REVIEW: Catalytic Mechanism and Substrate Specificity of HIF Prolyl Hydroxylases
N. A. Smirnova1, D. M. Hushpulian2, R. E. Speer1, I. N. Gaisina3, R. R. Ratan1, and I. G. Gazaryan1*
1Burke Medical Research Institute, 785 Mamaroneck Ave, White Plains NY 10605, USA; fax: (914) 597-2225; E-mail: nsmirnova@burke.org; respeer@gmail.com; rratan@burke.org; igazarya@burke.org2Department of Chemical Enzymology, Lomonosov Moscow State University, 119992 Moscow, Russia; fax: (495) 939-3208; E-mail: hushpulian@gmail.com
3Department of Medicinal Chemistry and Pharmacognosy, University of Illinois at Chicago, 833 South Wood Street, Chicago IL 60612, USA; fax: (312) 996-7107; E-mail: igaysina@uic.edu
* To whom correspondence should be addressed.
Received April 28, 2012
This review describes the catalytic mechanism, substrate specificity, and structural peculiarities of alpha-ketoglutarate dependent nonheme iron dioxygenases catalyzing prolyl hydroxylation of hypoxia-inducible factor (HIF). Distinct localization and regulation of three isoforms of HIF prolyl hydroxylases suggest their different roles in cells. The recent identification of novel substrates other than HIF, namely β2-adrenergic receptor and the large subunit of RNA polymerase II, places these enzymes in the focus of drug development efforts aimed at development of isoform-specific inhibitors. The challenges and prospects of designing isoform-specific inhibitors are discussed.
KEY WORDS: hypoxia-inducible factor, alpha-ketoglutarate-dependent iron dioxygenase, crystal structure, computer modeling, inhibitor binding modeDOI: 10.1134/S0006297912100033
Abbreviations: β2AR, β2-adrenergic receptor; bHLH, basic helix–loop–helix structural motif; CBP, CREB binding protein; DHB, dihydroxybenzoate; DMOG, dimethyloxalylglycine; FDA, Food & Drug Administration; FIH, factor inhibiting HIF; FRET, fluorescence resonance energy transfer; HIF, hypoxia-inducible factor; MIF, macrophage migration inhibitory factor; NTAD, N-terminal transactivation domain; ODD, oxygen degradable domain; PAS, a protein domain named after three proteins in which it occurs (Per, period circadian protein; Arnt, aryl hydrocarbon receptor nuclear translocator protein; Sim, single-minded protein); PHD, prolyl hydroxylase domain; TPEN, N,N,N′,N′-tetra(2-pyridyl)-1,2-ethanediamine; VHL, von Hippel–Lindau protein.
The iron chelator deferoxamine (DFO), an FDA approved drug for iron and
heavy metal overload, and some other iron chelators, are in human
trials for intracerebral hemorrhage, spinal cord injury, and
Alzheimer’s disease. Iron chelators have been shown to improve
functional recovery in animal models of hemorrhagic stroke [1, 2]. The rationale behind the
use of iron chelators in hemorrhagic stroke is easy to conceptualize:
bleeding into the parenchyma of the brain not only compresses
surrounding tissue to limit blood flow, but also results in the
breakdown of red blood cells leading to release of hemoglobin and its
cofactor, hemin [3, 4]. Hemin
exhibits significant peroxidase-like activity, and upon degradation by
heme oxygenase liberates biliverdin, iron, and carbon monoxide. Free
iron is believed to catalyze generation of hydroxyl radicals from
hydrogen peroxide via the Fenton reaction. Hydroxyl radicals covalently
modify biomolecules at diffusion-controlled rates, progressively
oxidizing cellular constituents and causing cell death [5]. However, for neurons cultured in vitro
hemin was shown to be more toxic than free iron [6]. Despite their movement forward to human testing,
two issues could potentially hinder the success of iron chelators at
the human bedside. First, iron is an important cofactor in many
proteins, particularly those in mitochondria, an organelle critical to
optimal synaptic activity and brain function [7, 8]. Second, iron chelators inhibit all iron-containing
proteins, and in the absence of a known target, appropriate dosing in
animals and ultimately humans may be problematic [9].
Iron chelators are equally protective in ischemic models. Neuroprotection in MCAO (Middle Cerebral Artery Occlusion) models was observed with the iron chelator DFO pre- and post-treatment [10, 11] and with α-ketoglutarate mimics not chelating iron such as dihydroxybenzoate (DHB) [12] and dimethyloxalylglycine (DMOG) [13]. Iron chelators and α-ketoglutarate mimics (DHB, DMOG) target iron-containing alpha-ketoglutarate dependent dioxygenases (more than 70 enzymes in the human genome). Among those, a group of prolyl hydroxylases (PHDs) catalyzes hydroxylation of hypoxia-inducible factor (HIF), a transcription factor responsible for cellular adaptation to hypoxia.
HIF is a transcriptional factor that regulates gene expression in mammalian development, physiology, and disease pathogenesis. HIF is a widespread transcription factor activating a battery of genes including those involved in glucose uptake and metabolism, extracellular pH control, angiogenesis, erythropoiesis, and mitogenesis acting to enhance the ability of cells to survive. HIF consists of two subunits, HIF-α being rapidly degraded under normoxic conditions (half-life less than 5 min at 21% O2), while the other subunit (β) is stable [14, 15]. HIF-α was a previously unidentified protein (in contrast to HIF-β, also known as aryl hydrocarbon nuclear translocator, ARNT), and its cDNA reveals 90% amino acid sequence identity for the human, mouse, and rat proteins.
In mammals, three genes have been shown to encode HIF-α subunits [16]: HIF-1α is ubiquitously expressed, while HIF-2α, also known as endothelial PAS-domain protein 1, and HIF-3α have more restricted expression patterns (Scheme 1). Nonetheless, HIF-2α is frequently detected in tumor cells along with HIF-1α. The levels of the α-subunits are acutely regulated in response to hypoxia. Although there is evidence for hypoxic induction of HIF-1α mRNA levels in some cell types, the predominant O2-dependent regulation of HIF-1α is mediated by posttranslational mechanisms. HIF-1α bound to HIF-1β represents the canonical form of HIF. Systemic disruption of the Hif-1a gene leads to embryonic lethality by day 11 of embryonic development (E11), which is accompanied by cardiovascular malformation and defective cephalic vascularization, indicating that HIF-1α is essential for embryonic vascularization. To investigate the function of HIF-1α in the central nervous system, a conditional knockout mouse was made: neural cell-specific HIF-1α-deficient mice exhibit hydrocephalus accompanied by a reduction in neural cells and an impairment of spatial memory, showing that expression of HIF-1α in neural cells is essential for normal development of the brain [17]. HIF-2α is essential for hematopoietic development in mice [18].
