Identification of Phosphorylation Sites in Aminoglycoside Phosphotransferase VIII from Streptomyces rimosus
S. M. Elizarov1,2, M. G. Alekseeva1,3, F. N. Novikov4, G. G. Chilov4, D. A. Maslov1,3, A. A. Shtil1, and V. N. Danilenko1*
1Vavilov Institute of General Genetics, Russian Academy of Sciences, ul. Gubkina 3, 119991 Moscow, Russia; fax: (499) 132-8962; E-mail: valerid@rutenia.ru2Bach Institute of Biochemistry, Russian Academy of Sciences, Leninsky pr. 33/2, 119071 Moscow, Russia; fax: (495) 954-2732
3Research Center of Biotechnology of Antibiotics and Other Biologically Active Substances “Bioan”, ul. Gubkina 2, 119991 Moscow, Russia; fax: (499) 135-4194
4Zelinsky Institute of Organic Chemistry, Russian Academy of Sciences, Leninsky pr. 47, 119991 Moscow, Russia; fax: (499) 135-5328
* To whom correspondence should be addressed.
Received March 11, 2012; Revision received May 3, 2012
We demonstrate for the first time the role of phosphorylation in the regulation of activities of enzymes responsible for inactivation of aminoglycoside antibiotics. The aminoglycoside phosphotransferase VIII (APHVIII) from the actinobacterial strain Streptomyces rimosus ATCC 10970 is an enzyme regulated by protein kinases. Two serine residues in APHVIII are shown to be phosphorylated by protein kinases from extracts of the kanamycin-resistant strain S. rimosus 683 (a derivative of strain ATCC 10970). Using site-directed mutagenesis and molecular modeling, we have identified the Ser146 residue in the activation loop of the enzyme as the key site for Ca2+-dependent phosphorylation of APHVIII. Comparison of the kanamycin kinase activities of the unphosphorylated and phosphorylated forms of the initial and mutant APHVIII shows that the Ser146 modification leads to a 6-7-fold increase in the kanamycin kinase activity of APHVIII. Thus, Ser146 in the activation loop of APHVIII is crucial for the enzyme activity. The resistance of bacterial cells to kanamycin increases proportionally. From the practical viewpoint, our results increase prospects for creation of highly effective test systems for selecting inhibitors of human and bacterial serine/threonine protein kinases based on APHVIII constructs and corresponding human and bacterial serine/threonine protein kinases.
KEY WORDS: aminoglycoside phosphotransferase, serine/threonine protein kinases, antibiotic resistance, phosphorylation sites, site-directed mutagenesisDOI: 10.1134/S0006297912110041
Abbreviations: APHVIII, aminoglycoside phosphotransferase VIII; DTT, dithiothreitol; EGTA, sodium ethylene glycol tetraacetate; MD, molecular dynamics; PKA, protein kinase A; PKC, protein kinase C; STPKs, serine/threonine protein kinases.
The family of aminoglycoside-3′-phosphotransferases (APHs)
includes seven main types of enzymes with different amino acid
sequences and substrate specificities [1-3].
We have found in Streptomyces rimosus and characterized a new enzyme – APH of type VIII (APHVIII) [4, 5]. Under usual conditions, the expression of the aphVIII gene is minimal in the S. rimosus wild-type strain. A step-like selection of bacteria by resistance to increasing concentrations of kanamycin is accompanied by an increase in the aph activity and amplification (to 50 additional copies) of the chromosomal region carrying the gene of kanamycin resistance [6]. The DNA fragment amplified in the resistant strain contains the open reading frame of APH. Based on analysis of the nucleotide sequence and biochemical substrates of the expression product, the detected gene was assigned to an earlier unknown type aphVIII (GenBank, Accession number AAG11411). This gene encodes the enzyme APHVIII, which inactivates kanamycin through an ATP-dependent transfer of phosphate onto the 3′-hydroxyl group of the antibiotic molecule. In the presence of Ca2+, the APHVIII-dependent resistance of streptomycetes to kanamycin increases [5].
The APH family enzymes are similar in structure and functions with serine/threonine protein kinases (STPKs) of eukaryotes [7], and according to the modern classification they are classified as kinases modulating resistance to antibiotics. Phylogenetic analysis has revealed five main groups of this family of kinases: APH(2′′), APH(3′) and APH(3′′), APH(4) and APH(7′′), APH(6), and APH(9) [8].
