Three-Dimensional Structure of Carboxypeptidase T from Thermoactinomyces vulgaris in Complex with N-BOC-L-leucine
V. I. Timofeev1, S. A. Kuznetsov1, V. Kh. Akparov2, G. G. Chestukhina2, and I. P. Kuranova1*
1Shubnikov Institute of Crystallography, Russian Academy of Sciences, Leninsky pr. 59, 119333 Moscow, Russia; fax: (499) 135-1011; E-mail: inna@ns.crys.ras.ru2Scientific Center of Russian Federation Research Institute for Genetics and Selection of Industrial Microorganisms, 1-yi Dorozhnyi Proezd 1, 117545 Moscow, Russia; fax: (495) 315-0501; E-mail: valery@akparov.ru
* To whom correspondence should be addressed.
Received October 15, 2012; Revision received November 9, 2012
The 3D structure of recombinant bacterial carboxypeptidase T (CPT) in complex with N-BOC-L-leucine was determined at 1.38 Å resolution. Crystals for the X-ray study were grown in microgravity using the counter-diffusion technique. N-BOC-L-leucine and SO42– ion bound in the enzyme active site were localized in the electron density map. Location of the leucine side chain in CPT–N-BOC-L-leucine complex allowed identification of the S1 subsite of the enzyme, and its structure was determined. Superposition of the structures of CPT–N-BOC-L-leucine complex and complexes of pancreatic carboxypeptidases A and B with substrate and inhibitors was carried out, and similarity of the S1 subsites in these three carboxypeptidases was revealed. It was found that SO42– ion occupies the same position in the S1′ subsite as the C-terminal carboxy group of the substrate.
KEY WORDS: carboxypeptidase T, S1 subsite, crystal structure, X-ray structural analysisDOI: 10.1134/S0006297913030061
Abbreviations: BOC-L-leucine, N-(tert-butoxycarbonyl)-L-leucine; CABS-Sepharose, p-aminobenzylsuccinyl-Sepharose 4B; CHAPS, 3-[(3-cholamidopropyl)-dimethylammonio]-1-propanesulfonate; CPA, carboxypeptidase A; CPB, carboxypeptidase B; CPT, carboxypeptidase T; DFP, diisopropyl fluorophosphate; IPTG, isopropyl-β-D-thiogalactopyranoside; MPD, 2-methyl-2,4-pentadiol.
Carboxypeptidase T (CPT) from Thermoactinomyces vulgaris (EC
3.4.17.18) isolated and characterized earlier [1]
belongs to zinc-dependent exopeptidases splitting out the C-terminal
amino acid residues of proteins and peptides. The 3D structure of CPT
is similar to that of such classical enzymology objects as pancreatic
metallocarboxypeptidases A (CPA) and B (CPB) (EC 3.4.17.1 and 3.4.17.2,
respectively) [2]. The amino acid sequence of CPT
is 23% identical to those of carboxypeptidases A and B, and their
active sites have similar structures, but specificity of CPT is
significantly wider than those of pancreatic carboxypeptidases: CPA
cleaves mainly hydrophobic and CPB the positively charged amino acid
residues, whereas CPT is able to split out both types of residues. That
is why CPT is a model suitable for revealing the elements of molecular
structure that impact the selectivity of the enzyme.
Being bound in the active center, the substrate interacts with the enzyme at several sites. According to the Schechter and Berger nomenclature, the subsites that bind the side chains of amino acid residues located before the cleaved bond are designated as subsites S1, S2, etc., the site number increasing with increasing distance from the cleaved bond [3]. The subsites that bind amino acid residues of the substrate located after the cleaved bond are designated as S1′, S2′, etc. The substrate amino acid residues bound in these sites are designated respectively as P1, P2 or P1′, P2′, etc.
The present view is that substrate specificity of metallocarboxypeptidases is defined by the composition and spatial structure of the S1′ subsite or the primary specificity subsite, which binds the C-terminal amino acid residue [4]. CPT differs from CPB by five amino acid replacements in the S1′ subsite. However, sequential replacement of these five residues in the primary specificity pocket of CPT by corresponding residues of CPB did not result in changes in the enzyme selectivity [4, 5]. That is why it was suggested that amino acid residues not belonging directly to the S1′ subsite can also participate in regulation of the primary specificity of CPT. For example, it was shown that CPT selectivity depends on Ca2+ concentration in solution [6]. In contrast to CPB, there are five Ca2+ binding sites in CPT [2]. Recently, the 3D structure of the calcium-free form of CPT was determined [7]. Superposition of the 3D structures of calcium-containing and calcium-free forms of the enzyme revealed differences in conformation of the Ca2+ binding sites and in location and number of water molecules in the active sites of these two enzyme forms. It was supposed that these differences can influence the rate of the catalyzed reaction. However, it is impossible to rationalize wide substrate specificity observed for CPT by this fact alone.
