REVIEW: Programmed Cell Death in Plants
A. S. Fomicheva1, A. I. Tuzhikov2, R. E. Beloshistov1, S. V. Trusova2, R. A. Galiullina2, L. V. Mochalova2, N. V. Chichkova2, and A. B. Vartapetian2*
1Faculty of Bioengineering and Bioinformatics, Lomonosov Moscow State University, 119991 Moscow, Russia2Belozersky Institute of Physico-Chemical Biology, Lomonosov Moscow State University, 119991 Moscow, Russia; E-mail: varta@genebee.msu.ru
* To whom correspondence should be addressed.
Received May 17, 2012; Revision received May 25, 2012
The modern concepts of programmed cell death (PCD) in plants are reviewed as compared to PCD (apoptosis) in animals. Special attention is focused on considering the potential mechanisms of implementation of this fundamental biological process and its participants. In particular, the proteolytic enzymes involved in PCD in animals (caspases) and plants (phytaspases) are compared. Emphasis is put on elucidation of both common features and substantial differences of PCD implementation in plants and animals.
KEY WORDS: apoptosis, caspase, programmed cell death, phytaspaseDOI: 10.1134/S0006297912130044
Abbreviations: AFC, 7-amino-4-trifluoromethyl-coumarin; AMC, 7-amino-4-methylcoumarin; AtMC, Arabidopsis thaliana metacaspase; CHO, aldehyde group; CMK, chloromethylketone; FMK, fluoromethylketone; HR, hypersensitive response; PCD, programmed cell death; TMV, tobacco mosaic virus; VPE, vacuolar processing enzyme.
Multicellular organisms require the ability to eliminate excessive or
damaged cells that are formed both during normal development and in the
interaction of the organism with the environment, e.g. under stress
conditions leading to irreversible cell damage, as well as in infection
with pathogens. The cellular process directed to successive
extermination (suicide) of undesirable cells is known as programmed
cell death (PCD). The most studied (although not the only) form of PCD
in animals is apoptosis, which is characterized by a distinct set of
morphological and biochemical features [1]. A
crucial role in programmed suicide of animal cells belongs to caspases,
a family of highly specific cysteine proteinases that are activated in
apoptosis, introducing single breaks in molecules of a restricted set
of cellular proteins [2]. Caspases have exclusive
specificity of hydrolysis: they introduce break after an aspartic acid
residue (D) localized within a certain amino acid context. Directed
fragmentation of target proteins by caspases eventually leads to the
ordered death of the cell. And, contrariwise, inhibition of caspases
counteracts apoptosis.
In correspondence with the name “apoptosis” (in Greek – the fall of the leaf), PCD also occurs in plants, playing the same role as in animals. Plants use PCD both in the course of development (for instance, during xylem formation, seed germination, prevention of self-pollination, and senescence) and in response to osmotic, thermal, and oxidative stresses and in defense from pathogens. Like in animals, PCD in plants takes various forms [3, 4], but a series of common PCD features can be traced in both kingdoms. These features include DNA fragmentation, cytochrome c release from mitochondria, cell shrinkage, generation of reactive oxygen species, exposure of phosphatidylserine, etc. [5].
It is worth noting that molecular mechanisms of plant PCD are much less studied than those of animal cell apoptosis. However, similar morphological features of animal and plant PCD imply the existence of similar fundamentals in the organisms of these two kingdoms used for PCD. In this regard, it is intriguing and significant that caspases, which generally fulfill PCD in animals, are absent in plants, as evident from sequencing of plant genomes. At the same time, much data suggests that inhibitors of animal caspases can suppress PCD development in plants. In connection with this, in plant PCD activation of unidentified caspase-like proteases is also observed, and these can hydrolyze various peptide substrates of caspases [6]. These data suggest that PCD in plants involves proteases that are functional analogs of animal caspases, but which are structurally different from caspases.
In this review, we focus on similarity and difference in PCD in plants and animals and give a modern view on plant proteases that might fulfill the role of caspases in PCD in plants.
CASPASES – APOPTOTIC PROTEASES OF ANIMALS
Structure and properties of caspases. Animal caspases differ from other proteases in a number of features. “Caspase” is an acronym from cysteine-dependent aspartate-specific proteases. Approximately 10 caspases having strict aspartate specificity of hydrolysis and differing in the preferred recognition site motif have been identified in mammals [7]. To avoid untimely triggering of the cell death mechanism, caspases are synthesized and stored in the cytoplasm as inactive precursors – procaspases. Procaspases are activated through their processing when the pro-domain is removed and the major part of the protein is cleaved into two subunits: p20 and p10. The active enzyme is a homodimer, where each monomer consists of one p20 chain and one p10 chain. The complex forms two symmetric active sites [8]. The catalytic dyad includes amino acid residues of the p20 chain and consists of an active-site cysteine residue that is a part of the conservative sequence QACXG and a histidine residue.
The absolute specificity of animal caspases to peptide bond hydrolysis after the D residue has already become “the talk of the town”. Caspases are very selective and usually make one, or rarely two, breaks per protein, i.e. are not degrading but processing proteases. This is due to the fact that caspases usually recognize in the substrate a tetrapeptide sequence with an aspartic acid residue at its C-terminus, after which the peptide bond is hydrolyzed (P1 substrate position). The preferred hydrolysis sequences for different caspases are determined by the amino acid motif of the recognition site [9, 10].
To date, about 400 caspase-cleaved substrates have been described (these target proteins are presented in the CASBAH database, http://www.casbah.ie). They include structural proteins, transcriptional and translational regulator proteins, kinases, signaling pathway components, pathogen proteins, etc. The limited proteolysis of cell proteins by caspases is aimed at successive switching of cellular pathways from life to programmed death. The notion that fragmentation of target proteins by caspases leads to inactivation of proteins vitally important to the cell and therefore results in its death is only partly true. Another objective of caspase hydrolysis is activation of proapoptotic mechanisms in a dying cell. So, fragmentation of the Bid protein from the Bcl-2 family provides the formed Bid fragment (so-called tBid, truncated Bid) the ability to be directed to mitochondria and to promote cytochrome c release from the mitochondria, which leads to a drastic increase in caspase activity in the cell (see below) [11, 12].
Another canonical example of activation through caspase hydrolysis is CAD nuclease. In healthy cells, the activity of this nuclease is suppressed due to its interaction with the inhibitor protein ICAD. Caspase-3 activated during the induction of apoptosis introduces two breaks in the ICAD molecule, which results in elimination of inhibition, activation of the nuclease through dimerization, and, finally, fragmentation of internucleosomes of cellular DNA with formation of a DNA “ladder” so typical of apoptotic cells [13-15].
However, it should be noted that the total number of caspase target proteins is relatively low – about 2% of all proteins of mammalian cells. Whether the hydrolysis of all targets by caspases is necessary for apoptosis or some proteins are merely “innocent bystanders” [16] is in most cases still an open question needing further investigation.
Caspases are divided into two groups by their functions in the apoptotic cascade. Initiator caspases (caspases-1, -2, -4, -5, -8, -9, -10, -11, -12) are activated in response to proapoptotic or other stimuli and participate in the processing (i.e. activation) of precursor proteins of other caspases, thereby forming a cascade of proteolytic enzymes. Effector, or executioner, caspases (-3, -6, -7) are activated by upstream initiator caspases and hydrolyze various cell proteins (see above), causing cell death [17, 18]. The structures of these enzymes are also different in accordance with this division. Initiator caspases have an extended pro-domain with one or two motifs responsible for the interaction with adaptor molecules. These are so-called DED (death effector domain) and CARD (caspase recruitment domain). Effector caspases have a shorter pro-domain.
Caspases can be also divided into proapoptotic and proinflammatory. Proapoptotic caspases (-2, -3, -6, -7, -8, -9, -10) are involved mainly in implementation of PCD. Proinflammatory caspases (-1, -4, -5, -11, -12) participate in the processing of cytokines during inflammation. However, since the activation of proinflammatory caspases can provoke apoptosis, this subdivision of caspases into groups is convenient but conditional. At the same time, more and more data demonstrate that caspases may be involved in different cellular processes unrelated to apoptosis or inflammation. It has been shown that caspase-8 participates in proliferation of immune cells [19-22] and in cell differentiation [23]. Caspase-3 is involved in differentiation of the long-living cells of skeletal muscles, osteoblasts, and neurons [24, 25]. A case in point is the involvement of proinflammatory caspase-1 in the processing of precursors of interleukins IL-1β and IL-18 [26, 27]. In addition, caspase-3 is able to process the precursor protein of IL-16 [28].
