Engineering of Substrate Specificity of D-Amino Acid Oxidase from the Yeast Trigonopsis variabilis: Directed Mutagenesis of Phe258 Residue
N. V. Komarova1,2, I. V. Golubev1,3, S. V. Khoronenkova1,3, T. A. Chubar’1,2,3, and V. I. Tishkov1,2,3*
1Innovations and High Technologies MSU Ltd., ul. Tsimlyanskaya 16, room 96, 109559 Moscow, Russia; fax: (495) 939-32082Bach Institute of Biochemistry, Russian Academy of Sciences, Leninsky pr. 33-2, 117234 Moscow, Russia; fax: (495) 954-2732; E-mail: vitishkov@gmail.com
3Chemistry Faculty, Lomonosov Moscow State University, 119992 Moscow, Russia; fax: (495) 939-2742
* To whom correspondence should be addressed.
Received June 3, 2012; Revision received June 21, 2012
Natural D-amino acid oxidases (DAAO) are not suitable for selective determination of D-amino acids due to their broad substrate specificity profiles. Analysis of the 3D-structure of the DAAO enzyme from the yeast Trigonopsis variabilis (TvDAAO) revealed the Phe258 residue located at the surface of the protein globule to be in the entrance to the active site. The Phe258 residue was mutated to Ala, Ser, and Tyr residues. The mutant TvDAAOs with amino acid substitutions Phe258Ala, Phe258Ser, and Phe258Tyr were purified to homogeneity and their thermal stability and substrate specificity were studied. These substitutions resulted in either slight stabilization (Phe258Tyr) or destabilization (Phe258Ser) of the enzyme. The change in half-inactivation periods was less than twofold. However, these substitutions caused dramatic changes in substrate specificity. Increasing the side chain size with the Phe258Tyr substitution decreased the kinetic parameters with all the D-amino acids studied. For the two other substitutions, the substrate specificity profiles narrowed. The catalytic efficiency increased only for D-Tyr, D-Phe, and D-Leu, and for all other D-amino acids this parameter dramatically decreased. The improvement of catalytic efficiency with D-Tyr, D-Phe, and D-Leu for TvDAAO Phe258Ala was 3.66-, 11.7-, and 1.5-fold, and for TvDAAO Phe258Ser it was 1.7-, 4.75-, and 6.61-fold, respectively.
KEY WORDS: D-amino acid oxidase, site-directed mutagenesis, substrate specificity, thermal stabilityDOI: 10.1134/S0006297912100100
D-Amino acids in higher organisms are involved in the regulation of a variety of processes such as aging, functioning of the nervous system, hormone secretion, etc. For example, the presence of excess amounts of certain D-amino acids in tissues of the mouse brain enhances synaptic transmission between neurons and spatial learning [1]. Reducing the concentration of D-serine leads to a decrease in functional activity of N-methyl-D-aspartate receptors, which is one of the causes of schizophrenia [2, 3]. One of the key regulators of hormone secretion is D-aspartic acid, which is responsible for the level of such hormones as melatonin [4], prolactin [5], testosterone [6], luteinizing hormone, and growth hormone [7], etc. In Alzheimer’s disease, D-Ala content in the gray matter of the brain is about 2.2 times higher than in healthy individuals [8]. In addition, the cerebrospinal fluid of such patients contains elevated total amount of D-amino acids, mostly D-Asp and D-Ser [9]. Development of highly sensitive methods for rapid selective determination of D-amino acids in various tissues will create new methods for diagnosing neurodegenerative and other diseases.
The content of D-amino acids in the body is regulated by the FAD-containing enzyme D-amino acid oxidase (EC 1.4.3.3, DAAO). Due to the high stereospecificity of DAAO to D-amino acids, this enzyme can be used for the analysis of D-amino acids in various biological samples.
Determination of D-amino acids in a variety of samples is an important task for medical diagnostics and also for food and chemical industries. Natural L-amino acids contained in foods can be converted into the D-isomers during processing and storage. To assess the quality of food, the total content of D-amino acids or ratio of isomers D/(D + L) can be used. Such D-amino acids as D-Ala, D-Glu, D-Asp, D-Lys, D-Ser, D-Pro, D-Phe, D-Arg, and D-Leu have been found in different foods. D-Amino acids have been found in bread, coffee, vegetables, fruits, chocolate, wine, and dairy products. The particularly high content of D-amino acids in cheese (e.g. cheese varieties Parmigiano Reggiano and Grana Padano) can be distinguished from each other based on the content of D-Ala, D-Asp, and D-Glu, and for determination of their age the ratio of the isomers D/(D + L) can be used [10]). Thus, the determination of D-amino acids in foods is an important task in the field of food biotechnology.
