Cap-Independent Translation Initiation of Apaf-1 mRNA Based on a Scanning Mechanism Is Determined by Some Features of the Secondary Structure of Its 5′ Untranslated Region
D. E. Andreev, S. E. Dmitriev, I. M. Terenin, and I. N. Shatsky*
Belozersky Institute of Physico-Chemical Biology, Lomonosov Moscow State University, 119991 Moscow, Russia; fax: (495) 939-3181; E-mail: shatsky@genebee.msu.su* To whom correspondence should be addressed.
Received September 6, 2012; Revision received October 18, 2012
We have earlier shown that the 5′-untranslated region (5′ UTR) of the mRNA coding for activation factor of apoptotic peptidase 1 (Apaf-1) can direct translation in vivo by strictly 5′ end-dependent way even in the absence of m7G-cap. Dependence of translational efficiency on the cap availability for this mRNA turned out to be relatively low. In this study we demonstrate that this surprising phenomenon is determined the 5′-proximal part (domains I and II) of highly structured Apaf-1 5′ UTR. Remarkably, domain II by itself was able to reduce dependence of the mRNA on the cap on its transferring to a short 5′ UTR derived from a standard vector. We suggest that the low cap-dependence inherent to some cellular mRNAs may have an important physiological significance under those stress conditions when the function of cap-binding factor eIF4E is impaired.
KEY WORDS: protein biosynthesis, translational control, cellular IRES-elements, cap-independent translation, Apaf-1 mRNADOI: 10.1134/S0006297913020041
Abbreviations: Apaf-1, apoptotic peptidase activating factor 1; CITEs, cap-independent translational enhancers; eIF, eukaryotic initiation factor; IRES, internal ribosome entry site; 5′ UTR, 5′-untranslated region.
Gene expression within cells is regulated at multiple levels, i.e.
transcription, pre-mRNA processing (splicing, polyadenylation), export
from nucleus accompanied by mRNA quality control, stability of mRNAs
and encoded proteins, translation, and post-translational modification.
In most cases global changes of translation represent the first
response of the cell to external stimuli. It is believed that the
global regulation of protein synthesis is effected at the level of
translation initiation by changing the activity of key initiation
factors that are involved either in recruitment of mRNAs onto ribosomes
(e.g. eukaryotic translation initiation factors eIF4E and eIF4G) or in
delivery of Met-tRNAiMet onto the 40S ribosomal
subunit (factor eIF2). The signal transduction pathways by which the
activity of these translation initiation factors is repressed or
activated are now under intense investigation [1-6].
Environmental changes or stresses affect the translation of individual mRNAs to different extents and the corresponding individual responses mainly depend on the structure of untranslated regions of mRNAs. In the case of 5′ UTRs, their secondary structure can influence the interaction of the cap with the three-subunit factor eIF4F and scanning efficiency. According to the current hypothesis, the more a 5′ UTR is structured, the more eIF4F is required for translation initiation.
The availability of eIF4E, and therefore formation of the whole three-subunit factor eIF4F, is regulated by a family of translation repressors, the eIF4E-binding proteins (4E-BPs) [7-9], which bind to eIF4E and prevent its association with eIF4G. The capacity of the 4E-BPs to bind eIF4E is determined by their phosphorylation status, which is enhanced by kinase mTOR. When 4E-BP1 is hypophosphorylated, it forms a complex with eIF4E and inhibits the cap-dependent translation initiation. However, when cells are stimulated with serum, growth factors, or hormones, 4E-BP1 becomes hyperphosphorylated, which releases eIF4E from 4E-BP1 and results in formation of the whole eIF4F heterotrimer. Several stress conditions (including serum deprivation, hypoxia, heat shock, viral infection, and apoptosis) can block the mTOR signal pathway and cause dephosphorylation of 4E-BP1, which results in the inhibition of cap-dependent translation [10].
It is logical to suppose that under stress conditions, when cap-dependent translation is impaired, the expression of some specific proteins is maintained at an appropriate level or at least suppressed less dramatically. The mRNA that codes for the apoptosome forming protein Apaf-1 is an example of such mRNAs [11]. One can expect (and this is experimentally supported) that during apoptosis, when eIF4E is blocked by 4E-BP1, the synthesis of pro- and anti-apoptotic proteins still occurs in a preferential manner. The question arises how these mRNAs continue to bind with ribosomes when the cap-binding factor is inactivated. Specific features of 5′ UTR of Apaf-1 mRNA have been subject of many studies [12-15], and this paper is also concerned with the mechanism of translation of Apaf-1 mRNA.