Caspase activation mechanisms. Caspases are activated upon the receipt of certain proapoptotic signals by the cell [29]. Two pathways of caspase activation during PCD induction have been described. One is associated with a group of transmembrane proteins, “death receptors”, which act as surface sensors locating external ligands signaling about the need for apoptosis. Among the best characterized “death receptors” are the tumor necrosis factor receptor (TNFR1), as well as Fas, DR3, TRAILR1 (TNF related apoptosis-inducing ligand receptor 1), TRAILR2, etc. [30]. Upon binding of the respective ligands, the death receptors multimerize with the formation of death-inducing signaling complexes (DISC complexes). Adaptor proteins are recruited to the DISC complex from the cytoplasmic side. For the Fas or TRAIL receptors, for example, it is a Fas-associated DD (FADD) protein, which is included in the complex via its C-terminal DD-domain, while its N-terminal death effector domain (DED) interacts with the same domain of caspase-8. Oligomerization of caspase-8 molecules in the DISC complex is considered to trigger autocatalytic activation of the caspase and, thereby, initiation of programmed cell death [17, 31]. Depending on the type of cells, caspase-8 can activate executive caspases -3 and -7 by cleaving the pro-domain, and this seems to be sufficient for apoptotic cell death. In other cases, the signal received by caspase-8 may be amplified via the mitochondrial apoptotic pathway [12].
The mitochondrial (“internal”) pathway is another pathway leading to cell death. It is switched on in the case of internal cell defects (DNA damage, various stresses, cytotoxic agents). Regulation of this pathway involves a large group of proteins from the Bcl-2 family. The latter includes both pro- and antiapoptotic proteins [32-39]. The perception of an apoptosis-inducing signal activates the proapoptotic proteins of this family, which form a Bak–Bax oligomeric complex in the outer mitochondrial membrane. This results in formation of channels through which cytochrome c is released from the mitochondria [40]. Cytochrome c, in turn, stimulates the assembly of a complex named the “apoptosome” [41] and triggers a sequence of events leading to the activation of procaspase-9. The apoptosome is a multi-protein complex comprising the following proteins: Apaf-1, cytochrome c, and dATP/ATP as a cofactor [42-44]. It serves as a “platform” for procaspase-9 binding and dimerization, which, in turn, leads to the autocatalytic processing of procaspase. This results in formation of two subunits of caspase-9, p35 and p12, combined into active dimers [45]. After activation in the apoptosome, caspase-9 triggers the processing of caspases-2, -3, -6, -7, -8, and probably caspase-1 [18, 46].
The two pathways of activation of apoptotic events are not independent. The proapoptotic protein Bid is directly cleaved by caspase-8, and the formed C-terminal fragment of this protein stimulates the release of cytochrome c from mitochondria [12], thereby increasing the apoptotic effect of the signal arriving through external receptors.
Regulation of caspase activity. Since the decision whether “to live or to die” is of vital importance for the cell and for the organism as a whole, it would be strange if caspase activity was not controlled in different ways. It is known that there is a multistep system of caspase activity control [47]. The induction of apoptosis is accompanied by abrupt increase of expression of the caspase genes [48-50]. The activity of caspases and, consequently, the development of apoptotic events are also regulated by various kinase signaling pathways. It has been shown, for example, that phosphorylation of caspase-9 leads to inhibition of its activity [51, 52] and suppression of apoptosis. In addition, animal cells contain endogenous inhibitor proteins capable of regulating the activity of mature caspases in vivo. The most significant of them are proteins of the IAP (inhibitor of apoptosis) family [53, 54]. They can bind caspases and not only neutralize the low level of caspase activity, but also create a barrier, above which a drastic activation of the caspase cascade begins [55]. Proteins that derepress IAP-bound caspases have also been characterized. Upon induction of PCD, these proteins (Smac, HtrA2, and some other proteins) are released from the intermembrane space of mitochondria with the aid of Bcl-2 proapoptotic proteins. Smac is able to displace IAP from its complex with caspase, thereby activating the apoptotic protease [56]. HtrA2 seems to act in an analogous manner. However, since HtrA2 is a protease itself, it has another way of eliminating caspase inhibition, this time irreversibly. HtrA2 can cleave most of the known IAPs, thereby activating caspases [57, 58]. In addition, some viral proteins (baculovirus protein p35, cowpox virus serpin CrmA) are able to inhibit caspases in the cells of the host organism [59-61], which is not very surprising because in many cases rapid cell death prevents replication of the virus.
PROGRAMMED CELL DEATH OF PLANTS
Forms and manifestations of cell death. As mentioned above, apoptosis in animals and PCD in plants have some similar morphological features [3-5]. However, PCD manifestations in plants have certain specificity. In some PCD models, DNA fragmentation in plant cells is accompanied by formation of extended DNA fragments but not an internucleosomal “ladder”. A substantial difference between animals and plants is also observed at the final stage of PCD development. In animals, the dying cell forms apoptotic bodies that are instantaneously phagocytized, thus allowing to avoid the lysis of dying cells and the inflammatory response of the organism. In plants, phagocytosis of dying cells is lacking not only due to the absence of professional phagocytizing cells, but also due to the presence of rigid cellulose walls separating the cells. The formation of apoptotic bodies has not been observed in plants either. Therefore, apoptotic plant cells must be eventually lysed and their contents are utilized.
The degree of cell wall degradation may vary depending on the type of tissue formed. Deep degradation is observed during aerenchyma formation, during leaf perforation formation, and when petals die off [62-64]. As a result, an empty space is left in the place of the dead cell. But every cloud has a silver lining. The triggering of the PCD mechanism that leads to the formation of aerenchyma (channels through which oxygen can be delivered to the roots from the above-ground parts of a plant) allows some plants (particularly rice) to survive on flooded soils. In other cases, the cell wall remains intact as, e.g. in the cases of xylem formation, rearrangement of leaf tissues, or fruit body formation [65, 66].
Recently an attempt has been made to classify plant cell death by a set of typical morphological characters. According to this classification, two main types of cell death are recognized: vacuolar cell death and necrotic cell death [67]. Vacuolar cell death is considered as a combination of autophagy performed by vacuoles and accompanied by increase in their sizes, followed by the release of hydrolases from the lytic vacuoles as a result of disruption of the vacuolar membrane (tonoplast). At the same time, the morphology of cell organelles and the integrity of cell plasma membrane are preserved till the moment of tonoplast disruption. Such type of cell death takes days and is typical of PCD occurring in the course of development of the organism.
On the contrary, necrotic death is accompanied by rapid disruption of the plasma membrane, shrinkage of the protoplast, disturbance of mitochondrial function, accumulation of active oxygen forms, and absence of characteristic features of vacuolar death. Necrotic death is believed to occur under conditions of abiotic stresses.
However, there are many cases of plant PCD not falling within either of the described categories. For example, hypersensitive response (HR) of plant cells to infection by pathogens (see below) is a well-described form of PCD but, at the same time, combines the signs of both vacuolar and necrotic death. In addition, shrinkage of the protoplast may not indicate disruption of the cell plasma membrane. For example, in one of the described HR variants the cell plasma membrane remains intact in spite of shrinkage of the protoplast [68], which shows the absence of a direct relationship between these phenomena.
It is clear that the proposed classification based solely on morphological features and assuming quite a number of non-classifiable exclusions is tentative. Therefore, it would be advisable to have a notion about the molecular mechanisms and basic components of the PCD apparatus in plants for its improvement.
Some data of that kind have been obtained in the study of hypersensitive response of plants. HR as a form of PCD is due to the fact that plants, in contrast to animals, have no immune system that could neutralize pathogens and infected cells. Therefore, plants use another strategy: induction of suicide of infected and surrounding cells. This prevents reproduction of the pathogen, on one hand, and creates a barrier of dead cells separating the pathogen from healthy tissue, on the other hand [69, 70]. The morphological features of cell death during HR in many respects coincide with those enlisted above, which are observed during PCD induced by other stimuli. HR induction requires recognition by a special protein of the plant (the R (resistance) gene product) of the respective protein of the pathogen (the product of the so-called avirulence (Avr) gene) [71, 72]. The R and Avr gene pairs can encode various proteins, or these genes may be absent. In the case of tobacco plants infected by the tobacco mosaic virus (TMV), the resistance gene is the so-called N gene, which is present not in all tobacco varieties. The product of this gene recognizes the viral protein replicase (which in this case is the product of the avirulence gene) and triggers HR. As a result, at the cost of death of a limited number of cells, the plant prevents the development of viral infection. Plants lacking the resistance gene do not respond to infection by induced cell death (HR); as a result, the pathogen spreads over the whole plant.
Vacuoles play a significant role in PCD that occurs not only during the development of a plant, but also during HR induced by infection of plants by viral, bacterial, or other pathogens. Two scenarios of PCD development have been described. During viral infection, the tonoplast is lysed with the release of the lytic enzymes of vacuoles into the cytosol [73, 74]. This is of biological significance because the overwhelming majority of plant viruses reproduce just in the cytosol.