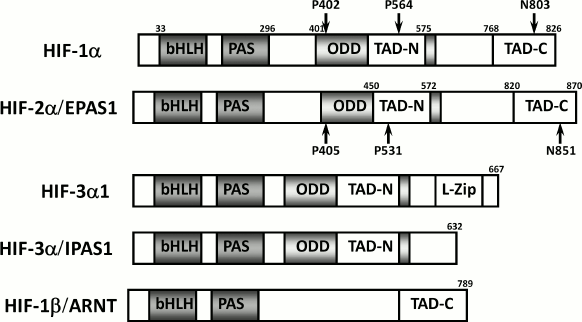
Comparative protein structure of HIF-α isoforms
Scheme 1
Initially, activation of HIF-1 was considered to be a major survival mechanism. However, several publications where HIF-1 has been upregulated by means other than iron chelators or α-ketoglutarate mimics have demonstrated pro-death functions of HIF-1 [19-22]. Very recently a two-phase HIF-1 response has been observed where HIF-1 stabilized by ischemia at early and late times lead to cellular apoptosis and survival, respectively [23]. In some scenarios of neuronal death HIF-2, but not HIF-1, was shown to be pro-survival [24]. Recently other protein substrates of HIF PHDs, such as β2-adrenergic receptor (β2AR) [25] and Rbp1 [26], have been identified. In the mid 1990s, Dr. Ratan’s laboratory outlined a model for the broad neuroprotective mechanism of iron chelators based on their ability to upregulate the hypoxia-inducible factor-1 (HIF-1) [27], rather than by inhibiting Fenton chemistry. The new model for neuroprotection has evolved by switching the focus from activating HIF-1 to inhibiting HIF prolyl hydroxylases (HIF PHDs) [28-30]. This review attempts to refine our understanding of the catalytic and structural features of HIF PHD isoforms with emphasis on the potential to design low molecular weight inhibitors specific for each isoform.
HYDROXYLATION: A MAJOR REGULATOR OF HIF PROTEIN STABILITY
The predominant O2-dependent regulation of HIF-1α is mediated by posttranslational mechanisms such as phosphorylation, acetylation, and hydroxylation followed by ubiquitination and proteasomal degradation. Hydroxylation is the major regulator and is catalyzed by nonheme iron α-ketoglutarate-dependent dioxygenases known as the HIF prolyl hydroxylases (HIF PHDs). Hydroxylation of Pro564 and/or 402 residues in HIF-1α is a prerequisite for the interaction with the tumor suppressor von Hippel–Lindau (VHL) protein, an E3 ubiquitin ligase that allows HIF-1α to be ubiquitinated and targeted for proteasomal degradation [31]. Pro564 is located within the oxygen degradable domain (ODD) and is considered to be the major site for hydroxylation catalyzed by all PHD isoforms. Hydroxylation of Pro564 occurs before that of Pro402 [32].
Hydroxylation of HIF-1α Asn803 is catalyzed by another enzyme of the same group, the factor inhibiting HIF-1α (FIH). FIH is an asparaginyl hydroxylase. Hydroxylation of HIF-α proteins by FIH blocks association of HIFs with the transcriptional coactivators CBP/p300 [33], thus inhibiting transcriptional activation. Mice with a null mutation in the FIH gene [34] show that FIH has no discernable role in mice in altering classical aspects of HIF function, e.g. angiogenesis, erythropoiesis, or development. Rather, it is an essential regulator of metabolism: mice lacking FIH exhibit reduced body weight, elevated metabolic rate, hyperventilation, and improved glucose and lipid homeostasis and are resistant to high-fat-diet-induced weight gain and hepatic steatosis. Neuron-specific loss of FIH phenocopied some of the major metabolic phenotypes of the global null animals: those mice have reduced body weight, increased metabolic rate, and enhanced insulin sensitivity and are also protected against high-fat-diet-induced weight gain. These results demonstrate that FIH acts to a significant degree through the nervous system to regulate metabolism.
While the two closely related HIF-α subunits HIF-1α and HIF-2α have been extensively studied, not much is known about the third variant, HIF-3α [35]. The HIF-1α subunit was characterized in 1995, HIF-2α in 1997, but HIF-3α was first discovered in the mouse only in 1998, and the identification of the human HIF-3α occurred in 2001 [36]. The human HIF-3α1 amino acid sequence is very similar to human HIF-1α and HIF-2α in the N-terminal bHLH region and the PAS domain, but it lacks the structures for transactivation found in the C-terminus of HIF-1α and HIF-2α and contains only the NTAD [36] (Scheme 1). It thus cannot bind p300 and is not regulated by FIH.
CATALYTIC MECHANISM
For both HIF PHDs and FIH, the general reaction mechanism includes activation of molecular oxygen to convert α-ketoglutarate into succinate and CO2 while hydroxylating the prime substrate, HIF-α subunit. The proposed mechanism (Scheme 2, adapted from [37]) is based on the reported incorporation of 18O into HIF-α substrate and succinate [38, 39], the available crystal structures for PHD2 [40] and FIH [41], and the recent stopped-flow kinetics for taurine/α-ketoglutarate dioxygenase [42] and prolyl-4-hydroxylase from Paramecium bursaria Chlorella virus 1 [43]. The mechanism suggests the initial binding of iron to the active site, then α-ketoglutarate coordination via C-1 carboxylate and ketone oxygen by iron, followed by the binding of HIF-α peptide (as a substrate), which results in displacement of the water molecule from the sixth coordination position (Step 1). This displacement guarantees oxygen binding and activation. The uncoordinated oxygen of the bound oxygen attacks the ketone carbonyl of α-ketoglutarate (Step 2) to form a bicyclic Fe(IV)-peroxyhemiketal complex (Step 3) in which decarboxylation occurs concomitantly with formation of an oxo-ferryl (Fe(IV)=O) intermediate (Step 4). The latter hydroxylates proline via a substrate radical intermediate (Step 5) as evidenced by the formation of prolyl radical [44]: i.e. oxo-ferryl attacks the proline residue to withdraw a hydrogen atom and then introduces the hydroxyl radical (Step 6).
