Characterization of a Cold-Active Lipase from Psychrobacter cryohalolentis K5T and Its Deletion Mutants
K. A. Novototskaya-Vlasova1*, L. E. Petrovskaya2*, E. M. Rivkina1, D. A. Dolgikh2,3, and M. P. Kirpichnikov2,3
1Institute of Physicochemical and Biological Problems in Soil Science, Russian Academy of Sciences, Institutskaya ul. 2, 142290 Pushchino, Moscow Region, Russia; fax: (496) 731-8155; E-mail: nksusha@gmail.com2Shemyakin and Ovchinnikov Institute of Bioorganic Chemistry, Russian Academy of Sciences, ul. Miklukho-Maklaya 16/10, 117997 Moscow, Russia; fax: (495) 335-0812; E-mail: lpetr65@yahoo.com
3Faculty of Biology, Lomonosov Moscow State University, 119234 Moscow, Russia; fax: (495) 939-4309
* To whom correspondence should be addressed.
Received December 7, 2012; Revision received December 23, 2012
A gene coding for cold-active lipase from the psychrotrophic Gram-negative bacterium Psychrobacter cryohalolentis K5T isolated from a Siberian cryopeg has been cloned and expressed in Escherichia coli. The recombinant protein Lip1Pc with a 6× histidine tag at its C-terminus was purified by nickel affinity chromatography. With p-nitrophenyl dodecanoate (C12) as a substrate, the purified recombinant protein displayed maximum lipolytic activity at 25°C and pH 8.0. Increasing the temperature above 40°C and addition of various metal ions and organic solvents inhibited the enzymatic activity of Lip1Pc. Most nonionic detergents, such as Triton X-100 and Tween 20, slightly increased the lipase activity, while SDS completely inhibited it. To investigate the functional significance of the Lip1Pc N-terminal domain, we constructed five deletion mutants of this protein. The ND1 and ND2 mutants displayed specific activity reduced by 30-35%, while other truncated proteins were completely inactive. Both mutants demonstrated increased activity towards p-nitrophenyl decanoate (C10) and impaired utilization of C16 substrate. Although optimum reaction temperature of ND2 lowered to 20°C, it displayed enhanced stability by 44% after incubation at 40°C. The results prove that the N-terminal domain of Lip1Pc has a fundamental impact on the activity and stability of the protein.
KEY WORDS: cryopeg, permafrost, Psychrobacter cryohalolentis, cold-active lipase, thermostability, deletion mutantsDOI: 10.1134/S000629791304007X
Abbreviations: CHAPS, 3[(3-cholamidopropyl)dimethylammonio]-propanesulfonic acid; EDTA, ethylenediamine tetraacetic acid; HSL, hormone sensitive lipase; IPTG, isopropyl β-D-1-thiogalactopyranoside; PMSF, phenylmethylsulfonyl fluoride; p-NPB, p-nitrophenylbutyrate; SDS, sodium dodecyl sulfate; SOE-PCR, splicing by overlapping extension PCR.
Lipases/esterases (triacylglycerol acyl hydrolases, EC 3.1.1.3) catalyze
the hydrolysis of acyl glycerols and are found in animal tissues,
plants, and microorganisms, including fungi, bacteria, and archaea [1, 2]. These enzymes belong to a
large superfamily of α/β-hydrolase proteins containing a
central α/β-sandwich domain and a catalytic triad (Ser, His,
Asp). The central domain usually includes β-sheet formed by 5 to
11 β-strands flanked on both sides by α-helical connections
[3]. The lipolytic enzymes are classified into
eight families based on their sequence similarities [4].
Lipases secreted by microorganisms are widely used in various industries including production of pharmaceuticals, food, detergents, and fine organic synthesis [1]. Cold-active lipases are of particular interest for industrial processes due to their high catalytic activity at low temperatures [5, 6]. Related perspectives of energy savings and possibility to perform reactions with thermolabile compounds pose the urgent task of discovery and studies of new cold-active enzymes including lipases for biotechnology and microbiology. Most known cold-active lipases have been isolated from psychrophilic and psychrotolerant microorganisms including bacteria of the Psychrobacter genus from cold marine environments [7-9].