The experimental findings presented in this work demonstrate that many inhibitors of STPKs (in particular, flavonol and quercetin, as well as an ATP-competitive inhibitor of kinase CKI – CKI-7) can act as inhibitors of aminoglycoside phosphotransferases [8, 9]. It is known that the STPK-dependent mechanism of inner signal transmission is active not only in higher organisms [10], but also in some prokaryotes, in particular, in actinobacteria of the Streptomyces genus [11, 12]. These bacteria can be used to study the influence of protein kinases on the role of APH in development of resistance to antibiotics. Studies on the posttranslational regulation in streptomycetes revealed Ca2+-dependent STPKs and established their role in the regulation of the resistance of Streptomyces strains to aminoglycoside antibiotics including kanamycin [13, 14]. We have found that phosphorylation of serine and/or threonine residues is important for modulation of the catalytic activity of APHVIII [15].
In the present work, we have identified STPKs modulating the activity of APHVIII in a kanamycin-resistant strain of S. rimosus and established that phosphorylation of a serine residue in position 146 activates the APH enzyme.
MATERIALS AND METHODS
Materials. NdeI and BamHI restrictases and T4 DNA-ligase were from Fermentas (Lithuania), a PCR kit from Dialat Ltd (Russia), the vector pET16b from Novagen (USA), and a kit of terminal primers AphN and AphC for cloning the aphVIII gene and a kit of primer pairs for preparing mutants of the aphVIII gene (Table 1) were synthesized by Sintol (Russia). The initial strain Streptomyces rimosus ATCC 10970 was from the Russian Collection of Industrial Microorganisms. The mutant strain S. rimosus 683 prepared by step-wise survival selection of the strain S. rimosus ATCC 10970 in the presence of kanamycin [6] was employed to analyze the phosphorylation of APHVIII. The E. coli strain DH5a (F–, Φ 80 ΔlacZΔM15, Δ(lacZYA-argF), U169) was from Promega (USA) [16]; the E. coli strain BL21(DE3) (F–, dcm, ompT, hsdS(rB–mB–), gal λ(DE3)) was from Novagen (USA) [17]. The initial construction pET16b–aphVIII contains the gene encoding the wild-type aminoglycoside phosphotransferase. The sequence of the full-size aphVIII gene of the S. rimosus strain is deposited in the NCBI/GenBank (Accession number AAG11411).
Table 1. Oligonucleotides used in this
work

* Recognition sites for restrictases NdeI (CATATG) and
BamHI (GGATCC) are printed in bold. Nucleotide substitutions are
underlined.
Phosphorylation sites of APHVIII were mapped with the programs NetPhos 2.0 (http://www.cbs.dtu.dk/services/NetPhos/) and NetPhosK (http://www.cbs.dtu.dk/services/NetPhosK/). Nucleotide sequences were analyzed with the BLAST program (http://www.ncbi.nlm.nih.gov/blast).
Molecular modeling. The full atomic model of APHVIII structure was prepared based on structures of APHII and APHIII enzymes available in PDB (PDB identifiers 1ND4 and 1L8T, respectively) with the Swiss-Model server [18]. The first step in optimization of the resulting APHVIII structure included molecular dynamics (MD) relaxation in vacuum at the temperature of 0 K for 1 ns with limitations on mobilities of the amino acid side radical atoms, which were smoothly taken away (from 500 kcal/mol/Å2 to zero) and with a constant limitation on the mobility of the protein main chain atoms. In the second step, the protein molecule was solvated (by submerging into a cubic cell of water with a retreat of 15 Å from coordinates of the protein atoms). The remaining limitations on the mobility were taken away during the molecular dynamics within 1 ns at the temperature of 0 K. Then the temperature of the molecular system was elevated to 300 K within the MD trajectory of 1 ns, and MD modeling was performed at 300 K for 10 ns without limitations on the atom mobilities. All MD calculations were performed with the Gromacs program [19, 20] (http://www.gromacs.org) in the full atomic force field OPLS using the model of water TIP3P and periodic boundary conditions. Motion equations were integrated with a time step of 4 fs (due to using constructions of atom-dummies and effective mass redistribution between atoms bound to hydrogen atoms). Electrostatic interactions were modeled using a reaction field approach at the boundary value of dielectric permeability 80 Fm at the cut-off radius of 15 Å.