The determination of enantiomeric composition of amino acids is usually performed using complex and labor intensive procedures such as liquid or gas chromatography or capillary electrophoresis in specific columns with a chiral phase [11]. For these purposes, a rather simple and rapid detection method utilizing biosensors based on D-amino acid oxidase can be used. The first biosensor for the determination of D-serine in brain tissues was developed in 2008 [12]. Now many research groups are developing biosensors based on DAAO.
A problem in the creation of biosensors based on DAAO is the wide range of substrate specificity of the natural enzymes, while the determination requires an enzyme that is highly specific to the target D-amino acids. In this case, the required specificity is determined by the type of analysis. For example, in early diagnosis of schizophrenia the main requirement is a high DAAO activity towards D-Ser, which is at least 10-fold higher than that towards D-Ala, and the absolute lack of DAAO activity towards D-Asp [12], i.e. for this analysis there is no need to use an enzyme that is specific to only one D-amino acid. Tuning of the substrate specificity of the enzyme can be achieved by the rational design approach.
Our laboratory previously cloned the gene of D-amino acid oxidase from the yeast Trigonopsis variabilis (TvDAAO) and developed a system of expression of the enzyme [13, 14]. In 2008, we solved the three-dimensional structure of one of the mutant forms of TvDAAO [15]. The enzyme is a dimer consisting of two identical subunits. In the dimer molecule, the subunits are linked by the second-order symmetry axis, i.e. contact between the subunits is of “head–tail” type. Each subunit has two domains – the catalytic and coenzyme-binding domain (Fig. 1). The coenzyme-binding domain contains an FAD molecule and is located in the lower part of the protein subunit. The catalytic domain is located in the upper part of the subunit. It is a large cavity with the FAD isoalloxazine ring located at the bottom, and the upper part of the cavity is limited by β-sheet. The entrance to the catalytic domain leading to amino acids of the active site is located in the anterior part of the globule (Fig. 1).
The knowledge of the three-dimensional structure of TvDAAO allows the use of the “rational design” approach for modification of the properties of the enzyme by site-directed mutagenesis. Introduction of a number of mutations have made directional changes in the spectrum of substrate specificity TvDAAO. For example, the replacement Cys108Phe or Cys108Ala led to an increase in the enzyme activity with cephalosporin C 3- and 4-fold, respectively [16, 17]. These mutant enzymes were also active towards D-Ser and were inactive towards D-Ala [16]. Site-directed mutagenesis of Arg169 and Arg220 residues yielded mutant TvDAAOs that were inactive towards D-Ser and active towards D-Ala [18]. Replacement of Phe54 residue by tyrosine or serine residues yielded mutants with improved catalytic efficiency towards D-amino acids with bulky side groups [19]. This work is a continuation of research on the role of the various residues in the structure of the enzyme and their effect on the catalytic properties.
MATERIALS AND METHODS
Materials. Molecular biology grade reagents were used for the genetic engineering experiments. In the microbial experiments, materials used were: bacto-tryptone, yeast extract, and agar (Difco, USA), glycerol (99.9%), calcium chloride (ultra pure), dipotassium phosphate, monosodium phosphate (pure for analysis), and lysozyme (Fluka/BioChemika, Switzerland); isopropyl-β-D-thiogalactopyranoside (IPTG), kanamycin, and chloramphenicol (Sigma, USA); and glucose and sodium chloride (pure for analysis) (Helicon, Russia). Restriction endonucleases, phage T4 DNA ligase, and Pfu DNA polymerase from Fermentas (Lithuania) were used for cloning of DNA fragments and site-directed mutagenesis. The DNA from agarose gels and plasmid from E. coli cells were isolated using reagent kits from Fermentas. Oligonucleotides for PCR and sequencing were synthesized by Syntol (Russia). MilliQ-purified water was used (Millipore, USA).
All reagents used for proteins electrophoresis were produced by BioRad (USA). For the purification and characterization of the enzyme, Tris (tris(hydroxymethyl)aminomethane, pure for analysis grade) from Merck (Germany), racemates of amino acids from Dia-M (Russia) and Reanal (Hungary), and horseradish peroxidase from BBY Enzymes (UK) were used.
Site-directed mutagenesis. Single-nucleotide substitutions were introduced using two-stage polymerase chain reaction (PCR) as described previously [19]. The plasmid pDAAO2 was used as the template, where the tvdaao gene was under the control of a strong promoter of phage T7 RNA polymerase [15]. To introduce the mutations, forward (DAO_For1) and reverse (T7_Rev) primers were used at the beginning and end of the gene, respectively, as well as forward and reverse (F258X_For and F258X_Rev, respectively, where X is Y, A, or S) primers carrying the desired replacement in the tvdaao gene. The sequences of the primers are shown below.