To explain the preferential synthesis of some proteins under conditions of inactivation of cap-binding protein eIF4E, a concept of cellular IRESs was put forward nearly two decades ago [16]. Even today this concept absolutely dominates. According to this hypothesis, some cellular mRNAs that encode proteins with regulatory functions harbor IRESs. It is believed that under normal conditions such mRNAs can be translated by both the 5′ end-dependent scanning and internal ribosome entry mechanisms, but under stresses the internal initiation mode becomes predominant. Although for none of the cellular mRNAs has the mechanism of internal ribosome entry been proven by direct experiments with the use of all necessary controls, no other explanations for stable translation under stress conditions are considered [17-21].
An alternative explanation for efficient mRNA translation under abnormal conditions has been suggested in our laboratory based on results of recent experiments. We found that the contribution of the m7G cap to the translation efficiency was remarkably different for various individual 5′ UTRs of cellular mRNAs, though in none of them the presence of an IRES was found. In other words, these 5′ UTRs did not pass the crucial test for the presence of an IRES: they were unable to support expression of the second reporter gene in bicistronic mRNAs with a reasonably significant efficiency, even when compared to the corresponding uncapped monocistronic mRNAs. Notably, contrary to conventional wisdom, there was no correlation between the magnitude of the stimulation by the 5′-cap (cap-dependence) and the overall stability of the secondary structure of 5′ UTR.
Our studies [22] showed that the lowest cap-dependence was characteristic of the 5′ UTR of Apaf-1 mRNA. It has the length of 577 nucleotide residues and a highly developed secondary structure in which one can define four structural domains [14]. Its translation was stimulated by the cap from 5- to 7-fold, whereas for the β-globin 5′ UTR the stimulation was 40-60-fold [22]. However, in our previous publication we did not address the question of which elements in the 5′ UTR of Apaf-1 mRNA made it “unusual”, i.e. conferred to it a reduced dependence on the cap.
In this paper, we show that the reduced cap-dependence of the 5′ leader of Apaf-1 mRNA is mostly accounted for by the 5′-proximal domains, I and II, of its secondary structure, whereas the next domains, III and IV, affect the cap-dependence very little. (It should be noted that domains III and IV that are proximal to the start codon were claimed to harbor an IRES [14]). Moreover, one of the 5′-proximal domains, domain II, in the context of a short unrelated 5′ UTR is capable alone of decreasing the mRNA dependence on the cap. It should be mentioned that the low contribution of the m7G cap to the translation initiation of mRNA with such a hybrid 5′ UTR containing domain II is mainly accounted for by a relatively high translation initiation efficiency of the corresponding uncapped (A-capped) mRNA.
Thus, we demonstrate here for the first time that a specific secondary structure within a 5′ UTR is capable of stimulating rather than inhibiting translation initiation of uncapped mRNAs, which is directed by a 5′ end-dependent scanning mechanism. We propose a mechanism of cap-independent scanning as an alternative explanation of efficient translation of some cellular mRNAs under conditions of suppression of cap-binding factor eIF4E.
MATERIALS AND METHODS
Plasmids. All dicistronic DNA constructs are based on the pGL3R vector [22]. pGL3R-Apaf-1, its four mutants pGL3R-Apaf-1Δdom I-pGL3R-Apaf-1Δdom IV, and pGL3R-β-globin constructs were described in [21, 23]. pGL-Apaf-1 Fluc was prepared by removing the Rluc sequence to obtain a monocistronic construct. pGL-Apaf-1 Rluc was prepared by replacing the Fluc sequence with Rluc sequence by digestion of the corresponding vector with NcoI and XbaI and incorporation of Rluc from the PCR product of pRluc containing an NcoI site (this site was inserted into the sequence of the corresponding primer for PCR since NcoI is absent from the construct pRluc). The pRluc plasmid was described in [24]. There are 49 nt between the T7 promoter and AUG initiation codon of pRluc, and EcoRV cleaves between nt 32 and 33 resulting in blunt ends. The corresponding PCR-fragments of the 5′ UTR of Apaf-1 mRNA were inserted in correct orientation in vector pRluc treated with EcoRV. In this way, we obtained constructions Rluc dom I (positions 1-105 of the 5′ UTR of Apaf-1 mRNA), Rluc dom II (99-353), Rluc dom III (346-454), and Rluc dom IV (positions 427-577). For subsequent mutagenesis of the construction Rluc dom II, we used the appropriate primers to obtain nucleotide deletions 126-169, 198-256, 273-308, 198-308, and 126-256 plus double deletion 126-169 and 273-308.