Preparation of aphVIII gene mutants. Mutants of the aphVIII gene were prepared by introducing point mutations [21] according to the two-step scheme with terminal primers AphN and AphC for cloning the aphVIII gene and primers with nucleotide sequences complementary to the initial aphVIII gene except for the region of the inserted mutation. The site-directed mutagenesis substituted the detected phosphorylated serine residues by neutral alanines. In the first stage of PCR, the left or 5′-terminal part of the gene was amplified using the primer pair AphN + b, and the right or 3′-terminal part of the gene was amplified using primer the pair c + AphC in separate tubes. This resulted in two DNA fragments overlapping in the regions that contained the inserted primers (where B and C are inner primers suggesting the required nucleotide substitutions and fully complementary to each other) (Table 1). In the second stage of PCR, the resulting fragments were mixed at equimolar ratio, and after a cycle of heat denaturation and renaturation were used as a template in another PCR with external primers AphN and AphC. The resulting mutant genes were cloned into the pET16b vector at sites of restriction endonucleases NdeI and BamHI. In the mutant variant A95 the AGT codon is substituted by the GCT codon, in the mutant variant A146 the AGC codon is substituted by the gcc codon, in the mutant variant A160 the TCG codon is substituted by the gcG codon, in the mutant variant A215 the TCC codon is substituted by the GCC codon (Table 2). The presence of mutations was confirmed by sequencing the gene.
Study of APHVIII aminoglycoside phosphotransferase expression in E. coli. The initial and mutant variants of the aphVIII gene were expressed in E. coli BL21(DE3) cells. To study the protein expression and biomass production, the E. coli cells containing the constructed plasmids were grown for ~2 h on a shaker in a liquid medium (L-broth) supplemented with ampicillin at 34°C to optical density of 0.6, then the expression was induced by addition of IPTG (isopropyl-β-D-1-thiogalactopyranoside) to the final concentration of 1.3 mM/ml medium. Then the samples were cultured at 28°C for 5 h, and the biomass was collected and suspended in buffer containing 62.5 mM Tris-HCl (pH 6.8), 5% glycerol, 2% mercaptoethanol, 0.1% SDS, 0.01% Bromophenol Blue. The cells were destroyed by boiling for 10 min. The protein fraction was analyzed by Laemmli electrophoresis in polyacrylamide gel. The protein fraction from E. coli BL21(DE3) strain containing the insert-free plasmid pET16b was analyzed as a control.
Preparation of His10-APHVIII variants and purification of native APHVIII from E. coli extracts were performed as described in [14, 22]. The enzymatic activity of APHVIII was analyzed in 30 µl of reaction mixture containing APHVIII (0.3 µg), kanamycin (1.2 mg/ml), 7.5 mM [γ-32P]ATP (0.01 Bq/pmol; Fosfor, Moscow), 4 mM NaHCO3, 20 mM Tris-HCl (pH 7.8), 10 mM MgCl2, 60 mM KCl, and 3 mM DTT [14].
Study of phosphorylation and enzymatic activity of APHVIII. A partially purified preparation of STPK was obtained from S. rimosus extracts in buffer A (20 mM triethanolamine hydrochloride, pH 7.8, 200 mM NaCl, 5 mM MgCl2, 1 mM CaCl2, 3 mM DTT, 0.5 mM phenylmethylsulfonyl fluoride, 10% glycerol). The extracts were placed onto Cibacron-Sepharose CL-6B (Pharmacia, USA), washed in buffer A, and proteins were eluted in the same buffer supplemented with 1.5 M NaCl and 20 mM ATP. The kinase activity of the eluates was analyzed by phosphorylation of APHVIII in the presence of 0.5 mM [γ-32P]ATP (0.005 Bq/pmol) and 20 µg/ml APHVIII. The fractions were precipitated with 5% TCA, placed on GF/A glass fiber filters, and the radioactivity was determined with a Perkin-Cultier 2000 counter. The fractions containing STPKs capable of phosphorylating APHVIII were combined and desalted on Sephadex G-10.
His10-APHVIII was phosphorylated with partially purified STPKs in 50 µl of reaction mixture containing 2 µg total protein from the mixture of STPKs, 40 µg His10-APHVIII, 0.5 mM [γ-32P]ATP, 50 mM Tris-HCl (pH 7.8), 125 mM NaCl, 5 mM MgCl2, 10% glycerol, 0.5 mM DTT, and 1 mM CaCl2. The reaction was initiated by addition of the STPKs and terminated after 15 min of incubation at 28°C by adding 17 µl of a fourfold buffer for loading protein lysates (SDS-PAGE sample buffer) and heating for 5 min to 95°C. The proteins were analyzed by electrophoresis in the presence of SDS or by inclusion of radiolabeled phosphate into APHVIII after purification of His10-APHVIII on a column with Ni-containing agarose and counting radioactivity of the eluate. The proteins were subjected to one-dimensional electrophoresis in 10% polyacrylamide gel. For radioautography, the gels were fixed in 50% TCA and stained with 0.1% Coomassie R-250, dried, and exposed to a radiation-sensitive film [5]. Two-dimensional electrophoresis of proteins was performed in 15% gel [23].