DAO_For5 ′-ATA TAC CAT GGC TAA AAT CGT TGT TAT TGG TGC-3′
T7_Rev5 ′-GCT AGT TAT TGC TCA GCG G-3′
F258Y_For5 ′-GGC GGT TGT TAC CAA CCC AAC AAC TGG TCA TC-3′
F258Y_Rev5 ′-GTT GTT GGG TTG GTA ACA ACC GCC AAT GAT AGA AGT AC-3′
F258A_For5 ′-GGC GGT TGT GCC CAA CCC AAC AAC TGG TCA TC-3′
F258A_Rev5 ′-GTT GTT GGG TTG GGC ACA ACC GCC AAT GAT AGA AGT AC-3′
F258S_For5 ′-GGC GGT TGT TCC CAA CCC AAC AAC TGG TCA TC-3′
F258S_Rev5 ′-GTT GTT GGG TTG GGA ACA ACC GCC AAT GAT AGA AGT AC-3′
The reaction mixture for PCR contained 2.5 µl 10× buffer for Pfu DNA polymerase (200 mM Tris-HCl (pH 8.8 at 25°C), 100 mM (NH4)2SO4, 100 mM KCl, 1 mg/ml BSA, 1% (v/v) Triton X-100, 20 mM MgSO4), 2.5 µl of a mixture of dNTP (2.5 mM each of dATP, dGTP, dTTP, and dCTP), 1 µl of DNA template (~10 ng/µl), 2 µl of primers (10 nmol/ml), 0.5 µl of Pfu DNA polymerase (2.5 U/µl), and deionized water to a total sample volume of 25 µl. PCR reactions were performed in 0.5-ml thin-walled plastic tube (SSI, USA) in the Tertsik device (DNA-Technology, Russia). To prevent evaporation of the reaction mixture, 30 µl of mineral oil was added to the PCR tube prior to the reaction. The tube was heated for 5 min at 95°C, and then the PCR reaction was initiated by the addition of the enzyme. The reaction was carried out according to the following program: first stage, 95°C for 30 s; second stage, 54-58°C for 60 s; third stage, 72°C for 2 min; 25-35 cycles total. After the last cycle, the reaction mixture was further kept for 10 min at 72°C. The temperature was chosen in the second stage to be 3-5°C below the melting temperature of duplexes (Tm) formed by the primers. The Tm was determined using the empirical formula:
Tm = 2(nA + nT) + 4(nG + nC),
where nX – the number of the nucleotides of the type X (X = A, T, C, G) in the primer.
To obtain fragments containing the desired substitution, two-step PCR using the primer pairs DAO_For/F258X_Rev (fragment 1) and F258X _For/T7_Rev (fragment 2) was performed. The PCR products, fragment 1 and fragment 2, were purified by electrophoresis in 1% agarose gel. Then a third combining PCR was performed with primers DAOFor1 and DAORev5, where fragments 1 and 2 were used as the DNA template. The product of the third PCR was purified as described above for fragments 1 and 2 and treated with restriction endonucleases NcoI and Bsp119I. Then the DNA fragments were purified by electrophoresis in 1% agarose gel followed by extraction from the gel and ligation to plasmid pDAAO2 treated with the same restriction endonucleases. After the ligation, the reaction mixture was used to transform E. coli Mach1 cells. The cells were then plated on Petri dishes with agar medium containing kanamycin (30 µg/ml) and incubated for 16 h at 37°C. For each mutant, three colonies were taken from a plate and used for plasmid isolation. To monitor the introduction of the desired mutations, the DNA was sequenced at the Center for Collective Use Genome (Engelhardt Institute of Molecular Biology, Russian Academy of Sciences).
Expression of mutant forms of TvDAAO in E. coli cells. Wild-type TvDAAO and its mutants were expressed in E. coli BL21(DE3) CodonPlus/pLysS cells. To obtain a producing strain, the cells were transformed with the appropriate plasmid and plated on Petri dishes with agar medium containing kanamycin (30 µg/ml). To prepare the inoculum, a single colony was taken from a plate and cultured for 16 h at 30°C in 10 ml of 2YT medium (bacto-tryptone, 16 g/liter; yeast extract, 10 g/liter; sodium chloride, 5 g/liter, pH 7.5) in the presence of 30 µg/ml kanamycin and 25 µg/ml chloramphenicol. The next morning, the cells were seeded into fresh medium (dilution 1 : 1000) and cultured at 30°C until A600 ≈ 0.6-0.8. Inoculum was introduced into a culture flask at 10% of the total volume of the medium (modified LB medium: yeast extract, 10 g/liter; bacto-tryptone, 5 g/liter; glucose, 5 g/liter; monosodium phosphate, 1.5 g/liter, and dipotassium phosphate, 1 g/liter, pH 7.5) containing 30 µg/ml kanamycin. Cultivation was carried out in 1-liter conical flasks (the volume of the medium was no more than 10-15% of the flask volume). The cultivation temperature and shaker rotation speed were 20°C and 120-160 rpm, respectively. After A600 reached ~0.7-0.8, expression of the enzyme was induced by adding IPTG to the medium to a final concentration of 0.1 mM. After the induction, the cells were cultured for an additional 24 h and then precipitated in an Eppendorf 5403 centrifuge (5 min, 5000 rpm, 4°C). The resulting pellet was resuspended in 0.02 M Tris-HCl, pH 8.0, at a ratio of 1 : 4 (by weight). The resulting suspension was frozen and stored at –20°C.
