Mitochondria-Targeted Plastoquinone Antioxidant SkQR1 Decreases Trauma-Induced Neurological Deficit in Rat
N. K. Isaev1,2,3*, S. V. Novikova2,3, E. V. Stelmashook2,3, I. V. Barskov2, D. N. Silachev1,3, L. G. Khaspekov2, V. P. Skulachev1,3, and D. B. Zorov1,3
1Belozersky Institute of Physico-Chemical Biology, Lomonosov Moscow State University, 119991 Moscow, Russia; fax: (495) 939-3181; E-mail: isaev@genebee.msu.ru2Research Center of Neurology, Russian Academy of Medical Sciences, Volokolamskoe Shosse 80, 125367 Moscow, Russia
3Institute of Mitoengineering, Lomonosov Moscow State University, 119991 Moscow, Russia
* To whom correspondence should be addressed.
Received March 14, 2012; Revision received June 18, 2012
A protective effect of a mitochondria-targeted antioxidant, a cationic rhodamine derivative linked to a plastoquinone molecule (10-(6′-plastoquinonyl)decylrhodamine-19, SkQR1) was studied in the model of open focal trauma of rat brain sensorimotor cortex. It was found that daily intraperitoneal injections of SkQR1 (100 nmol/kg) for 4 days after the trauma improved performance in a test characterizing neurological deficit and decreased the volume of the damaged cortical area. Our results suggest that SkQR1 exhibits profound neuroprotective effect, which may be explained by its antioxidative activity.
KEY WORDS: brain trauma, neuroprotection, mitochondria-targeted antioxidants, SkQR1DOI: 10.1134/S0006297912090052
Abbreviations: MMP, mitochondrial membrane permeabilization (nonspecific permeability of the inner mitochondrial membrane); ROS, reactive oxygen species; SkQR1, 10-(6′-plastoquinonyl)decylrhodamine-19.
In the late 1960s and early 1970s our group and the group of E. A.
Liberman published results of our joint study indicating that
lipophilic ions with delocalized charge screened by spacial substituent
can easily penetrate into mitochondria and submitochondrial particles
driven by electric field on the inner mitochondrial membrane [1-4]. Later this observation was
realized in practice in the development of new mitochondria-targeted
antioxidants of the SkQ family. To create such targeted antioxidants,
plastoquinone residue was linked to penetrating lipophilic cations.
Plastoquinone is a powerful antioxidant and a component of the
photosynthetic electron transport chain in plant chloroplasts and
cyanobacteria, i.e. in oxygen-generating structures existing under
conditions of a persistent oxidative stress. Historically, the compound
SkQ1 was synthesized first, and later it was supplemented with various
analogs carrying different components as the penetrating cation. The
antioxidative efficiency of these compounds occurred to be much higher
than the efficiency of the analog based on the initial quinone CoQ10
having animal origin [5, 6].
Considering the important role of reactive oxygen species (ROS) in
pathogenesis of various diseases, it seems promising to use the
extremely low, nanomolar concentrations of mitochondria-targeted
antioxidants for treatment of many age-related diseases, such as
Alzheimer’s disease, cardiac arrhythmias, myocardial and renal
infarction, and brain stroke [5, 7-9].
Skull–brain trauma is another cerebral pathology that has severe medical, social, and economic consequences. Initial stages of the development of skull–brain trauma are associated with a deterioration of brain blood flow and metabolism, decrease in oxygen consumption, and exhaustion of energy-rich phosphates. The deficit in the cell energetics leads, in turn, to an increase in the intracellular contents of Na+ and Ca2+, an excessive production and accumulation of glutamate, and triggering the cell death mechanism. It was supposed that pathological consequences could be mainly caused by the formation of nonspecific permeability of the inner mitochondrial membrane (MMP) due to decrease in oxidative phosphorylation of the nervous tissue [10, 11]. The increase in the MMP induced by ROS in mitochondria in turn triggers MMP [12]. Thus, a vicious circle is produced resulting in the massive death of brain cells.
