Approaches to the Design of Selective Ligands for Membrane Progesterone Receptor Alpha
O. V. Lisanova1, T. A. Shchelkunova1, I. A. Morozov2, P. M. Rubtsov2, I. S. Levina3, L. E. Kulikova3, and A. N. Smirnov1*
1Biological Faculty, Lomonosov Moscow State University, 119899 Moscow, Russia; fax: (495) 939-4309; E-mail: smirnov__an@mail.ru2Engelhardt Institute of Molecular Biology, Russian Academy of Sciences, ul. Vavilova 32, 119991 Moscow, Russia; fax: (499) 135-1405; E-mail: rubtsov@eimb.ru
3Zelinsky Institute of Organic Chemistry, Russian Academy of Sciences, Leninsky pr. 47, 117913 Moscow, Russia; fax: (499) 135-5328; E-mail: li@ioc.ac.ru
* To whom correspondence should be addressed.
Received September 20, 2012; Revision received November 2, 2012
A number of progesterone derivatives were assayed in terms of their affinity for recombinant human membrane progesterone receptor alpha (mPRα) in comparison with nuclear progesterone receptor (nPR). The 16α,17α-cycloalkane group diminished an affinity of steroids for mPRα without significant influence on affinity for nPR, thus rendering a prominent selectivity of ligands for nPR. On the contrary, substitution of methyl at C10 for ethyl or methoxy group moderately increased the affinity for mPRα and significantly lowered the affinity for nPR. A similar but even more prominent effect was observed upon substitution of the 3-oxo group for the 3-O-methoxyimino group. A significant preference towards mPRα was also rendered by the 17α-hydroxy group and additional C6–C7-double bond. The data suggest that the modes of ligand interaction with mPRα and nPR in the C3 region of the steroid molecule are different. One can speculate that combination of the above substitutions at C17, C10, C6, and C3 may give ligand(s) with high specificity towards mPRα over nPR.
KEY WORDS: membrane progesterone receptor alpha, steroid ligand binding, selectivityDOI: 10.1134/S0006297913030048
Abbreviations: Bmax, concentration of binding sites; CBG, corticosteroid-binding globulin; Kd, equilibrium dissociation constant; mPR, membrane progesterone receptor; nPR, nuclear progesterone receptor; RBA, relative binding affinity; YNB, yeast nitrogen bases.
After initial findings on prolonged accumulation of
[3H]estradiol in reproductive organs [1], the main research efforts were directed to genomic
effects of steroid hormones mediated by a group of nuclear receptors.
It is well recognized, however, that steroid hormones can also elicit
rapid, non-genomic effects (reviewed in [2-4]). Progestin induction of oocyte maturation in
fishes and amphibians (reviewed in [5]) stimulated
investigations on identification of receptors that mediate
surface-initiated actions of progestins. These studies culminated in
cloning of three membrane progesterone receptors (mPRs), α,
β, and γ, that belong to the recently defined progestin and
adiponectin Q receptor family (PAQR), both in fish and humans [6, 7]. Expression of mPRs has been
documented in many mammalian tissues, including reproductive organs,
brain, immune cells, bone, etc. (reviewed in [8]).
However, with the possible exclusion of stimulation of spermatozoa
maturation and motility, physiological functions of mPRs in mammals
remain to be elucidated. The main barrier is the absence of selective
agonists and antagonists for mPRs. A major step toward creation of such
tools was made by Kelder et al. [9] who found that
substitution of angular methyl groups of the progesterone molecule for
ethyl or ethenyl groups gives rise to increase in affinity for
mPRα and decrease in affinity for nuclear progesterone receptor
(nPR). The object of the present study was to reveal additional
determinants of steroid ligands that render preferential binding to
membrane progesterone receptor α. Given that the 3-oxo group of
progesterone plays a crucial role in forming a hydrogen bond network
with nPR [10], we decided to check whether this
group is also necessary for interaction with mPRα. In addition,
we assayed the influence of other modifications of a steroid molecule
on the affinity for mPRα in comparison with nPR. Human mPRs have
been found to be functional in yeast [11]. Hence,
here we used yeast transformed with human mPRα for direct
measurements of ligand competition for [3H]progesterone
binding.