The S1 subsite, which binds the amino acid residue preceding the cleaved residue, can be one of the molecule regions participating in regulation of the enzyme selectivity. Change in the nature of the side groups of amino acid residues interacting with the S1 subsite is known to have a significant effect on activity of carboxypeptidases B and T [8, 9]. It was also shown that the S1 subsite affects the substrate specificity and efficiency of catalysis by metallocarboxypeptidase M [10]. The location of the S1 subsite in the CPT molecule was still unclear.
In this work, we obtained a crystalline CPT complex with N-(tert-butoxycarbonyl)-L-leucine (N-BOC-L-leucine) and performed the X-ray study of its 3D structure at 1.38 Å resolution. The bound N-BOC-L-leucine and SO42– ion were localized in the enzyme active site. Location of the leucine side chain in the CPT–N-BOC-L-leucine complex allowed us to identify the S1 subsite of the enzyme and to describe its structure. The structures of CPT–N-BOC-L-leucine complex and complexes of pancreatic carboxypeptidases A and B with peptide ligands and selective inhibitors were superimposed, and the similarity of the S1 subsites in these three carboxypeptidases was revealed. It was also found that the SO42– ion occupies the same position in the S1′ subsite as the C-terminal carboxylic group of the substrate.
MATERIALS AND METHODS
Reagents. We used the following reagents in this study: MilliQ deionized water from Millipore (USA) with resistance 19 MΩ·cm; glycerol from Khimmed (Russia) and chemically pure urea of Russian production; Protosubtilin G10× from Ladyzhinsky Plant of Bio-/Enzyme Preparations (Ukraine) (subtilisin 72 was isolated from Protosubtilin G10× according to [11]); 2-methyl-2,4-pentadiol (MPD), L-cystine and L-cysteine, diisopropyl fluorophosphate (DFP), isopropyl-β-D-thiogalactopyranoside (IPTG), N-BOC-L-leucine, 3-[(3-cholamidopropyl)-dimethylammonio]-1-propanesulfonate (CHAPS), 4-morpholinoethanesulfonic acid (MES) from Sigma (USA); mercaptoethanol, CaCl2·2H2O, ZnSO4, NaCl from Merck (Germany); YM10 ultrafiltration membranes and ultrafiltration cells from Amicon (USA); Centripack microfiltration holder from Millipore (USA). For CPT crystallization, glass capillaries 60 mm in length and inner diameter of 0.5 mm from Confocal Science Inc. (Japan) were used.
The chromogenic substrate ZAALpNA for subtilisin determination and colored substrate DNP-AAR for determination of CPT activity were synthesized in the Laboratory of Protein Chemistry of the GOSNIIGENETIKA [12]. Affinity sorbent p-aminobenzylsuccinyl-Sepharose 4B (CABS-Sepharose) was prepared according to [13].
Recombinant CPT. The pro-CPT gene was expressed in Escherichia coli BL21(DE3) pLysS cells according to the instructions provided by Novagen (USA) [14]. After induction of expression by IPTG, the cells were ultrasonically disintegrated.
Native CPT was isolated from inclusion bodies as described earlier [15]. Inclusion bodies were precipitated by centrifugation and washed with 0.05% CHAPS, 2 M NaCl, and water. The pellet containing inclusion bodies was dissolved in 8 M urea containing 1 mM mercaptoethanol to the final protein concentration 5 mg/ml. Then this solution was added dropwise to 100-fold volume of 50 mM Tris-HCl buffer, pH 8.0, containing 30% glycerol (v/v), 0.5 M NaCl, 10 mM CaCl2, 3 mM cysteine, and 10 mM cystine. The reaction mixture was incubated for 16 h, and the non-renatured protein was removed by centrifugation. The solution was concentrated by ultrafiltration using an YM10 membrane to volume of 200 ml, and then the medium was replaced by buffer of the same composition but without glycerol using ultrafiltration. To obtain mature enzyme, subtilisin 72 was added to the proCPT solution in the ratio CPT/subtilisin = 200 : 1 (w/w) and the mixture was incubated for 1 h at 37oC. Subtilisin was inactivated by addition of DFP. Solution with the activated CPT was again concentrated by ultrafiltration to 2 ml volume and centrifuged.