Isolation and purification of mutant enzymes. To isolate mutant TvDAAO, a 20% cell suspension in 20 mM Tris-HCl, pH 8.0, was twice freeze–thawed, and then the cells were sonicated in a Branson 250 Sonifier (Germany) under constant cooling. The precipitate was removed by centrifugation in an Eppendorf 5804 R centrifuge (11,000 rpm, 30 min).
Purification of the enzyme included ion-exchange chromatography on a MonoQ HR 10/10 column using an FPLC instrument from Biotech (Sweden) and desalting on Sephadex G-25 [20]. The purity of the resulting preparations was monitored by analytical electrophoresis in 12% polyacrylamide gel in the presence of 0.1% SDS using a MiniProtean III device (BioRad, Austria) according to the manufacturer’s instructions.
Measurement of activity and catalytic parameters. The DAAO–horseradish peroxidase two-enzyme system was used to determine the activity of D-amino acid oxidase. D-Methionine was used as a substrate for the first enzyme, and 2,2′-azino-bis(3-ethylbenzothiazoline-6-sulfonate) (ABTS) (Sigma, USA) was used as a substrate for the second enzyme. The activity was determined at 30°C by following the accumulation of ABTS oxidation product (absorbance at 414 nm, ε414 = 36,600 liter·mol–1·cm–1) using a Shimadzu UV-1601PC spectrophotometer (Japan). The spectrophotometer cuvette (working volume of 1 ml, optical path 1 cm) contained 870 µl of 50 mM potassium phosphate buffer (PBS), pH 8.0, previously saturated with air, 100 µl of 100 mM D-Met in 50 mM PBS, 20 µl of ABTS in water (16 mg/ml), and 10 µl of peroxidase in 50 mM PBS (5 mg/ml). After incubation of the cell for 10 min at 30°C, wild-type TvDAAO or one its mutants (30 µl) was added to the sample.
To determine the values of the maximum reaction rate and Michaelis constant, concentration of the corresponding D-amino acids ranged from 0.5 to 5 KM [16]. The apparent KM value was determined in a separate experiment by measuring the reaction rate at concentrations of the corresponding D-amino acids of 0.1, 0.5, 1, 5, 10, and 50 mM.
Kinetic parameters were calculated by nonlinear regression using the program OriginPro 8.5 SR1 (OriginLab).
Investigation of thermal stability. The temperature stability of the mutant TvDAAO and wild-type enzyme was studied in 0.1 M potassium phosphate buffer, pH 8.0. For each experiment, a series of 0.5-ml plastic tubes containing 100 µl of enzyme solution were used. The tubes were placed in a water bath pre-heated to the desired temperature (temperature control accuracy of ±0.1°C). At regular time intervals, tubes were taken, rapidly cooled for 1-2 min on ice, and the activity of the enzyme was measured as described above. The interval between sampling was adjusted so that during the experiment the enzyme activity decreased to 10-15% of the original value. To determine the inactivation rate constants, the residual activity was plotted against time, and the resulting dependence was treated by nonlinear regression using the OriginPro 8.5 SR1 program.
Computer simulation. The structures of the TvDAAO enzymes were analyzed using the Accelrys Discovery Studio 2.1 software package. The same software was used to obtain images of the protein globule.