MATERIALS AND METHODS
Chemicals. All reagents were of analytical grade. [1,2,6,7-3H]Progesterone with specific radioactivity of 86 Ci/mmol was purchased from Isotope (Russia). Progesterone, 17α-hydroxyprogesterone, 5α- and 5β-dihydroprogesterone, 11-deoxycorticosterone, corticosterone, testosterone, phenylmethylsulfonyl fluoride (PMSF), EDTA, dithiothreitol (DTT), propylene glycol, glycerol, and Trizma base were from Sigma (USA); activated charcoal from Serva (Germany); dextran 70 from Fluka (Switzerland); BSA from Dia-M (Russia). 16α,17α-Cycloalkane derivatives of progesterone were synthesized as described previously [12-15]. The following components of media for bacteria and yeast were used: bacto-tryptone and bacto-peptone from Pronadisa (Spain); yeast extract, casamino acids, and yeast nitrogen bases YNB from Difco (USA); bacto-agar from Becton Dickinson (USA); glucose and galactose from Panreac (Spain) and ChimMed (Russia); amino acids and adenine from Sigma (USA), Serva (Germany), and Calbiochem (USA); PEG 3350, DMSO, and LiAc from Sigma; salts and agarose from Merck (USA) and Serva (Germany). Solutions were prepared using MilliQ deionized water.
Plasmid design and cell transformation. DNA manipulations (restriction, ligation, and electrophoresis) were performed using standard methods [16] with enzymes from Fermentas (Lithuania) and Promega (USA). Oligonucleotide primers were synthesized by Syntol (Russia). Plasmids and DNA fragments for cloning were purified using kits from Qiagen (Germany). Genomic human DNA was isolated from peripheral blood leucocytes using a Wizard Genomic DNA purification kit (Promega). The PAQR7 (mPRα) gene was amplified using specific primers containing an Eco321 site at the 5′-end (PARQ7-F 5′-GATATCATGGCCATGGCCCAGAAACTCA-3′) and an XhoI site at the 3′-end (PARQ7-R 5′-CTCGAGCCCTTCACTTGGTCTTCTGATCA-3′) and high fidelity PCR Enzyme Mix from Fermentas. PCR was conducted using following regimen: denaturation for 2 min at 95ºC, then 35 cycles (denaturation 95ºC, 20 sec; primer annealing 60ºC, 30 sec; elongation 72ºC, 1 min), and final extension 72ºC, 4 min. Fragments thus obtained were ligated with pGEM-T Easy vector (Promega) and cloned in E. coli JM109 strain. The sequence of the insertion was confirmed by sequencing in the Genome Interinstitutional Collective Use Center of the Engelhardt Institute of Molecular Biology of the Russian Academy of Sciences. The PAQR7 gene was inserted under control by the GAL1 promoter into yeast 2 μm DNA based pPDX2 vector kindly provided by Dr. D. S. Karpov (Engelhardt Institute of Molecular Biology, Moscow). After restriction analysis, transformation of Saccharomyces cerevisiae strain 334t wt (MATα pep4-3 pvb1-1122 uva 3-52 leu2-3,112 veg1-501 gal1) was performed as described [17]. Transformants were selected using PCR, and the correct insert length was confirmed by electrophoresis in 2% agarose gel.
Yeast cultivation. An individual colony of transformants from fresh agar plate was inoculated into 4 ml of selective medium containing 0.67% (w/v) YNB, 2% (w/v) glucose, amino acid mix, adenine, and 0.5% casamino acids, without uracil. Cells were grown in 15 ml vials at 30ºC for 8 h and agitation at 200 rpm until optical density measured at 600 nm reached 1.5-2.5. Then the content was transferred into 170 ml of the same medium in 1000 ml vessel, and cultivation was continued overnight under the same conditions until optical density reached 1.5-2.5. In some experiments, glucose was substituted for galactose partially or completely. Then, cells were harvested by centrifugation, frozen in liquid nitrogen, and kept at –20ºC until RNA or membrane fraction isolation.