There is still no sufficiently effective treatment of the consequences of skull–brain trauma. Therefore, it is vital to create new agents with neuroprotective properties for correcting pathological processes caused by brain trauma.
Using the model of an open focal trauma of the rat brain sensorimotor cortex, in the present study we explored the neuroprotective effect of the mitochondria-targeted cationic antioxidant 10-(6′-plastoquinonyl)decylrhodamine-19 (SkQR1), which is a conjugate of a rhodamine derivative linked to a plastoquinone molecule.
MATERIALS AND METHODS
In the present work, we employed our modification of the earlier used model of focal open severe brain trauma in rats [13]. The study was performed on male Wistar rats with body weight of 180-250 g. Before the surgery, the animals were anesthetized by an intraperitoneal injection of 3% chloral hydrate (330 mg/kg). To create the trauma, the left frontal part of the skull was trepanized above the sensorimotor cortex zone, and a movable Teflon piston 4 mm in diameter with depth of insertion of 2.5 mm was placed into it; this piston was struck from the height of 10 cm with a 50-g load sliding along a directing rail. Localization of the sensorimotor cortex zone was determined based on data described in [14].
SkQR1 (100 nmol/kg) was injected intraperitoneally 1 h after the trauma and then daily in the same dose during the following four days.
A magnetic resonance imaging (MRI) study was performed as described in [15] using a BioSpec 70/30 (Bruker, Germany) with magnetic field induction of 7 T and gradient of 105 mT/m. The damaged focal volume was determined morphometrically on the 7th day after the trauma. For this, the brain of the animal was fixed by submerging it in a mixture of formalin–ethanol–acetic acid in the ratio 2 : 7 : 1 for 24 h; then the brain was placed in 70% ethanol for 24 h; then serial sections were prepared using a NVSLM1 vibratome (World Precision Instruments) with a step of 100 µm. Every second section was sequentially mounted onto glasses covered with gelatin and stained with 0.2% Methylene Blue. Then the sections were treated routinely (dehydrated with increasing ethanol concentration, cleared with xylene, and mounted in balsam) and scanned on a slide-device of an Epson Perfection V100 PHOTO scanner. As a result, files were obtained with a picture of light-blue section of the brain with a clearly shown damage area. The volume of the lesion was determined as a cylinder with V = ΣSn × d, where d is thickness of the section pair (200 µm); Sn is the area of the lesion in the section in mm2; Σ is the sum of volumes of ischemic damage in the sections using an Image J program (Bethesda MD, USA).
Behavioral “limb-placing test” was performed 24 h before the operation and then on the third and seventh days after the trauma. Neurologic deficit caused by the skull–brain trauma was estimated using a 12-score scale [16] in modification [17]. The resulting score on this scale is determined as the sum of points obtained in six tests assessing the response of the forelimbs and hindlimbs to tactile and proprioceptive stimulation in the presence of obvious reflexes. Malfunctioning of the limb was estimated using the following system: 2 points corresponds to complete performance of the test; 1 point corresponds to performing the test with a delay of 2 sec or incompletely; 0 point corresponded to the lack of response to the stimulation of the limb.
The results are expressed as the mean ± standard error of mean. The results of behavioral tests were compared using the Mann–Whitney test for independent samples. The statistical significance of differences in the damaged volumes was assessed using Student’s t-test at the significance level of p < 0.05.
The animals were treated and subjected to experimental procedures in accordance with requirements of the Counsel of the European Community 86/609/EEC on use of animals for experimental studies.
RESULTS
Magnetic resonance images obtained on the first day after the skull–brain trauma allowed us to clearly identify the lesion area in the sensorimotor cortex of the left hemisphere (figure, panel (a)). The localization of the damaged area was in complete correlation with data on behavioral performance based on the sensitivity and motor activity of the limbs.