During bacterial or fungal infection, a pathogen is located outside the plant cell, in the intercellular fluid (apoplast). Such pathogens affect plant cells by means of so-called “effector” proteins secreted by pathogens into plant cells. For controlling some extracellular pathogens, the vacuolar membrane can be fused with the plasma membrane, permitting the hydrolytic enzymes of the vacuole to be released into the extracellular space [75]. Membrane fusion is induced by the interaction between the plant R-gene product and the pathogen avirulence factor and ends not only with neutralization of the pathogen, but also with induction of PCD in the infected plant cells. The process of fusion of the vacuolar and plasma membranes during PCD caused by certain pathogenic strains was shown to require the functioning of plant cell proteasomes. Inhibition of proteasome activity by peptide inhibitors suppresses membrane fusion and the release of vacuolar proteins into the intercellular space [75]. RNA silencing of any of the genes encoding the subunits of Arabidopsis thaliana proteasome also inhibits membrane fusion.
Proteasome activity is important not only for membrane fusion, but also for the development of HR during bacterial infection. The authors [75] measured the percentage of dead cells (by their ability to be stained with trypan blue and by the electrical conductivity of tissues increasing during cell death) and thereby showed that inhibition of the activity of any of the proteasome subunits prevents HR development.
It is interesting that the peptide inhibitor of human caspase-3, Ac-DEVD-FMK (see the next section for more detailed information about the structure of peptide inhibitors of caspases), prevented HR in the case of bacterial infection, which suggested to the authors [75] that membrane fusion requires an activity similar to the activity of caspase-3, and this caspase-like activity might be typical of the plant proteasome.
It should be noted that the fact that animal and yeast proteasomes possess a caspase-like activity has been known for a long time [76]. Moreover, the animal proteasome inhibitor Ac-APnLD-CHO (nL = norleucine) proved to be an inhibitor of the plant proteasome subunit PBA1 as well, indicating the possible presence of caspase-like activity in this subunit. The silencing of the Arabidopsis PBA1 gene reduced the DEVDase activity found in extracts, which did not contradict the above suggestion but, however, was not strict evidence for it. The inhibition of other proteasome subunits possessing trypsin- and chymotrypsin-like activities was observed as well. Moreover, cell death was also suppressed by the common proteasome inhibitor clasto-lactacystin β-lactone, as well as by silencing of the genes of other proteasome subunits. It seems that the determining factor in implementation of this type of PCD is the activity of the proteasome as a whole but not the supposed caspase-like activity of subunit PBA1. Hence, the application of biotinylated inhibitor DEVD-FMK resulted in the inhibition of activity of not only PBA1, but also, strange as it may seem, of other proteasome subunits. Nevertheless, the PBA1 subunit bound the DEVD-FMK inhibitor. However, the traditional proteasome inhibitor MG132 (LLL-CHO) containing no D residue was also able to modify PBA1 [77]. It seems that the question whether any plant proteasome subunit displays a specific caspase-like activity and whether this activity is necessary for plant PCD implementation needs further elucidation.
Approaches for detection of caspase-like plant proteases. In spite of some similar features of PCD in animals and plants, the question about the similarity of molecular mechanisms of PCD in the two kingdoms is still open. For example, the absence of caspases (the key apoptotic enzymes of animals) in plants is a striking example of difference (at least technically) between animal and plant PCD. Therefore, it is highly relevant to determine whether any plant proteases perform the functions of caspases during PCD and, if so, what these proteases are.
An argument in favor of the assumption that caspase-specific proteases of plants exist and participate in PCD was the fact that the protein inhibitors of animal caspases (baculovirus proteins p35 and Op-IAP), which are produced in plants, were reported to prevent the development of PCD. Transgenic tomato plants carrying the p35 gene proved to be more resistant to toxin-induced PCD and towards infection with various pathogenic fungi [78], while HR development in transgenic (by the p35 gene) tobacco leaves caused by the Pseudomonas syringae infection was partially suppressed [79]. In both cases transgenic plants, possessing the gene of mutant protein p35, which is not an inhibitor of animal caspases, had no antiapoptotic properties. The expression of the p35 gene in the embryonic callus of maize also suppressed PCD [80]. Transient expression of the p35 gene in A. thaliana protoplasts prevented DNA fragmentation and cell death upon UV radiation [81]. Transgenic tobacco plants carrying the gene of protein Op-IAP, a caspase inhibitor, demonstrated higher resistance to PCD exhibited by suppression of the formation of dead cell areas during infection with viral or bacterial pathogens [82]. These results suggested that caspase-specific proteases may exist in plant cells, and that these enzymes may be involved in PCD and the protective reactions of plants.
Since the natural protein substrate of hypothetical plant caspases was unknown, the most straightforward way of finding the caspase-like activity in plants consisted in using peptide fluorogenic substrates and peptide inhibitors of animal caspases. The canonical peptide substrates of caspases are tetrapeptides with a XXXD-AFC (AFC, 7-amino-4-trifluoromethyl-coumarin) sequence, where the motif preceding the amino acid residue D, after which the bond is hydrolyzed by the enzyme, is typical of each (or several) animal caspase(s). As a result of enzymatic hydrolysis, AFC is cleaved from the C-terminal aspartate and starts to emit fluorescence (at 505 nm), allowing the fluorometric detection of substrate cleavage. The specific peptide inhibitors of animal caspases have the same XXXD amino acid sequences, but the aspartate residue is modified by the aldehyde (CHO), fluoromethyl ketone (FMK), or chloromethyl ketone (CMK) group that modifies the amino acid residues of the active site (cysteine in case of caspases) during substrate binding to the enzyme [83].
The peptide substrates of caspases were used for the first time to reveal the activity of plant apoptotic proteases in 1998 in the classic publication of del Pozo and Lam [84]. They used extracts from the NN-genotype tobacco leaves infected with the tobacco mosaic virus (TMV), where HR was developing, as well as extracts from uninfected plants. The proteolytic activity hydrolyzing the specific caspase-1 substrate Ac-YVAD-AFC was detected in the extracts from apoptotic leaves but not in the extracts from healthy leaves. Moreover, the specific caspase inhibitors Ac-YVAD-CMK and Ac-DEVD-CMK could both suppress this YVADase activity and prevent PCD induced by P. syringae bacterial infection in tobacco plants. The latter circumstance is particularly significant because it suggests that the revealed YVADase activity may relate to implementation of the cell death program.
Dozens of works have been published since that time where various peptide substrates and animal caspase inhibitors have been tested for the presence of caspase-like activities in plants. Depending on the peptide at hand, authors named the revealed activities DEVDase, VEIDase, YVADase, etc. (see review of Bonneau et al. [6]). Interestingly, the spectrum of detected caspase-like activities that are revealed during plant PCD caused by various biotic and abiotic stimuli can be substantially different. For example, DEVDase activity is registered rather frequently, but in some cases this activity is absent and another is observed (e.g. YVADase activity).
Summarizing the results obtained by the described approach (the application of peptide substrates and animal caspase inhibitors), one should note that they afforded solid grounds to believe that caspase-specific proteases are activated in different model systems of plant PCD, and that the activities of these proteases may be important for implementation of PCD.
Metacaspases. Soon after the Arabidopsis and rice genomes were sequenced, it became obvious that plants (at least those with the sequenced genomes) do not contain caspase genes that could be detected by simple homology search. Therefore, the above-described caspase-like activities observed during plant PCD and involved in PCD implementation seem to be typical of proteases that are structurally different from caspases. This conclusion was not quite obvious; therefore, the detection by high-sensitive bioinformatics analysis of two families of proteases that distantly resembled caspases caused general enthusiasm [85]. One of these families which is present in animals and myxomycetes (and, hence, of no immediate interest for us now) was named paracaspases, and the other one typical for plants, fungi, and protozoa was named metacaspases.
According to their structure, metacaspases belong to the clan of cysteine-dependent (CD) proteinases, which also includes caspases (that cleave peptide bond after a D amino acid residue), legumains (cleavage after the N residue and, more rarely, N and D residues), separases (cleavage after an R residue), and bacterial proteases clostripains and gingipains (cleavage after R and K). These proteolytic enzymes are combined into a single clan because they have a common type of three-dimensional structure, the so-called “caspase/hemoglobinase fold” [86], and they contain a typical dyad of Cys and His catalytic residues. Plant genomes contain approximately 10 metacaspase genes. Metacaspases are synthesized as precursor proteins and can be subdivided into two types [87]. Type I plant metacaspases (predominant in some plants) possess an N-terminal pro-domain that is absent from type II metacaspases. In the analogy with caspases, metacaspase structure comprises large (20 kDa) and small (~10 kDa) subunits. In the case of the precursor protein of type II metacaspases, the small subunit is separated from the large one by a relatively long linker sequence. The large metacaspase subunit comprises, as in the case of caspases, the His and Cys catalytic dyad. Mature metacaspase is formed through the autocatalytic processing of the zymogen [88, 89]. Metacaspases are localized in plant cell cytosol (figure).