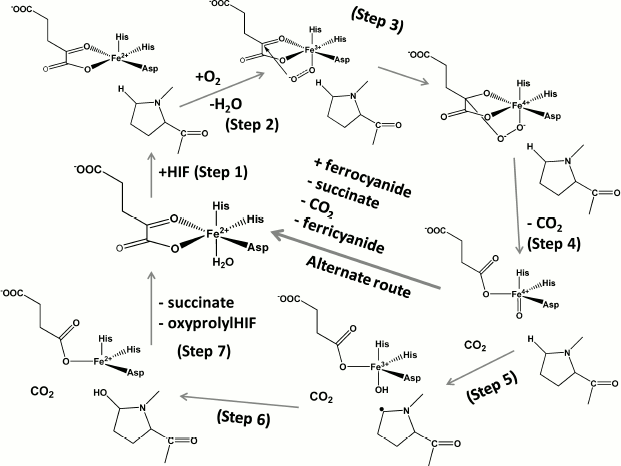
Catalytic cycle of HIF prolyl hydroxylase (Steps 1-6, see explanation in
text) and side-reaction of ferrocyanide oxidation catalyzed by HIF
prolyl hydroxylase in the presence of its specific substrate HIF (or
HIF peptide)
Scheme 2
The presence of the water molecule in the sixth coordination position is marked in the resolved crystal structures of PHD2 [40] in complex with the enzyme inhibitors (PDB 2G19, 2HBU), while the absence of this water molecule is clearly seen in the deposited crystal structure of FIH in complex with Asn803 containing HIF peptide (PDB 1H2K, 1H2M). These crystal structures provide direct evidence for the role played by the coordinated water in the catalytic mechanism (Fig. 1 (see color insert) and Scheme 2). The presence of the water molecule in the sixth position does not prevent oxygen from binding and activation completely: in the absence of the substrate the enzymes of this class are known to catalyze the so-called uncoupled reaction. However, the rate of the reaction is rather slow as one may judge from the very recent stopped-flow kinetics for prolyl-4-hydroxylase [43]: α-ketoglutarate binding to the active site Fe elicits a visible absorption feature in the 500 nm region assigned to a metal-to-ligand charge-transfer transition arising from Fe chelation; the decay of this band upon addition of oxygen in the absence of peptide substrate is negligible at 5°C compared to the loss of absorbance within 0.1 sec upon the addition of both oxygen and peptide [43]. This “substrate triggering”, which is thought to be due, at least in part, to dissociation of a water ligand seems to be a general feature of enzymes in this family. For instance, for taurine/α-ketoglutarate dioxygenase, binding of taurine enhances O2 reactivity by three orders of magnitude [42]. Thus, substrate analogs that bind in a similar fashion and displace the water but cannot be hydroxylated or oxidized will also result in an open coordination position, thus facilitating the uncoupled reaction. In the absence of reducing agents and in the presence of ferrocyanide as an alternative substrate, the enzyme catalyzes the oxidation of the latter using HIF (or HIF peptide) as a cofactor, the binding of which activates the active site iron as shown in Scheme 2, alternate route. This is the simplest continuous HIF prolyl hydroxylase activity assay as exemplified in Fig. 2 for recombinant PHD2 produced from E. coli inclusion bodies. The problem with the assay is the need to remove dithiothreitol, which results in fast inactivation of the enzyme upon storage (80% inactivation on ice within 2 h). The assay is good for enzyme purification protocol development, since the enzyme activity in this reaction is proportional to the enzyme hydroxylation activity. However, development of PHD inhibitors bearing an iron chelation motif is impossible using this assay.
Fig. 1. Displacement of active site water (left, PHD2 active center, PDB 2G19) upon binding of HIF peptide (right, modeling using FIH-HIF peptide structure, PDB 1H2K).
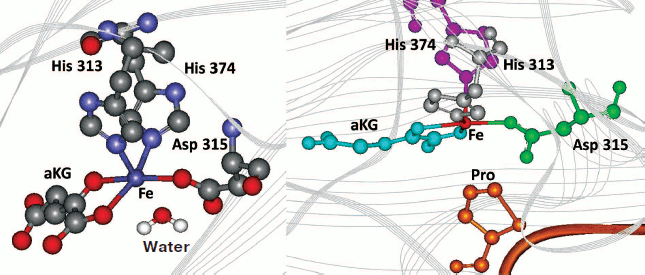
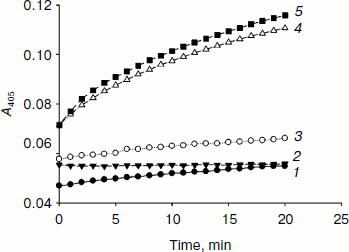
Fig. 2. Ferrocyanide oxidation catalyzed by HIF prolyl hydroxylase in the presence of HIF. Assay conditions: 50 µg/ml PHD2, 50 µM peptide substrate, 100 µM α-ketoglutarate, 8 µM FeSO4, 1 mM K4Fe(CN)6, and 50 mM Tris-HCl buffer, pH 7.5 at 25°C. The reaction was carried out at 37°C. 1) Control (peptide); 2) control (PHD2 no substrate); 3) control (GST-HIF); 4) PHD2 (peptide); 5) PHD2 (GST-HIF).
THREE ISOFORMS OF HIF PROLYL HYDROXYLASE: LOCALIZATION AND REGULATION
PHDs 1, 2, and 3 (also known as EGLN 2, 1, and 3, respectively) in humans are represented by three isozymes. The catalytic domains are highly homologous to each other. PHD3 contains the catalytic domain only, in contrast to PHD1 and PHD2 that have additional N-terminal domains. The role of the N-terminal domain of PHD1 is unknown, whereas the N-terminal domain of PHD2 contains a Zn-finger motif [45]. Zinc binding was shown to inhibit the enzyme activity in vitro [45], while the addition of the Zn chelator TPEN stimulated the enzyme activity as compared to the activity of PHD3 [46]. Since TPEN is also known to inhibit ubiquitination, its addition to human liver cells induced the accumulation of nonfunctional hydroxylated HIF-1α: HIF-1α was asparaginyl-hydroxylated and therefore failed to recruit cAMP-response element-binding protein (CBP) [46]. Thus, HIF-1α is regulated by two separate processes, namely PHD2/VHL/ubiquitin-dependent degradation and FIH-1/CBP-dependent transactivation.
PHD1 from C. elegans gives two expression bands in different cell cultures that correspond to the full-size protein and an alternative initiation variant (AUG encodes the 34th amino acid residue and is an alternative initiation site) [47]. These alternative forms exhibit similar catalytic activities, but the shorter variant is less stable with respect to proteolytic degradation [47].
PHD3, in addition to the full-size product, was shown to form a spliced variant omitting exon 4 (corresponding to PHD2 amino acid sequence numbers Tyr384-Arg408) that exhibited the catalytic activity and was found in primary cancer tissues [48]. This is in contrast to PHD2, where alternative splicing protein variants were shown to be catalytically inactive [49].
Human PHD1 is predominantly localized in the nucleus, whereas PHD2 is present in the cytoplasm. Human PHD3 distributes evenly in both compartments. The expression of PHD2 and PHD3, but not PHD1, is induced by hypoxia, suggesting a role for these enzymes in a negative feedback pathway responsible for enhanced degradation of HIF-1α after re-oxygenation. On the other hand, the half-life of PHD1 and PHD3 is regulated by O2 concentration, i.e. their stability is decreased in hypoxic cells [50]. These enzymes are also degraded by the ubiquitin-proteasome system [50].
The silencing of PHD1 and PHD3 using RNA interference in various cell lines revealed no effect on HIF-1α accumulation, in contrast to PHD2, which was identified as a key oxygen sensor in normoxia [51]. These differences in enzyme properties imply distinct functions and physiological impact for all three PHDs that await elucidation. In addition to its role in HIF hydroxylation, PHD3 has been recently shown to induce neuronal apoptosis downstream of c-Jun [52]. This ability is linked to PHD3 hydroxylase activity, which can be feedback inhibited by succinate [52].