The results of the limb-placing test revealed the development after the trauma of a functional deficit in the right limbs, whereas it was absent in the left limbs. In the intact rats before the trauma, the test gave 12 points for both the right and left limbs (figure, panel (b)), and after the skull–brain trauma in the rats not treated with SkQR1 this score for the right limbs was 4.5 ± 0.9 points on the third day (n = 14) and 4.3 ± 0.8 points on the seventh day (n = 14) (figure, panels (c) and (d)).
Effect of SkQR1 on the neurological deficit and volume of brain damage caused by focal trauma of the sensorimotor cortex. a) Localization of the damage area in the rat brain 24 h after the trauma. MR-imaging. The damaged area is indicated by the arrow in serial section. b) Neurological deficit in the “limb-placing test” in intact rats before the trauma was evaluated as 12 points for both right and left limbs. SkQR1 decreased the neurological deficit on the third (c) and on the seventh (d) day after the trauma. SkQR1 also decreased the volume of the damaged area on the seventh day after the trauma (e). * p < 0.05 comparing the group of animals treated with SkQR1 after the trauma (white columns) with the group not treated with SkQR1 (black columns).
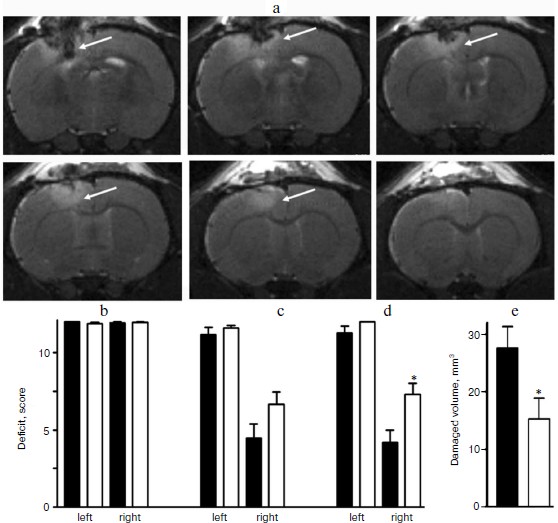
The intraperitoneal injections of SkQR1 significantly decreased the neurological deficit. In this case the right limbs of the rats showed on the third day 6.6 ± 0.8 (n = 14) and 7.4 ± 0.8 points (n = 14) on the seventh day after the trauma (figure, panels (c) and (d)). The morphometric analysis after completing the behavioral test revealed that on the seventh day after the trauma the injection of SkQR1 decreased the damaged volume nearly twofold (to 15 ± 4 mm3 (n = 14)), whereas without treatment with SkQR1 the damaged area was 28 ± 4 mm3 (n = 13) (figure, panel (e)).
Thus, in our work the injection of SkQR1 to animals after skull–brain trauma was shown for the first time to significantly decrease the neurological posttraumatic deficit and the volume of lesion.
DISCUSSION
It has been earlier shown that extremely low, nanomolar concentrations of mitochondria-targeted antioxidants, plastoquinone derivatives SkQ1 and SkQR1, could be used on animal models of various pathologies including Alzheimer’s disease, cardiac arrhythmia, and myocardial and renal infarction [5, 7-9], the pathogenesis of which is significantly determined by increased production of ROS in mitochondria. On various models of ischemic brain stroke, we have demonstrated that mitochondria-targeted antioxidants are efficient in preventing the consequences of brain ischemia such as destruction of nervous tissue and neurological deficit [7, 8]. Skull–brain trauma is another serious medical problem. In the present work, this pathology was studied on our modified model of open focal brain trauma [13]. This model allowed to obtain a standard in the size and localization of cortical damage accompanied by a pronounced neurological deficit corresponding to clinical manifestations of the brain trauma. This model improves perspectives in the search and experimental base for pharmacological correction of this cerebral pathology.