Real-time polymerase chain reaction. Total RNA was extracted with TRIzol Reagent (Invitrogen, USA), treated with DNase I (Promega) for disruption of plasmid DNA, and used for cDNA synthesis. The synthesis of cDNA was performed using 1 μg of total RNA and a Promega ImProm_IITM Reverse Transcription System kit (Promega). The expression of human mPRα was evaluated by quantitative real-time polymerase chain reaction (qRT-PCR) on a Rotor-Gene 3000 amplifier (Corbett Research, Australia) with a kit of reagents including the intercalating dye SYBR Green I (Syntol) as recommended by the manufacturer. For amplification of a fragment of the PAQR7 gene, the following primers were used: forward 5′-TGCCTTCTTCTCTACCTTCATGCC-3′; reverse 5′-GCCTCATAGTCCAGTGCCACAG-3′. The data were normalized by expression of S. cerevisiae housekeeping gene TAF10 whose fragment was amplified using primers: forward 5′-ATATTCCAGGATCAGGTCTTCCGTAGC-3′; reverse 5′-GTAGTCTTCTCATTCTGTTGATGTTGTTGTTG-3′.
Preparation of membrane fraction. Yeasts were thawed, weighed, and mixed with homogenizing buffer (50 mM Tris-HCl, pH 7.5, 10 mM KCl, 1 mM EDTA, 0.5 mM PMSF, 1 mM DTT, 10% (v/v) glycerol) and acid-washed glass beads 425-600 µm (Sigma) in proportion 1 : 1 : 2 (v/v). The mixture was subjected to eight cycles of intensive agitation for 30 sec and cooling on ice for 30 sec and then was centrifuged at 3000g for 5 min, 4ºC. The supernatant was collected and diluted with homogenizing buffer up to protein concentration 1-2 mg/ml.
Preparation of uterine cytosol. Mongrel albino adult female rats (180-220 g) were administered intramuscularly with estradiol (10 μg) in propylene glycol (200 μl) for 3-4 consecutive days and sacrificed by decapitation a day later. Uteri from 3-4 animals were placed on ice, minced, and homogenized using a glass homogenizer in 10 mM Tris-HCl buffer, pH 7.5, containing 10 mM KCl, 1.5 mM EDTA, 30% (v/v) glycerol, 1 mM DTT, and 0.5 mM PMSF at tissue/buffer ratio (w/v) 1 : 6. After centrifugation at 105,000g for 1 h, the supernatant (cytosol) with protein concentration of 4-6 mg/ml was collected and used immediately. All these and subsequent procedures were performed at 0-4°C.
Measurement and analysis of [3H]progesterone binding. The procedure was essentially the same as used by us previously for nPR [18]. Briefly, yeast membrane fraction (100 μl) or uterine cytosol (100 μl) was incubated at 0-4ºC for 3 or 20 h, respectively, with 100 μl of buffer containing [3H]progesterone (final concentration 4-6 nM) and unlabeled competitor (final concentration 0-6.3 μM). Then unbound ligand was removed by adding dextran-coated charcoal (100 μl of 2% suspension) for 5 min followed by sedimentation at 1500g for 5 min. Aliquots of supernatant (200 μl) were used for measurements of radioactivity. All measurements were performed in duplicate. The value of nonspecifically bound [3H]progesterone measured in presence of an excess of cold progesterone (6.3 μM) was subtracted from the value of total [3H]progesterone binding for each experimental point. Kd1 and Bmax values for progesterone as reference control were obtained by fitting to experimental points in a “one protein–one ligand” model. Then Kd2 value for a competitor under study was obtained by fitting to respective experimental points according to a “one protein–two ligands” model [18]. Relative binding affinity (RBA) values were calculated as the ratio Kd1/Kd2. The results were presented as mean ± standard deviation from 3-4 experiments. For comparisons of the impact of a substitution on the affinity for nPR and mPRα, we used “discrimination index” calculated as the ratio RBAmPRα/RBAnPR relative to the corresponding reference compound.
RESULTS
In control experiments with empty vector, neither mPRα mRNA nor specific [3H]progesterone binding was found in cells. In preliminary experiments, when a mixture of glucose and galactose was used in the growth medium, the best induction of mPRα mRNA (2.5-fold compared with glucose only) was obtained at galactose/glucose ratio 1 : 9. Higher proportions of galactose/glucose did not provide benefits in mPRα mRNA contents, while it inhibited cell growth.