After concentration, the protein solution was acidified to pH 6.2 by adding 100 mM MES-NaOH buffer, pH 6.0. Then the protein solution was applied on a column (volume 20 ml) with CABS-Sepharose [15] equilibrated with 10 mM MES-NaOH buffer, pH 6.0, containing 0.5 M NaCl and 10 mM CaCl2, and the CPT was eluted by the same buffer. Fractions containing the active protein were pooled, concentrated to 1 ml, and the buffer was replaced by buffer for crystallization (0.01 M MES-NaOH, pH 6.0, containing 1 mM CaCl2, 0.1 mM ZnSO4 and 0.25 M NaCl) by three ultrafiltrations using an Amicon cell with 10-fold dilution, and then protein was concentrated to 10 mg/ml and sterilely filtered using Centripack centrifuge holders. Protein concentration was determined according to Bradford [16]. The final solution was used for protein crystallization. The absence of subtilisin activity was proved using specific chromogenic substrate ZAALpNA. SDS-PAGE was performed according to Laemmli [17].
Crystal growth. Crystals of CPT–N-BOC-L-leucine complex were grown in microgravity in a capillary using the counter-diffusion technique. Equipment and technology developed by the Aerospace Agency of Japan JAXA were used for crystal growth [18, 19]. The protein concentration in 10 mM MES-NaOH buffer, pH 6.0, containing 0.25 M NaCl, 0.1 mM ZnSO4, and 1 mM CaCl2 was 11 mg/ml. A 2.4 M N-BOC-L-leucine solution was added to precipitant solution containing 1.6 M (NH4)2SO4, 5% MPD in 50 mM MES-NaOH buffer, pH 6.0.
A drop of protein solution (10 µl) was placed on siliconized glass. A capillary was filled with the protein solution, touching the drop by the end of the capillary, and then the opposite end of capillary was hermetically sealed with plasticine. The end of the capillary with solution was placed in a silicone tube with 1% agarose gel. The tube was shortened to 5 mm and immersed in a plastic cylinder 180 µl in volume filled with precipitant solution containing 2.4 M N-BOC-L-leucine (Fig. 1). The bottom of the cylinder was closed by a stopper with holes for removal of excess solution, and both ends of the cylinder were hermetically isolated with glue. Six crystallization units thus equipped and united together were placed in a plastic bag. The bags were packed in containers and delivered to the International Space Station where containers were placed in a thermostat at 20°C. After the end of the experiment and delivery of the samples back to the laboratory, the quality of crystals grown in microgravity was estimated visually using a microscope (Fig. 2). To collect diffraction data using a synchrotron source, the crystal was taken off the capillary to precipitant solution, then placed in cryosolution and frozen in nitrogen vapor flow. Along with the precipitant components, the cryosolution also contained 20% glycerol (v/v).
Fig. 1. Scheme of device for crystallization in a capillary using the counter-diffusion technique [18].
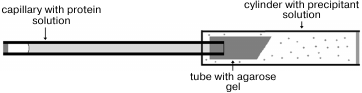
Fig. 2. Capillary with a crystal of CPT–N-BOC-L-leucine complex.

Data collection and processing. Diffraction data sets from grown crystals were collected using Spring 8 (Japan) synchrotron at the temperature 100 K on BL41XU station with CCD detector MAR225HE. Diffraction data for one crystal were obtained by the rotation method at wavelength 0.8 Å, crystal–detector distance 125 mm, the oscillation angle 0.3°, the rotation angle 180°. Processing of experimental data set was performed with HKL-2000 program package [20] to 1.38 Å resolution. There is one enzyme molecule per asymmetric unit (space group 6322). The data processing statistics are given in the table.
Structure determination and refinement. The structure of CPT–N-BOC-L-leucine complex was determined at 1.38 Å resolution by the molecular replacement method using the Phaser program [21] and CPT atomic coordinates (PDB ID: 3QNV) as the starting model. The Refmac program was used for structure refinement [22]. Models were manually corrected using the Coot program [23], and electron density maps were calculated with 2|Fo|–|Fc| and |Fo|–|Fc| coefficients. Water molecules were localized on the electron density maps, and difference Fourier synthesis revealed electron density in the active site, which was identified as the N-BOC-L-leucine ligand and SO42– ion. Refinement of ligands with complete occupancies of atomic positions gave B factor 19.9 Å2, close to the average B factor value for the structure 17.1 Å2. Refinement statistics are presented in the table.