RESULTS AND DISCUSSION
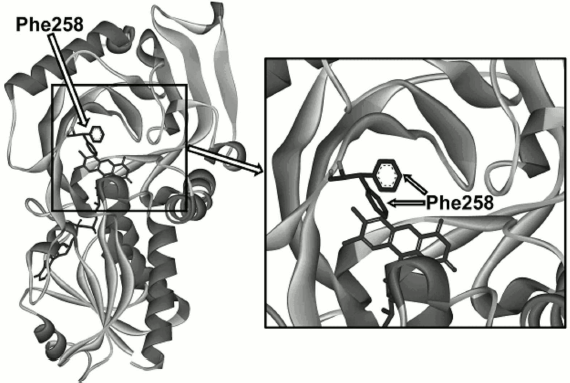
Analysis of the TvDAAO structure, search for positions for mutagenesis, and selection of amino acid substitutions. To alter the substrate specificity of the enzyme, it was decided to subject to site-directed mutagenesis one of the residues of TvDAAO that is located near the active site but is not directly involved in catalysis. Figure 1 shows residue Phe258 located at the entrance (left side) to the active site. This residue has high mobility since according to the X-ray data it adopts two conformations even in the crystal state (Fig. 1). Such positioning of the Phe258 residue suggests that it may influence the binding of the substrate in the active site of the enzyme, both due to steric factors and to hydrophobicity.
Fig. 1. Location of the Phe258 residue in the TvDAAO structure.
Comparison of 52 amino acid sequences of D-amino acid oxidases found in the databases and exhibiting the highest homology with the amino acid sequence of TvDAAO shows that the position corresponding to TvDAAO residue Phe258 is most frequently occupied by Phe (17 times out of 52). Also in this position, Tyr (11), Ala (8), Ser (4), His (3), Met (2), Glu (2), Asp (2), Gln (1), Asn (1) and Cys (1) were found. Thus, the total frequency of occurrence of residues in this position of Phe, Tyr, Ala, and Ser is 76%. Therefore, to clarify the role of residue Phe258 in the binding of substrates and to study its effect on the catalytic properties of the enzyme, three mutant forms of TvDAAO with amino acid substitutions Phe258Tyr, Phe258Ala, and Phe258Ser were prepared. Replacement of the Phe258 residue with serine or alanine can increase the size of the entrance to the active site, while its substitution with tyrosine, on the contrary, reduces the size of the entrance.
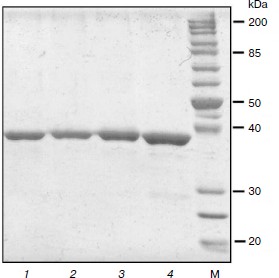
Preparation of TvDAAO mutants Phe258Tyr, Phe258Ala, and Phe258Ser. The nucleotide substitutions providing the desired mutations were introduced using polymerase chain reaction. For all three mutants, three plasmids were selected for sequencing. The results of sequencing showed that all the plasmids in the tvdaao gene were only the required mutations, i.e. other nucleotide substitutions were absent. Plasmids containing the gene replacement tvdaao Phe258Tyr, Phe258Ala, and Phe258Ser were transformed into E. coli strain BL21(DE3) Codon Plus/pLysS. The resulting recombinant strains were cultured for 24 h as described in Materials and Methods. Culturing revealed that all three mutant enzymes are synthesized in active and soluble form. The TvDAAO Phe258Tyr, Phe258Ala, and Phe258Ser mutants were purified to near homogeneity as evidenced by analytical polyacrylamide gel electrophoresis in the presence of SDS (Fig. 2, lanes 1-3).
Fig. 2. Analytical SDS-PAGE of preparations of mutant TvDAAO with substitutions Phe258Tyr, Phe258Ser, and Phe258Ala (lanes 1-3, respectively), and the wild-type enzyme (lane 4). M, protein molecular weight standards.
Temperature stability of TvDAAO with substitutions Phe258Tyr, Phe258Ala, and Phe258Ser. The temperature stability of the TvDAAO Phe258Tyr, Phe258Ala, and Phe258Ser mutants was studied in the temperature range of 52-60, 52-58, and 48-56°C, respectively.
Earlier studies of the wild-type TvDAAO enzyme thermostability showed that thermal inactivation proceeds via a two-stage dissociative mechanism: in the first stage dimers are dissociated into monomers, and in the second stage irreversible inactivation of the monomeric form of the enzyme occurs [15, 19]. The main criteria for thermal inactivation by this mechanism is the presence of an inflection point on the dependences of decreasing activity versus time in semilogarithmic coordinates and a decrease in the slope of the linear portion of the second part of the thermal inactivation curve by increasing the initial concentration of the enzyme [21]. The same dependences are characteristic of the two mutant forms of TvDAAO with amino acid substitutions Phe258Tyr and Phe258Ser. Figure 3a shows the dependence of the residual activity on time in semilogarithmic coordinates for the Phe258Tyr mutant; the dependence can be approximated by two straight lines with a characteristic breakpoint. Inactivation curves for TvDAAO Phe258Ser had similar form (data not shown). In the case of the Phe258Ala mutant, the dependence of the residual activity on time in semilogarithmic coordinates is linear and has no breakpoints (Fig. 3b). Thus, this amino acid substitution leads to a change in the mechanism of thermal inactivation.
