. 4). This assumption is supported by the results of modeling the structure of the donor region of PS2 core complexes depleted of Mn4Ca cluster and three peripheral proteins [55]. Removal of these subunits from PS2 structure creates a cavity on the donor side of the enzyme. It has been shown that the TMPD molecule rather tightly adjoins the cavity bottom, with a distance of ~17 Å between the edges of molecular π-orbitals of YZ and TMPD (the oxygen atom of YZ and the nearest nitrogen atom of TMPD).
It should be noted that electron transfer inside the protein is a rather complicated process determined by many factors. According to theory [56], the rate of electron transfer depends on the distance between the donor and the acceptor, the difference of their redox potentials, and the energy of reorganization (the energy needed for changing the equilibrium geometry of the initial state into the equilibrium geometry of the product). Electron transfer inside the protein is supposed to occur through specific tunneling and may be modulated by conformational changes in the secondary protein structure on the protein–water interface [57]. As mentioned in work [58], effective tunneling is not limited by any specifically formed pathway inside the protein but rather occurs via several trajectories inside the protein matrix.Fig. 4. Scheme of electron transport in the donor region of PS2 depleted of Mn4Ca cluster in the presence of artificial donors DPC, TMPD, DAD, DCPIP, NH2NH2, and NH2OH. Solid arrows show the electrogenic stages of electron transfer; the dashed arrow shows the hypothetical diffusion of NH2NH2 (NH2OH) through the hydrophilic channel leading from the protein surface to the binding site of the Mn4Ca cluster. The scheme was adapted from work [59].
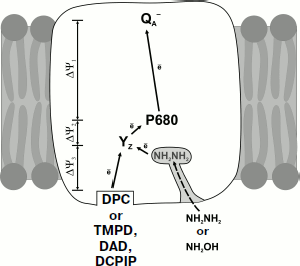
As regards the mechanism of electron transfer in the presence of low molecular weight donors in PS2 preparations depleted of Mn4Ca cluster and peripheral proteins, let us note that the modeling of structure of the donor side of such preparations with Caver 2.0 v0.003 [59] has shown three channels with a minimal diameter of about 2.0-3.0 Å that link the binding site of the manganese cluster to the water–protein interface. It is obvious that the sizes of TMPD, DAD, DCPIP, and DPC molecules (4 × 14 Å) noticeably exceed the diameter of these channels, while the hydrophilic, low molecular weight electron donors NH2NH2 and NH2OH (2.0 × 2.4 Å) can diffuse through these channels to the binding site of the Mn4Ca cluster (Fig. 4).
The results obtained by the direct electrometric method with a single turnover of the enzyme make it possible to follow the transfer of charges (electron and proton) inside the protein in real time. It has been shown that electrogenesis observed during the catalytic cycle of the OEC in dark-adapted PS2 samples is determined by the electron transfer between manganese bound in the high-affinity site of subunit D1 and tyrosine radical YZ (transition S1→S2), as well as with the transfer of protons in the opposite direction from the manganese complex to the water phase (transitions S2→S3 and S4→S0) (Fig. 2).
The data obtained with the preparations of PS2 core complexes depleted of Mn4Ca cluster and peripheral proteins show that the effective reduction of oxidized YZ• from artificial electron donors can be both electrogenic and non-electrogenic. Hydrophobic artificial electron donors (PMS, TMPD, DAD, DCPIP, and DPC) and synthetic trinuclear manganese complex reduce the tyrosine radical YZ electrogenically due to vectorial electron transfer from the binding site on the protein–water interface, while more hydrophilic and low molecular weight donors (NH2OH, NH2NH2) can diffuse through the channels with minimum diameter of 2.0-3.0 Å passing from the water (lumen) surface of the protein to the Mn4Ca cluster, followed by non-electrogenic reduction of tyrosine radical YZ• (Fig. 4). All these results are important for understanding the mechanism of interaction between the artificial electron donors and PS2 RC.
The cardinal problem that arises during the study of artificial photosynthesis based on light-dependent water cleavage into hydrogen and oxygen (2 H2O + 4 hν → 2 H2 + O2) is the coupling of photoinduced charge separation (a one-electron one-photon process) with the release of molecular oxygen (a four-electron process). Nature has solved this problem by creating the Mn4Ca cluster – the catalytic component capable of donating electrons step-by-step and oxidizing water molecules in a way so as to avoid the formation of high-energy intermediates. Creation of specific multi-electron redox catalysts is an important problem of modern chemistry [60, 61]. In recent years, works on the synthesis of various manganese compounds for simulation of PS2 OEC have become very relevant.
The findings extend modern concepts of the mechanisms of electrogenic reactions on the donor side of PS2 and may serve as a basis for creation of effective solar energy conversion systems.
This work was supported by the Russian Foundation for Basic Research (project Nos. 11-04-00818-a, 12-04-00821-a, 11-04-92503-a, 11-04-91330-a), CRDF (RUB1-7029-MO-11), and the Ministry of Education and Science (State Contract No. 16.512.12.2010).