Cryopegs – the lenses of overcooled saline water brines derived from ancient marine sediments and sandwiched within permafrost – represent one of the specific cold habitats [10]. Previous studies have shown that they are inhabited by numerous and diverse microorganisms [11] including producers of lipolytic enzymes [12]. In particular, the Gram-negative psychrotrophic bacterium Psychrobacter cryohalolentis K5T was isolated from a Siberian cryopeg and characterized [13]. Standard biochemical tests have demonstrated that it possesses lipolytic activity [13]. We have previously cloned the gene coding for cold-adapted esterase from P. cryohalolentis K5T and expressed it in Escherichia coli cells [14]. Study of the recombinant protein revealed its high activity at low temperatures and its relative thermostability [14].
In the current work we continued the study of lipolytic enzymes from P. cryohalolentis K5T. We have overproduced in E. coli and characterized the Lip1Pc protein belonging to the hormone-sensitive lipase (HSL) family. It was found that Lip1Pc is a typical cold-active enzyme with decreased thermostability. To investigate the role of the Lip1Pc N-terminal domain, several deletion mutants of the protein were constructed and studied.
MATERIALS AND METHODS
Reagents from Bio-Rad (USA), Merck (USA), Panreac (Spain), bacteria cultivation media from Difco (USA), p-nitrophenyl ester substrates (Sigma, USA), organic solvents (Chimmed, Russia) were used in the study. Solutions were prepared using MilliQ water. Psychrobacter cryohalolentis strain K5T (B-2378) was provided by the All-Russian Collection of Microorganisms. The culture was grown on agar plates with TSB medium (Difco) and 2% NaCl for 16 h.
Homology search in the P. cryohalolentis K5T genome (GenBank CP000323) was performed using BLAST [15]. Multiple protein alignments were produced with ClustalW2 (http://www.ebi.ac.uk/Tools/msa/clustalw2/). DNA and protein sequences were analyzed with Sequence Manipulation Suite (http://www.bioinformatics.org/sms2). Secondary structure prediction was performed by PsiPred (http://bioinf.cs.ucl.ac.uk/psipred/).
Recombinant DNA cloning. DNA manipulations were performed by standard methods [16] in E. coli strain XL-1 Blue (Stratagene, USA) using enzymes from Fermentas (Lithuania). Primers were synthesized by Evrogen (Russia).
Genes were amplified by PCR using Pfu and Taq DNA-polymerases under conditions recommended by the supplier. To amplify the Lip1Pc gene the cell lysate from P. cryohalolentis K5T was prepared by boiling of 10 μl of strain biomass for 5 min in 50 μl of PCR buffer for Taq DNA polymerase and used as a template. The reaction mixture contained Pfu DNA polymerase buffer with 2 mM MgSO4, 0.2 mM each of dNTP, primers (50 pmol each of LIP1PCf and LIP1PCr; Table 1), 2.5 units of Pfu DNA polymerase, 0.5 unit of Taq DNA polymerase, and cell lysate (5 μl) in a total volume of 50 μl. PCR was performed with an initial denaturation at 95oC for 5 min followed by 30 cycles of 95oC for 45 s, 55oC for 45 s, and 72oC for 45 s; the final extension was at 72oC for 10 min.
Table 1. Nucleotide sequences of primers
used for construction of genes for Lip1Pc and mutants ND1-ND5

The PCR product was digested for 2 h at 37°C with NdeI and SalI restrictases, purified from 1% agarose (Bio-Rad) with a MinElute Gel Extraction Kit (Qiagen, USA), and cloned into pET-32a vector (Novagen, USA) cut by NdeI and XhoI and purified the same way. Insertion in the resulting plasmid pLip1Pc was verified by restriction analysis and sequencing (Genome Center, Russia).
To produce mutant Lip1Pc genes encoding N-terminally truncated proteins ND1-ND4, PCR was conducted with pLip1Pc as a template, one of the forward primers ND1f-ND4f (Table 1) correspondingly, and T7term reverse primer. The reaction mixture contained Pfu DNA polymerase buffer with 2 mM MgSO4, 0.2 mM dNTP mix, primers (50 pmol each), 10 ng of template DNA and 2.5 units of Pfu DNA polymerase. After initial denaturation at 95oC for 3 min, 25 cycles of 95oC for 40 s, 55oC for 40 s, 72oC for 1 min were conducted followed by a final extension step at 72oC for 10 min.