Fig. 3. Kinetics of thermal inactivation of the TvDAAO Phe258Tyr (a) and Phe258Ala (b) mutants. Initial enzyme concentration 15 µg/ml, 0.1 M potassium phosphate buffer, pH 8.0.
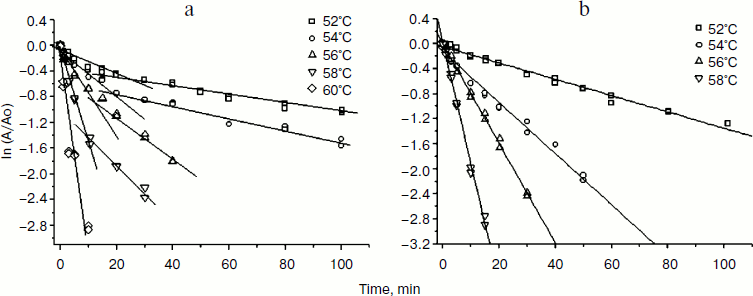
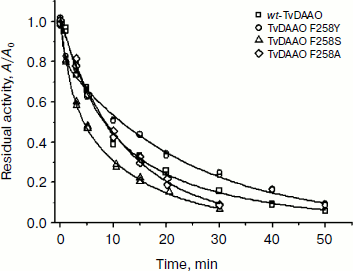
Figure 4 shows time dependences of residual activity of the mutant and wild-type enzymes under the same conditions. The figure shows that the replacement of residue Phe258 by serine and tyrosine leads to a slight change in the stability of the enzyme. The least stable mutant is TvDAAO Phe258Ser, its half-life being decreased almost twofold compared to the wild-type enzyme. The half-life period of the Phe258Ala mutant did not change, but at lower levels of activity its stability is slightly lower than that of wt-TvDAAO due to the change in the mechanism of thermal inactivation. The opposite picture is observed in the case of the Phe258Tyr mutant. The initial rate of its inactivation is slightly higher than for wild-type enzyme, but at the level of residual activity of 60% and below it is more stable than wt-TvDAAO. This is apparently due to the fact that the Phe258Tyr replacement decreases the inactivation rate constant of the second stage [15, 19].
Fig. 4. Dependence of residual activity on time for the mutants and the wild-type TvDAAO at 56°C; 0.1 M potassium phosphate buffer, pH 8.0, enzyme concentration 15 µg/ml.
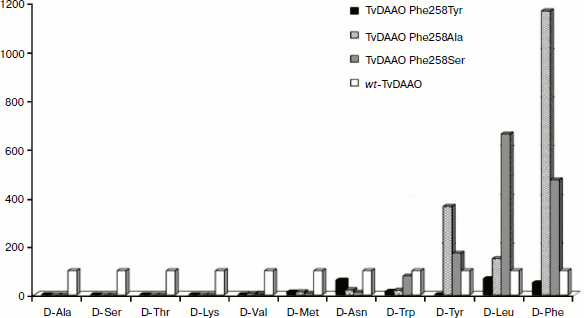
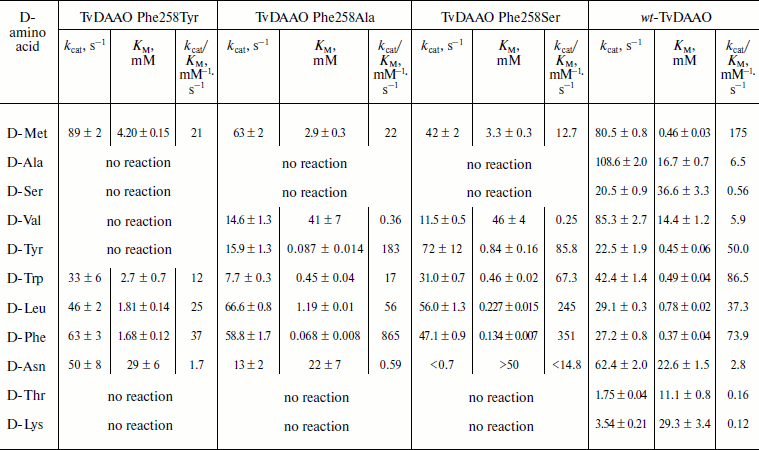
Catalytic properties of TvDAAO mutants Phe258Tyr, Phe258Ala, and Phe258Ser. The results of measurements of the kinetic parameters of mutant forms TvDAAO with different D-amino acids are shown in the table. Figure 5 shows the data obtained for the catalytic efficiency of mutant enzymes normalized to that of wt-TvDAAO ((kcat/KM)mut/(kcat/KM)wt × 100%). The table and Fig. 5 show that the introduction of substitutions Phe258Tyr, Phe258Ser, and Phe258Ala sharply increases the substrate specificity of the enzyme. All three mutant enzymes are inactive with D-Ala, D-Ser, D-Lys, and D-Thr. In addition, the Phe258Tyr mutant is also inactive with D-Val and D-Tyr, and the catalytic efficiency of the Phe258Ser and Phe258Ala mutants with several other D-amino acids has fallen more than 10-fold. For the Phe258Ala and Phe258Ser mutants, the activity with D-Tyr, D-Leu and especially with D-Phe significantly increases.