ENZYME ASSAYS AND CATALYTIC PARAMETERS
The distinct localization and regulation of HIF prolyl hydroxylases strongly suggest their distinct substrate specificity and distinct roles in the cell. There is no consensus on relative activity of PHD isoforms with respect to each HIF isoform as a result of the use of the enzyme in in vitro assays. There are three different ways to assay PHD activity in vitro [53]: 1) assay of the first half-reaction substrates and products (O2 and α-ketoglutarate consumption; CO2 evolution, succinate production); this format should account for the uncoupled reaction, which is nonspecific; 2) assay of the second half-reaction oxidized product (mass-spectrometry of hydroxylated peptides); mass-spectrometric analysis is difficult to make suitable for high-throughput screening (HTS) format; 3) the so-called “capture assay” monitoring the interaction of the hydroxylated product with VHL; requires recombinant HIF, reticulocyte-produced VHL, protein/peptide labeling and corresponding antibodies.
The most reliable way to monitor the formation of hydroxylated product is either by mass spectrometry or using the “capture assay”. The latter is known in three different formats: 1) end-point immunochemical assay is based on the tight interaction between pVHL and the hydroxylated Pro564 of HIF-1α. After hydroxylation, immobilized HIF-peptide is recognized by the thioredoxin-labeled VHL–elongin B–elongin C complex (expressed in E. coli and purified), which is in turn detected by anti-thioredoxin antibodies by the method of double antibodies with peroxidase-labeled secondary antibodies; 2) continuous fluorescence polarization assay: HIF peptide modified with a fluorescent label upon capture by VHL–elongin B–elongin C shows higher polarization signal [54]; 3) continuous homogeneous time-resolved fluorescence resonance energy transfer (TR-FRET) assay: europium-labeled heterotrimeric complex of Von Hippel–Lindau protein, elongin B, and elongin C (VCB-Eu) recognizes hydroxylated P564 on a biotinylated HIF-1α peptide, which interacts with streptavidin-labeled allophycocyanin. This assay, developed by the Amgen team, is the best one in terms of sensitivity and applicability for HTS. The assay utilizes europium in addition to other expensive reagents [55].
All enzymatic assays are based on the use of PHD2 recombinant enzyme produced in either baculovirus or an E. coli expression system. High throughput screening for PHD inhibitors using an enzyme assay is a challenge both in terms of the enzyme source and the assay format. The enzymatic activity and stability of purified PHD is very low [56], and enzyme assays require large quantities of recombinant enzyme supplemented with iron. The binding constant for iron is not established, but it is supposed to be extremely good under in vitro conditions.
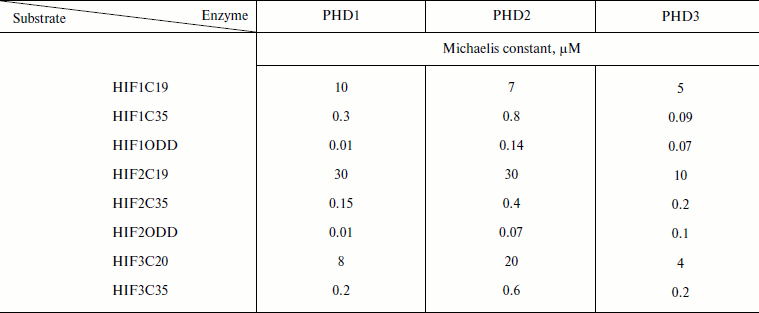
No specific peptide substrate has been developed for each PHD: the 19-mer HIF-1α peptide is an equally good substrate for all PHDs (Km 7-8 µM) [49]; PHD2 is 2-fold less specific for HIF-3α peptide than the others, and PHD3 exhibits a 3-fold higher affinity for HIF-2α peptide than the others [49]. A switch to longer peptide substrates, 35-mers, allowed the affinity for the peptide substrate to be improved by orders of magnitude (Table 1), while the values of the maximum rates were changed insignificantly [57]. With the oxygen degradable domain as a substrate, the decrease in the Michaelis constants was even more pronounced (Table 1 [49, 57]) as compared to the 4-15 µM range for 19-mer peptide substrates [57]. Based on the numbers presented in Table 1, it is still difficult to make conclusions regarding substrate specificity of either enzyme, except that PHD1 has a definite advantage over the other two in terms of high affinity for HIF-ODD.
Table 1. Improvement in Michaelis constants
(µM) with increasing peptide length (adapted from [49])
The high values of the Michaelis constant for oxygen in the case of PHD (Table 2 [37, 58]) show that oxygen actually regulates the enzyme activity, especially under in vivo conditions, where oxygen tension is an order of magnitude lower than that in vitro (ca. 220 µM, pH 7.0, RT).
Table 2. Kinetic parameters of PHD for
oxygen and ketoglutarate (adapted from [37, 58])
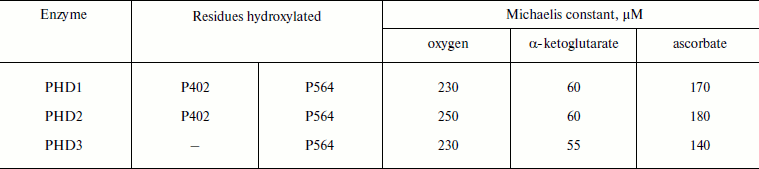
The co-purification of α-ketoglutarate with recombinant PHD2 [59] indicates that the binding constant for α-ketoglutarate must be much better than the Michaelis constants reported elsewhere (Table 2, ca. 60 µM). Ascorbate is not an obligatory substrate, and it is actually used in vitro to help iron to complete the cycle and return into the catalytically active ferro-form.
RESOLUTION OF HIF PHD2 CRYSTAL STRUCTURE
The crystal structure of the catalytic domain of PHD2 was resolved by two independent groups (PDB 2G19 [40]; PDB 2HBT & 2HBU resolved and deposited by Evdokimov et al.). PHD2 crystallizes as a homotrimer, although it exists as a monomer in solution [40]. The active site comprises a relatively deep pocket compared to other α-ketoglutarate-dependent oxygenases. Iron is coordinated in an octahedral manner by His313, His374, and Asp315, an inhibitor (occupying two sites) and a water molecule. The active site is predominantly lined with hydrophobic residues, with Ile256 and Trp258 leading to the opening of the active site. The hydrophobic nature of residues at the active site may reflect a requirement for enzyme protection from potential oxidation by active oxygen species generated by the active site iron in the absence of the protein substrate. Participation of Arg383 in α-ketoglutarate binding is clearly seen from the inhibitor binding in the crystal structure [40] and thus supports previous mutagenesis work showing the complete loss of catalytic activity in the Arg367Ala (equivalent of Arg383 in PHD2) mutation of PHD1 [60]. The narrow active site opening in PHD2 compared to FIH and other human and bacterial enzymes of this class may have significance for PHD isozymes as sensors: it may explain the fact of tight binding of iron and αKG in the enzyme active site and thus explain the enzyme purification with the cofactors bound [61]. There is significant difference in amino acid residues coordinating α-ketoglutarate in PHD2 versus FIH, which guarantees the possible development of specific inhibitors for either one. As we noted in our earlier paper [62], a PHD2 inhibitor should slide into the active site, and this imposes specific restrictions on its structure, while the FIH inhibitor accesses the active site from the side of the bound active site water perpendicular to the plane of bound ketoglutarate. The similarity in the structure of catalytic domains of PHD1-3 and identical α-ketoglutarate binding residues (according to homology modeling, see Fig. 3) makes the task of developing PHD isoform-specific inhibitors based on αKG analogs unlikely to succeed.