To determine the kinase activity in the extracts from S. rimosus, the proteins were renatured in gel using the method of Kameshita and Fujisawa [24] in our modification [25]. Phosphoamino acids resulting from protein phosphorylation were identified after the separation of hydrochloric hydrolysates of labeled polypeptides by thin-layer chromatography on cellulose plates (Merck, Germany) [25].
Immunoprecipitation and immunoblotting of APHVIII. Rabbit serum containing antibodies to APHVIII was a gift from M. Fuhrmann and P. Hegemann (Regensburg, Germany). The serum was treated with 40% solution of ammonium sulfate, and the precipitate was dialyzed against 0.01 M sodium phosphate buffer (pH 6.6) supplemented with 0.05 M KCl. IgG was purified by chromatography on DEAE-Sephadex A-50. The APHVIII protein was immunoprecipitated at 4°C for 10 h in buffer B (10 mM sodium phosphate, pH 7.4, 0.2 M NaCl, 0.2% Triton X-100) supplemented with 1% bovine serum albumin, 97.7 µg extract of the labeled proteins, and 12.3 µg pre-immune rabbit serum (the control) or antibodies to APHVIII (IgG). Immune complexes were adsorbed in buffer B onto the staphylococcal protein A conjugated with Sepharose, washed in the same buffer supplemented with 1% Triton X-100 and 0.5 M NaCl, washed in buffer B, and eluted in SDS-PAGE sample buffer. After electrophoresis in the presence of SDS, the proteins were transferred onto nitrocellulose. The APHVIII protein was detected with rabbit serum, goat IgG conjugated with horseradish peroxidase being used as secondary antibodies. Solutions for chemiluminescence were from Pharmacia.
RESULTS AND DISCUSSION
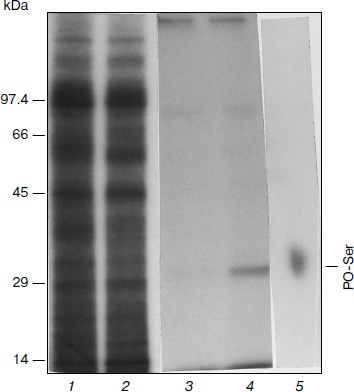
Phosphorylation of APHVIII in vitro. After radiolabeling, electrophoretic separation of proteins, and radioautography of extracts of kanamycin-sensitive and kanamycin-resistant strains of S. rimosus 683, nine main labeled polypeptides of the following molecular weights were found: 95.5, 92.5, 80, 55, 45, 35, 29, 17.5, and 14 kDa (Fig. 1). In the kanamycin-resistant strain, the 29-kDa fraction displayed higher radioactivity than in the kanamycin-sensitive strain, and the molecular weight of this fraction was the most like the molecular weight of 29,196 Da calculated for a product of the aphVIII gene [5]. These calculations were confirmed by data of immunoprecipitation and immunoblotting of the phosphorylated proteins with antiserum to APHVIII. Figure 1 (lane 4) shows that the 29-kDa phosphorylated protein from the kanamycin-resistant strain can be precipitated with antibodies to APHVIII. Elimination of this protein from the extract was accompanied by a 92-97% decrease in the kanamycin-phosphorylating activity. This protein was absent in extracts from the wild-type strain; the kanamycin-phosphorylating activity of the extract from the parental strain was minimal. These experiments show that the 29-kDa phosphoprotein detected in the kanamycin-resistant bacteria is APHVIII. In this protein only serine residues are phosphorylated (Fig. 1, lane 5). Thus, STPKs of the S. rimosus 683 strain are able to phosphorylate the 29-kDa protein (APHVIII).
Fig. 1. Phosphorylation of endogenous APHVIII in extracts from S. rimosus. Autoradiograms of proteins from the initial (1) and kanamycin-resistant (2) strains preincubated with [γ-32P]ATP and separated by SDS-PAGE. In each well of the gel, 5.2 µg protein was introduced. Autoradiograms of the gels after SDS-PAGE of the labeled proteins precipitated by anti-APHVIII IgG from extracts from the initial (3) and kanamycin-resistant (4) strains. 5) Autoradiogram of phosphoamino acids after high-voltage electrophoresis of the phosphorylated 29-kDa protein hydrolysate. Typical results of three independent experiments are presented.