Fig. 5. Relative catalytic efficiency (kcat/KM)mut/(kcat/KM)wt for TvDAAO with substitutions Phe258Tyr, Phe258Ala, and Phe258Ser. The catalytic efficiency of wild-type TvDAAO is taken as 100%.
Catalytic parameters of the mutant TvDAAO with substitutions Phe258Tyr,
Phe258Ala, and Phe258Ser and of wild-type TvDAAO with different D-amino
acids
These data suggest that the amino acid residue at position 258 does have a significant effect on the binding of substrate in the active site of TvDAAO. One might think that this is first of all due to role of steric access of the substrate to the active center. Introduction of a Tyr residue into position 258 leads to an additional hydroxyl group, resulting in a decrease in catalytic efficiency towards all D-amino acids that are substrates for the TvDAAO Phe258Tyr mutant. However, the steric factor cannot explain the loss of activity of all three mutant enzymes with D-amino acids that have a small side group, as in the case of substitutions Phe258Ala and Phe258Ser, and the role of steric factor disappears. The most likely explanation is a change in the redox potential of FAD by increasing the polarity of the environment of the flavin. For the Phe258Tyr substitution, this may occur due to the appearance of the hydroxyl group, while for the Phe258Ala and Phe258Ser substitutions it could be due to increased access of solvent to the active center. Binding at the active site of the Phe258Ala and Phe258Ser mutants of D-amino acids with a small side group are not able to screen the flavin cofactor from water molecules. At the same time, hydrophobic amino acids with a bulky side group provide sufficiently effective shielding of the active center from the solvent, which results in reaching the redox potential required for oxidation of the substrate. Furthermore, the decrease in steric hindrance for access of the bulky substrate to the active site due to these substitutions leads to an increase in catalytic efficiency with such substrates.
As noted above, in this position in D-amino acid oxidases, after Phe, the most common residue is Tyr. In D-amino acid oxidase from Rhodotorula gracilis (RgDAAO) it is Tyr238 [22]. It has been replaced by Phe and Ser residues [23, 24]. In both cases mutant enzymes highly specific for D-Phe and D-Trp were also obtained, but the increase in specificity was achieved primarily due to a significant decrease in their catalytic efficiency with other D-amino acids. For the RgDAAO Tyr238Ser mutant, its catalytic efficiency towards D-Phe compared with the wild-type enzyme did not change, while it decreased by 2.3-fold with D-Trp. The RgDAAO Tyr238Phe mutant catalytic efficiency towards D-Phe increased by 1.4-fold, while its decreased by 3.1-fold with D-Trp. For both RgDAAO mutants the kcat value with all D-amino acids studied (with except for D-Pro) decreased from 3- to 9-fold. Unfortunately, the authors of those works did not present data on the catalytic efficiency of mutants RgDAAO with D-Leu and D-Tyr, which does not allow completion of the profile of substrate specificity.
An increased specificity of mutant TvDAAO to hydrophobic bulky amino acids is achieved due to increased catalytic efficiency (table). The greatest effect, 11.7-fold increase in catalytic efficiency, is observed with D-Phe for the Phe258Ala replacement. In fact, this mutant enzyme exhibits high specificity for only two D-amino acids with aromatic side-group – D-Phe and D-Tyr. The catalytic efficiency towards D-Leu is 16-fold lower than that with D-Phe, and the catalytic efficiency towards D-Met and D-Trp is 40- and 50-fold lower, respectively. The TvDAAO Phe258Ser mutant exhibits high catalytic activity towards four amino acids: D-Phe, D-Leu, D-Tyr, and D-Trp (for the first three amino acids the catalytic efficiency increased 4.75-, 6.6-, and 1.7-fold, respectively).
Thus, the results of our experiments have shown that even one point amino acid substitution can dramatically change the spectrum of substrate specificity of D-amino acid oxidase from the yeast T. variabilis. In this study we succeeded in obtaining mutant enzymes specific towards D-amino acids with a bulky hydrophobic radical, an increase in specificity being achieved both due to increase in activity and due to decrease in KM. Given the fact that a significant increase in catalytic efficiency does not affect the thermal stability of TvDAAO Phe258Ala, which is very close to that of wild-type enzyme, the mutant enzyme is the best candidate for selective determination of total concentration of D-Phe and D-Tyr in samples.