Fig. 3. Alignment of amino acid sequences of catalytic domains of HIF prolyl hydroxylases based on structural modeling of PHD1 and PHD3 with PHD2 as a template.

HIF PHD SUBSTRATE SPECIFICITY PROBLEM
At the level of HIF–PHD molecular interactions, the question of specificity of proline hydroxylation can be divided into two aspects: 1) what is the location of HIF amino acid residues recognized by PHD?; 2) what is the location of the HIF substrate recognition site within PHD?
PHDs were long supposed to recognize and hydroxylate proline residues found in the conserved sequence LXXLAP without additional requirements for flanking regions. Both prolines (564 and 402) satisfy this requirement. Only very recently, by means of yeast two-hybrid screening, a Spanish group mapped permitted replacements ranging from –6 to +10 outside the LXXLAP sequence that were relevant to recognition preferences by PHD2 and PHD3 (Scheme 3). Those authors concluded that PHDs are sensitive to the region –9 to +12 around the target proline and that PHDs exhibit low activity towards peptides shorter than 20 amino acids (based on the absence of stop-codons at any positions of the interacting constructs) [63]. Close examination of the data show that despite similar substrate requirements of PHD2 and PHD3, there are significant differences in their substrate preferences. The major difference is in the preference of PHD2 for Tyr or Phe in the +1 position (i.e. LXXLAPY): PHD2 does not bind the mutant version with Tyr565 replaced with amino acid residues other than Phe, while PHD3 binds a mutant version with Ala in this position [63]. On the other hand, PHD3 shows a strong preference for a Pro residue at position +3 (i.e. LAAPYIP), while PHD2 does not. This finding is consistent with the fact that PHD3, the isoform containing the catalytic domain only, is capable of hydroxylating Pro564 only, not Pro402 in HIF-1α. Additionally, PHD3 is extremely sensitive to the presence of negative charge at and around position +6 (D570) compared to PHD2. The authors conclude that these differences may be used to design inhibitors specific for each isoform.

Comparison of HIF1-3 sequences recognized by PHDs
Scheme 3
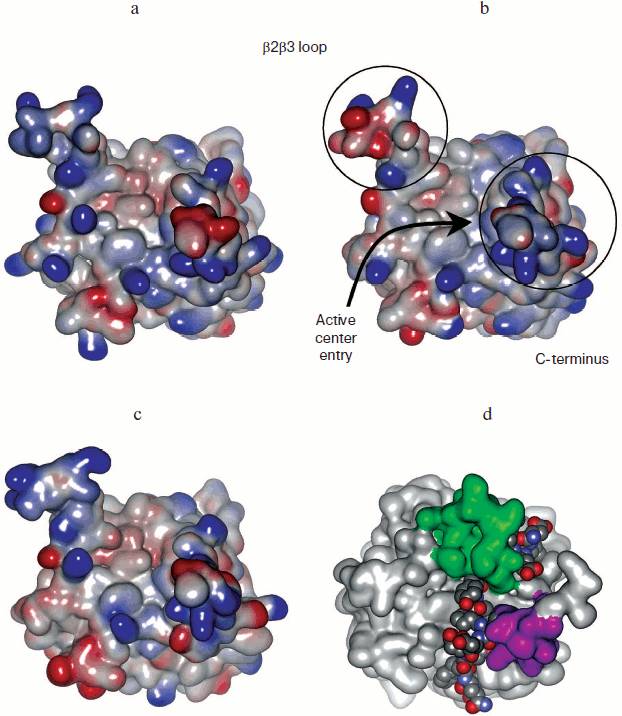
The yeast two-hybrid approach and regular mutagenesis work on purified PHDs confirmed that the PHD sequence responsible for substrate recognition is relatively far from the active site residues (Fig. 4; see color insert). The β2β3 loop has to interact with HIF close to the LXXLAP region to provide specific recognition. It is supposed to move down to hug the HIF sequence in the region of Asp570. However, in the crystal structure of the PHD2-HIF peptide [64] the β2β3 loop (green color in Fig. 4d) becomes disordered, although it actually does hug the peptide. The peptide goes from top to bottom interacting with the sequence of the C-terminal domain (Arg209, Tyr216) (pink in Fig. 4d). Computer models of PHD1 and PHD3 obtained by homology modeling using PHD2 as a template show the qualitative electrostatic surface potential, which is barely different for the PHD1/PHD3 pair, in contrast to PHD2/PHD1 or PHD2/PHD3 pairs (Fig. 4): in other words, design of inhibitor discriminating between PHD1 and PHD3 is problematic unless the N-terminal portion of PHD1 plays a role in PHD1 conformation and binding to the protein substrate. The so-called β2β3 loop has one and the same fold for all isoforms, except PHD3 has an arginine residue that pops up from the loop (shown in red in Scheme 4). The loop differs in charge for individual isoforms and truly participates in HIF peptide binding, thus giving hope for design of isoform-specific inhibitors.
Fig. 4. Qualitative electrostatic potentials of PHD1 computer model (a), PHD2 crystal structure (b), and PHD3 computer model (c): red and blue shades represent negative and positive potentials, respectively. d) Location of β2β3 loop (green) and C-terminal domain (pink) with respect to bound HIF peptide in the PHD2-HIF crystal structure (PDB 3HQR).

Variable portion of β2β3 loop sequences for PHDs
Scheme 4
NEW SUBSTRATES OF HIF PROLYL HYDROXYLASES
The data from the literature and that obtained in our laboratory unequivocally demonstrate that PHDs are important targets for medical intervention. The challenge is to develop inhibitors specific for each isoform, since very recently it became clear that the PHD isozymes have specific endogenous substrates.
HIF1 and HIF2 are established substrates for PHD2. PHD1 apparently is specific for Rpb1, the large subunit of RNA polymerase II, which carries the fundamental enzymatic activity of the complex synthesizing all cellular mRNAs [26]. Rpb1 is ubiquitinylated and degraded in response to DNA lesions induced by UV light and high millimolar concentrations of H2O2. Phosphorylation of Ser5 in Rpb1 is a prerequisite for ubiquitylation of Rpb1. It has been found that Pro1465 hydroxylation catalyzed by PHD1 is necessary for subsequent phosphorylation of Ser5 of Rpb1 in response to oxidative stress. PHD2, in contrast, has an inhibitory effect on this modification [26]. Recently, the Kaelin group [65] demonstrated a link between PHD1 and cyclin D1: PHD1 is estrogen-inducible in breast carcinoma cells, and PHD1 inactivation decreases cyclin D1 levels and suppresses mammary gland cell proliferation in vivo. Regulation of cyclin D1 is a specific attribute of PHD1 among the PHD proteins and is HIF-independent. Loss of PHD1 (but not PHD2) catalytic activity inhibits estrogen-dependent breast cancer tumorigenesis and can be rescued by exogenous cyclin D1. PHD1 depletion also impairs the fitness of lung, brain, and hematopoietic cancer lines. These findings support the exploration of PHD1 inhibitors as therapeutics for estrogen-dependent breast cancer and other malignancies. PHD1 appears to be an attractive drug target because it is not essential in mammals.