The gene encoding for ND5 deletion form was constructed using two-step SOE-PCR [17] with ND5f and ND5r (Table 1) as mutagenic, T7prom, and T7rev as flanking primers. The reaction conditions were the same as for the ND1-ND4 genes. During the next round, the resulting fragments were combined in PCR with primers T7prom and T7rev.
The resulting products were treated and cloned as described above for wild-type Lip1Pc gene.
Protein purification. Escherichia coli BL21(DE3) cells (Novagen) harboring the recombinant plasmid pLip1Pc or plasmids containing the mutant genes were grown at 37°C and 250 rpm in 200 ml of LB medium supplemented with ampicillin (100 μg/ml). After the cell culture optical density at 560 nm reached 0.8, expression was induced with 0.1 mM IPTG. After incubation at 27°C and 200 rpm for 4 h, the induced cells were harvested by centrifugation (7000 rpm, 15 min at 4°C) and resuspended in buffer A (50 mM Tris-HCl, 200 mM NaCl, pH 8.0). The cell suspension was sonicated using a Branson Sonifier 450 on an ice bath for 5 min (10 sec on, 1 min off). Cell debris was removed by centrifugation at 17,000 rpm for 15 min. The supernatant solution was loaded on a column with 2 ml of Ni-Sepharose Fast Flow resin (GE Healthcare, USA) that had already been equilibrated with buffer A containing 10 mM imidazole. The column was extensively washed with buffer B (20 mM Tris-HCl, 400 mM NaCl, 20 mM imidazole, pH 8.0). The target proteins were eluted with buffer C (20 mM Tris-HCl, 200 mM NaCl, 300 mM imidazole, pH 8.0). Combined fractions containing the target proteins with purity more than 90% were dialyzed against buffer D (50 mM HEPES-NaOH, pH 7.5) and sterilized. Protein concentration was measured with a Protein Assay Kit (Bio-Rad) with BSA as a standard.
Lipolytic activity assay. Assay was performed using p-nitrophenyl butyrate (p-NPB; Sigma) as a substrate [18]. To the reaction mixture containing 50 mM Tris-HCl, pH 8.0, 100 mM NaCl, 0.5% (w/v) Triton X-100, 0.25 mM p-NPB in 1 ml, 1 μg of purified protein was added. After incubation at 25°C for 15 min, the reaction was stopped by adding 200 μl of 10% SDS solution. Two hundred microliters of the mixture after centrifugation (13,000 rpm, 5 min) was placed in the wells of 96-well assay plates (Deltalab). Absorbance at 415 nm was determined with a Model 680 Microplate Reader (Bio-Rad). One unit of lipase activity was defined as the amount of enzyme releasing 1 μmol of p-nitrophenol per minute.
The optimum reaction temperature was determined by measuring the lipase activity at different temperatures (5-50°C) in 5°C increments. The reaction mixture was incubated at each temperature for 10 min without enzyme and substrate, and 15 min after their addition. Then the activity was measured as described above. The thermostability of the lipase was evaluated by measuring residual lipase activity under standard conditions after incubating the protein at different temperatures (from –20 to 50°C) for 45 min.
To determine the optimum pH for the activity of Lip1Pc, 50 mM potassium phosphate (pH 5.6-8.0) or 50 mM Tris-HCl (pH 8.0-9.5) buffers were used. The effect of salt on the activity of the lipase was evaluated by addition of a specific NaCl concentration (0-1.75 M) to the reaction mixture. The effect of various metal ions (Zn2+, Ca2+, Ni2+, Cu2+, Co2+, Mg2+ and Mn2+) and other additives (PMSF, EDTA) on the lipase activity was assessed after incubating the lipase at 5°C for 30 min in the buffer supplemented with 1 mM solution of each of the additives. Detergent resistance of Lip1Pc was estimated by determining its residual activity after 30 min incubation in buffer containing 0.5 and 0.05% (w/v) of various detergents (SDS, Triton X-100, Tween 20, and CHAPS) at 5°C. The effects of various organic solvents (methanol, ethanol, acetonitrile, DMSO, and DMFA) on Lip1Pc were estimated by determining residual activity after 30 min incubation at 5°C in the buffer containing 5 and 10% (w/v) of each solvent. Substrate specificity of the lipase and its mutants was studied in the reactions with p-nitrophenyl ester substrates with variable chain length (C2 to C16).