Mapping probable phosphorylation sites of APHVIII. Analysis of phosphorylation sites of APHVIII using the NetPhos and NetPhosK programs revealed three canonic sites for STPKs of eukaryotes. The first site includes Ser95, which is a probable target for phosphorylation by Ca2+/calmodulin-dependent protein kinase II, Ca2+/phospholipid-dependent protein kinase C (PKC), and cAMP-dependent protein kinase A (PKA), as well as the residues Ser160 and Ser215, which are probable sites for phosphorylation by PKA.
There are literature data indicating that the activation of protein kinases of eukaryotes is the most significantly due to phosphorylation of serine, threonine, and tyrosine residues in the activation loop of the enzyme [10, 26, 27].
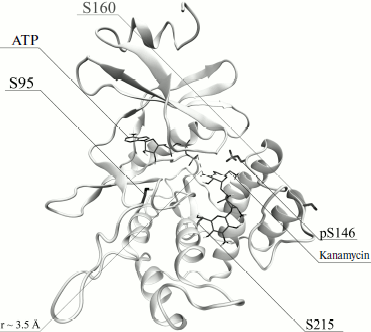
Using the structures of the enzymes APHII and APHIII available in the PDB base, we constructed a molecular model of the APHVIII structure. The modeling revealed the phosphorylation site Ser146 in the enzyme activation loop. This site is an analog of phosphoserine in the region homologous to the ribose pocket of the PKA type serine/threonine protein kinases [28]. Molecular dynamics (MD) of the unphosphorylated APHVIII complex with kanamycin, bound ATP molecules, and two Mg2+ ions revealed significant changes in the APHVIII structure, such as weakening of the contact between the N- and C-terminal domains (Fig. 1; see Supplement to this paper on the site of Biochemistry (Moscow) (http://protein.bio.msu.ru/biokhimiya)). The mobility of domains promotes the release of ATP from the substrate bound with the C-terminal part and the interaction of phosphate with the N-terminal part. These changes result in a catalytically inactive conformation of APHVIII, and phosphorylation of kanamycin becomes impossible (Fig. 2; see Supplement at http://protein.bio.msu.ru/biokhimiya) because of a significant distance (~5 Å) between its hydroxyl group and γ-phosphate of ATP. This change in the conformation occurs within the first 7 ns of the MD simulation and probably would continue further decreasing the phosphotransferase activity of APHVIII. However, under these conditions the binding of kanamycin with APHVIII weakens less markedly because the antibiotic mainly interacts with the C-terminus of the enzyme.
The model of APHVIII phosphorylated on Ser146 revealed the following features. The structure retains its stability during 10 ns of the MD trajectory as can be concluded based on absence of significant conformational changes in the major chain packing. The enzyme has a catalytically active conformation because the mean distance between the phosphorus atom in the γ-phosphate residue of ATP and the oxygen atom of the hydroxyl group of kanamycin is about 3.5 Å, the hydroxyl group forms a hydrogen bond with the D184 residue of the catalytic chain, and a nucleophilic attack can occur due to proton transfer (Fig. 3; see Supplement at http://protein.bio.msu.ru/biokhimiya). Thus, phosphoserine in position 146 located in a region similar to the activation loop of eukaryotic protein kinases is responsible for the catalytically competent conformation and for preventing the opening of the protein parts. A salt bridge is found between pS146 and R207 produced at the beginning of the protein relaxation in vacuum and retained on MD simulation in aqueous medium. This bridge functions as a “lock” between the activation loop and the DFG region (DMG for APHVIII) in which R207 directly follows G206. This region is highly conservative for protein kinases, and here ATP binds with Mg2+. Moreover, this region determines the orientation of the loop, which is required for the catalytic function due to approaching of the α2-curl via hydrophobic interactions. Thus, the pS146–R207 salt bridge stabilizes the APHVIII conformation in which the activation loop involved in kanamycin binding, the DFG region involved in ATP binding and approaching to the N-terminal region, and the N-terminal domain itself constitute an integrity responsible for the catalytic function. Such a salt bridge is found in protein kinases of eukaryotes; phosphorylated serine or threonine residues in the activation loop interact with lysine or arginine residues located before the DFG region of the catalytic loop. Thus, in PKA pS197 interacts with K189 located before the DFG region and with R165 in the catalytic loop. Although the arginine residue is located rather distantly from the DFG region, the spatial position of the guanidine group of this residue is close to K189 similarly to residue R207 in the APHVIII structure.