PHD3 was shown to accumulate with age in different tissues [66]. In a recent study evidence was provided that HIF-1 plays a critical role in delaying the onset of senescence in rodent cells via transcriptional activation of macrophage migration inhibitory factor (MIF) and thereby inhibition of the p53-mediated pathway [67]. PHD3 was found to form subcellular aggregates [68]. The most intriguing finding was that inhibition of PHD3 prevented it from forming aggregates [68]. Given that PHD3 expression is upregulated with aging, it may contribute to the reduced cell tolerance to hypoxia–reoxygenation and other pathological scenarios. It is worth mentioning again that PHD3 in mice has a mitochondria-targeting leader sequence and that PHD3 seems to be the most flexible PHD isoform regarding stimuli-induced changes in expression. PHD3 has been shown to hydroxylate β2-adrenergic receptor (β2AR) [25], the prototypic GPCR that plays an important role in the regulation of cardiovascular and pulmonary function, and sustained β2AR down-regulation and dysfunction is associated with diseases such as heart failure and asthma. In particular, β2AR enhances bronchodilation and alveolar fluid clearance (which increase O2 uptake), enhances cardiac output and peripheral vasodilation (which increase O2 delivery), and enhances cardioprotection and angiogenesis under ischemic conditions, thereby effectively recapitulating the integrated physiological response to hypoxia. Up-regulation of β2AR in response to hypoxia puts the function of the receptor in a new light. The ability of the PHD3-pVHL hydroxylation and ubiquitylation pathway to regulate the β2AR and the implications of that regulation for the response to ischemia and hypoxia suggest previously unidentified targets in the treatment of cardiovascular and respiratory diseases. PHD3 is most abundant in cardiac and smooth muscle, where the β2AR is highly abundant in vivo.
CONCLUSION: NOVEL APPROACH TO STUDY OF HIF PROLYL HYDROXYLASE
SUBSTRATE SPECIFICITY AND DESIGN OF ISOFORM-SPECIFIC INHIBITORS
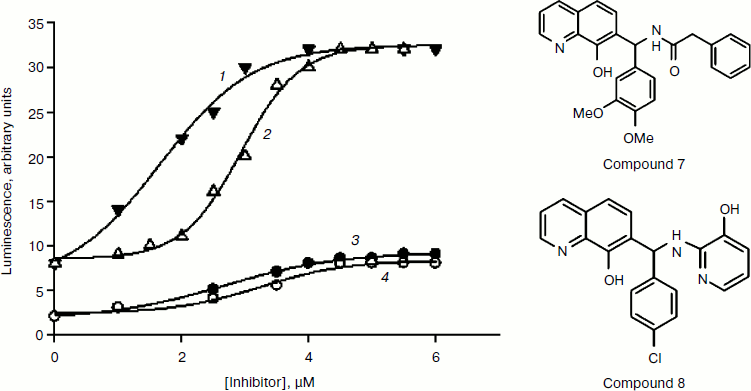
Taking into account the low specific activity of recombinant enzymes and the inadequacy of interpretation of the inhibition constant generated using different types of in vitro enzyme assays, we developed a novel approach to measuring PHD activity that is a variant of the cell-based “capture” assay [62]. The HIF-1 ODD-luc reporter system permits monitoring of consumption of a labeled substrate (ODD-luciferase) in real time. It is equally suited for substrate specificity studies (mutagenesis), determination of enzyme kinetic parameters, and high-throughput screening of multi-thousand and multi-million libraries of chemical compounds. The construction, performance, and advantages of this novel reporter system are described in detail in our recent open-access review [69]. It is worth noting that this system is sensitive to minor changes in the structure of the protein substrate because it is based on protein–protein recognition between the enzyme and substrate, which precedes catalysis and subsequent degradation of its hydroxylated product. Only due to this novelty, we successfully identified experimentally “branching tail” inhibitors specific to PHD only and holding promise for future development of isoform-specific PHD inhibitors. Docking of the best hit, compound 8, into the active site of PHD2 [62] closely resembles the fold of the 564PYIP567 sequence of the HIF peptide, in particular, the hydroxypyridyl ring of compound 8 occupies the position of the Tyr565 hydroxyphenyl ring. Compound 8 is now sold by Calbiochem as a specific HIF PHD inhibitor (ref. 400084, HIF Prolyl Hydroxylase Inhibitor, EMD Millipore USA) based on the results we published in [62]. In addition to this reporter cell line, we developed its mutant variants, which can be used to refine inhibition patterns. A mutant reporter provides better discrimination between PHD inhibitors:Fig.9B the advantage of the mutant PAIP line (Tyr565Ala HIF ODD-luc) is that it lowers the ability of HIF to compete with the inhibitors and permits better discrimination between the properties of inhibitors by providing higher activation numbers (Fig. 5): compounds 7 and 8 behave similarly in wild-type HIF 1 ODD-luc reporter assay, whereas in the mutant line, the IC50 for compound 8 becomes ca. 1 µM lower, i.e. compound 8 better competes with PAIP for PHD2, which is a proof of its specificity for PHD2.
We were the first to predict the possibility of developing isoform-specific PHD inhibitors employing variations in the branching motif adjacent to the iron-binding core [62]. Our conclusion on the role of the branched portion in recognition of different PHD isoforms has been experimentally confirmed in a recent publication from the Amgen group: the triple branching peptide-like tail attached to the quinolone core resulted in up to 10-fold difference in magnitude of inhibition between PHD2 and PHD1/PHD3 [70], but not between PHD1 and PHD3, which is a difficult task considering their surface potentials as we discussed above.Fig. 5. Activation of wild-type and PAIP mutant HIF ODD-luc reporter by compounds 7 and 8. 1) PAIP mutant and compound 8; 2) PAIP mutant and compound 7; 3) wild-type and compound 8; 4) wild-type and compound 7.
We are currently working on construction of HIF2, HIF3, and β2AR-based luciferase reporters, which will allow us to launch the screening program for PHD isoform-specific inhibitors. The novel approach we develop may principally yield inhibitors that target protein–protein interaction without entering into the active site of PHD: we expect that only compounds of this mode of action may discriminate between PHD1 and PHD3.
REFERENCES
1.Hua, R., and Walz, W. (2006) Crit. Rev.
Neurobiol., 18, 5-11.
2.Okauchi, M., Hua, Y., Keep, R. F., Morgenstern, L.
B., Schallert, T., and Xi, G. (2010) Stroke, 41,
375-382.
3.Regan, R. F., Chen, J., and Benvenisti-Zarom, L.
(2004) BMC Neurosci., 5, 34.
4.Dang, T. N., Bishop, G. M., Dringen, R., and
Robinson, S. R. (2011) Glia, 59, 1540-1550.