Diverse localization of proteases involved in programmed cell death in plants. Metacaspases exist in the cytosol, VPE locates in the vacuoles. Phytaspases are secreted from the plant cell (by the canonical pathway including endoplasmic reticulum (ER) and Golgi apparatus) into the intercellular fluid (apoplast) of healthy plants. On induction of cell death, the phytaspases are rapidly transferred from the apoplast into the cytosol
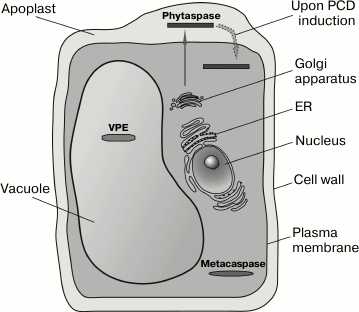
The discovery of metacaspases in plants gave reasonable grounds to anticipate that metacaspases are the sought caspase analogs in plants with the specificity and functions of caspases. At first it seemed that plant metacaspases actually possessed the specificity of animal caspase hydrolysis. Caspase activity was observed to increase upon superexpression of the metacaspase genes in plants and to be suppressed upon RNA interference of metacaspases, as was shown through the use of fluorogenic peptide substrates of animal caspases [90, 91]. However, everything fell into place when plant metacaspases were isolated and recombinant enzymes were obtained, and the autocatalytic processing of metacaspase precursor proteins was investigated. It turned out that metacaspases did not hydrolyze the peptide substrates of caspases, but they possessed strict Arg- and Lys-specificity [88, 92-94]. The subsequent longstanding discussion in the literature on whether metacaspases could be considered caspases [95-97] has recently come to an end with the conclusion that metacaspases are not caspases because they have no aspartate substrate specificity.
Nevertheless, though metacaspases turned out not to be the aspartate-specific apoptotic plant proteases that had been sought, what have we learned about plant PCD during the study of metacaspases? The modern point of view on plant metacaspases is that metacaspases are involved in many plant cell processes including PCD. The silencing of the gene of one of the metacaspases resulted in disturbance of terminal cell differentiation and embryo development in spruce [91]. Knockouts of the genes of some Arabidopsis metacaspases displayed no marked disturbance in PCD implementation, probably due to redundancy (overlapping functions) of metacaspases. At the same time, metacaspase-8 (AtMC8) superproduction intensified and RNA interference suppressed the level of PCD caused by UV radiation and hydrogen peroxide in protoplasts [98]. Seeds and shoots of Arabidopsis plants with AtMC8 knockout showed enhanced resistance to the herbicide methyl viologen. The involvement of Arabidopsis metacaspase AtMC4 in implementation of cell death caused by oxidative stress and Fumonisin B-1 was described [99]. Recently, it was shown that type I Arabidopsis metacaspase AtMC1 is a positive regulator of HR, while another metacaspase (AtMC2) suppresses the proapoptotic effect of the former [100]. Interestingly, this suppression does not require the presence of proteolytic activity in AtMC2, because the active site Cys residue mutant retained the antiapoptotic properties. Thus, metacaspases seem to be multifunctional cell enzymes.
The only known protein substrate of plant metacaspases is the evolutionally conservative protein TSN (Tudor staphylococcal nuclease), the function of which in plants has not yet been established [101]. The recombinant TSN protein is hydrolyzed by spruce metacaspase mcII-Pa at four sites in accordance with metacaspase specificity (after the R and K residues). Analogous fragmentation of this protein is observed during PCD caused by oxidative stress or occurring during embryo development. In is interesting to note that the human TSN protein is a target of caspase-3; however, in this case only one break of regulatory significance is introduced into the TSN molecule, and it does not coincide with the sites of metacaspase hydrolysis.
Vacuolar protease VPE. The vacuolar processing enzyme (VPE) of plants is a cysteine-dependent protease and is localized in plant cell vacuoles (see figure), where it participates in the processing of vacuolar proteins. VPE is related to legumains, which belong to the clan of CD proteases, which includes caspases, metacaspases, and a number of other proteases (see above). The affiliation of VPE with the CD clan is demonstrated by three-dimensional fold of the proteolytic domain and the presence of catalytic residues His and Cys in characteristic positions. However, the similarity between the amino acid sequences of VPE and other members of the clan is very low. Like most proteases, Arabidopsis VPE is synthesized as an inactive precursor, and the cleavable pro-domains are present at both N- and C-termini of the protease domain. The precursor protein can be processed autocatalytically [102, 103]. At the very N-terminus of the precursor protein, a signal peptide resides that directs the synthesized protein to the vacuole.
VPE possesses a substrate specificity typical of legumains, and it hydrolyzes a peptide bond after an asparagine (N) residue; common inhibitor of this protease is Ac-ESEN-CHO. It has been shown through the use of a number of synthetic peptides that correspond to some plant protein sequences that VPE can hydrolyze peptide bonds also after some D residues, though the efficiency of such cleavage is lower than that occurring after an N residue [102]. Moreover, one of the caspase inhibitors (biotinylated VAD-FMK) when introduced into leaves became covalently bound to VPE [104]. It is interesting that Ac-YVAD-CHO could act as a competitor of VAD-FMK, while Ac-DEVD-CHO had no such ability. The binding of Arabidopsis VPE to VAD-FMK and YVAD-CMK was confirmed by another research group [105]. Based on these findings, the authors concluded that plant VPE has caspase-1 activity. The “partially purified” recombinant γ-VPE which had been produced in insect cells (one of four VPE forms existing in plants) [106] was able to hydrolyze a fluorogenic substrate of VPE, Ac-ESEN-AMC (AMC, 7-amino-4-methylcumarin), and caspase-1, Ac-YVAD-AMC, but it did not hydrolyze Ac-DEVD-AMC (the substrate of caspase-3).
Another example relates to the Papaver VPE. The recombinant enzyme was produced in E. coli cells and purified by affinity chromatography. The enzyme was able to bind biotinylated DEVD-CHO but not YVAD-CHO. Surprisingly, such activity was common for the precursor enzyme and the enzyme that preserved the N-terminal pro-domain, but not mature VPE [107]. Nevertheless, the Papaver VPE exhibited hydrolytic activity not only towards the Ac-DEVD-AMC substrate, but also towards the YVAD-derivative, as well as the derivative of IETD and, to a lesser degree, LEVD and VEID. In these cases as well, only the precursors but not the mature VPE displayed proteolytic activity. Another unexpected result was that the proVPE that displayed the activity did not undergo autocatalytic processing.
Thus, at present it is not quite clear which caspase-like activity VPE possesses and whether the processing of the precursor protein is required for the activation of the enzyme and how it may occur. The existing discrepancies are probably associated with differences between species or with the methods of isolation of the enzyme for the analysis of its activity. Nevertheless, is VPE related to plant PCD? The available data demonstrate that VPE may be involved in implementation of PCD, and it is associated not least with the vacuolar localization of this enzyme (see figure). VPE has been shown to take part in PCD that occurs in tobacco leaves during viral (TMV) infection and includes the breakage of the tonoplast, DNA fragmentation, and formation of dead cell areas. The silencing of four VPE genes suppresses the collapse of the vacuole, DNA fragmentation, and formation of necroses [74, 104, 108]. How exactly VPE may be involved in the described processes remains unknown. Interestingly, expression of the VPE genes increases in the beginning of HR and then declines rapidly. This effect is probably indicative of the role of VPE at the early stages of cell death. VPE is also involved in PCD induced by some fungal toxins. Morphologically, this cell death is similar to TMV-induced HR [106]. In this case, inactivation of all four VPE genes also suppresses PCD.
It should be noted that inhibition of VPE (by Ac-YVAD-FMK, in particular) had no effect on development of PCD in response to bacterial infection, in contrast to PCD caused by viral infection [75]. At the same time, it is worth mentioning that the fusion of the vacuolar and plasma membranes that takes place during PCD caused by certain pathogenic strains does not depend on VPE either.
Thus, a relationship between VPE activity and some forms of plant PCD is evident. Elucidation of the extent to which this involvement of VPE in cell death may be associated with the assumed caspase-like activity of the enzyme and finding of the apoptotic protein targets of VPE is in prospect.