RESULTS
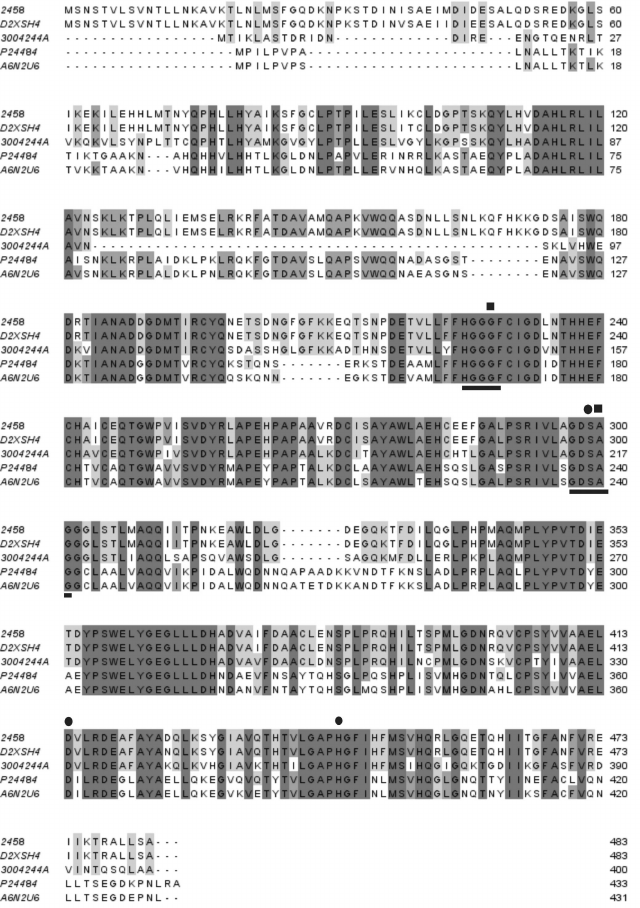
Lipase gene sequence analysis and design of deletions. The Lip1Pc coding sequence was identified in P. cryohalolentis K5T genome as a homolog of Psychrobacter sp. Ant300 esterase [18]. The level of similarity between these two proteins comprises 72%, with 61% identical residues. Later the sequence of lipase D2XSH4 from Psychrobacter sp. G with higher similarity (99%) to Lip1Pc was published, but this protein was not functionally expressed in E. coli cells [19]. Other proteins with considerable amino acid homology to Lip1Pc are a cold-active lipase from Psychrobacter sp. 2-17 [20] and Lip2 from Moraxella sp. TA144 [21] (Fig. 1).
Fig. 1. Alignment of deduced amino acid sequence of Lip1Pc from Psychrobacter cryohalolentis K5T (2458) with sequences of homologous lipases/esterases: D2XSH4, Psychrobacter sp. G lipase; 3004244A, Psychrobacter sp. Ant300 esterase; P24484, Moraxella sp. strain TA144 lipase 2; A6N2U6, Psychrobacter sp. 2-17 lipase. Motifs characteristic of the HSL lipase family are underlined. Amino acid residues presumably comprising the catalytic triad and the oxyanion hole are indicated by circles and squares, correspondingly.
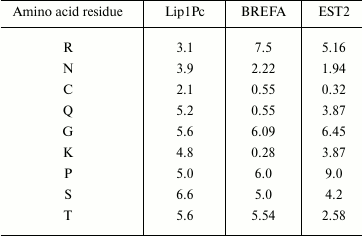
Analysis of the deduced protein sequence of Lip1Pc with ProtParam (http://web.expasy.org/protparam/) revealed clear signatures of cold-adapted phenotype including increased number of polar residues (S, T, N, Q, C) and decrease in proline, glutamic acid, and arginine in comparison with brefeldin A from mesophilic B. subtilis and EST2 carboxylesterase from thermophilic A. acidocaldarius [22]. For example, the content of asparagine residues in Lip1Pc is 3.9% in comparison with 2.22 and 1.94% in brefeldin A and EST2, correspondingly. Prevalence of lysine residue content over arginine (4.8 versus 3.1%) is also typical for cold-adapted proteins (Table 2).