mRNA preparation. mRNAs were prepared exactly as in [24]: we performed PCR with the universal reverse primer 5′-(T)50AACTTGTTTATTGCAGCTTATAATGG-3′ and either 5′ UTR-specific primers containing the T7-promotor, or the universal forward primer 5′-CTAGCAAAATAGGCTGTCCC-3′ for those constructs that already had the T7 promoter. The PCR products were used then as templates for in vitro RNA transcription using a T7 RiboMAX Large Scale RNA Production kit (Promega, USA). For preparation of m7G- or A-capped transcripts, the analog of m7G cap, the 3′-O-Me-m7GpppG, or ApppG (NEB, USA) were added to the transcription mix in a proportion of 10 : 1 to GTP. The resulting RNAs were purified by LiCl precipitation and checked for integrity by denaturing PAGE.
Cell culture and transfection procedures. HEK293T cells were cultivated in DMEM supplemented with 10% FBS as described [24]. The day before transfection, actively proliferating cells were replated to 24-well plates. After 12-16 h of growth, when the cell density reached 60-80%, the transfection was performed using Unifectin-56 (RusBioLink, Russia) [24]. The protocol was slightly modified to obtain the maximal yield of transfection: a mixture of 0.2 µg of Fluc mRNA to be tested and 0.01 µg of a reference reporter mRNA (m7G-capped Rluc-poly(A)) was incubated with 0.42 μl of Unifectin in 125 μl DMEM for 15 min and then added to cells. Two hours later, the cells were lysed and luciferase activities were measured with the Dual Luciferase Assay kit (Promega). All the transfections were repeated several times with different cell passages. Some of the most important experiments were repeated using another transfection reagent, Lipofectamine 2000 (Invitrogen, USA), and produced the same results.
For the experiments assessing the mRNA stability, another method of transfection, Magnet Assisted Transfection (MATra), was also employed along with lipofection. The protocol suggested by manufacturer (IBA, USA) for transfection of DNA and siRNAs was adapted to mRNA transfection. Briefly, 0.6 μg of reporter mRNAs (0.5 μg of m7G/A-capped Rluc or dom II-Rluc supplemented with 0.1 μg of normalizing m7G/β-globin Fluc) were mixed with 50 μl of DMEM without serum and 1.2 μl of MATra-A Reagent (IBA), incubated at room temperature for 20 min, then added to the growth medium, and the 24-well cultural plate was placed on a Universal Magnet Plate (IBA) for 1 h. The Magnet Plate was then removed, and after the time indicated in the text cells were harvested and activities of reporter proteins were determined.
RESULTS
Reduced cap-dependence of translation initiation at the 5′ UTR of Apaf-1 mRNA is determined by its 5′-proximal domains. We have recently demonstrated [22] that the 5′ UTR of Apaf-1 mRNA possesses a significantly reduced cap-dependence as compared with 5′ UTRs from several other mammalian mRNAs. We thought that this observation deserved our special consideration since the 5′ UTR of Apaf-1 mRNA is sufficiently long and has a highly developed secondary structure. According to the currently dominating idea, highly structured 5′ leaders should require more factor eIF4F (and hence more factor eIF4E) since they should recruit more helicase eIF4A to unfold their secondary structure during the scanning process than simple unstructured 5′ UTRs. We demonstrated that this reduced cap-dependence is not accounted for by the presence of an IRES-element in the 5′ UTR of Apaf-1 mRNA, since this 5′ UTR proved to be inactive in the translation initiation when placed between two reporters in a dicistronic mRNA. Moreover, it was even inactive if compared with the A-capped monocistronic mRNA. (A-capped variants are mRNAs where the 5′ end is protected from exonuclease degradation by a non-functional analog of m7G, ApppN …). We decided to determine which structural properties of the Apaf-1 5′ UTR are responsible for this reduced m7G cap requirement. To this end, we deleted each of the four structural domains of the Apaf-1 5′ UTR [14]. Translation activities of both m7G- and A-capped mRNAs were determined in lysates of transfected cells by the Fluc luciferase assay. To take into account transfection efficiencies, these Fluc activities were normalized to that of co-transfected m7G-capped reporter Rluc mRNA. The cap-dependence, which is quantitatively expressed as fold-stimulation of translation by m7G cap as compared with non-functional A-cap, is shown in Fig. 1a. Deletions of two 5′-proximal domains, I and II, strongly decreased the cap-dependence, whereas removal of domains III and IV affected the cap-dependence of reporter translation less significantly. It should be mentioned that the IRES-activity earlier revealed on the base of transfection of cells with cDNAs was ascribed exactly to domains III and IV [14], rather than to domain I and II.