The mechanisms of development of brain trauma and brain ischemia are similar in common, and the propagation of these pathologies significantly depends on mitochondria and the MMP [11, 18], which leads to an increased production of ROS by mitochondria [10, 12]. These active molecules directly damage lipids, proteins, and nucleic acids in the cell. ROS also activate different molecular signaling pathways associated with cell death [19]. The increased production of ROS by mitochondria under conditions of both ischemia and trauma is the most important pathogenetic detail in the mechanism of the neurodestruction. Therefore, it was reasonable to suggest that the trauma-induced development of neurological distortion could be decreased using mitochondria-targeted antioxidants [20]. In the present work, for the first time we were able to demonstrate that the injection of SkQR1 in nanomolar concentrations during four days after skull–brain trauma reliably decreases the neurological deficit, whereas in the absence of SkQR1 this deficit did not decrease during the same period. The morphometric analysis of the damaged area revealed that in the SkQR1-treated rats the volume of the lesion in the brain cortex was significantly lower than in the untreated animals. As it has been shown earlier, the mechanism of the protective effect can be mediated through the ability of SkQR1 to decrease the level of mitochondrial ROS and also to induce in vivo ischemic tolerance due to initiating an increased production of erythropoietin that, in turn, decreases the activity of glycogen synthase kinase (GSK-3β) [21], which is involved in the apoptotic cascade in neurons under conditions of ischemia, trauma, and in Alzheimer’s disease. There have been attempts to treat ischemia and trauma using common, non-targeted antioxidants, but their efficiency was rather limited, especially in clinical studies [22]. Therefore, our findings on the possibility of curing trauma using mitochondria-targeted antioxidants are very promising for further studies of the pharmacological potential of the agents.
This work was supported by the Russian Foundation for Basic Research (the projects 12-04-00025-a and 11-04-01307-a) and by the Russian Federation President project MK-729.2012.4.
REFERENCES
1.Liberman, E. A., Topali, V. P., Tsofina, L. M.,
Jasaitis, A. A., and Skulachev, V. P. (1969) Nature, 222,
1076-1078.
2.Grinius, L. L., Jasaitis, A. A., Kadziauskas, Yu.
L., Liberman, E. A., Skulachev, V. P., Topali, V. P., Tsofina, L. M.,
and Vladimirova, M. A. (1970) Biochim. Biophys. Acta,
216, 1-12.
3.Bakeeva, L. E., Grinius, L. L., Jasaitis, A. A.,
Kuliene, V. V., Levitsky, D. O., Liberman, E. A., Severina, I. I., and
Skulachev, V. P. (1970) Biochim. Biophys. Acta, 216,
12-21.
4.Liberman, E. A., and Skulachev, V. P. (1970)
Biochim. Biophys. Acta, 216, 30-42.
5.Skulachev, V. P., Anisimov, V. N., Antonenko, Y.
N., Bakeeva, L. E., Chernyak, B. V., Erichev, V. P., Filenko, O. F.,
Kalinina, N. I., Kapelko, V. I., Kolosova, N. G., Kopnin, B. P.,
Korshunova, G. A., Lichinitser, M. R., Obukhova, L. A., Pasyukova, E.
G., Pisarenko, O. I., Roginsky, V. A., Ruuge, E. K., Senin, I. I.,
Severina, I. I., Skulachev, M. V., Spivak, I. M., Tashlitsky, V. N.,
Tkachuk, V. A., Vyssokikh, M. Y., Yaguzhinsky, L. S., and Zorov, D. B.
(2009) Biochim. Biophys. Acta, 1787, 437-461.
6.Skulachev, M. V., Antonenko, Y. N., Anisimov, V.
N., Chernyak, B. V., Cherepanov, D. A., Chistyakov, V. A., Egorov, M.
V., Kolosova, N. G., Korshunova, G. A., Lyamzaev, K. G., Plotnikov, E.