Statistical characteristics of experimental data set and refinement of
the structure of the CPT–N-BOC-L-leucine complex

*Values for the last shell are given in parentheses.
Structures of complexes CPT–N-BOC-L-Leu and CPA–Gly-Tyr (PDB ID: 3CPA) [24], CPA–potato inhibitor (PDB ID: 4CPA) [25], CPB–inhibitor BX528 (PDB ID: 2PIY) [26] were compared by superposition of the structures via atoms of the following amino acid residues that form the environment of the bound ligands: 166, 167, 206, 255, 287 (numeration as in CPT); 163, 164, 198, 248, 279 (numeration as in CPA and CPB). The root-mean-square deviations of Cα atoms of the superimposed residues in pairs CPT/CPA–Gly-Tyr, CPT/CPA–potato inhibitor, CPT/CPB–inhibitor BX528 were 0.428, 0.348, and 0.289 Å, respectively.
RESULTS AND DISCUSSION
The purity of the carboxypeptidase T preparation after renaturation, activation, and purification by affinity chromatography was not less than 95% according to the SDS-PAGE data. The yield of protein after renaturation was ~1%. The purified preparation was used for growth of crystals of CPT–N-BOC-L-leucine complex.
Crystals of CPT–N-BOC-L-leucine complex grown in microgravity by the counter-diffusion technique belong to space group P6322 and are 0.20-0.35 mm in size (Fig. 2). The crystal used for collection of the diffraction data set using the Spring-8 synchrotron diffracted at 1.38 Å resolution, which is 0.3-0.4 Å better than the resolution of control crystals grown on the Earth. The higher quality of crystals grown in microgravity is rationalized by the absence of convectional flow in the crystallization solution. In the absence of convection a stable zone depleted of protein arises around a crystal, and substance is transported by diffusion to the growing crystal [27, 28]. Diffusion transport provides a low degree of precipitant oversaturation, and this promotes the higher degree of crystal lattice ordering.
Using the collected diffraction data set, the 3D structure of the CPT–N-BOC-L-leucine complex was determined by the molecular replacement method and refined to 1.38 Å resolution to Rcryst 13.9% and Rfree 15.2% (the table). The refined atomic coordinates were deposited in Protein Data Bank (PDB ID: 4FBZ).
The structure of the complex is almost identical to that of the free CPT molecule (PDB ID: 1OBR): the root-mean-square deviation at superposition via Cα atoms is 0.241 Å. Difference Fourier synthesis revealed electron density in the active site, which was identified as the N-BOC-L-leucine molecule and localized with occupancy close to 100%. As in the structure of the free enzyme [1], a SO42– ion was also localized in the active site. These ions were present in the crystallization solution at high concentration as one of the precipitant components. The CPT molecule with the ligands bound in the active site is shown in Fig. 3.
Fig. 3. CPT molecule with ligands N-BOC-L-leucine and SO42– ion bound in the active site. The zinc ion in the active site is shown as gray sphere. The electron density map for ligands was calculated with coefficients 2|Fo|–|Fc| and contoured at σ = 1.
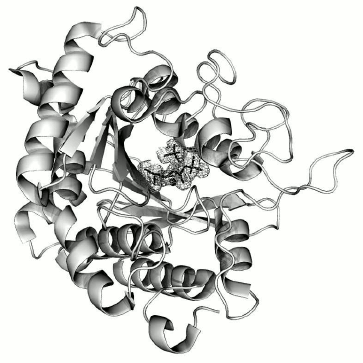
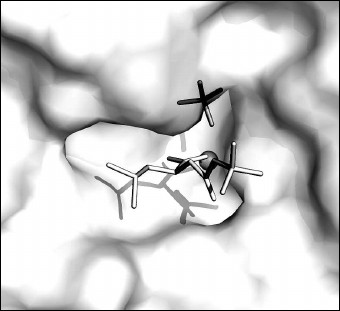
The bound ligands on the surface of the pocket of the CPT active site are shown in Fig. 4. A part of the active site where the N-BOC-L-leucine molecule is located is opened and accessible to solvent. In contrast, the SO42– ion is partly shielded by amino acid residues bound with it.
Fig. 4. N-BOC-L-leucine and SO42– ion in the pocket of the CPT active site. The zinc ion in the active site is shown as gray sphere.