REFERENCES
1.Ferreira, K. N., Iverson, T. M., Maghlaoui, K.,
Barber, J., and Iwata, S. (2004) Science, 303,
1831-1838.
2.Guskov, A., Kern, J., Gabdulkhakov, A., Broser, M.,
Zouni, A., and Saenger, W. (2009) Nat. Struct. Mol. Biol.,
16, 334-342.
3.Umena, Y., Kawakami, K., Shen, J.-R., and Kamiya,
N. (2011) Nature, 473, 55-60.
4.Styring, S., Sjoholm, J., and Mamedov, F. (2012)
Biochim. Biophys. Acta, 1817, 26-87.
5.Shinkarev, V. P. (2004) in Chlorophyll
Fluorescence: a Signature of Photosynthesis (Papageorgiou, G. C.,
and Govindjee) Kluwer Academic Publishers, Amsterdam, pp. 197-229.
6.Shelaev, I. V., Gostev, F. E., Vishnev, M. I.,
Shkuropatov, A. Ya., Ptushenko, V. V., Mamedov, M. D., Sarkisov, O. M.,
Nadtochenko, V. A., Semenov, A. Yu., and Shuvalov, V. A. (2011)
Photochem. Photobiol., 104, 44-50.
7.Novoderezhkin, V. I., Dekker, J. P., and van
Grondelle, R. (2007) Biophys. J., 93, 1293-1311.
8.Rappaport, F., and Lavergne, J. (1997)
Biochemistry, 36, 15294-15302.
9.Sproviero, E. M., Gascon, J. A., McEvoy, J. P.,
Brudvig, G. W., and Batista, V. S. (2008) J. Am. Chem. Soc.,
130, 3428-3442.
10.Renger, G., and Renger, T. (2008) Photosynth.
Res., 98, 53-80.
11.Gernot, R., and Kuhn, P. (2007) Biochim.
Biophys. Acta, 1767, 458-471.
12.Haumann, M., Liebisch, P., Muller, C., Barra, M.,
Grabolle, M., and Dau, H. (2005) Science, 310,
1019-1021.
13.Dau, H., and Haumann, M. (2007) Biochim.
Biophys. Acta, 1767, 472-483.
14.Semenov, A. Yu., Mamedov, M. D., and Chamorovsky,
S. K. (2006) in Photosystem I: the Light-Driven,
Plastocyanine:Ferredoxin Oxidoreductase (Golbeck, J. H., ed.)
Springer, Dordrecht, pp. 319-424.
15.Trissl, H. W., and Leibl, W. (1989) FEBS
Lett., 244, 85-88.
16.Pokorny, A., Wulf, K., and Trissl, H.-W. (1994)
Biochim. Biophys. Acta, 1184, 65-70.
17.Semenov, A., Cherepanov, D., and Mamedov, M.
(2008) Photosynth. Res., 98, 121-130.
18.Haumann, M., Mulkidjanian, A., and Junge, W.
(1997) Biochemistry, 36, 9304-9315.
19.Mamedov, M. D., Beshta, O. P., Gurovskaya, K. N.,
Mamedova, A. A., Neverov, K. D., Samuilov, V. D., and Semenov, A. Yu.
(1999) Biochemistry (Moscow), 64, 504-509.
20.Kurashov, V. N., Lovyagina, E. R., Shkolnikov, D.
Y., Solntsev, M. K., Mamedov, M. D., and Semin, B. K. (2009)
Biochim. Biophys. Acta, 1787, 1492-1498.
21.Shimada, Y., Suzuki, H., Tsuchiya, T., Mimuro,
M., and Noguchi, T. (2011) J. Am. Chem. Soc., 133,
3808-3811.
22.Ishikita, H., Saenger, W., Loll, B., Biesiadka,
J., and Knapp, E. W. (2006) Biochemistry, 45,
2063-2071.
23.Renger, G. (2004) Biochim. Biophys. Acta,
1655, 195-204.
24.Muh, F., and Zouni, A. (2011) Front.
Biosci., 17, 3072-3132.
25.Ho, F. M. (2012) Biochim. Biophys. Acta,
1817, 106-120.
26.Belevich, I., and Verkhovsky, M. I. (2008)
Antioxid. Redox Signal., 10, 1-29.
27.Murray, J. W., and Barber, J. (2007) J.
Struct. Biol., 159, 228-237.
28.Ho, F. M., and Styring, S. (2008) Biochim.
Biophys. Acta, 1777, 140-153.
29.Gabdulkhakov, A., Guskov, A., Broser, M., Kern,
J., Muh, F., Saenger, W., and Zouni, A. (2009) Structure,
17, 1223-1234.
30.Swanson, J. M. J., and Simons, J. (2009) J.
Phys. Chem. B., 113, 5149-5161.
31.McEvoy, J. P., and Brudvig, G. W. (2004) Phys.