5.Robinson, S. R., Dang, T. N., Dringen, R., and
Bishop, G. M. (2009) Redox Rep., 14, 228-235.
6.Dang, T. N., Robinson, S. R., Dringen, R., and
Bishop, G. M. (2011) Neurochem. Int. (Epub ahead of
print).
7.Texel, S. J., Zhang, J., Camandola, S., Unger, E.
L., Taub, D. D., Koehler, R. C., Harris, Z. L., and Mattson, M. P.
(2011) PLoS One, 6, e25077;
doi:10.1371/journal.pone.0025077.
8.Yoon, H., Zhang, Y., Pain, J., Lyver, E. R.,
Lesuisse, E., Pain, D., and Dancis, A. (2011) Biochem. J.,
440, 137-146.
9.Fredenberg, J. P., Berglund, P. T., and Dieken, H.
A. (1996) J. Am. Diet. Assoc., 96, 64-65.
10.Mu, D., Chang, Y. S., Vexler, Z. S., and
Ferriero, D. M. (2005) Exp. Neurol., 195, 407-415.
11.Freret, T., Valable, S., Chazalviel, L.,
Saulnier, R., Mackenzie, E. T., Petit, E., Bernaudin, M., Boulouard,
M., and Schumann-Bard, P. (2006) Eur. J. Neurosci., 23,
1757-1765.
12.Baranova, O., Miranda, L. F., Pichiule, P.,
Dragatsis, I., Johnson, R. S., and Chavez, J. C. (2007) J.
Neurosci., 27, 6320-6332.
13.Nagel, S., Papadakis, M., Chen, R., Hoyte, L. C.,
Brooks, K. J., Gallichan, D., Sibson, N. R., Pugh, C., and Buchan, A.
M. (2011) J. Cereb. Blood Flow Metab., 31, 132-143.
14.Wang, G. L., Jiang, B. H., Rue, E. A., and
Semenza, G. L. (1995) Proc. Natl. Acad. Sci. USA, 92,
5510-5514.
15.Wang, G. L., and Semenza, G. L. (1995) J.
Biol. Chem., 270, 1230-1237.
16.Hirota, K., and Semenza, G. L. (2005) Biochem.
Biophys. Res. Commun., 338, 610-616.
17.Tomita, S., and Castillo, P. E. (2012) J.
Physiol. (Epub ahead of print).
18.Scortegagna, M., Morris, M. A., Oktay, Y.,
Bennett, M., and Garcia, J. A. (2003) Blood, 102,
1634-1640.
19.Aminova, L. R., Chavez, J. C., Lee, J., Ryu, H.,
Kung, A., Lamanna, J. C., and Ratan, R. R. (2005) J. Biol.
Chem., 280, 3996-4003.
20.Chen, C., Ostrowski, R. P., Zhou, C., Tang, J.,
and Zhang, J. H. (2011) J. Neurosci. Res., 88,
2046-2055.
21.Higashida, T., Kreipke, C. W., Rafols, J. A.,
Peng, C., Schafer, S., Schafer, P., Ding, J. Y., Dornbos, D., 3rd, Li,
X., Guthikonda, M., Rossi, N. F., and Ding, Y. (2011) J.
Neurosurg., 114, 92-101.
22.Higashida, T., Peng, C., Li, J., Dornbos, D.,
3rd, Teng, K., Li, X., Kinni, H., Guthikonda, M., and Ding, Y. (2011)
Curr. Neurovasc. Res., 8, 44-51.
23.Yeh, S. H., Ou, L. C., Gean, P. W., Hung, J. J.,
and Chang, W. C. (2011) Brain Pathol., 21, 249-262.
24.Lomb, D. J., Desouza, L. A., Franklin, J. L., and
Freeman, R. S. (2009) Mol. Pharmacol., 75, 1198-1209.
25.Xie, L., Xiao, K., Whalen, E. J., Forrester, M.
T., Freeman, R. S., Fong, G., Gygi, S. P., Lefkowitz, R. J., and
Stamler, J. S. (2009) Sci. Signal., 2, 1-10.
26.Mikhaylova, O., Ignacak, M. L., Barankiewicz, T.
J., Harbaugh, S. V., Yi, Y., Maxwell, P. H., Schneider, M., van Geyte,
K., Carmeliet, P., Revelo, M. P., Wyder, M., Greis, K. D., Meller, J.,
and Czyzyk-Krzeska, M. F. (2008) Mol. Cell Biol., 28,
2701-2717.
27.Ratan, R. R., Siddiq, A., Aminova, L., Lange, P.
S., Langley, B., Ayoub, I., Gensert, J., and Chavez, J. (2004)
Stroke, 35, 2687-2689.
28.Zaman, K., Ryu, H., Hall, D., O’Donovan,
K., Lin, K. I., Miller, M. P., Marquis, J. C., Baraban, J. M., Semenza,
G. L., and Ratan, R. R. (1999) J. Neurosci., 19,
9821-9830.
29.Siddiq, A., Aminova, L. R., Troy, C. M., Suh, K.,
Messer, Z., Semenza, G. L., and Ratan, R. R. (2009) J.
Neurosci., 29, 8828-8838.
30.Siddiq, A., Ayoub, I. A., Chavez, J. C., Aminova,
L., Shah, S., LaManna, J. C., Patton, S. M., Connor, J. R., Cherny, R.
A., Volitakis, I., Bush, A. I., Langsetmo, I., Seeley, T., Gunzler, V.,
and Ratan, R. R. (2005) J. Biol. Chem., 280,
41732-41743.
31.Kaelin, W. G., Jr. (2005) Biochem. Biophys.
Res. Commun., 338, 627-638.
32.Chan, D. A., Sutphin, P. D., Yen, S. E., and
Giaccia, A. J. (2005) Mol. Cell Biol., 25, 6415-6426.
33.Lando, D., Peet, D. J., Whelan, D. A., Gorman, J.
J., and Whitelaw, M. L. (2002) Science, 295, 858-861.
34.Zhang, N., Fu, Z., Linke, S., Chicher, J.,
Gorman, J. J., Visk, D., Haddad, G. G., Poellinger, L., Peet, D. J.,
Powell, F., and Johnson, R. S. (2012) Cell Metab., 11,
364-378.
35.Lendahl, U., Lee, K. L., Yang, H., and
Poellinger, L. (2009) Nat. Rev. Genet., 10, 821-832.
36.Hara, S., Hamada, J., Kobayashi, C., Kondo, Y.,
and Imura, N. (2001) Biochem. Biophys. Res. Commun., 287,
808-813.
37.Dann, C. E., 3rd, and Bruick, R. K. (2005)
Biochem. Biophys. Res. Commun., 338, 639-647.
38.Schofield, C. J., and Ratcliffe, P. J. (2005)
Biochem. Biophys. Res. Commun., 338, 617-626.
39.Welford, R. W., Kirkpatrick, J. M., McNeill, L.
A., Puri, M., Oldham, N. J., and Schofield, C. J. (2005) FEBS
Lett., 579, 5170-5174.