Y., Roginsky, V. A., Savchenko, A. Y., Severina, I. I., Severin, F. F.,
Shkurat, T. P., Tashlitsky, V. N., Shidlovsky, K. M., Vyssokikh, M. Y.,
Zamyatnin, A. A., Jr., Zorov, D. B., and Skulachev, V. P. (2011)
Curr. Drug Targets, 12, 800-826.
7.Bakeeva, L. E., Barskov, I. V., Egorov, M. V.,
Isaev, N. K., Kapelko, V. I., Kazachenko, A. V., Kirpatovsky, M. I.,
Kozlovsky, S. V., Lakomkin, V. L., Levina, S. V., Pisarenko, O. I.,
Plotnikov, E. Y., Saprunova, V. B., Serebryakova, L. I., Skulachev, M.
V., Stelmashuk, E. V., Studneva, I. M., Tskitishvili, O. V.,
Vasil’eva, A. K., Viktorov, I. V., Zorov, D. B., and Skulachev,
V. P. (2008) Biochemistry (Moscow), 73, 1288-1299.
8.Plotnikov, E. Y., Silachev, D. N., Chupyrkina, A.
A., Danshina, M. I., Yankauskas, S. S., Morosanova, M. A., Stelmashook,
E. V., Vasil’eva, A. K., Goryacheva, E. S., Pirogov, Yu. A.,
Isaev, N. K., and Zorov, D. B. (2010) Biochemistry (Moscow),
75, 145-150.
9.Kapay, N. A., Isaev, N. K., Stelmashook, E. V.,
Popova, O. V., Zorov, D. B., Skrebitsky, V. G., and Skulachev, V. P.
(2011) Biochemistry (Moscow), 76, 1367-1370.
10.Andriessen, T. M., Jacobs, B., and Vos, P. E.
(2010) J. Cell Mol. Med., 14, 2381-2392.
11.Veech, R. L., Valeri, C. R., and VanItallie, T.
B. (2012) IUBMB Life, 64, 203-207.
12.Zorov, D. B., Filburn, C. R., Klotz, L. O.,
Zweier, J. L., and Sollott, S. J. (2000) J. Exp. Med.,
192, 1001-1014.
13.Feeney, D. M., Boyeson, M. G., Linn, R. T.,
Murray, H. M., and Dail, W. G. (1981) Brain Res., 211,
67-77.
14.Hicks, S. P., and D’Amato, C. J. (1977) Exp.
Neurol., 56, 410-420.
15.Silachev, D. N., Uchevatkin, A. A., Pirogov, Y.
A., Zorov, D. B., and Isaev, N. K. (2009) Byul. Eksp. Biol.
Med., 147, 232-237.
16.De Ryck, M., van Reempts, J., Borgers, M.,
Wauquier, A., and Janssen, P. A. (1989) Stroke,
20, 1383-1390.
17.Jolkkonen, J., Puurunen, K., Rantakomi, S.,
Harkonen, A., Haapalinna, A., and Sivenius, J. (2000) Eur. J.
Pharmacol., 400, 211-219.
18.Gouriou, Y., Demaurex, N., Bijlenga, P., and De
Marchi, U. (2011) Biochimie, 93, 2060-2067.
19.Niizuma, K., Yoshioka, H., Chen, H., Kim, G. S.,
Jung, J. E., Katsu, M., Okami, N., and Chan, P. H. (2010) Biochim.
Biophys. Acta, 1802, 92-99.
20.Skulachev, V. P. (2012) J. Alzheimer’s
Dis., 28, 283-289.
21.Plotnikov, E. Y., Chupyrkina, A. A., Jankauskas,
S. S., Pevzner, I. B., Silachev, D. N., Skulachev, V. P., and Zorov, D.
B. (2011) Biochim. Biophys. Acta, 1812, 77-86.
22.Slemmer, J. E., Shacka, J. J., Sweeney, M. I.,
and Weber, J. T. (2008) Curr. Med. Chem., 15,
404-414.