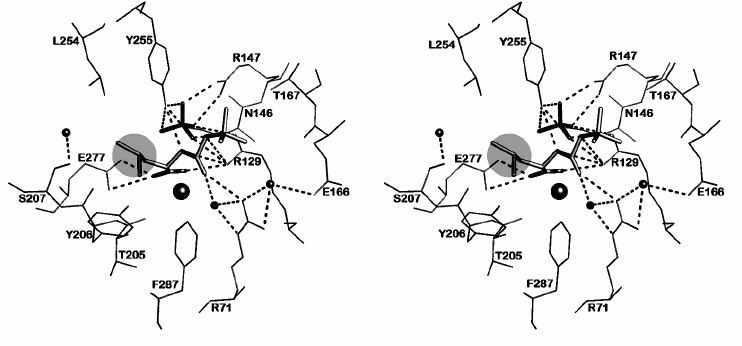
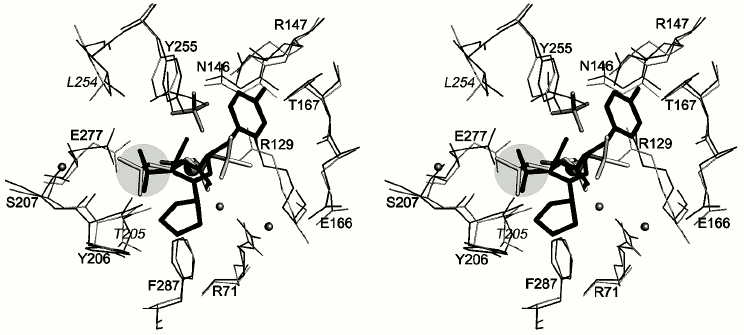
The environment of N-BOC-L-leucine and the SO42– ion in a radius of ≤4.5 Å is presented in Fig. 5. Both oxygen atoms of the carboxy group of N-BOC-L-leucine are coordinated by the zinc atom in the active site. Along with this, one of the atoms via water molecule is H-bonded with Glu72, a constituent of the coordination sphere of the zinc atom; the second oxygen atom forms a hydrogen bond with the guanidine group of Arg129. Amino acid residues Tyr255, Tyr206, Glu277, Thr205, and Ser207 are located within a radius of 4 Å from the side chain of L-leucine. Tyr255 forms a hydrogen bond with the nitrogen atom of the imino group of N-BOC-L-leucine. The distance between the side chain of leucine in N-BOC-L-leucine and the side chain of Leu254 of CPT is ~4.2 Å. The homologous tyrosine residue (Tyr248) in CPB participates in binding the C-terminal amino acid residue of the substrate in the S1′ subsite, and Ile247 residue homologous to Leu254 is a constituent of a hydrophobic cluster limiting the S′ subsite in carboxypeptidases A and B. The methyl groups of the tert-butoxycarbonyl residue are surrounded by side chains of the Thr167, Arg71, and Arg129 residues. Arg129 (127 in CPA numeration) is known to directly participate in the reaction catalyzed by the enzyme [25]. The carbonyl oxygen of the BOC group forms a hydrogen bond with the guanidine group of Arg71; the phenyl group of Phe287 is also near the carbonyl group.
Fig. 5. Stereo view of the nearest environment of N-BOC-L-leucine and the SO42– ion. Amino acid residues within the range ≤4.5 Å are presented. Ligands are colored gray. The zinc ion is shown as a large radius sphere, and water molecules as spheres of the smaller radii. Hydrogen bonds are drawn as dashed lines. The side chain of leucine is in the light-gray circle.
The SO42– ion forms hydrogen bonds with the imino group of N-BOC-L-leucine and with the following amino acid residues of the enzyme: Arg129, Asn146, Arg147, Tyr255, and Thr257. In pancreatic carboxypeptidases A and B, homologous residues (Arg127, Asn144, Arg145, and Tyr248) participate in binding the C-terminal carboxyl group of the substrate, so that SO42– in the CPT–N-BOC-L-leucine complex occupies the same place as the carboxyl group of substrate in complexes with CPA and CPB [25, 26].
To be sure that it is the S1 subsite that is occupied by the side chain of leucine in the CPT–N-BOC-L-leucine complex, the structure of this complex was superimposed with complexes of pancreatic carboxypeptidases A and B containing the substrates or specific inhibitors bound in their active centers. These are CPA complexes with peptide inhibitor from potato [25] and with Gly-Tyr substrate [24] and CPB complex with a selective inhibitor – peptide mimetic BX528 [26]. The structures were superimposed via the atoms of five homologous amino acid residues in the nearest environment of the bound ligands.