40.McDonough, M. A., Li, V., Flashman, E.,
Chowdhury, R., Mohr, C., Lienard, B. M., Zondlo, J., Oldham, N. J.,
Clifton, I. J., Lewis, J., McNeill, L. A., Kurzeja, R. J., Hewitson, K.
S., Yang, E., Jordan, S., Syed, R. S., and Schofield, C. J. (2006)
Proc. Natl. Acad. Sci. USA, 103, 9814-9819.
41.Elkins, J. M., Hewitson, K. S., McNeill, L. A.,
Seibel, J. F., Schlemminger, I., Pugh, C. W., Ratcliffe, P. J., and
Schofield, C. J. (2003) J. Biol. Chem., 278,
1802-1806.
42.Price, J. C., Barr, E. W., Hoffart, L. M., Krebs,
C., and Bollinger, J. M., Jr. (2005) Biochemistry, 44,
8138-8147.
43.Hoffart, L. M., Barr, E. W., Guyer, R. B.,
Bollinger, J. M., Jr., and Krebs, C. (2006) Proc. Natl. Acad. Sci.
USA, 103, 14738-14743.
44.Wu, M., Moon, H. S., Begley, T. P., Myllyharju,
J., and Kivirikko, K. I. (1999) J. Am. Chem. Soc., 121,
587-588.
45.Choi, K. O., Lee, T., Lee, N., Kim, J. H., Yang,
E. G., Yoon, J. M., Lee, T. G., and Park, H. (2005) Mol.
Pharmacol., 68, 1803-1809.
46.Choi, S. M., Choi, K. O., Lee, N., Oh, M., and
Park, H. (2006) Biochem. Biophys. Res. Commun., 343,
1002-1008.
47.Tian, Y. M., Mole, D. R., Ratcliffe, P. J., and
Gleadle, J. M. (2006) Biochem. J., 397, 179-186.
48.Cervera, A. M., Apostolova, N., Luna-Crespo, F.,
Sanjuan-Pla, A., Garcia-Bou, R., and McCreath, K. J. (2006) Cancer
Lett., 233, 131-138.
49.Hirsila, M., Koivunen, P., Gunzler, V.,
Kivirikko, K. I., and Myllyharju, J. (2003) J. Biol. Chem.,
278, 30772-30780.
50.Nakayama, K., Frew, I. J., Hagensen, M., Skals,
M., Habelhah, H., Bhoumik, A., Kadoya, T., Erdjument-Bromage, H.,
Tempst, P., Frappell, P. B., Bowtell, D. D., and Ronai, Z. (2004)
Cell, 117, 941-952.
51.Berra, E., Benizri, E., Ginouves, A., Volmat, V.,
Roux, D., and Pouyssegur, J. (2003) Embo J., 22,
4082-4090.
52.Lee, S., Nakamura, E., Yang, H., Wei, W., Linggi,
M. S., Sajan, M. P., Farese, R. V., Freeman, R. S., Carter, B. D.,
Kaelin, W. G., Jr., and Schlisio, S. (2005) Cancer Cell,
8, 155-167.
53.Hewitson, K. S., Schofield, C. J., and Ratcliffe,
P. J. (2007) Methods Enzymol., 435, 25-42.
54.Cho, H., Park, H., and Yang, E. G. (2005)
Biochem. Biophys. Res. Commun., 337, 275-280.
55.Dao, J. H., Kurzeja, R. J., Morachis, J. M.,
Veith, H., Lewis, J., Yu, V., Tegley, C. M., and Tagari, P. (2009)
Anal. Biochem., 384, 213-223.
56.Tuckerman, J. R., Zhao, Y., Hewitson, K. S.,
Tian, Y. M., Pugh, C. W., Ratcliffe, P. J., and Mole, D. R. (2004)
FEBS Lett., 576, 145-150.
57.Koivunen, P., Hirsila, M., Kivirikko, K. I., and
Myllyharju, J. (2006) J. Biol. Chem., 281,
28712-28720.
58.Ehrismann, D., Flashman, E., Genn, D. N.,
Mathioudakis, N., Hewitson, K. S., Ratcliffe, P. J., and Schofield, C.
J. (2007) Biochem. J., 401, 227-234.
59.McNeill, L. A., Flashman, E., Buck, M. R.,
Hewitson, K. S., Clifton, I. J., Jeschke, G., Claridge, T. D.,
Ehrismann, D., Oldham, N. J., and Schofield, C. J. (2005) Mol.
Biosyst., 1, 321-324.
60.McNeill, L. A., Hewitson, K. S., Claridge, T. D.,
Seibel, J. F., Horsfall, L. E., and Schofield, C. J. (2002) Biochem.
J., 367, 571-575.
61.Mole, D. R., Schlemminger, I., McNeill, L. A.,
Hewitson, K. S., Pugh, C. W., Ratcliffe, P. J., and Schofield, C. J.
(2003) Bioorg. Med. Chem. Lett., 13, 2677-2680.
62.Smirnova, N. A., Rakhman, I., Moroz, N., Basso,
M., Payappilly, J., Kazakov, S., Hernandez-Guzman, F., Gaisina, I. N.,
Kozikowski, A. P., Ratan, R. R., and Gazaryan, I. G. (2010) Chem.
Biol., 17, 380-391.
63.Landazuri, M. O., Vara-Vega, A., Viton, M.,
Cuevas, Y., and del Peso, L. (2006) Biochem. Biophys. Res.
Commun., 351, 313-320.
64.Chowdhury, R., McDonough, M. A., Mecinovic, J.,
Loenarz, C., Flashman, E., Hewitson, K. S., Domene, C., and Schofield,
C. J. (2009) Structure, 17, 981-989.
65.Zhang, Q., Gu, J., Li, L., Liu, J., Luo, B.,
Cheung, H. W., Boehm, J. S., Ni, M., Geisen, C., Root, D. E., Polyak,
K., Brown, M., Richardson, A. L., Hahn, W. C., Kaelin, W. G., Jr., and
Bommi-Reddy, A. (2009) Cancer Cell, 16, 413-424.
66.Rohrbach, S., Simm, A., Pregla, R., Franke, C.,
and Katschinski, D. M. (2005) Biogerontology, 6,
165-171.
67.Welford, S. M., Bedogni, B., Gradin, K.,
Poellinger, L., Broome Powell, M., and Giaccia, A. J. (2006) Genes
Dev., 20, 3366-3371.
68.Rantanen, K., Pursiheimo, J., Hogel, H., Himanen,
V., Metzen, E., and Jaakkola, P. M. (2008) Mol. Biol. Cell,
19, 2231-2240.
69.Smirnova, N. A., Hushpulian, D. M., Ratan, R. R.,
and Gazaryan, I. G. (2011) in Drug Discovery and Development –
Present and Future (Kapetanovic, I. M., ed.) ISBN
978-953-307-615-7, InTechOpen, pp. 295-322.
70.Murray, J. K., Balan, C., Allgeier, A. M.,
Kasparian, A., Viswanadhan, V., Wilde, C., Allen, J. R., Yoder, S. C.,
Biddlecome, G., Hungate, R. W., and Miranda, L. P. (2010) J. Comb.
Chem., 12, 676-686.