Chem. Chem. Phys., 6, 4754-4763.
32.Hoganson, C. W., and Babcock, G. T. (1997)
Science, 277, 1953-1956.
33.Paddock, M. L., Graige, M. S., Feher, G., and
Okamura, M. Y. (1999) Proc. Natl. Acad. Sci. USA, 96,
6183-6188.
34.Kuznetsova, S. S., Atsarkina, N. V., Vygodina, T.
V., Siletskiy, S. A., and Konstantinov, A. A. (2005) Biochemistry
(Moscow), 70, 128-136.
35.Klishin, S. S., Junge, W., and Mulkidjanian, A.
Y. (2002) Biochim. Biophys. Acta, 1553, 177-182.
36.Giachini, L., Francia, F., Veronesi, G., Lee,
D.-W., Daldal, F., Huang, L.-S., Berry, E. A., Cocco, T., Papa, S.,
Boscherini, F., and Venturoli, G. (2007) Biophys. J., 93,
2934-2951.
37.Wydrzynski, T., Hillier, W., and Messinger, J.
(1996) Physiol. Plant., 96, 342-350.
38.Babcock, G. T., and Sauer, K. (1975) Biochim.
Biophys. Acta, 396, 48-62.
39.Conjeaud, H., and Mathis, P. (1980) Biochim.
Biophys. Acta, 590, 353-359.
40.Blubaugh, D. J., and Cheniae, G. M. (1990)
Biochemistry, 29, 5109-5118.
41.Chroni, S., and Ghanotakis, D. F. (2001)
Biochim. Biophys. Acta, 1504, 432-437.
42.Semin, B. K., Ghirardi, M. L., and Seibert, M.
(2002) Biochemistry, 41, 5854-5864.
43.Dasgupta, J., Ananyev, G. M., and Dismukes, G. C.
(2008) Coord. Chem. Rev., 252, 347-360.
44.Gopta, O. A., Tyunyatkina, A. A., Kurashov, V.
N., Semenov, A. Yu., and Mamedov, M. D. (2008) Eur. Biophys. J.,
37, 1045-1050.
45.Toth, S. Z., Puthur, J. T., Nagy, V., and Garab,
G. (2009) Plant Physiol., 149, 1568-1578.
46.Kurashov, V. N., Allakhverdiev, S. I.,
Zharmukhamedov, S. K., Nagata, T., Klimov, V. V., Semenov, A. Yu., and
Mamedov, M. D. (2009) Photochem. Photobiol. Sci., 8,
162-166.
47.Nagata, T., Zharmukhamedov, S. K., Khorobrykh, A.
A., Klimov, V. V., and Allakhverdiev, S. I. (2008) Photosynth.
Res., 98, 277-284.
48.Mobius, K. (2000) Chem. Soc. Rev.,
29, 129-134.
49.Beatty, J. T., Gest, H., and Govindjee (2003)
Photosynth. Res., 76, 1-11.
50.Drachev, L. A., Kaminskaya, O. P., Konstantinov,
A. A., Semenov, A. Yu., and Skulachev, V. P. (1985) FEBS Lett.,
189, 45-49.
51.Drachev, L. A., Kaminskaya, O. P., Konstantinov,
A. A., Kotova, E. A., Mamedov, M. D., Samuilov, V. D., Semenov, A. Yu.,
and Skulachev, V. P. (1986) Biochim. Biophys. Acta, 848,
137-146.
52.Gourovskaya, K. N., Mamedov M. D., Vassiliev, I.
R., Golbeck, J. H., and Semenov, A. Y. (1997) FEBS Lett.,
414, 193-196.
53.Mamedov, M. D., Gadzhieva, R. M., Gourovskaya, K.
N., Drachev, L. A., and Semenov, A. Yu. (1996) J. Bioenerg.
Biomembr., 28, 517-522.
54.Mamedov, M. D., Mamedova, A. A., Chamorovsky, S.
K., and Semenov, A. Yu. (2001) FEBS Lett., 500,
172-176.
55.Mamedov, M. D., Kurashov, V. D., Cherepanov, D.
A., and Semenov, A. Yu. (2010) Front. Biosci., 15,
1007-1017.
56.Marcus, R. A., and Sutin, N. (1985) Biochim.
Biophys. Acta, 811, 265-322.
57.Gray, H. B., and Winkler, J. R. (1996) Annu.
Rev. Biochem., 65, 537-561.
58.Moser, C. C., Keske, J. M., Warncke, K., Farid,
R. S., and Dutton, P. L. (1992) Nature, 355, 796-802.
59.Mamedov, M. D., Kurashov, V. N., Petrova, I. O.,
Zaspa, A. A., and Semenov, A. Yu. (2010) Biochemistry (Moscow),
75, 579-584.
60.Semenov, N. N. (1981) Science and Society
[in Russian], Nauka, Moscow.
61.Balzani, V., Credi, A., and Venturi, M. (2008)
Chem. Sus. Chem., 1, 26-58.