Table 2. Selected amino acid residues
content (in %) in the protein sequences of Lip1Pc, brefeldin A from
B. subtilis (BREFA), and carboxylesterase EST2 from A.
acidocaldarius. The data were obtained using the ProtParam
program
The secondary structure of Lip1Pc predicted by PsiPred contains elements typical for a protein belonging to the α/β hydrolase family. The enzyme core is composed of eight β-strands and nine α-helices. The N-terminal domain (1-177) consists of 10 α-helices (Fig. 2).
Fig. 2. Lip1Pc secondary structure predicted by PsiPred. Starting positions of ND1-4 are marked with arrows. The ND5 deletion is designated by the gray bar.
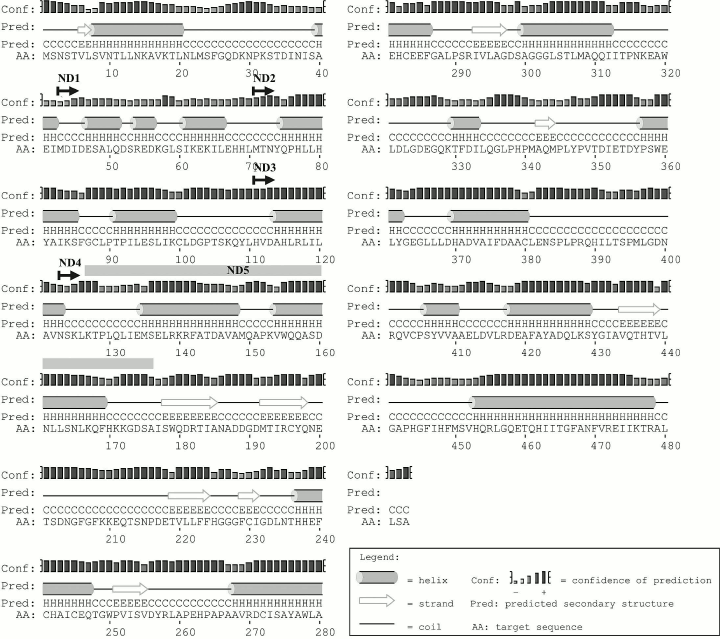
We planned construction and characterization of five Lip1Pc deletion variants with consecutive truncation of its N-terminus in ND1-4 (ND1, Δ1-42; ND2, Δ1-70; ND3, Δ1-110; ND4, Δ1-122) and an internal deletion of 50 residues in ND5 (Δ127-176). Making a choice for the new N-termini in ND1-4 we took into account the borders of predicted α-helices in the N-terminal domain (Fig. 2). The design of the ND5 deletion was inspired by the absence of a corresponding amino acid sequence in Psychrobacter sp. Ant300 esterase (Fig. 1).
Expression in E. coli and characterization of Lip1Pc and mutant proteins. The lip1Pc gene was amplified from the genomic DNA of P. cryohalolentis K5T with gene-specific oligonucleotide primers and then cloned into the expression vector pET-32a under the control of a strong regulated T7lac promoter. The 3′-end of the resulting gene contains a sequence encoding a hexahistidine fragment for subsequent purification of the recombinant protein by metal-affinity chromatography. The genes for mutants ND1-4 were produced and cloned as the full-length gene. To construct the ND5 deletion variant, we utilized SOE-PCR method with overlapping internal primers and a pair of standard flanking primers.
To obtain recombinant Lip1Pc protein, E. coli BL21(DE3) cells were transformed with the resulting plasmid pLip1Pc. Induction was performed with 0.1 mM IPTG at 27°C promoting the maximal yield of the expression product in the soluble form according to protein electrophoresis and activity measurement data. Upon induction, the expression level of Lip1Pc in BL21(DE3) cells reached 10% of total cellular protein. Proteins ND1-5 were produced under the same conditions (Fig. 3a). It should be mentioned that ND3 and ND4 expression level was substantially higher (up to 30% of total cellular protein); nevertheless, the majority of the expression products was found in inclusion bodies.