Fig. 1. a) Effect of deletions of individual domains of the Apaf-1 5′ UTR on the cap-dependence of translation of the corresponding Fluc mRNAs in transfected HEK293T cells. The cap-dependence is expressed as the ratio of translational activities for m7G-capped vs. uncapped (A-capped) mRNAs. These activities were first normalized to that obtained for the co-transfected capped Renilla luciferase mRNA to take into account variations in transfection efficiency. The constructs used are shown. The secondary structure of the Apaf-1 5′ UTR is schematically depicted in the left corner of the figure. b) Effect of insertion of individual structural domains of Apaf-1 5′ UTR into the 5′ UTR of Rluc on the cap-dependence of resulting transcripts. The cap-dependence was determined as in (a); c) though expressed in arbitrary units, the columns in this diagram correctly reflect absolute translational activities of the corresponding mRNAs. All measurements were routinely performed 2 h after transfection.
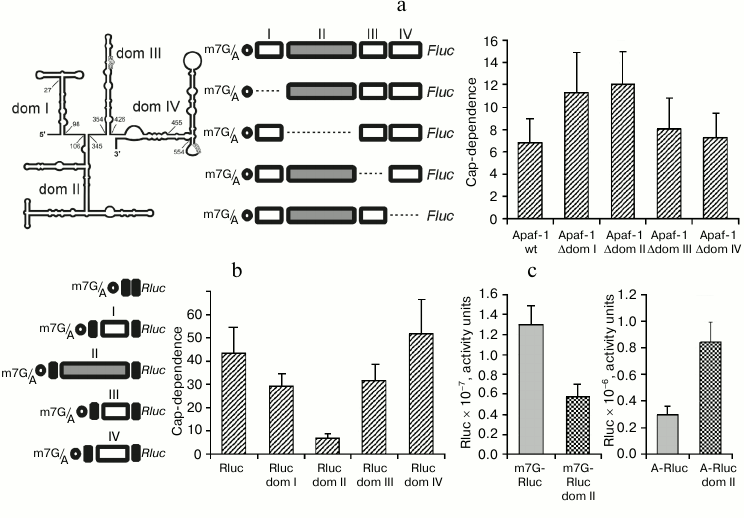
Domain II of the Apaf-1 5′ UTR alone provides the reduced requirement for the cap. The above results showed that the reduced cap-dependence of the Apaf-1 5′ UTR is accounted for by some structural elements present within domains I, II, or both. Therefore, it was interesting to find out whether this reduced dependence on the m7G cap would be preserved in isolated domains and could be transferred with them to another unrelated 5′ UTR. For this purpose to create chimeric constructions we used the simple 5′ UTR from plasmid pGL3R [23], in which each of the four principal structural domains of the Apaf-1 5′ UTR was inserted separately at position 32. In this way, the chimeric mRNAs with the same sequence adjacent to the 5′ end and with the same context of AUG initiation codon (see constructs in Fig. 1b) were obtained.
As seen from Fig. 1b, insertion of domain II into irrelevant 5′ UTR greatly reduced cap-dependence. The effect on the cap-dependence of the insertion in the same position of domains I or III was substantially less significant, whereas domain IV even increased this dependence.
Figure 1c explains the origin of reduced cap-dependence, which was conferred by the insertion of domain II in the 5′ UTR of Rluc mRNA. In this figure the data, albeit expressed in arbitrary units, represent absolute, rather than relative, translation activities of the control Rluc 5′ UTR and the transcript with the inserted domain II both in the m7G- and A-capped form. One can see that domain II in the context of the 5′ leader of Rluc mRNA with m7G cap at the 5′ end reduces the translation efficiency slightly more than twice (as could be expected from the classical model of scanning mechanism of translation initiation). However, when the corresponding mRNAs were A-capped, the activity of the 5′ UTR with the inserted domain II proved to be much higher than the activity of the initial unstructured 49-nucleotide-long leader of Rluc mRNA. This example shows how a specific structural domain can enhance the ability of mRNA to function in the absence of the interaction of eIF4E with m7G cap, even when this domain is positioned at a large distance from the 5′ end.
It should be emphasized that the excision of domain II from the Apaf-1 5′ UTR and its transferring to the vector 5′ UTR did not significantly change its secondary structure (data not shown).