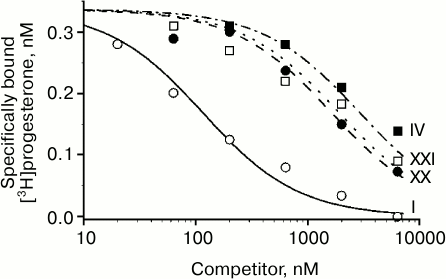
Characteristic competition curves for [3H]progesterone displacement from its complexes with mPRα by cold progesterone and steroid ligands under study are depicted in Fig. 1. Combined results from 3-4 similar experiments for each steroid ligand are presented in the table. It should be noted that RBA values for nPR and several progesterone derivatives as measured in this study are in reasonable correspondence with previously reported values (compound VI: 3.4 and 4.0% [21]; compound VII: 44.0 and 79.0% [22]; compound VIII: 1.7 and 0.3% [23]; compound IX: 0.7 and 0.2% [23]; compound XV: 26.5 and 32.0% [21]; compound XVI: 13.1 and 7.0% [21]).
Fig. 1. Competition between [3H]progesterone and the studied compounds for mPRα in yeast membranes. Numbers near curves correspond to the numbers of the compounds in the table.
As shown in the table, an additional 16α,17α-carbocycle (compounds II-V) significantly reduced the affinity of steroids for mPRα with little if any influence on their affinity for nPR, thus resulting in discrimination index mPRα/nPR below 0.1. The data confirm previously revealed [24] unfavorable influence of α-substituent in steroid D-ring on affinity for mPRα. The 17α-hydroxy group in compound VI also decreased affinity for mPRα. However, this effect was much less prominent than the drastic drop in affinity for nPR. As a result, the 17α-hydroxy group provides discrimination index mPRα/nPR of 18.8. While the 21-hydroxy group in compound VII only weakly affected the binding to nPR, this substituent significantly lowered affinity for mPRα, resulting in discrimination index mPRα/nPR of 0.3. The additional 11β-hydroxy group in compound IX induced further reduction in affinity for mPRα.
Relative binding affinities (RBA) of steroid ligands for nuclear and
membrane progesterone receptors and impact of substituents on
mPRα/nPR preference compared with respective reference compounds
(discrimination index)
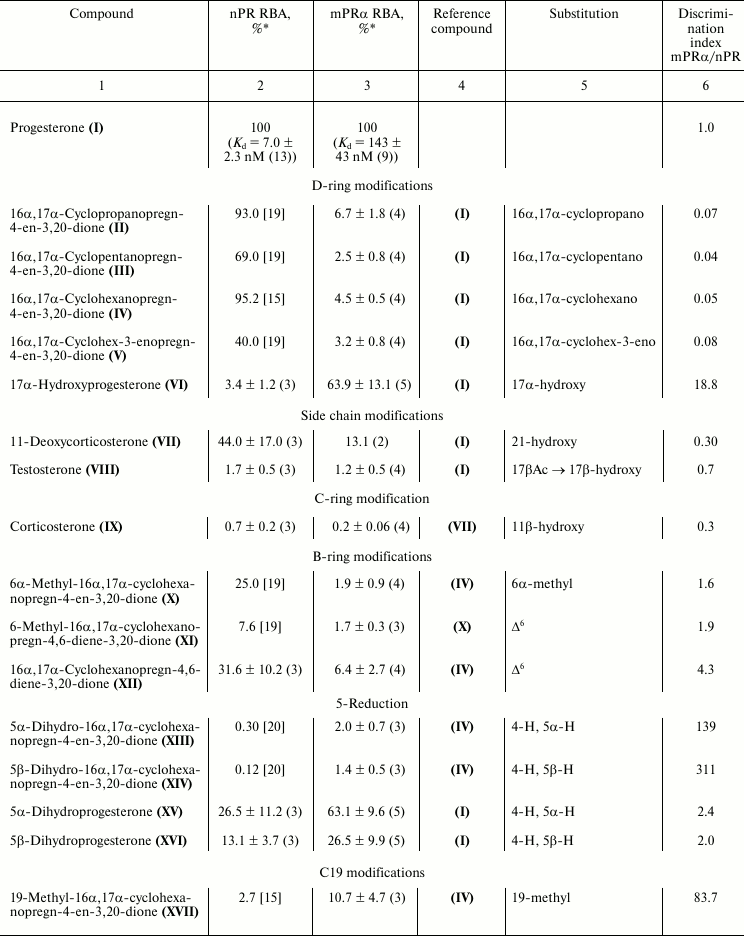

* Mean ± standard deviation and n values are shown.