Subtilisin-like plant proteases with aspartate specificity. The alternative approach to the search of PCD-related plant proteases based on identification of a plant protease that hydrolyzes the native target protein at the same site as the animal caspase proved to be efficient. The VirD2 protein of the agrobacterium Agrobacterium tumefaciens – a plant pathogen, was shown to be specifically fragmented by human caspase-3. It then was shown that induction of HR in Nicotiana tabacum plants of the NN genotype caused by TMV infection is accompanied by activation of a plant protease with a similar specificity in hydrolysis of the VirD2 protein (protein cleavage after the D residue in the TATD motif) [109]. The revealed protease was named phytaspase (from “plant aspartate-specific protease”) [110]. Phytaspase activity may also be registered during mechanical destruction of plant tissue, which has revealed phytaspase activity in quite different plants [111] including dicotyledons and monocotyledons.
Identification of tobacco and rice phytaspases demonstrated that phytaspases are subtilisin-like proteases (subtilases) of plants [110]. Although it had been assumed that caspase-like (in the functional sense) plant proteases could be structurally different from animal caspases (otherwise, phytaspases would be identified long ago by homology), it was hard to expect that the difference would be so drastic. Indeed, the structure of subtilisin-like proteases is dramatically different in structure from caspases. Subtilases are Ser-dependent proteases, while caspases are Cys-dependent. An active caspase is a tetramer consisting of two large and two small subunits, while phytaspase is a monomer. The presence of a potential signal peptide in the precursor protein of phytaspases (see below) could be indicative of extracellular localization of the enzyme, while caspases are intracellular proteins (see table).
Comparison of properties of aspartate-specific apoptotic animal and
plant proteases: caspases and phytaspases
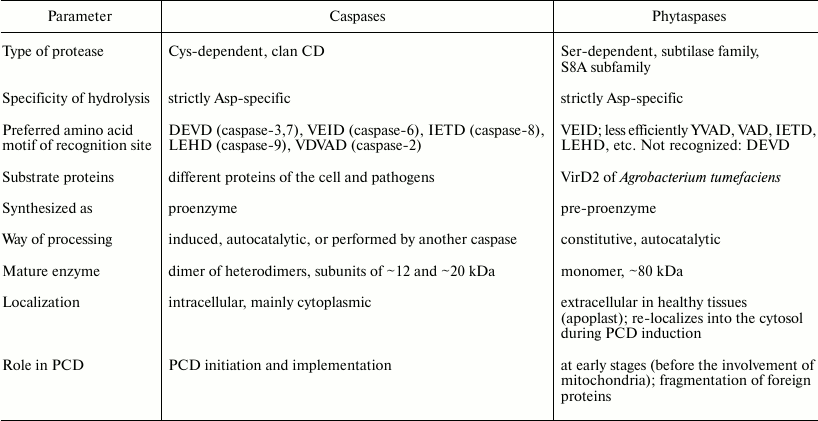
Nevertheless, the study of the substrate specificity of tobacco and rice phytaspases has shown that these enzymes, just as caspases, hydrolyze substrates strictly after a D residue in a certain amino acid context [110]. The optimal substrate of phytaspases is the Ac-VEID-AFC peptide (substrate of caspase-6), though a comparable (2-4 times lower) rate of hydrolysis was observed for the derivatives of VAD (substrate of various caspases), YVAD (substrate of caspase-1), VDVAD (the substrate of caspase-2), IETD (the substrate of caspase-8), and LEHD (substrate of caspase-9) (table). The only exception was Ac-DEVD-AFC (substrate of caspase-9), which was not hydrolyzed by phytaspases at all.
The ability of phytaspases from different plant organisms to hydrolyze various peptide substrates may create an illusion of a relatively low selectivity of the plant enzyme. However, such conclusion seems to be erroneous on taking into consideration the fact that phytaspases exceed the human caspase-3 in selectivity at the level of protein substrates. Phytaspase is undoubtedly a “processing”, not a “digestive” proteolytic enzyme. However, this statement concerns also animal caspases.
Phytaspase with its broad spectrum of hydrolyzed peptide substrates of caspases alone is able to explain almost the entire diversity of caspase-like activities revealed during plant PCD on such substrates. Therefore, the number of various caspase-like enzymes in plants is probably not oppressively large, as it was commonly supposed. Yet, there is an exception. Phytaspase has no DEVDase activity (table), which is revealed quite often during PCD in plants. Therefore, it may be anticipated that at least one caspase-like protease of plants has not yet been discovered.
Phytaspases were shown to participate in plant PCD caused by biotic and abiotic stresses. Cell death was stimulated by the enhanced level of phytaspase activity during superproduction of the enzyme, while reduction of phytaspase activity by a specific inhibitor or through RNA interference suppressed PCD [110].
Thus, phytaspases are similar to caspases both in their specificity and in the role of these proteases in PCD. However, phytaspases are fundamentally different from caspases in structure, and this difference has important functional consequences. It has been shown that phytaspases are synthesized as inactive precursor proteins that contain an N-terminal signal peptide, pro-domain, and the protease domain. The N-terminal signal peptide as a part of the proenzyme directs phytaspase secretion from a plant cell. Active phytaspase is formed through pro-domain cleavage. This process is autocatalytic and constitutive, i.e. it occurs even in the absence of PCD. Pro-domain cleavage is required for the formation and secretion of the proteolytically active enzyme.
In healthy plant tissues, phytaspase accumulates in the intercellular fluid (apoplast) (see figure). Thereby, spatial uncoupling of the enzyme and its intracellular substrates is achieved. However, upon the induction of PCD, the phytaspase is re-localized from the apoplast into the dying plant cell and gains access to its intracellular target proteins [110]. The mechanism of this absolutely novel phenomenon is unknown, but there are grounds to believe that “retrograde” transport of phytaspase occurs specifically.
Localization of the processed phytaspase in the apoplast may imply that the enzyme has protective functions associated with proteolysis (neutralization) of the effector proteins of bacterial pathogens. The only natural target protein of apoptotic plant subtilases known at present is the protein VirD2 of the phytopathogenic bacterium A. tumefaciens. When infecting plants, the bacterial protein VirD2 with attached bacterial DNA (T-DNA) enters the cytoplasm and then is imported into the nucleus of the plant cell. This provides the integration of bacterial DNA into the plant genome and plant cell transformation [112]. For active transport into the nucleus, the VirD2 protein, despite being bacterial, is equipped with a nuclear localization signal [113]. It has been shown that the hydrolysis of agrobacterial VirD2 protein by phytaspase is a protective mechanism of plant cells limiting the delivery into the nucleus and expression of foreign (agrobacterial) DNA [114]. This is due to the fact that VirD2 cleavage by phytaspase at the TATD400 site results in removal of a short C-terminal fragment of the VirD2 protein. Since the nuclear localization signal of VirD2 is located in this very region, VirD2 loses the ability to be imported into the plant cell nucleus and to carry there the attached bacterial T-DNA.
Subtilisin-like proteases of the S8A subfamily which includes phytaspases and all other plant subtilases are usually not characterized by high specificity of hydrolysis [115, 116]. The ability to specifically hydrolyze substrates strictly after the D residue is generally uncommon for proteases. In this respect, it is worth noting that each plant species has dozens of subtilase genes (more than 50 family members for A. thaliana and more than 60 for rice Oryza sativa [117, 118]). Just a few representatives of these families have been characterized, and none of the previously identified subtilases had a phytaspase activity. It is an interesting question how many members of the subtilase family in each plant species have aspartate specificity: one or several? If several such proteases exist, it would be interesting to know whether these enzymes differ in the preferred sites of hydrolysis, localization, expression in tissues, and involvement in the process of PCD during development and in response to stresses.
Phytaspases display similarity with the animal and yeast subtilisin-like proteases, so-called proprotein convertases, which belong to the S8B subfamily (which apparently is absent from plants), in the high selectivity of substrate hydrolysis [119, 120]. Convertases are involved in the processing of precursor proteins, which results in formation of biologically active peptides and proteins, and one may assume an existence of a similar function of phytaspase in the “out-of-PCD” hours. It should be noted, however, that convertases introduce a break into target proteins after the basic amino acid residues (K, R) but not after D.
Plant subtilisin-like proteases apparently include also the saspase of oat [121]. The PCD model that helped to discover this activity was the oat Avena sativa plant infected by the fungus Cochliobolus victoria. The pathogen secretes an unusual toxin named victorin. Victorin-induced PCD is a HR to the infection [68, 122, 123]. The protease isolated from the oat leaves treated with victorin was named saspase (serine-dependent aspartate-specific protease). Saspase, just as phytaspase, is able to hydrolyze many peptide substrates of caspases [121, 124] and, at the same time, lacks the DEVDase activity. Several peptides of the enzyme were sequenced showing that the protein must be a plant subtilisin-like protease (subtilase). One may think that saspase is an oat phytaspase, but there is an interesting difference between the two classes of enzymes. Whereas phytaspases of two evolutionally distant plants (tobacco and rice) prefer the VEID derivative as a substrate, saspase does not hydrolyze VEID derivative at all [121]. The reason for this difference can be elucidated after identification of saspase.