The reduced cap-dependence of translation initiation of the mRNA with the hybrid Rluc dom II 5′ UTR does not result from changes in the mRNA stability. It was necessary to rule out the possibility that the observed difference in the cap-dependence for the control Rluc mRNA and hybrid Rluc dom II mRNA is accounted for by their different stability. As before [22], we used for this purpose a simple approach: we analyzed the translation kinetics of m7G- and A-capped mRNAs in transfected cells and assessed the time course of cap-dependence (Fig. 2). If, for instance, for some reason the A-capped form of mRNA turns out to be unstable as compared with the m7G-capped version, then the expression of a reporter from the A-capped mRNA should stop with time as a consequence of degradation of this mRNA. As a result, the cap-dependence of such an mRNA (i.e. the ratio of translation levels m7G/A) should go up with time. To verify the validity of such a criterion, we used completely uncapped mRNA, i.e. not protected with anything from exonuclease degradation from the 5′ end. Moreover, we employed two principally different transfection methods: lipofection (Fig. 2, left part) and Magnet Assisted Transfection (MATra) (Fig. 2, right part). In case of the second method, nucleic acids are first adsorbed on magnetic nanoparticles. Then, exploiting magnetic force, the nucleic acids are drawn towards and delivered into the cells.
Fig. 2. Time course of cap-dependence for the Rluc mRNA containing either its standard 5′ UTR or the 5′ UTR with the inserted domain II (Rluc dom II) from the 5′ UTR of Apaf-1 mRNA. For its determination, both m7G- and A-capped forms were tested for each construct. As a control, uncapped mRNAs (i.e. unprotected with Appp at their 5′ ends) were employed. Transfection of HEK293T cells was performed using two different methods: by lipofection (left part of the figure) or by Magnet Assisted Transfection (right part). For experimental details see the text in sections “Materials and Methods” and “Results”. a-d) Kinetics of translation of m7G-capped, A-capped and uncapped Rluc and Rluc dom II mRNAs; e, f) time course of cap-dependence calculated on the base of data obtained in experiments shown in (a)-(d).
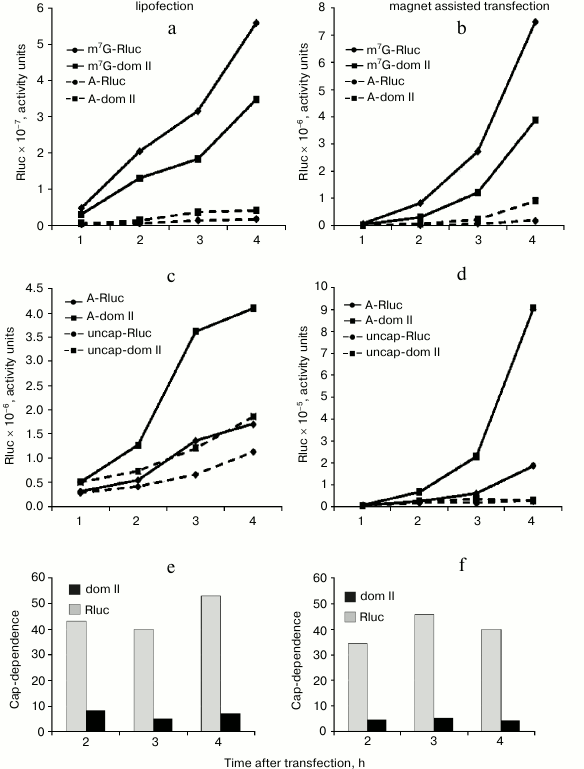
As clear from Fig. 2 (a-d), the reporter expression from both m7G-capped Rluc and Rluc dom II mRNAs permanently increased during the time of experiment. Importantly, the same was true for the corresponding A-capped mRNAs. Consequently, cap-dependence for these mRNAs did not change substantially with time (Fig. 2, e and f). In contrast, the Renilla luciferase expression from the 5′ end unprotected mRNAs completely stopped as early as 1 h post transfection: this most likely indicates their degradation (Fig. 2d); some increase in the reporter expression from uncapped mRNAs in lipofection experiments (Fig. 2c) is most probably accounted for by a continued penetration of mRNA from liposomes in the course of the experiment. Both methods of transfection clearly showed that the cap-dependence for the standard Rluc 5′ UTR and the hybrid dom II Rluc 5′ UTR did not vary with time (Fig. 2, e and f). We believe that the approach we employed can be a good alternative to routinely used methods to assess the mRNA stability (Q-PCR, Northern blotting) as these conventional methods are poorly suitable for mRNA transfection: a significant portion of transfected mRNA does not enter the cytoplasm: it sticks to the cellular membrane and remains intact for a rather long period. This does not allow one to estimate the actual stability of transfected mRNA in the cell (data are not shown, see also [25]).