Fig. 3. SDS-PAGE analysis of expression (a) and purification (b) of recombinant Lip1Pc and its deletion variants. a) Lanes: 2) uninduced cells; 3-8) total protein samples of BL21(DE3) cells expressing Lip1Pc (3), ND1 (4), ND2 (5), ND3 (6), ND4 (7), ND5 (8) after 4 h induction. b) Purified proteins after Ni-affinity column. Lanes: 2) Lip1Pc; 3) ND1; 4) ND2; 5) ND3; 6) ND4; 7) ND5; 1) protein molecular weight markers, kDa (Fermentas).

Due to high expression level and solubility of the recombinant Lip1Pc, its purification using Ni-affinity chromatography resulted in a 17-fold increase in protein purity in comparison with the sonicated lysate (according to activity measurement data). The purified Lip1Pc appeared on SDS-PAGE as a single band with an apparent molecular mass of about 54 kDa, consistent with the predicted value for 6×His-tagged protein (54.6 kDa; Fig. 3b, lane 2). Electrophoretic mobilities of the ND1-5 mutant proteins purified by the same protocol also corresponded to their calculated molecular weights (50.1, 46.9, 42.5, 41.1, and 50 kDa, respectively) (Fig. 3b, lanes 3-7). As a result, the recombinant lipase Lip1Pc was obtained with 15.7% yield and specific activity against p-NPB of 1410 U/mg. Specific activity of purified ND1 and ND2 constituted 896 and 1000 U/mg, respectively. Proteins ND3-ND5 showed no lipolytic activity against all tested substrates.
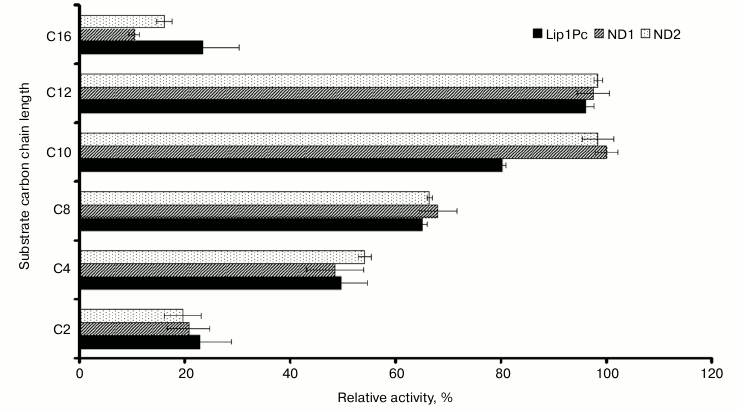
Effects of substrates, pH, and temperature of the reaction mixture on the lipolytic activity of Lip1Pc. Our results showed that recombinant Lip1Pc displays broad substrate specificity. The highest lipolytic activity was observed with p-nitrophenyl dodecanoate (C12), and the activity with C10 and C8 esters reached 80 and 65% of the maximum, respectively (Fig. 4). The purified Lip1Pc was active in different buffers (pH 7.2-9.5) with a maximum in Tris-HCl buffer at pH 8.5.
Fig. 4. Substrate specificity of purified Lip1Pc and its mutants ND1 and ND2. Lipase assay was performed using p-nitrophenyl esters of fatty acid with varying carbon chain length (C2, C4, C8, C10, C12, and C16). The activity towards p-nitrophenyl decanoate of ND1 was taken as 100%. Mean results of three independent experiments ± standard deviation are presented.
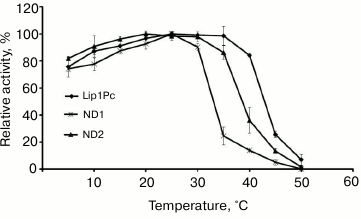
The influence of the reaction temperature on Lip1Pc activity was assayed in the range 5-50°C using p-NPB as the substrate. Maximum activity toward this ester was observed at 25°C. At temperatures in the range 5-40°C the lipase activity amounted to 60-95% of maximum, but it decreased sharply above 45°C (Fig. 5).
Fig. 5. Temperature dependence of lipase activity of Lip1Pc (rhombs), ND1 (crosses), and ND2 (triangles). The enzymes were incubated in the reaction mixture containing 50 mM Tris-HCl, pH 8.0, and 0.25 mM p-NPB for 15 min. Activity values obtained at 25°C (for Lip1Pc and ND1) and at 20°C (for ND2) were taken as 100%.