Interestingly, saspase activity became detectable in the apoplast under PCD induction by victorin or heat shock, though before PCD induction such activity had not been observed in the apoplast [121]. Presumably upon PCD induction either rapid secretion of saspase into the apoplast or unmasking of the protease already present in the apoplast may occur.
CONCLUSION
PCD in plants displays quite a number of common features with PCD in animals but, on the other hand, it has a number of substantial differences. Proteolytic enzymes that are involved in PCD are localized in different compartments of plant cells: the cytoplasm (metacaspases), the vacuoles (VPE), and the intercellular fluid (phytaspases) (figure). However, the example of phytaspase which is being transferred from the apoplast into the cytoplasm upon PCD induction shows that localization of the enzyme in a certain compartment of healthy cells (tissues) does not exclude the functioning of this protease in a quite different compartment of dying cells. It is of considerable interest how the functions are distributed among the plant apoptotic proteases and whether they can influence each other’s function.
Among the proteolytic enzymes of plants described above which are involved in PCD, phytaspases most closely correspond to animal caspases by substrate specificity (table). At the same time, the apoptotic proteases of animals and plants are totally different in structure and, moreover, phytaspases are Ser-dependent, while caspases are Cys-dependent enzymes. The comparison of caspase and phytaspase properties gives the impression that animals and plants may have followed different tactics that eventually resulted in a similar policy decision: creation of proteases with similar function and specificity.
Animals and plants use different strategies with respect to their apoptotic proteases. Both caspases and phytaspases are synthesized as inactive precursor proteins; however, further their paths diverge. Procaspases are stored within animal cells. They are activated through the processing and association of subunits in response to PCD-inducing stimuli, which results in fragmentation of intracellular target proteins and cell death. In contrast to this scenario, prophytaspases are processed constitutively and autocatalytically, forming active enzyme even in the absence of a PCD-inducing stimuli. However, mature phytaspases are secreted from the cell into the apoplast (due to the presence of signal peptide in the precursor protein). It allows the active proteolytic enzyme to be spatially separated from intracellular target proteins so that unauthorized proteolysis and cell death can be avoided. Under PCD induction, phytaspase is transported from the apoplast into the cells, which results in fragmentation of intracellular proteins.
Thus, intracellular caspase activity is controlled at the level of processing of precursor proteins, while phytaspase activity is controlled at the level of enzyme transport from the apoplast into the cytoplasm. It may be concluded that plants have developed their own mechanism to control apoptotic proteases absent from (or not yet discovered) in animals [124, 125]. Thus, animal and plant cells demonstrate both common features and substantial differences in how they treat their apoptotic proteases.
One of the basic approaches for elucidation of the mechanism of action and new functions of PCD-related plant proteases is the identification of the target proteins of these enzymes. The study of cellular protein substrates will disclose important features in the molecular mechanisms of plant PCD, reveal novel signaling pathways, and allow more thorough comparison of the animal and plant machineries responsible for the key processes of cell life and death.
Work in the authors laboratory was supported by the Russian Foundation for Basic Research (projects No. 11-04-01120 and No. 11-04-00984) and by the Ministry of Education and Science of the Russian Federation (P334, 14.740.11.0168).
REFERENCES
1.Kerr, J. F., Wyllie, A. H., and Currie, A. R.
(1972) Br. J. Cancer, 26, 239-257.
2.Nicholson, D. W., and Thornberry, N. A. (1997)
Trends Biochem. Sci., 22, 299-306.
3.Williams, B., and Dickman, M. (2008) Mol. Plant
Pathol., 9, 531-544.
4.Reape, T. J., Molony, E. M., and McCabe, P. F.
(2008) J. Exp. Bot., 59, 435-444.
5.Reape, T. J., and McCabe, P. F. (2010)
Apoptosis, 15, 249-256.
6.Bonneau, L., Ge, Y., Drury, G. E., and Gallois, P.
(2008) J. Exp. Bot., 59, 491-499.
7.Degterev, A., Boyce, M., and Yuan, J. (2003)
Oncogene, 22, 8543-8567.
8.Walker, N. P., Talanian, R. V., Brady, K. D., Dang,
L. C., Bump, N. J., Ferenz, C. R., Franklin, S., Ghayur, T., Hackett,
M. C., Hammill, L. D., et al. (1994) Cell, 78,
343-352.
9.Thornberry, N. A., Rano, T. A., Peterson, E. P.,
Rasper, D. M., Timkey, T., Garcia-Calvo, M., Houtzager, V. M.,
Nordstrom, P. A., Roy, S., Vaillancourt, J. P., Chapman, K. T., and
Nicholson, D. W. (1997) J. Biol. Chem., 272,
17907-17911.
10.Talanian, R. V., Quinlan, C., Trautz, S.,
Hackett, M. C., Mankovich, J. A., Banach, D., Ghayur, T., Brady, K. D.,
and Wong, W. W. (1997) J. Biol. Chem., 272,
9677-9682.
11.Luo, X., Budihardjo, I., Zou, H., Slaughter, C.,
and Wang, X. (1998) Cell, 94, 481-490.
12.Li, H., Zhu, H., Xu, C. J., and Yuan, J. (1998)
Cell, 94, 491-501.
13.Liu, X., Zou, H., Slaughter, C., and Wang, X.
(1997) Cell, 89, 175-184.
14.Enari, M., Sakahira, H., Yokoyama, H., Okawa, K.,
Iwamatsu, A., and Nagata, S. (1998) Nature, 391,
43-50.
15.Sakahira, H., Enari, M., and Nagata, S. (1998)
Nature, 391, 96-99.
16.Timmer, J. C., and Salvesen, G. S. (2007) Cell
Death Differ., 14, 66-72.
17.Juo, P., Kuo, C. J., Yuan, J., and Blenis, J.
(1998) Curr. Biol., 8, 1001-1008.
18.Slee, E. A., Harte, M. T., Kluck, R. M., Wolf, B.
B., Casiano, C. A., Newmeyer, D. D., Wang, H. G., Reed, J. C.,
Nicholson, D. W., Alnemri, E. S., Green, D. R., and Martin, S. J.
(1999) J. Cell Biol., 144, 281-292.
19.Chun, H. J., Zheng, L., Ahmad, M., Wang, J.,
Speirs, C. K., Siegel, R. M., Dale, J. K., Puck, J., Davis, J., Hall,
C. G., Skoda-Smith, S., Atkinson, T. P., Straus, S. E., and Lenardo, M.
J. (2002) Nature, 419, 395-399.
20.Salmena, L., Lemmers, B., Hakem, A.,
Matysiak-Zablocki, E., Murakami, K., Au, P. Y., Berry, D. M., Tamblyn,
L., Shehabeldin, A., Migon, E., Wakeham, A., Bouchard, D., Yeh, W. C.,
McGlade, J. C., Ohashi, P. S., and Hakem, R. (2003) Genes Dev.,
17, 883-895.
21.Beisner, D. R., Ch’en, I. L., Kolla, R. V.,
Hoffmann, A., and Hedrick, S. M. (2005) J. Immunol., 175,
3469-3473.
22.Su, H., Bidere, N., Zheng, L., Cubre, A., Sakai,
K., Dale, J., Salmena, L., Hakem, R., Straus, S., and Lenardo, M.
(2005) Science, 307, 1465-1468.
23.Kang, T. B., Ben-Moshe, T., Varfolomeev, E. E.,
Pewzner-Jung, Y., Yogev, N., Jurewicz, A., Waisman, A., Brenner, O.,
Haffner, R., Gustafsson, E., Ramakrishnan, P., Lapidot, T., and
Wallach, D. (2004) J. Immunol., 173, 2976-2984.
24.Fernando, P., Kelly, J. F., Balazsi, K., Slack,
R. S., and Megeney, L. A. (2002) Proc. Natl. Acad. Sci. USA,
99, 11025-11030.
25.Miura, M., Chen, X. D., Allen, M. R., Bi, Y.,
Gronthos, S., Seo, B. M., Lakhani, S., Flavell, R. A., Feng, X. H.,
Robey, P. G., Young, M., and Shi, S. (2004) J. Clin. Invest.,
114, 1704-1713.
26.Howard, A. D., Kostura, M. J., Thornberry, N.,
Ding, G. J., Limjuco, G., Weidner, J., Salley, J. P., Hogquist, K. A.,
Chaplin, D. D., Mumford, R. A., et al. (1991) J. Immunol.,
147, 2964-2969.
27.Sleath, P. R., Hendrickson, R. C., Kronheim, S.
R., March, C. J., and Black, R. A. (1990) J. Biol. Chem.,
265, 14526-14528.