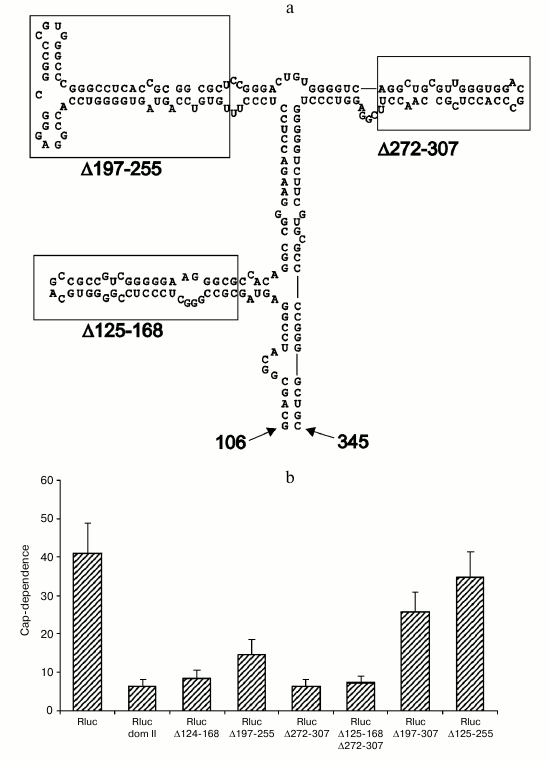
Deletions within domain II of the Apaf-1 5′ UTR affect cap-dependence of translation initiation. To get an idea which features of domain II determine its ability to reduce cap-dependence, we tested the effect of various deletions within this domain in the context of unrelated 5′ UTR. As seen in Fig. 3b, deletions that remove several stem–loop structures (Δ197-307, Δ125-255 and, to some extent, Δ197-255) resulted in a dramatic shift to the enhanced cap-dependence, whereas smaller deletions were not significant for the cap-dependence. Results of these experiments on mutagenesis show that deletions of some elements in the apical part of domain II exert the largest effect on the cap-dependence (obviously deletion Δ125-255 should result in a complete disruption of domain structure). In the future we plan to perform a point mutagenesis for a more precise determination of specific elements that are responsible for the reduced cap-dependence. It is important to emphasize once again that in the case of derivatives of domain II, the presence of m7G cap at the 5′ end is more critical for less structured 5′ UTRs. This fact is in obvious disagreement with the current point of view on the cap-dependent mechanism of translation of eukaryotic mRNAs. The results thus obtained nicely demonstrate a nontrivial fact: changes within the secondary structure of a domain that is positioned rather far from the 5′ end of mRNA considerably increase the requirement for m7G cap, i.e. the structure that is situated at the very 5′ end.
Fig. 3. Deletions within the 5′ UTR of Rluc mRNA with the inserted domain II affect the cap-dependence of its translation in transfected cells HEK293T. a) Secondary structure of domain II (adapted from [14]). The boundaries of deletions are designated with numbers (numbering is from the 5′ end of the complete 5′ UTR of Apaf-1 mRNA). Small deletions of three stem–loop structures are boxed; b) cap-dependence of Rluc mRNA containing the intact domain II and its various deletions versions. The cap-dependence was estimated as in Fig. 1.