Because the pS146–R207 salt bridge is responsible for the catalytically competent conformation of protein APHVIII, this structural feature suggests that the activation of the enzyme is mediated through phosphorylation. Based on the model of the tertiary structure of APHVIII (Fig. 2) together with data obtained with the NetPhos and NetPhosK programs, we supposed that residues Ser95, Ser146, Ser160, and Ser215 should be functionally important phosphorylation targets. To elucidate the role of each of these residues in the catalytic function of APHVIII, they were subjected to directed point mutagenesis.
Fig. 2. Model of tertiary structure of APHVIII. Positions of serine residues (S95, S146, S160, S215) that seem to be possible phosphorylation sites of APHVIII are indicated.
Identification of phosphorylation sites functionally important for APHVIII activity. By directed mutagenesis, nine variant aphVIII mutant genes were prepared providing from one to four substitutions of phosphorylated serine residues with neutral alanines in positions 95, 146, 160, and 215 (Table 2). The initial variant (the wild-type sequence) and mutant variants were cloned and expressed in E. coli. The resulting recombinant proteins were purified by affinity chromatography on a Ni-agarose column and subjected to phosphorylation by partially purified STPKs from S. rimosus (see “Materials and Methods”). Results of the experiments revealed that the Ser146Ala substitution decreased the phosphorylation of APHVIII by 3-4-fold (Table 2). The Ser160Ala substitution decreased the Ca2+-dependent phosphorylation of APHVIII less significantly (to the level of 74-85% of the control). The phosphorylation of Ser146 mainly depended on the presence of Ca2+, whereas the phosphorylation of Ser160 changed little in the presence of Ca2+. Mutations of residues Ser95 and Ser215 failed to significantly influence the phosphorylation of APHVIII. These data indicate that residue Ser146 is the most significant result of the in silico modeling of this process. The direct analysis of phosphorylation of the APHVIII molecule does not exclude that the Ser160 residue can also be a possible modulator of APHVIII activity.
Table 2. STPK-dependent phosphorylation of
initial and mutant variants of APHVIII from S. rimosus

Notes: Here and in Table 3 Ser-Ala substitutions
are printed in bold in the corresponding positions in APHVIII. Averaged
results of three independent determinations with mean square deviations
are presented.
Influence of phosphorylation on enzymatic activity of APHVIII. The comparison of kanamycin kinase activities of the phosphorylated and unphosphorylated initial and mutant variants of the APHVIII protein has shown that phosphorylation of the Ser146 residue in APHVIII increases 6-7-fold the studied activity. Without phosphorylation of this residue, the kanamycin kinase activity is not stimulated. Thus, the Ser146 residue located in the activation loop of APHVIII (Fig. 2) is crucial for the activity of the enzyme. The bacterial cell resistance to kanamycin increases proportionally.
The activities of unphosphorylated and phosphorylated APHVIII were compared under conditions of kanamycin excess in the reaction mixture, which also contained partially purified STPKs from S. rimosus, labeled ATP, and Ca2+ or its chelator EGTA. The Ser146Ala substitution abolished the stimulation of the kanamycin phosphotransferase activity of APHVIII in the presence of Ca2+ and STPKs from S. rimosus (Table 3). But the Ser160Ala substitution failed to significantly change this activity. The Ca2+-independent phosphorylation of APHVIII (Table 2) also failed to influence its ability to phosphorylate kanamycin. Consequently, the Ca2+-dependent phosphorylation of APHVIII mainly occurs by residue Ser146 in the activation loop, and this posttranslational modification is necessary for the enzymatic activity of APHVIII.
Table 3. Kanamycin phosphotransferase
activity of initial and mutant variants of His10-APHVIII
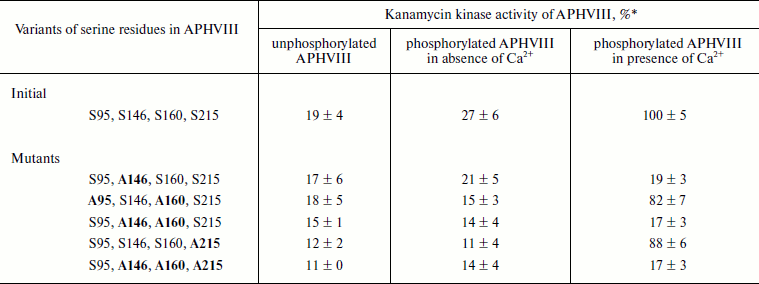
* % of wild-type APHVIII activity in the presence of Ca2+
(taken as 100%). Averaged results of three independent determinations
and mean square deviations are presented.