The 6α-methyl group in compound X produced moderate, approximately proportional decrease in affinity for nPR and mPRα resulting in discrimination index mPRα/nPR slightly higher than 1.0. The Δ6-bond in compounds XI and XII seems to be more promising, since it did not affect significantly the affinity for mPRα while it reduced affinity for nPR, resulting in discrimination index mPRα/nPR between 1.9 and 4.3.
Saturation of the Δ4-bond in compounds XIII-XVI moderately decreased affinity for mPRα, cis-coupling of the A and B rings (5βH) being less favorable for binding compared with trans-coupling (αΗ). The magnitude of the negative effect on binding to nPR depended very significantly on the presence of an additional D′-cycloalkane ring: in its presence (compounds XIII-XIV), the affinity dropped to values more than two orders lower than that of reference compound, while in the absence of a D′-ring (compounds XV-XVI) the effect was much less prominent. These facts are reflected in respective values of mPRα/nPR discrimination indexes.
All three tested modifications at C19 (compounds XVII-XIX) significantly diminished affinity for nPR. An additional methylene group (compound XVIII) also moderately reduced affinity for mPRα, while methyl (compound XVII) and hydroxy (compound XIX) groups gave moderate increase in affinity values. As a result, mPRα/nPR discrimination indexes for these two compounds reached values of 80-90. Two differences between our data and those reported in [9] should be noted. First, unlike Kelder’s study [9], in our experiments a 19-methylene group moderately decreased affinity for mPRα. Second, the 19-hydroxyl group showed opposite influences on affinity for mPRα in two studies. These discrepancies might be attributed to differences in steroid backbones used, as noted above for the case of 5H-steroids.
Replacement of the 3-oxo group by a 3-hydroxyimino group (compound XX) and particularly by a 3-O-methoxyimino group (compound XXI) gave the highest values of mPRα/nPR discrimination indexes (more than 500 for compound XXI) due to decrease in affinity for nPR and rise in affinity for mPRα (compare curves for compounds XX, XXI, and IV in Fig. 1). The data indicate that the modes of involvement of C3-substituents in ligand–receptor interaction for nPR and mPRα are quite different. The 3-O-methoxyimino group supports half-chair geometry of the A-ring, which is characteristic for steroids containing a Δ4-3-ketone. However, it cannot form the respective hydrogen bond network with amino acids in the nPR ligand-binding pocket that drives conformation switch in the protein structure [10]. Obviously, such hydrogen bonds do not play a role in interactions of Δ4-3-ketosteroids with mPRα. Since the 3-hydroxyimino and 3-O-methoxyimino derivatives have similar affinities for mPRα, one can suggest the presence of a cavity in the protein neighboring the C3 of the ligand. Taking into account the inhibitory effect of enlargement of the C3-substituent on affinity for nPR [15], this cavity in mPRα could serve as an additional reserve for further improvement of ligand selectivity towards mPRα.
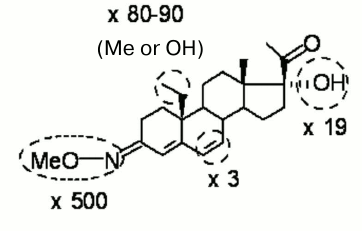
The simultaneous presence of two substituents in a steroid molecule often leads to additive effects on the affinity for the receptor. However, as demonstrated here in the case of 5H-derivatives of progesterone and its 16α,17α-cycloalkane derivative, this is not a general rule. Nevertheless, one can expect that double or even triple derivatization will give a benefit in the context of selectivity improvement as compared with the effect of a single substituent. Figure 2 summarizes modifications of the progesterone molecule favorable for selective binding to mPRα with potential impact of each substituent. Only those modifications, which increase or at least do not diminish significantly the affinity for mPRα were included. Ideally (in the case of full additivity), a ligand bearing all four modifications might have a preference toward mPRα over nPR equal (500 × 80 × 19 × 3) = 2,280,000.
Fig. 2. Modifications that significantly increase discrimination index (shown as ×fold values) and moderately increase or only slightly decrease binding affinity of ligands for mPRα.