The CPA complex with the peptide inhibitor from potato is formed after breaking of the C-terminal peptide bond between the C-terminal amino acid residues of the inhibitor, Val38 and Gly39. After cleavage of Gly39, Val38 becomes the C-terminal residue. The complex thus formed can be considered as a complex of enzyme with the reaction product. In this complex the side chain of Val38 occupies the S1 subsite, whereas the penultimate side chain of Tyr37 occupies the S2 subsite.
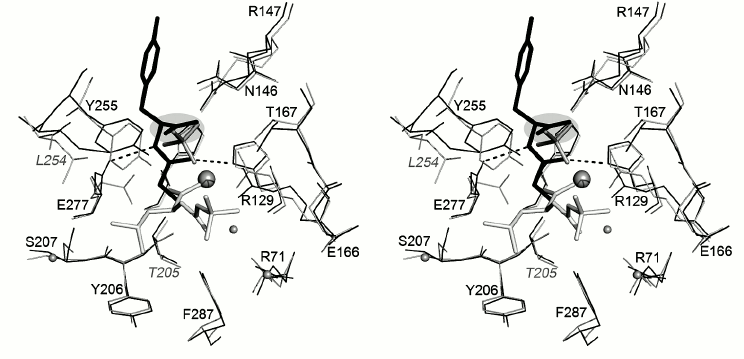
The results of superposition of structures of CPA and CPB complexes indicate that the isobutyl chain of N-BOC-L-leucine in the CPT–N-BOC-L-leucine complex and the side chain of the C-terminal Val38 of the inhibitor molecule in the CPA–potato inhibitor complex occupy similar positions and are in similar environment (Fig. 6). The difference in position of the Cβ atoms of Val and Leu in corresponding complexes is less than 0.49 Å. The fact that the side chain environments in both complexes differ somewhat is rationalized by replacement of Leu254 and Thr205 in CPT for Ile247 and Ser197 in CPA and CPB. Earlier it was found [6] that Leu254 affects the substrate selectivity of CPT: the CPT L254N mutant less efficiently catalyzes splitting of the negatively charged C-terminal amino acid residues than the wild type enzyme. The position of the butoxy group of N-BOC-L-leucine almost completely coincides with the position of the side group of Tyr37 of the potato inhibitor occupying the S2 subsite.
Fig. 6. Superposition of the structures of complexes CPT–N-BOC-L-leucine and CPA–potato inhibitor (stereo view). N-BOC-L-leucine and the SO42– ion in CPT are shown in gray; three C-terminal residues of potato inhibitor (Pro36, Tyr37, and Val38) are shown in black; the environment of these ligands is shown within the radius 4.5 Å. Amino acid residues are numerated as in CPT. The numbers of amino acid residues that differ in the two proteins are shown in italic and light-gray. The side chains in the S1 subsite are shown on a gray background. The zinc ion is shown by a sphere of large radius and water molecules by spheres of smaller radii.
Comparison of positions of N-BOC-L-leucine and SO42– ion in the CPT–N-BOC-L-leucine complex with the substrate position in the CPA–Gly-Tyr complex demonstrates that the carboxyl group of tyrosine and the sulfate anion occupy similar positions (Fig. 7). Amino acid residues Asn144, Arg147, and Tyr248 participate in binding the C-terminal carboxyl group of tyrosine in the CPA–Gly-Tyr complex. These residues are analogous to those mentioned above that bind the SO42– ion in the CPT–N-BOC-L-leucine complex. The data indicate that the SO42– ion is located in the same center where the C-terminal carboxyl group of the substrate is bound.
Fig. 7. Superposition of the structures of CPT–N-BOC-L-leucine and CPA–Gly-Tyr complexes (stereo view). Gly-Tyr is shown in black, and other designations are as in Fig. 6.
It should be mentioned that the CPT–N-BOC-L-leucine and CPA–Gly-Tyr complexes represent the state of the active site at different stages of the reaction catalyzed by the enzyme. In the complex of CPA with substrate Gly-Tyr, when the peptide bond is not yet cleaved, the carbonyl oxygen of the peptide group is at the distance of H-bond length from the zinc atom ligands His69 and His196. The nitrogen atom of the peptide group forms a hydrogen bond with Glu270 (Glu277 in CPT) that is directly involved in catalysis by the enzyme [25]. In the CPT–N-BOC-L-leucine complex, the ligand simulates the N-terminal fragment of substrate after cleavage of peptide bond and removal of the C-terminal amino acid. Both oxygen atoms of the carboxyl group of N-BOC-L-leucine are coordinated by the zinc atom of the active site. This complex can be considered as an enzyme complex with one of the reaction products.