28.Zhang, Y., Center, D. M., Wu, D. M., Cruikshank,
W. W., Yuan, J., Andrews, D. W., and Kornfeld, H. (1998) J. Biol.
Chem., 273, 1144-1149.
29.Riedl, S. J., and Shi, Y. (2004) Nat. Rev.
Mol. Cell. Biol., 5, 897-907.
30.Lavrik, I., Golks, A., and Krammer, P. H. (2005)
J. Cell Sci., 118, 265-267.
31.Varfolomeev, E. E., Schuchmann, M., Luria, V.,
Chiannilkulchai, N., Beckmann, J. S., Mett, I. L., Rebrikov, D.,
Brodianski, V. M., Kemper, O. C., Kollet, O., Lapidot, T., Soffer, D.,
Sobe, T., Avraham, K. B., Goncharov, T., Holtmann, H., Lonai, P., and
Wallach, D. (1998) Immunity, 9, 267-276.
32.Hockenbery, D., Nunez, G., Milliman, C.,
Schreiber, R. D., and Korsmeyer, S. J. (1990) Nature,
348, 334-336.
33.Boise, L. H., Gonzalez-Garcia, M., Postema, C.
E., Ding, L., Lindsten, T., Turka, L. A., Mao, X., Nunez, G., and
Thompson, C. B. (1993) Cell, 74, 597-608.
34.Adams, J. M., and Cory, S. (1998) Science,
281, 1322-1326.
35.Oltvai, Z. N., Milliman, C. L., and Korsmeyer, S.
J. (1993) Cell, 74, 609-619.
36.Chittenden, T., Harrington, E. A.,
O’Connor, R., Flemington, C., Lutz, R. J., Evan, G. I., and
Guild, B. C. (1995) Nature, 374, 733-736.
37.Kiefer, M. C., Brauer, M. J., Powers, V. C., Wu,
J. J., Umansky, S. R., Tomei, L. D., and Barr, P. J. (1995)
Nature, 374, 736-739.
38.Yang, E., Zha, J., Jockel, J., Boise, L. H.,
Thompson, C. B., and Korsmeyer, S. J. (1995) Cell, 80,
285-291.
39.Wang, K., Yin, X. M., Chao, D. T., Milliman, C.
L., and Korsmeyer, S. J. (1996) Genes Dev., 10,
2859-2869.
40.Chipuk, J. E., and Green, D. R. (2008) Trends
Cell Biol., 18, 157-164.
41.Riedl, S. J., and Salvesen, G. S. (2007) Nat.
Rev. Mol. Cell. Biol., 8, 405-413.
42.Zou, H., Henzel, W. J., Liu, X., Lutschg, A., and
Wang, X. (1997) Cell, 90, 405-413.
43.Liu, X., Kim, C. N., Yang, J., Jemmerson, R., and
Wang, X. (1996) Cell, 86, 147-157.
44.Li, P., Nijhawan, D., Budihardjo, I.,
Srinivasula, S. M., Ahmad, M., Alnemri, E. S., and Wang, X. (1997)
Cell, 91, 479-489.
45.Yuan, S., Yu, X., Topf, M., Ludtke, S. J., Wang,
X., and Akey, C. W. (2010) Structure, 18, 571-583.
46.Lakhani, S. A., Masud, A., Kuida, K., Porter, G.
A., Jr., Booth, C. J., Mehal, W. Z., Inayat, I., and Flavell, R. A.
(2006) Science, 311, 847-851.
47.Shi, Y. (2002) Mol. Cell, 9,
459-470.
48.Wang, S., Miura, M., Jung, Y., Zhu, H.,
Gagliardini, V., Shi, L., Greenberg, A. H., and Yuan, J. (1996) J.
Biol. Chem., 271, 20580-20587.
49.Lin, X. Y., Choi, M. S., and Porter, A. G. (2000)
J. Biol. Chem., 275, 39920-39926.
50.Eckhart, L., Ban, J., Fischer, H., and
Tschachler, E. (2000) Biochem. Biophys. Res. Commun.,
277, 655-659.
51.Cardone, M. H., Roy, N., Stennicke, H. R.,
Salvesen, G. S., Franke, T. F., Stanbridge, E., Frisch, S., and Reed,
J. C. (1998) Science, 282, 1318-1321.
52.Allan, L. A., Morrice, N., Brady, S., Magee, G.,
Pathak, S., and Clarke, P. R. (2003) Nat. Cell. Biol., 5,
647-654.
53.Deveraux, Q. L., and Reed, J. C. (1999) Genes
Dev., 13, 239-252.
54.Fesik, S. W., and Shi, Y. (2001) Science,
294, 1477-1478.
55.Shiozaki, E. N., Chai, J., Rigotti, D. J., Riedl,
S. J., Li, P., Srinivasula, S. M., Alnemri, E. S., Fairman, R., and
Shi, Y. (2003) Mol. Cell, 11, 519-527.
56.Chai, J., Shiozaki, E., Srinivasula, S. M., Wu,
Q., Datta, P., Alnemri, E. S., and Shi, Y. (2001) Cell,
104, 769-780.
57.Yang, Q. H., Church-Hajduk, R., Ren, J., Newton,
M. L., and Du, C. (2003) Genes Dev., 17, 1487-1496.
58.Srinivasula, S. M., Gupta, S., Datta, P., Zhang,
Z., Hegde, R., Cheong, N., Fernandes-Alnemri, T., and Alnemri, E. S.
(2003) J. Biol. Chem., 278, 31469-31472.
59.Bump, N. J., Hackett, M., Hugunin, M., Seshagiri,
S., Brady, K., Chen, P., Ferenz, C., Franklin, S., Ghayur, T., Li, P.,
et al. (1995) Science, 269, 1885-1888.
60.Xue, D., and Horvitz, H. R. (1995) Nature,
377, 248-251.
61.Zhou, Q., Snipas, S., Orth, K., Muzio, M., Dixit,
V. M., and Salvesen, G. S. (1997) J. Biol. Chem., 272,
7797-7800.
62.Drew, M. C., He, C. J., and Morgan, P. W. (2000)
Trends Plant Sci., 5, 123-127.
63.Gunawardena, A. H. (2008) J. Exp. Bot.,
59, 445-451.
64.Rubinstein, B. (2000) Plant Mol. Biol.,
44, 303-318.
65.Fukuda, H. (1996) Annu. Rev. Plant Physiol.
Plant Mol. Biol., 47, 299-325.
66.Greenberg, J. T. (1996) Proc. Natl. Acad. Sci.
USA, 93, 12094-12097.
67.Van Doorn, W. G., Beers, E. P., Dangl, J. L.,
Franklin-Tong, V. E., Gallois, P., Hara-Nishimura, I., Jones, A. M.,
Kawai-Yamada, M., Lam, E., Mundy, J., Mur, L. A., Petersen, M.,
Smertenko, A., Taliansky, M., Van Breusegem, F., Wolpert, T.,
Woltering, E., Zhivotovsky, B., and Bozhkov, P. V. (2011) Cell Death
Differ., 18, 1241-1246.
68.Curtis, M. J., and Wolpert, T. J. (2004) Plant
J., 38, 244-259.
69.Dangl, J. L., and Jones, J. D. (2001)
Nature, 411, 826-833.
70.Lam, E., Kato, N., and Lawton, M. (2001)
Nature, 411, 848-853.
71.Hammond-Kosack, K. E., and Jones, J. D. (1997)
Annu. Rev. Plant Physiol. Plant Mol. Biol., 48,
575-607.
72.Hutcheson, S. W. (1998) Annu. Rev.
Phytopathol., 36, 59-90.
73.Hara-Nishimura, I., and Hatsugai, N. (2011)
Cell Death Differ., 18, 1298-1304.
74.Hatsugai, N., Kuroyanagi, M., Nishimura, M., and
Hara-Nishimura, I. (2006) Apoptosis, 11, 905-911.
75.Hatsugai, N., Iwasaki, S., Tamura, K., Kondo, M.,
Fuji, K., Ogasawara, K., Nishimura, M., and Hara-Nishimura, I. (2009)
Genes Dev., 23, 2496-2506.
76.Kisselev, A. F., Garcia-Calvo, M., Overkleeft, H.
S., Peterson, E., Pennington, M. W., Ploegh, H. L., Thornberry, N. A.,
and Goldberg, A. L. (2003) J. Biol. Chem., 278,
35869-35877.
77.Gu, C., Kolodziejek, I., Misas-Villamil, J.,
Shindo, T., Colby, T., Verdoes, M., Richau, K. H., Schmidt, J.,
Overkleeft, H. S., and van der Hoorn, R. A. (2010) Plant J.,
62, 160-170.
78.Lincoln, J. E., Richael, C., Overduin, B., Smith,
K., Bostock, R., and Gilchrist, D. G. (2002) Proc. Natl. Acad. Sci.