DISCUSSION
The nPR naturally expressed in mammalian cells and mPRα expressed in heterologous yeast cells were used for revealing structural determinants of steroid ligands that have an effect on interactions with nuclear and membrane progesterone receptors. Such comparison between two progesterone receptors needs special comment. The nature of nPR (soluble protein) and mPR (integral membrane protein) is quite different. The folding, processing, and intracellular transfer will be needed in specific sets of proteins. It is not clear whether such yeast proteins can substitute completely for their mammalian orthologs. Thus, when human mineralocorticoid receptor was expressed in yeast, it partially lost its ligand specificity in signal transduction on a reporter gene [25]. Similarly, mPRs expressed in yeast did not require G-proteins for progesterone-dependent signaling [11], while, in mammalian cells, mPRs are apparently coupled with G-proteins [24]. Therefore, heterologous expression of both progesterone receptors in the same type of cells does not guarantee the absence of possible anomalies in its characteristics.
The functionality of mPRs in yeast has been shown using the FET3-lacZ reporter [11]. In this system, human mPRs rendered an ability of cells to respond to physiological concentrations of progesterone similarly to the effect of adiponectin via its receptors, AdipoR1 and AdipoR2, expressed in these cells. It is not yet clear which messengers are involved in progesterone signal transduction on expression of the FET3 gene. However, another member of the PARQ family, Izh2p, which is endogenous for yeast, produces sphingoid bases that probably function as the second messenger responsible for the effect of Izh2p on FET3 [26]. In yeast, the topology of heterologically expressed mPRs may be opposite to that in mammalian cells as well as involvement or no G-protein coupling in signaling (compare [11] and [24]). For the aims of the present study, these issues do not have great significance since in the used conditions progesterone can equally easily reach its binding sites located on extracellular, cytoplasmic, or intravesicular surfaces of yeast membranes.
In our hands, under very similar although not identical experimental conditions (see “Materials and Methods”), Kd values for progesterone interactions with nPR and mPRα were 7.0 and 143 nM, respectively (see table), i.e. nPR binds progesterone with 20-fold higher affinity compared to mPRα. Similarly, 10-fold difference between nPR and mPRα in their affinities for progesterone was reported in the study of Kelder at al. [9], where mPRα was expressed in mammalian MDA-MB-231 cells. In that study [9], unbound ligand was removed by filtration for mPR and by charcoal adsorption for nPR. Also, similarly to our data, ovine mPRα expressed in CHO cells demonstrated Kd for [3H]progesterone of 122 nM and IC50 for unlabeled progesterone competition of 174 nM when measured using charcoal adsorption [27]. To the best of our knowledge, direct measurements of progesterone affinity for nPR expressed in yeast have not been performed. However, in a yeast reporter system with human nPR, progesterone demonstrated EC50 of 7.38 nM [28], i.e. at least one order of magnitude lower than the observed Kd for mPR and similar to values reported for nPR expressed in mammalian cells.
These data suggest that the observed difference in affinities for progesterone reflects differences in intrinsic properties of two receptors and is not a consequence of conditions of their expression in mammalian or yeast cells or methods of measurements. Accordingly, a ligand with discrimination index of 20 will equally bind to both receptors. Thus, to be really selective for mPRα, a ligand has to have a discrimination index of at least 2000.
At this point, it is impossible to predict which ones from suggested progesterone derivatives will be agonists or antagonists of mPRα. Moreover, it is still unclear how such activities might be examined. Currently used transfection of mammalian cells with mPR vectors may give rise to unnatural coupling of a receptor with downstream effectors. On the other hand, it is difficult to find cells that naturally express exceptionally a receptor of interest. Apparently, the problem will be solved gradually using both approaches. Thus, one can contemplate experiments with knockdown or knockout of a receptor of interest and comparisons between the effects of suggested progesterone derivatives before and after receptor inactivation.
Many other pitfalls on the path to creation of effective ligands for mPRs, both agonists and antagonists, are expected. First, such ligands should be stable enough for use in vivo. Second, they should not be effective competitors for other steroid-binding proteins such as various nuclear receptors and transport proteins. For example, progesterone has rather high affinity for corticosteroid-binding globulin (CBG) [29]. As such, suggested progesterone derivatives even at pharmacological concentrations should not disturb the equilibrium between bound and unbound corticosteroids in the bloodstream.