Superposition of the structures of the CPB complex with selective inhibitor BX528 and the CPT–N-BOC-L-leucine complex is shown in Fig. 8. Inhibitor BX528 is a mimetic of tripeptide Phe-Val-Lys [26]. The Phe–Val peptide bond in the former is replaced by the sulfonamide group, phosphinate simulates the amide group of Val–Leu, and the side chain of lysine is replaced by the aminomethylphenyl group. The isopropyl radical of the ligand is located in the S1 subsite of this complex, the S2 subsite is occupied by the phenyl radical, and the aminomethylphenyl group simulating the positively charged side chain of the substrate is bound in the S1′ subsite. The positions of the side chain of leucine in the CPT–N-BOC-L-leucine complex and the isopropyl radical in the CPB–BX528 complex along with the environment of both residues practically coincide in the two complexes. The position of the phenyl radical occupying the S2 subsite in the CPB–BX528 complex coincides with the position of the butoxy group in the CPT–N-BOC-L-leucine complex.
Fig. 8. Superposition of the structures of the CPT–N-BOC-L-leucine and CPA–inhibitor BX528 complexes (stereo view). The BX528 inhibitor is shown in black. The side groups of ligands located in subsites S1, S2, and S1′ are shown on gray background. Other designations as in Fig. 6.
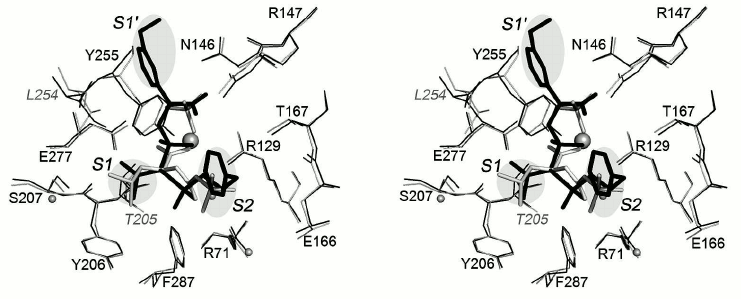
The Tyr248 residue (255 in CPT) participating in binding the terminal carboxyl group of the BX528 ligand in the S1′ subsite contacts with the aminomethylphenyl group that simulates the positively charged side chain of the substrate, and along with this it is close to the phenyl radical in the S2 subsite. Hence, Tyr248 playing a significant role in the S1′ subsite, can simultaneously interact with the residues that account for the S1 and S2 subsites. This example demonstrates that the considered subsites of carboxypeptidases are spatially close and can influence each other.
Our comparison shows that the S1 subsite of CPT is a very conservative structure and negligibly differs from corresponding sites of CPA and CPB in the composition and the 3D structure. In all the structures of carboxypeptidases, the S1 subsite is close to the catalytic zinc ion and to the residues of arginine and glutamic acid (Arg71, Arg147, Arg129, and Glu277 according to CPT numeration) important for catalysis. Such residues as Leu254 and Tyr255 important for substrate binding and enzyme selectivity are located between the S1 and S1′ subsites and can provide interaction between them. The data suggest that amino acid replacements among the residues contacting with Leu254 and Tyr255 (e.g. Leu209, Leu211, Ile256) can affect the kinetic constants of hydrolysis as well as selectivity of carboxypeptidases.
The authors are grateful to colleagues from the Japanese Space Agency (JAXA) H. Tanaka, K. Inaka, and K. Ohta for providing the equipment for crystallization and help in obtaining the diffraction data.
This work was financially supported by the Russian Foundation for Basic Research (grant No. 04-10-01541) and Central Scientific Research Institute of Mechanical Engineering of the Russian Federal Space Agency (Roscosmos).
REFERENCES
1.Teplyakov, A., Polyakov, K., Obmolova, G.,
Strokopytov, B., Kuranova, I., Osterman, A., Grishin, N., Smulevitch,
S., Zagnitko, O., Galperina, O., Matz, M., and Stepanov, V. M. (1992)
Eur. J. Biochem., 208, 281-288.
2.Osterman, A. L., Grishin, N. V., Smulevitch, S. V.,
Matz, M. V., Zagnitko, O. P., Revina, L. P., and Stepanov, V. M. (1992)
J. Protein Chem., 11, 561-570.
3.Schechter, I., and Berger, A. (1967) Biochem.