Thus, we conclude that aminoglycoside phosphotransferase from APHVIII can be a representative of a new group of bacterial enzymes capable of being activated by protein kinases. This phenomenon opens prospects for studies on the role of STPKs in the regulation of resistance to antibiotics in bacteria, including pathogenic ones [29, 30]. In fact, the resistance of S. coelicolor and S. lividans to some antibiotics can be lowered in the presence of an inhibitor of STPK, bis-indolyl maleimide I and its derivatives [14, 31]. This means that the STPK-regulated mechanism of resistance to antibiotics can be rather widely distributed in microorganisms of the genus Actinobacteria.
The results of the present work are the first experimental proof of the STPK role in the regulation of activities of enzymes of the aminoglycoside phosphotransferase family through phosphorylation of a serine residue in the activation loop of APHVIII. Our findings extend the role of actinomycetes as the natural reservoir (“resistome”) of drug resistance genes [3, 32, 33] due to the suggestion of epigenetic mechanisms (phosphorylation) of this phenomenon. The key role of APHVIII in the regulation of bacterial resistance to aminoglycoside antibiotics, i.e. the dependence of enzymatic inactivation of kanamycin on phosphorylation of a specific amino acid residue in the activation loop of the enzyme, seems promising for creation of test systems for screening of STPK inhibitors. In such test systems, the STPK-dependent phosphorylation of APHVIII is accompanied by an increase in the growth of actinobacteria in the presence of kanamycin, whereas using an inhibitor of STPK induces lysis of the bacteria preventing the phosphorylation of APHVIII and proportionally decreasing the kanamycin kinase activity of this element of the test system.
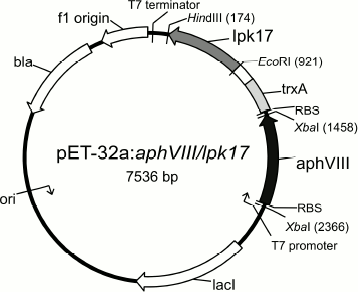
From the practical standpoint, the findings provide a scientific basis and extend prospects for creating a panel of highly effective test systems for choosing STPK inhibitors based on the APHVIII protein construct and targeted serine/threonine human and bacterial protein kinases. We have already constructed some models of such test systems, and they are used for prescreening STPK inhibitors [34, 35]. In particular, the E. coli aphVIII/pk17 system for prescreening PK17 inhibitors of the S. lividans strain that is supposed to be involved in programmed cell death [36] and its structural homologs (Fig. 3).
Fig. 3. Map of expression plasmid pET-32a:aphVIII/lpk17. The construct contains the gene of the type VIII aminoglycoside phosphotransferase from S. rimosus (aphVIII) and the nucleotide sequence of the STPK catalytic domain from LPK17 S. lividans (lpk17). EcoRI, HindIII, and XbaI are recognition sites of the corresponding restriction endonucleases.
We are grateful to E. S. Dmitrieva and T. A. Seregina for their help in performing the experiments.
This work was supported by the State contract No. 16.512.11.2236 of July 12, 2011 in the framework of the Federal Purpose Program “Investigations and Elaborations in Priority Lines of the Development of the Scientific-Technical Complex of Russia for 2007-2013”.
REFERENCES
1.Kannan, N., Taylor, S. S., Zhai, Y., Venter, J. C.,
and Manning, G. (2007) PLoS Biol., 5, 0466-0478.
2.Davies, J., and Wright, G. D. (1997) Trends
Microbiol., 5, 234-240.
3.Wright, G. D. (2007) Nat. Rev. Microbiol.,
5, 175-186.
4.Sizova, I. A., Lapina, T. V., Frolova, O. N.,
Alexandrova, N. N., Akopiants, K. E., and Danilenko, V. N. (1996)
Gene, 181, 13-18.
5.Sizova, I. A., Hegemann, P., Furman, M., and
Danilenko, V. N. (2002) Mol. Biol. (Moscow), 36,
18-25.
6.Danilenko, V. N., and Akopiants, K. E. (1995) in
Proc. 9th Int. Symp. on Biology of Actinomycetes, Moscow, pp.
104-112.
7.Daigle, D. M., McKay, G. A., Thompson, P. R., and
Wright, G. D. (1999) Chem. Biol., 6, 11-18.
8.Shakya, T., Stogios, P. J., Waglechner, N.,
Evdokimova, E., Ejim, L., Blanchard, J. E., McArthur, A. G., Savchenko,
A., and Wright, G. D. (2011) Chem. Biol., 18,
1591-1601.