As shown in the table, corticosterone was a very weak competitor for [3H]progesterone binding in our assay with uterine cytosol. Therefore, the binding to CBG from blood contamination can be responsive for a negligible part if any of the observed [3H]progesterone binding. We were unable to check the interaction of our 3-hydroxyimino and 3-O-methoxyimino derivatives with CBG since these compounds bear also the additional D′ carbocycle that render high affinity for a yet unidentified serum protein [30]. The data on crystal structure of CBG suggests that the 3-oxo group of a ligand does not form hydrogen bonds with the protein [31]. Thus, one can expect that 3-hydroxyimino and 3-O-methoxyimino derivatives of progesterone will have affinities for CBG comparable with affinities of respective 3-oxo derivatives.
The expression of mPRs has been documented in many cell types in both reproductive and non-reproductive mammalian tissues [8]. These data suggest that mPRs participate in many functions in mammals. However, there is some controversy surrounding whether or not mPR is a true receptor for progesterone. The main obstacle for resolving this issue is simultaneous presence of several potential progesterone sensors in the same cell. In addition to two isoforms of nPR and three (or five) mPRs, progesterone receptor membrane component 1 (PGRMC1) [32], the α-subunit of Na/K-ATPase [33], and presynaptic receptors and ion channels like σ-1 receptor, α(1) receptor, nicotine receptor, D1 receptor, NMDA receptor, GABA(A) receptor, and L-type Ca2+ channels [34] have been implicated in progesterone action. The design of selective progesterone analogs, both agonists and antagonists, for each of these potential progesterone sensors will be helpful for probing the biology of mPRs and other progesterone sensors and for important practical applications such as immunosuppression [35], neuroprotection [36], or anticancer therapy [37]. Four modifications in the progesterone molecule (substitution 3-oxo → 3-O-methoxyimino or 3-oxo → 3-hydroxyimino; introduction of 19-methyl, 19-methylene, or 19-hydroxy group; introduction of 17α-hydroxy group; and double bond C6–C7) were found here to be favorable for preferential binding of progesterone to mPRα over nPR. Previously, stimulatory effect for the binding to mPRα has been shown for an 18-methyl or 18-methylene group, thought the effect of these modifications on the binding to nPR was not investigated [9]. Thus, a multitude of possible combinations of such modifications may give rise a broad spectrum of progesterone derivatives with desired properties of selective agonists or antagonists for mPRs.
REFERENCES
1.Jensen, E., and Jacobson, H. (1962) Rec. Progr.
Horm. Res., 18, 387-414.
2.Luconi, M., Francavilla, F., Porazzi, I.,
Macerola, B., Forti, G., and Baldi, E. (2004) Steroids,
69, 553-559.
3.Evanson, N. K., Herman, J. P., Sakai, R. R., and
Krause, E. G. (2010) J. Neuroendocrinol., 22,
846-861.
4.Davis, P. J., Lin, H. Y., Mousa, S. A., Luidens, M.
K., Hercbergs, A. A., Wehling, M., and Davis, F. B. (2011)
Steroids, 76, 829-833.
5.Thomas, P. (2012) Gen. Comp. Endocrinol.,
175, 367-383.
6.Zhu, Y., Rice, C. D., Pang, Y. F., Pace, M., and
Thomas, P. (2003) Proc. Natl. Acad. Sci. USA, 100,
2231-2236.
7.Zhu, Y., Bond, J., and Thomas, P. (2003) Proc.
Natl. Acad. Sci. USA, 100, 2237-2242.
8.Dressing, G. E., Goldberg, J. E., Charles, N. J.,
Schwertfeger, K. L., and Lange, C. A. (2011) Steroids,
76, 11-17.
9.Kelder, J., Azevedo, R., Pang, Y., de Vlieg, J.,
Dong, J., and Thomas, P. (2010) Steroids, 75,
314-322.
10.Williams, S. P., and Sigler, P. B. (1998)
Nature, 393, 392-396.
11.Smith, J. L., Kupchak, B. R., Garitaonandia, I.,
Hoang, L. K., Maina, A. S., Regalla, L. M., and Lyons, T. J. (2008)
Steroids, 73, 1160-1173.
12.Levina, I. S., and Kamernitzky, A. V. (1990)
Khim.-Farm. Zh., 24, 31-39.
13.Levina, I. S., Nikitina, G. V., Kulikova, L. E.,
and Kamernitzky, A. V. (1995) Rus. Chem. Bull., 44,
547-550.
14.Levina, I. S., Kulikova, L. E., Kamernitskii, A.