USA, 99, 15217-15221.
79.Del Pozo, O., and Lam, E. (2003) Mol. Plant
Microbe Interact., 16, 485-494.
80.Hansen, G. (2000) Mol. Plant Microbe
Interact., 13, 649-657.
81.Danon, A., Rotari, V. I., Gordon, A., Mailhac,
N., and Gallois, P. (2004) J. Biol. Chem., 279,
779-787.
82.Dickman, M. B., Park, Y. K., Oltersdorf, T., Li,
W., Clemente, T., and French, R. (2001) Proc. Natl. Acad. Sci.
USA, 98, 6957-6962.
83.Shaw, E. (1990) Adv. Enzymol.,
63, 271-347.
84.Del Pozo, O., and Lam, E. (1998) Curr.
Biol., 8, 1129-1132.
85.Uren, A. G., O’Rourke, K., Aravind, L. A.,
Pisabarro, M. T., Seshagiri, S., Koonin, E. V., and Dixit, V. M. (2000)
Mol. Cell, 6, 961-967.
86.Aravind, L., and Koonin, E. V. (2002)
Proteins, 46, 355-367.
87.Tsiatsiani, L., Van Breusegem, F., Gallois, P.,
Zavialov, A., Lam, E., and Bozhkov, P. V. (2011) Cell Death
Differ., 18, 1279-1288.
88.Vercammen, D., van de Cotte, B., De Jaeger, G.,
Eeckhout, D., Casteels, P., Vandepoele, K., Vandenberghe, I., Van
Beeumen, J., Inze, D., and Van Breusegem, F. (2004) J. Biol.
Chem., 279, 45329-45336.
89.Watanabe, N., and Lam, E. (2011) J. Biol.
Chem., 286, 10027-10040.
90.Bozhkov, P. V., Filonova, L. H., Suarez, M. F.,
Helmersson, A., Smertenko, A. P., Zhivotovsky, B., and von Arnold, S.
(2004) Cell Death Differ., 11, 175-182.
91.Suarez, M. F., Filonova, L. H., Smertenko, A.,
Savenkov, E. I., Clapham, D. H., von Arnold, S., Zhivotovsky, B., and
Bozhkov, P. V. (2004) Curr. Biol., 14, R339-R340.
92.Bozhkov, P. V., Suarez, M. F., Filonova, L. H.,
Daniel, G., Zamyatnin, A. A., Jr., Rodriguez-Nieto, S., Zhivotovsky,
B., and Smertenko, A. (2005) Proc. Natl. Acad. Sci. USA,
102, 14463-14468.
93.Watanabe, N., and Lam, E. (2005) J. Biol.
Chem., 280, 14691-14699.
94.Vercammen, D., Belenghi, B., van de Cotte, B.,
Beunens, T., Gavigan, J. A., De Rycke, R., Brackenier, A., Inze, D.,
Harris, J. L., and Van Breusegem, F. (2006) J. Mol. Biol.,
364, 625-636.
95.Vercammen, D., Declercq, W., Vandenabeele, P.,
and Van Breusegem, F. (2007) J. Cell Biol., 179,
375-380.
96.Carmona-Gutierrez, D., Frohlich, K. U., Kroemer,
G., and Madeo, F. (2010) Cell Death Differ., 17,
377-378.
97.Enoksson, M., and Salvesen, G. S. (2010) Cell
Death Differ., 17, 1221.
98.He, R., Drury, G. E., Rotari, V. I., Gordon, A.,
Willer, M., Farzaneh, T., Woltering, E. J., and Gallois, P. (2008)
J. Biol. Chem., 283, 774-783.
99.Watanabe, N., and Lam, E. (2011) Plant J.,
66, 969-982.
100.Coll, N. S., Vercammen, D., Smidler, A.,
Clover, C., Van Breusegem, F., Dangl, J. L., and Epple, P. (2010)
Science, 330, 1393-1397.
101.Sundstrom, J. F., Vaculova, A., Smertenko, A.
P., Savenkov, E. I., Golovko, A., Minina, E., Tiwari, B. S.,
Rodriguez-Nieto, S., Zamyatnin, A. A., Jr., Valineva, T., Saarikettu,
J., Frilander, M. J., Suarez, M. F., Zavialov, A., Stahl, U., Hussey,
P. J., Silvennoinen, O., Sundberg, E., Zhivotovsky, B., and Bozhkov, P.
V. (2009) Nat. Cell. Biol., 11, 1347-1354.
102.Hiraiwa, N., Nishimura, M., and Hara-Nishimura,
I. (1999) FEBS Lett., 447, 213-216.
103.Kuroyanagi, M., Nishimura, M., and
Hara-Nishimura, I. (2002) Plant Cell Physiol., 43,
143-151.
104.Hatsugai, N., Kuroyanagi, M., Yamada, K.,
Meshi, T., Tsuda, S., Kondo, M., Nishimura, M., and Hara-Nishimura, I.
(2004) Science, 305, 855-858.
105.Rojo, E., Martín, R., Carter, C.,
Zouhar, J., Pan, S., Plotnikova, J., Jin, H., Paneque, M.,
Sanchez-Serrano, J. J., Baker, B., Ausubel, F. M., and Raikhel, N. V.
(2004) Curr. Biol., 14, 1897-1906.
106.Kuroyanagi, M., Yamada, K., Hatsugai, N.,
Kondo, M., Nishimura, M., and Hara-Nishimura, I. (2005) J. Biol.
Chem., 280, 32914-32920.
107.Bosch, M., Poulter, N. S., Perry, R. M.,
Wilkins, K. A., and Franklin-Tong, V. E. (2010) Plant Mol.
Biol., 74, 381-393.
108.Hara-Nishimura, I., Hatsugai, N., Nakaune, S.,
Kuroyanagi, M., and Nishimura, M. (2005) Curr. Opin. Plant
Biol., 8, 404-408.
109.Chichkova, N. V., Kim, S. H., Titova, E. S.,
Kalkum, M., Morozov, V. S., Rubtsov, Y. P., Kalinina, N. O., Taliansky,
M. E., and Vartapetian, A. B. (2004) Plant Cell, 16,
157-171.
110.Chichkova, N. V., Shaw, J., Galiullina, R. A.,
Drury, G. E., Tuzhikov, A. I., Kim, S. H., Kalkum, M., Hong, T. B.,
Gorshkova, E. N., Torrance, L., Vartapetian, A. B., and Taliansky, M.
(2010) EMBO J., 29, 1149-1161.
111.Chichkova, N. V., Galiullina, R. A., Taliansky,
M. E., and Vartapetian, A. B. (2008) Plant Stress, 2,
89-95.
112.Pitzschke, A., and Hirt, H. (2010) EMBO
J., 29, 1021-1032.
113.Howard, E. A., Zupan, J. R., Citovsky, V., and
Zambryski, P. C. (1992) Cell, 68, 109-118.
114.Reavy, B., Bagirova, S., Chichkova, N. V.,
Fedoseeva, S. V., Kim, S. H., Vartapetian, A. B., and Taliansky, M. E.
(2007) Plant Cell Rep., 26, 1215-1219.
115.Schaller, A. (2004) Planta, 220,
183-197.
116.Ottmann, C., Rose, R., Huttenlocher, F.,
Cedzich, A., Hauske, P., Kaiser, M., Huber, R., and Schaller, A. (2009)
Proc. Natl. Acad. Sci. USA, 106, 17223-17228.
117.Rautengarten, C., Steinhauser, D., Bussis, D.,
Stintzi, A., Schaller, A., Kopka, J., and Altmann, T. (2005) PLoS
Comput. Biol., 1, e40.
118.Tripathi, L. P., and Sowdhamini, R. (2006)
BMC Genomics, 7, 200.
119.Steiner, D. F. (1998) Curr. Opin. Chem.
Biol., 2, 31-39.
120.Seidah, N. G., Mayer, G., Zaid, A., Rousselet,
E., Nassoury, N., Poirier, S., Essalmani, R., and Prat, A. (2008)
Int. J. Biochem. Cell. Biol., 40, 1111-1125.
121.Coffeen, W. C., and Wolpert, T. J. (2004)
Plant Cell, 16, 857-873.
122.Curtis, M. J., and Wolpert, T. J. (2002)
Plant J., 29, 295-312.
123.Navarre, D. A., and Wolpert, T. J. (1999)
Plant Cell, 11, 237-249.
124.Vartapetian, A. B., Tuzhikov, A. I., Chichkova,
N. V., Taliansky, M., and Wolpert, T. J. (2011) Cell Death
Differ., 18, 1289-1297.
125.Chichkova, N. V., Tuzhikov, A. I., Taliansky,
M., and Vartapetian, A. B. (2012) Physiol. Plant., 145,
77-84.