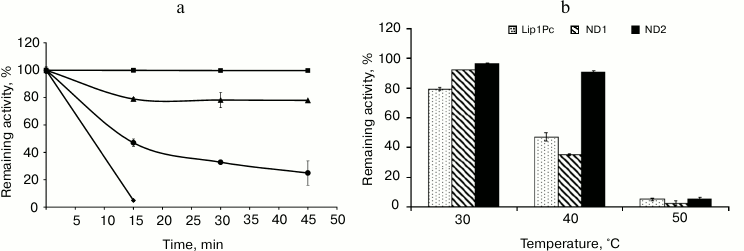
Thermostability of the recombinant Lip1Pc was determined by preincubation of the enzyme at different temperatures for 45 min. The remaining lipase activity was measured at 25°C. The recombinant lipase was very stable in the interval from –20 to 20°C and rapidly inactivated at temperatures above 40°C (Fig. 6a).
Fig. 6. Effect of temperature on stability of Lip1Pc and its mutants. a) Lip1Pc was incubated in 50 mM Tris-HCl, pH 8.0, at 20 (square), 30 (triangle), 40 (rhombus), and 50°C (circle) for 15, 30, and 45 min, respectively. Remaining lipase activity was measured with 0.25 mM p-NPB as the substrate. Remaining activities after incubation at –20, 5, 10, and 20°C were 100%. b) Lip1Pc, ND1, and ND2 were incubated in 50 mM Tris-HCl, pH 8.0, at 30, 40, and 50°C for 45 min, and the remaining lipase activity was measured with 0.25 mM p-NPB as the substrate. Activity values obtained before heating were taken as 100%.
Effects of salinity, metal ions, detergents, and organic solvents on lipase activity. Studies of Lip1Pc activity in the presence of different salt concentrations showed that addition of NaCl at concentrations up to 1 M does not influence the enzymatic activity. Higher concentrations slightly inhibited the enzyme (by 10%). Activity of Lip1Pc was partially inhibited by the presence of NaN3 and Mg2+, Mn2+, Ca2+ ions and strongly inhibited by Zn2+, Co2+, Ni2+, and Cu2+ ions and PMSF. EDTA had a slight enhancing effect on the lipase activity (Table 3).
Table 3. Effect of metal ions and other
additives on Lip1Pc activity*
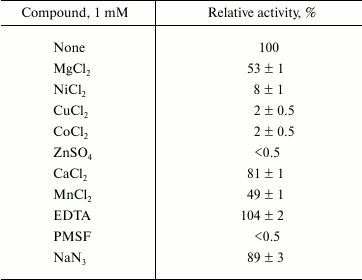
* Lip1Pc (1 µg/ml) was incubated in 50 mM Tris-HCl
(pH 8.0) containing each compound for 30 min at 5°C.
Remaining activity was determined with 0.25 mM p-NPB at
25°C. Data are given as mean ± SD, n = 3.
The presence of nonionic detergents Triton X-100, Tween 20, and CHAPS did not influence Lip1Pc activity, while the harsh ionic detergent SDS completely inhibited the recombinant protein. All tested organic solvents present at 5 and 10% concentration reduced the activity of Lip1Pc with only the exception of DMSO, which was found to have no effect on it (Table 4).
Table 4. Effect of organic solvents on
Lip1Pc activity*
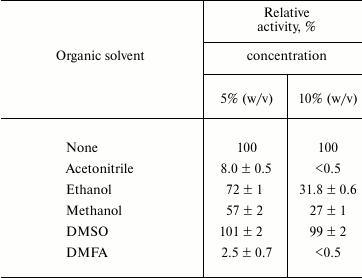
* For conditions see note for Table 3.
Effects of N-terminal truncations on Lip1Pc substrate specificity, activity, and thermostability. The following characteristics were determined for ND1 and ND2 proteins, which preserved the catalytic activity in contrast to ND3-5, which completely lacked activity. The activities of ND1 and ND2 towards p-nitrophenyl palmitate (C16) decreased by 13 and 7%, respectively, compared with Lip1Pc; however, both mutants displayed enhanced by 20% activity with p-nitrophenyl decanoate (C10; Fig. 4). The temperature optimum of ND1 activity was 25°C, the same as for the wild-type protein. For ND2 it decreased to 20°C. The range of temperatures at which enzymes displayed more than 80% activity became narrower (5-30°C for ND1 and 5-35°C for ND2 in comparison with 5-40°C for Lip1Pc) (Fig. 5). Both deleted forms displayed enhanced thermostability after incubation for 45 min at 30°C by 13 and 17%, respectively. Stability of ND2 was also 43% higher in comparison with Lip1Pc after 45 min incubation at 40°C. Nevertheless, both mutant proteins along with the full length Lip1Pc were almost completely inactivated after incubation at 50°C (Fig. 6b).