This work was supported by the Ministry of Education and Science of the Russian Federation (state contract 16.512.11.2253) and the Russian Foundation for Basic Research (grant 11-04-00959-a).
REFERENCES
1.Maekawa, M., Watanabe, M., Yamaguchi, S., Konno,
R., and Hori, Y. (2005) Neurosci. Res., 53, 34-38.
2.Chumakov, I., M. Blumenfeld, O., Guerassimenko, L.,
Cavarec, M., Palicio, M., et al. (2002) Proc. Natl. Acad. Sci.
USA, 99, 13675-13680.
3.Harrison, P. J., and Owen, M. J. (2003)
Lancet, 361, 417-419.
4.Takigawa, Y., Homma, H., Lee, J. A., Fukushima, T.,
Santa, T., Iwatsubo, T., and Imai, K. (1998) Biochem. Biophys. Res.
Commun., 248, 641-647.
5.D’Aniello, G., Tolino, A., D’Aniello,
A., Errico, F., Fisher, G. H., and Di Fiore, M. M. (2000)
Endocrinology, 141, 3862-3870.
6.D’Aniello, A., Di, C. A., Di, C. C.,
Annunziato, L., Petrucelli, L., and Fisher, G. (1996) Life Sci.,
59, 97-104.
7.D’Aniello, A., Di Fiore, M. M., Fisher, G.
H., Milone, A., Seleni, A., D’Aniello, S., Perna, A. F., and
Ingrosso, D. (2000) FASEB J., 14, 699-714.
8.Fisher, G. H., D’Aniello, A., Vetere, A.,
Padula, L., Cusano, G. P., and Man, E. H. (1991) Brain Res.
Bull., 26, 983-985.
9.Fisher, G., Lorenzo, N., Abe, H., Fujita, E., Frey,
W. H., Emory, C., Di Fiore, M. M., and D’Aniello, A. (1998)
Amino Acids, 15, 263-269.
10.Sacchi, S., Rosini, E., Caldinelli, L., and
Pollegioni, L. (2012) Methods Mol. Biol., 794,
313-324.
11.Hamase, K., Konno, R., Morikawa, A., and Zaitsu,
K. (2005) Biol. Pharm. Bull., 28, 1578-1584.
12.Pernot, P., Mothet, J. P., Schuvailo, O.,
Soldatkin, A., Pollegioni, L., Pilone, M., Adeline, M. T., Cespuglio,
R., and Marinesco, S. (2008) Anal. Chem., 80,
1589-1597.
13.Tishkov, V. I., and Khoronenkova, S. V. (2005)
Biochemistry (Moscow), 70, 40-54.
14.Savin, S. S., Chernyshev, I. V., Tishkov, V. I.,
and Khoronenkova, S. V. (2006) Mosc. Univ. Chem. Bull..,
47, 25-30.
15.Khoronenkova, S. V. (2008) Recombinant D-Amino
Acid Oxidase: Isolation and Structure-Activity Studies:
Candidate’s dissertation [in Russian], Lomonosov Moscow State
University, Moscow.
16.Tishkov, V. I., Khornenkova, S. V., and Savina,
L. I. RF Patent No. RU 2,362,806 from 23.07.2007; Byull. Izobret.
Poleznye Modeli (2009) No. 21.
17.Khoronenkova, S. V., and Tishkov, V. I. (2008)
Chin. J. Biotechnol., 24, 2122-2124.
18.Cherskova, N. V., Choronenkova, S. V., and
Tishkov, V. I. (2010) Russ. Chem. Bull. Int. Ed., 59,
269-275.
19.Komarova, N. V., Golubev, I. V., Choronenkova, S.
V., and Tishkov, V. I. (2012) Russ. Chem. Bull. Int. Ed.,
61, No. 7.
20.Khoronenkova, S. V., and Tishkov, V. I. (2008)
Anal. Biochem., 374, 405-410.
21.Chukhrai, E. S. (1981) Mosc. Univ. Chem.
Bull., 22, 331-340.
22.Umhau, S., Pollegioni, L., Molla, G., Diederichs,
K., Welte, W., Pilone, M. S., and Ghisla, S. (2000) Proc. Natl.
Acad. Sci. USA, 97, 12463-12468.
23.Boselli, A., Sacchi, S., Job, V., Pilone, M. S.,
and Pollegioni, L. (2002) Eur. J. Biochem., 269,
4762-4771.
24.Frattini, L., Rosini, E., Pollegioni, L., and
Pilone, M. S. (2011) J. Chromatogr. B, 879,
3235-3239.