9.Fong, D. H., Xiong, B., Hwang, J., and Berghuis,
A. M. (2011) PLoS One, 6, e19589.
10.Scheeff, E. D., and Bourne, P. E. (2005) PLoS
Comput. Biol., 1, 0359-0381.
11.Kennely, P. (2002) FEMS Microbiol. Lett.,
206, 1-8.
12.Narayan, A., Sachdeva, P., Sharma, K., Saini, A.,
Tyagi, A., and Singh, Y. (2007) Physiol. Genomics,
29, 66-75.
13.Pereira, S. F., Goss, L., and Dworkin, J. (2011)
Microbiol. Mol. Biol. Rev., 75, 192-212.
14.Bekker, O., Elizarov, S. M., Alekseeva, M. G.,
Lyubimova, I., and Danilenko, V. (2008) Mikrobiologiya,
77, 630-638.
15.Elizarov, S. M., Sergienko, O. V., Sizova, I. A.,
and Danilenko, V. N. (2005) Mol. Biol. (Moscow), 39,
255-263.
16.Inoue, H., Nojima, H., and Okayama, H. (1990)
Gene, 96, 23-28.
17.Mierendorf, R., Yeager, K., and Novy, R. (1994)
Newslett. Novagen Inc., 1, 1-3.
18.Arnold, K., Bordoli, L., Kopp, J., and Schwede,
T. (2006) Bioinformatics, 22, 195-201.
19.Berendsen, H. J. C., van der Spoel, D., and van
Drunen, R. (1995) Comp. Phys. Commun., 91, 43-56.
20.Lindahl, E., Hess, B., and van der Spoel, D.
(2001) J. Mol. Model., 7, 306-317.
21.Nelson, R. M., and Long, G. L. (1989) Anal.
Biochem., 180, 147-151.
22.Elizarov, S. M., Mironov, V. A., and Danilenko,
V. N. (2000) Life, 50, 139-143.
23.Hutchcroft, J. E., Anostario, M., Harrison, M.
L., and Geahlen, R. L. (1991) Methods Enzymol., 200,
417-422.
24.Kameshita, I., and Fujisawa, H. (1989) Anal.
Biochem., 183, 139-143.
25.Elizarov, S. M., and Danilenko, V. N. (2001)
FEMS Microbiol. Lett., 202, 135-138.
26.Azucena, E., and Mobashery, S. (2001) Drug.
Resist. Updat., 4, 106-117.
27.Nurizzo, D., Shewry, S. C., Perlin, M. H., Brown,
S. A., Dholakia, J. N., Fuchs, R. L., Deva, T., Baker, E. N., and
Smith, C. A. (2003) J. Mol. Biol., 327, 491-506.
28.Taylor, S. S., Yang, J., Wu, J., et al. (2004)
Biochim. Biophys. Acta, 1697, 259-269.
29.Prisic, S., Dankwa, S., Schwartz, D., Chou, M.
F., Locasale, J. W., Kang, C. M., Bemis, G., Church, G. M.,
Steen, H., and Husson, R. N. (2010) Proc. Natl. Acad. Sci. USA,
107, 7521-7526.
30.Danilenko, V. N., Osolodkin, D. I., Lakatosh, S.
A., Preobrazhenskaya, M. N., and Shtil, A. A. (2011) Curr. Topics
Med. Chem., 11, 1352-1369.
31.Danilenko, V., Simonov, A., Lakatosh, S.,
Kubbutat, M., Totzke, F., Schaechtele, C., Elizarov, S., Bekker, O.,
and Printsevskaya, S. (2008) J. Med. Chem., 51,
7731-7736.
32.D’Costa, V. M., McGrann, K. M., Hughes, D.
W., and Wright, G. D. (2006) Science,
311, 374-377.
33.D’Costa, V. M., Griffiths, E., and Wright,
G. D. (2007) Curr. Opin. Microbiol., 10, 481-489.
34.Anizon, F., Shtil, A. A., Danilenko, V. N., and
Moreau, P. (2010) Curr. Med. Chem., 17, 4114-4133.
35.Bekker, O. B., Alekseeva, M. G., Osolodkin, D.
I., Palyulin, V. A., Elizarov, S. M., Zefirov, N. S., and Danilenko, V.
N. (2010) Acta Naturae, 2, 126-139.
36.Manteca, A., Mader, U., Connolly, B. A., and
Sanchez, J. (2006) Proteomics, 6, 6008-6022.