DISCUSSION
The current predominant concept claims that any secondary structure within the 5′ UTR of eukaryotic mRNAs causes an adverse effect on the translation of mRNA unless the corresponding stem–loop structures form an IRES-element. The well-known inhibitory effect of a stable secondary structure placed at the very 5′ end of an mRNA is often automatically extended by many researchers to natural stem–loop structures that occur in internal positions of 5′ UTRs. As shown in our previous investigations [22, 26, 27] and in this paper, the real situation is far more complex. The secondary structure within the 5′ UTR is not necessary to be harmful and real stem–loops in natural 5′ UTRs are easily overcome by the scanning ribosome [27]. Therefore, a specific secondary structure can play a positive role even in the case of 5′ end-dependent scanning mechanism of translation. Moreover, under conditions that result in inactivation of eIF4F, it can enhance rather than decrease the translational efficiency. Thus, widely adopted identification of cap-independent initiation as internal initiation directed by IRES-elements does not appear to be justified, since some mRNAs that do not have a functional cap at their 5′ ends can be translated relatively efficiently using a scanning mechanism. Indeed, although the translation level of the A-capped mRNA with the 5′ UTR of Apaf-1 mRNA is substantially lower than that for analogous m7G-capped mRNA, it is not negligibly small and makes up 15-20% of that for the m7G-capped variant, whereas for mRNAs with standard 5′ UTRs (e.g. 5′ UTRs from β-globin, β-actin mRNAs, or from standard vectors) this level does not exceed 2-3%. A principally new aspect about our concept is the point that the translation directed by the 5′ UTR of Apaf-1 mRNA, even in the absence of the functional m7G-cap remains 5′ end-dependent. This conclusion inevitably follows from the fact that the 5′ UTR of Apaf-1 mRNA is not able to work in the intercistronic position, and insertion of an additional AUG-codon in the cap-proximal part of monocistronic mRNA almost completely blocks translation of such an mRNA (both m7G- and A-capped) [22]. The requirement for a free 5′ end of mRNA for translation initiation even in the absence of interaction of the cap with eIF4E was noted in several papers of other authors. Indeed, fragments of factor eIF4G lacking the eIF4E-binding site can support in vitro the 5′ end-dependent translation, and availability of a free 5′ end is indispensable [28, 29]. Furthermore, it has been shown that uncapped mRNAs generated in vivo by RNA polymerase III transcription are nevertheless translated by means of a 5′ end-dependent scanning mechanism [30]. Thus, the possibility of a 5′ end-dependent but cap-independent translation initiation does not cause any doubt.
How can such a mode of initiation be relatively efficient as we see in the case of 5′ UTR of Apaf-1 mRNA? We speculate that some components of the translation apparatus, for example eIF4G, eIF3 (or their analogs with a similar function), are able to be directly or indirectly recruited onto 5′ UTRs of some mRNAs via RNA–protein interactions with concomitant recruitment of other components of the scanning apparatus. To attract these components, a 5′ UTR should possess corresponding binding sites playing the role of Cap-Independent Translational Enhancers (CITEs) [26]. Owing to their presence, an elevated concentration of initiation factors is created in vicinity of the 5′ UTR of mRNA. This helps to overcome the competition for factors with other cellular mRNAs upon inactivation of eIF4F. We propose that whether this recruitment of factors takes place at the very 5′ end or at some distance from it, the only point of the ribosome entry where the scanning can be initiated is the 5′ terminus of mRNA. This hypothesis was described in detail in [31], and the feasibility of such a model has been recently demonstrated by direct experiments in our lab [32].
An alternative way to provide a relative resistance of translation of some mRNAs to stress conditions has been proposed in [33]: according to the authors’ idea, mRNAs can utilize some specific elements, “zip-codes”, to be targeted to special cellular compartments where the translation apparatus activity is maintained in some way under stress conditions. For example, it has been shown that upon Coxsackie B virus infection and subsequent eIF4G cleavage, suppression of mRNA translation in the endoplasmic reticulum compartment was less pronounced relative to that in the cytosol.
Thus, in this paper we have shown that two 5′-proximal domains in the 5′ UTR of Apaf-1 mRNA are responsible for a reduced cap-dependence of translation of a reporter mRNA. Moreover, domain II alone in the context of unrelated 5′ UTR proved to be capable of reducing cap-dependence of a reporter mRNA to the extent comparable to that of the full-length Apaf-1 5′ UTR. It is obvious, however, that domain II in the context of the whole 5′ UTR of Apaf-1 mRNA is not fully responsible for the cap-dependence reduction since its deletion does not result in a complete loss of Apaf-1 5′ UTR ability to direct the cap-independent 5′ end-dependent translation. One can make a conclusion on a complex modular organization of this 5′ UTR, which obviously has more than one regulatory element. However, we have now the possibility to work with a much simpler model mRNA – hybrid Rluc dom II mRNA. The first results presented in this paper suggest the important role of the apical part of domain II to allow the corresponding reporter mRNA to efficiently direct the cap-independent scanning. In the future we plan to use this model to study the mechanism of action of enhancer(s) of cap-independent initiation (CITEs).
This work was supported in part by the Russian Foundation for Basic Research (grants 11-04-01010_a and 12-04-32039-mol_a) and by State Contract of Ministry of Education and Science of Russian Federation No. 14.740.11.0168.
REFERENCES
1.Cully, M., and Downward, J. (2009) Biochem. Soc.
Trans., 37, 284-288.
2.Polunovsky, V. A., and Bitterman, P. B. (2006)
RNA Biol., 3, 10-17.
3.Averous, J., and Proud, C. G. (2006)
Oncogene, 25, 6423-6435.
4.Robert, F., and Pelletier, J. (2009) Expert
Opin. Ther. Targets, 13, 1279-1293.