V., Pokrovskaya, E. V., and Smirnov, A. N. (2005) Russ. Chem. Bull.
Int. Ed., 54, 2664-2670.
15.Levina, I. S., Pokrovskaya, E. V., Kulikova, L.
E., Kamernitzky, A. V., Kachala, V. V., and Smirnov, A. N. (2008)
Steroids, 73, 815-827.
16.Sambrook, J., Fritsch, E. F., and Maniatis, T.
(1989) Molecular Cloning. A Laboratory Manual, 2nd Edn., Cold
Spring Harbor Laboratory Press.
17.Matchmaker, T. M. (2007) Library Construction
and Screening Kits User Manual. Clontech Laboratories, Inc. A
Takara Bio Company. Cat. No. 630445 PT3955-1 (PR792376).
18.Smirnov, A. N., Pokrovskaya, E. V., Kogteva,
G. S., Shevchenko, V. P., Levina, I. S., Kulikova, L. E., and
Kamernitzky, A. V. (2000) Steroids, 65,
163-170.
19.Pokrovskaia, E. V., Levina, I. S., Kulikova, L.
E., Kamernitskii, A. V., and Smirnov, A. N. (2004) Russ. J. Bioorg.
Chem., 30, 268-274.
20.Smirnov, A. N., Pokrovskaya, E. V., Levina, I.
S., Kulikova, L. E., Kamernitskii, A. V., and Shevchenko, V. P. (2001)
Bull. Exp. Biol. Med., 131, 245-247.
21.Kasid, A., Buckshee, K., Hingorani, V., and
Laumas, K. R. (1978) Biochem. J., 176, 531-539.
22.Atkins, D. T., Kraemer, D. C., Harms, P. G., and
Fleeger, J. L. (1980) Biol. Reprod., 23, 317-323.
23.Vacas, M. I., Lowenstein, P. R., and Cardinali,
D. P. (1979) Neuroendocrinology, 29, 84-89.
24.Thomas, P., Pang, Y., Dong, J., Groenen, P.,
Kelder, J., de Vlieg, J., Zhu, Y., and Tubbs, C. (2007)
Endocrinology, 148, 705-718.
25.Bureik, M., Bruck, N., Hubel, K., and
Bernhardt, R. (2005) FEMS Yeast Res., 5, 627-633.
26.Villa, N. Y., Kupchak, B. R., Garitaonandia, I.,
Smith, J. L., Alonso, E., Alford, C., Cowart, L. A., Hannun, Y. A., and
Lyons, T. J. (2009) Mol. Pharmacol., 75, 866-875.
27.Ashley, R. L., Arreguin-Arevalo, J. A., and Nett,
T. M. (2009) Reprod. Biol. Endocrinol., 7, 42.
28.Wang, J., Xie, P., Kettrup, A., and Schramm, K.
W. (2005) Sci. Total Environ., 349, 120-128.
29.Cameron, A., Henley, D., Carrell, R., Zhou, A.,
Clarke, A., and Lightman, S. J. (2010) Clin. Endocrinol. Metab.,
95, 4689-4695.
30.Smirnov, A. N., Pokrovskaya, E. V., Levina,
I. S., Kulikova, L. E., Kamernitzky, A. V., and Shevchenko, V. P.
(2001) Biochemistry (Moscow), 66, 688-692.
31.Klieber, M. A., Underhill, C., Hammond, G. L.,
and Muller, Y. A. (2007) J. Biol. Chem., 282,
29594-29603.
32.Rohe, H. J., Ahmed, I. S., Twist, K. E., and
Craven, R. J. (2009) Pharmacol. Ther., 121, 14-19.
33.Morrill, G. A., Erlichman, J., Gutierrez-Juarez,
R., and Kostellow, A. B. (2005) Steroids, 70,
933-945.
34.Zheng, P. (2009) Prog. Neurobiol.,
89, 134-152.
35.Ndiaye, K., Poole, D. H., Walusimbi, S., Cannon,
M. J., Toyokawa, K., Maalouf, S. W., Dong, J., Thomas, P., and Pate, J.
L. (2012) J. Reprod. Immunol., 95, 15-26.
36.Thomas, P., and Pang, Y. (2012)
Neuroendocrinology, 96, 162-171.
37.Dressing, G. E., Alyea, R., Pang, Y., and Thomas,
P. (2012) Horm. Cancer, 3, 101-112.