Biophys. Res. Commun., 27, 157-162.
4.Akparov, V. Kh., Grishin, A. M., Yusupova, M. P.,
Ivanova, N. M., and Chestukhina, G. G. (2007) Biochemistry
(Moscow), 72, 416-423.
5.Trachuk, L. A., Bushueva, A. M., Shevelev, A. B.,
Novgorodova, S. A., Akparov, V. Kh., and Chestukhina, G. G. (2002)
Voprosy Med. Khim., 48, 577-580.
6.Grishin, A. M., Akparov, V. Kh., and Chestukhina,
G. G. (2008) Biochemistry (Moscow), 73, 1140-1145.
7.Akparov, V. Kh., Timofeev, V. I., and Kuranova, I.
P. (2011) Kristallografiya, 56, 641-647.
8.Sukenaga, Y., Akanuma, H., and Yamasaki, M.
(1980) J. Biochem., 87, 1691-1701.
9.Stepanov, V. M. (1995) in Methods in Enzymology
(Barrett, A. J., ed.) Academic Press, Inc., N. Y., Vol. 248, pp.
675-683.
10.Deiteren, K., Surpateanu, G., Gilany, K.,
Willemse, J. L.,, Hendriks, D. F., Augustyns, K., Laroche, Y., Scharpe,
S., and Lambeir, A. M. (2007) Biochim. Biophys. Acta,
1774, 267-277.
11.Akparov, V. Kh., Belyanova, L. P., Baratova, L.
A., and Stepanov, V. M. (1975) Biokhimiya, 44,
886-891.
12.Yusupova, M. P., Kotlova, E. K., Timokhina, E.
A., and Stepanov, V. M. (1995) Bioorg. Khim., 21,
33-38.
13.Cueni, L. B., Bazzone, T. J., Riordan, J. F., and
Vallee, B. L. (1980) Anal. Biochem., 107, 341-349.
14.Novagen pET System Manual TB055, 7th Edn.
(1997) NovagenMadison, W. I.
15.Trachuk, L., Letarov, A., Kudelina, I. A.,
Yusupova, M. P., and Chestukhina, G. G. (2005) Protein Expr.
Purif., 40, 51-59.
16.Bradford, M. M. (1976) Anal. Biochem.,
72, 248-254.
17.Laemmli, U. K. (1970) Nature, 277,
680-685.
18.Takahashi, S., Tsurumura, T., Aritake, K.,
Furubayashi, N., Sato, M., Yamanaka, M., Hirota, E., Sano, S.,
Kobayashi, T., Tanaka, T., Inaka, K., Tanaka, H., and Urade, Y. (2010)
Acta Cryst. F, 66, 846-850.
19.Kuranova, I. P., Smirnova, E. A., Abramchik, Yu.
A., Chupova, L. A., Esipov, R. S., Akparov, V. Kh., Timofeev, V. I.,
and Kovalchuk, M. V. (2011) Kristallografiya, 56,
941-948.
20.Otwinowski, Z., and Minor, W. (1997) in
Methods in Enzymology (Barrett, A. J., ed.) Academic Press,
Inc., N. Y., Vol. 276, pp. 307-326.
21.McCoy, A. J., Grosse-Kunstleve, R. W., Storoni,
L. C., and Read, R. J. (2005) Acta Cryst. D, 61,
458-464.
22.Murshudov, G. N., Vagin, A. A., and Dodson, E. J.
(1997) Acta Cryst. D, 53, 240-255.
23.Emsley, P., and Cowtan, K. (2004) Acta
Cryst. D, 60, 2126-2132.
24.Rees, D. C., and Lipscomb, W. N. (1980) Proc.
Natl. Acad. Sci. USA, 77, 4633-4637.
25.Cristianson, D. W., and Lipscomb, W. N. (1986)
Proc. Natl. Acad. Sci. USA, 83, 7568-7572.
26.Adler, M., Buckman, B., Bryant, J., Chang, Z.,
Chu, K., Emayan, K., Hrvatin, P., Islam, I., Morser, J., Sukovich, D.,
West, C., Yuan, S., and Whitlow, M. (2008) Acta Cryst. D,
64, 149-157.
27.McPherson, A. (1996) Crystallogr. Rev.,
6, 157-308.
28.Kuranova, I. P. (2004) Poverkhnost, No. 6,
6-14.