5.Silvera, D., Formenti, S. C., and Schneider, R. J.
(2010) Nat. Rev. Cancer, 10, 254-266.
6.Proud, C. G. (2009) Biochem. Soc. Trans.,
37, 227-231.
7.Lin, T. A., Kong, X., Haystead, T. A., Pause, A.,
Belsham, G., Sonenberg, N., and Lawrence, J. C., Jr. (1994)
Science, 266, 653-656.
8.Pause, A., Belsham, G. J., Gingras, A. C., Donze,
O., Lin, T. A., Lawrence, J. C., Jr., and Sonenberg, N. (1994)
Nature, 371, 762-767.
9.Poulin, F., Gingras, A. C., Olsen, H., Chevalier,
S., and Sonenberg, N. (1998) J. Biol. Chem., 273,
14002-14007.
10.Reiling, J. H., and Sabatini, D. M. (2006)
Oncogene, 25, 6373-6383.
11.Morley, S. J., Coldwell, M. J., and Clemens, M.
J. (2005) Cell. Death Differ., 12, 571-584.
12.Schafer, Z. T., and Kornbluth, S. (2006) Dev.
Cell., 10, 549-561.
13.Coldwell, M. J., Mitchell, S. A., Stoneley, M.,
MacFarlane, M., and Willis, A. E. (2000) Oncogene, 19,
899-905.
14.Mitchell, S. A., Spriggs, K. A., Coldwell, M. J.,
Jackson, R. J., and Willis, A. E. (2003) Mol. Cell., 11,
757-771.
15.Ungureanu, N. H., Cloutier, M., Lewis, S. M., de
Silva, N., Blais, J. D., Bell, J. C., and Holcik, M. (2006) J. Biol.
Chem., 281, 15155-15163.
16.Macejak, D. G., and Sarnow, P. (1991)
Nature, 353, 90-94.
17.Holcik, M., and Sonenberg, N. (2005) Nat. Rev.
Mol. Cell. Biol., 6, 318-327.
18.Komar, A. A., and Hatzoglou, M. (2005) J.
Biol. Chem., 280, 23425-23428.
19.Graber, T. E., and Holcik, M. (2007) Mol.
Biosyst., 3, 825-834.
20.Spriggs, K. A., Stoneley, M., Bushell, M., and
Willis, A. E. (2008) Biol. Cell, 100, 27-38.
21.Komar, A. A., and Hatzoglou, M. (2011) Cell.
Cycle, 10, 229-240.
22.Andreev, D. E., Dmitriev, S. E., Terenin, I. M.,
Prassolov, V. S., Merrick, W. C., and Shatsky, I. N. (2009) Nucleic
Acids Res., 37, 6135-6147.
23.Stoneley, M., Paulin, F. E., Le Quesne, J. P.,
Chappell, S. A., and Willis, A. E. (1998) Oncogene, 16,
423-428.
24.Dmitriev, S. E., Andreev, D. E., Ad’yanova,
Z. V., Terenin, I. M., and Shatsky, I. N. (2009) Mol. Biol.
(Moscow), 43, 108-113.
25.Barreau, C., Dutertre, S., Paillard, L., and
Osborne, H. B. (2006) RNA, 12, 1790-1793.
26.Dmitriev, S. E., Andreev, D. E., Terenin, I. M.,
Olovnikov, I. A., Prassolov, V. S., Merrick, W. C., and Shatsky, I. N.
(2007) Mol. Cell. Biol., 27, 4685-4697.
27.Vassilenko, K. S., Alekhina, O. M., Dmitriev, S.
E., Shatsky, I. N., and Spirin, A. S. (2011) Nucleic Acids Res.,
39, 5555-5567.
28.De Gregorio, E., Preiss, T., and Hentze, M. W.
(1998) RNA, 4, 828-836.
29.Ali, I. K., McKendrick, L., Morley, S. J., and
Jackson, R. J. (2001) EMBO J., 20, 4233-4242.
30.Gunnery, S., Maivali, U., and Mathews, M. B.
(1997) J. Biol. Chem., 272, 21642-21646.
31.Shatsky, I. N., Dmitriev, S. E., Terenin, I. M.,
and Andreev, D. E. (2010) Mol. Cells, 30,
285-293.
32.Terenin, I. M., Andreev, D. E., Dmitriev, S. E.,
and Shatsky, I. N. (2012) Nucleic Acids Res., doi:
10.1093/nar/gks 1282.
33.Lerner, R. S., and Nicchitta, C. V. (2006)
RNA, 12, 775-789.