DISCUSSION
In this work we report results of the study of a new lipolytic enzyme from the psychrotrophic bacterium P. cryohalolentis K5T. This strain was isolated from an 110,000-year-old Siberian cryopeg and was capable of growth over the wide temperature interval from –10 to 30°C. The genomic sequence of P. cryohalolentis was obtained and annotated by the DOE Joint Genome Institute (USA). Search for homologs of known lipases enabled us to identify in this sequence a putative lipase gene belonging to family IV of lipolytic enzymes (HSL family [4]).
Amino acid sequences of lipases and esterases comprising this family display similarity to the catalytic domain of mammalian HSL [23] and the presence of characteristic sequences HGGGF and GDSAG, the latter containing the catalytic serine residue. From the sequence alignment, the positions of amino acid residues forming the catalytic triad (S299, D414, H444) and oxyanion hole (G228, A300) in Lip1Pc could be predicted (Fig. 1).
The HSL family includes members produced by thermophilic (Alicyclobacillus acidocaldarius, Archeoglobus fulgidus), mesophilic (Escherichia coli, Alcaligenes eutrophus), and psychrophilic (Moraxella sp., Psychrobacter immobilis) microorganisms. It was shown that specific trends in amino acid composition were associated with thermal adaptation of lipases comprising this family [22]. Comparison of amino acid composition of Lip1Pc and homologous enzymes isolated from warmer environments confirmed these relationships (Table 2).
Examination of the Lip1Pc amino acid sequence with the SignalP tool (http://www.cbs.dtu.dk/services/SignalP/) revealed no presence of signal sequence on its N-terminus. Therefore, we amplified the entire lip1Pc gene from the genomic DNA of P. cryohalolentis K5T and cloned it into the pET-based vector to obtain expression product with C-terminally placed hexahistidine tag. The results of specificity study of the purified enzyme proved that Lip1Pc is a lipase rather than an esterase because it displayed higher activity with medium- to long-chain fatty acids (C10 – 82%, C12 – 100% maximum activity) in comparison with substrates with shorter chain length (C4 – 51.8% and C8 – 67.5%). This distinguishes Lip1Pc from homologous enzymes that exhibit maximum activity with p-nitrophenylbutyrate (C4) [18, 21] or p-nitrophenylhexanoate (C6) [20], and therefore they are esterases.
The optimal temperature for the activity of Lip1Pc was 25°C, which is typical for many cold-active enzymes. The Lip1Pc activity at lower temperatures remained also high and was 76 and 87% of its maximum level at 5 and 10°C, respectively. Low stability at elevated temperatures constitutes a distinctive feature of cold-active enzymes including lipases [24]. Lip1Pc along with homologous cold-active lipases/esterases [20, 25] demonstrated this property, whereas even brief heating at 50°C completely inactivated the protein. Therefore, from the amino acid sequence analysis and the study of functional characteristics of Lip1Pc, we conclude that it represents a typical cold-active enzyme possessing high activity at low temperatures and limited thermostability. Comparison of thermal profiles of Lip1Pc and previously studied EstPc protein [14] activity and stability allows us to speculate about the presence of enzymes with different temperature optimum and stability in the lipolytic system of P. cryohalolentis K5T that presumably provides this microorganism with an additional mechanism of adaptation to a wide range of environmental conditions.
Examination of the amino acid sequence of Lip1Pc revealed that it possesses an unusually long N-terminus (177 amino acid residues) with predominantly α-helical structure (Fig. 2). It is interesting to note that during BLAST alignment, this domain displayed no significant homology with other known protein sequences in public databases. In proteins sharing α/β hydrolase fold, N-terminal α-helices often contribute to the formation of a cap structure, which determines access of the substrate to the active site [26, 27]. The prototype member of the family, human HSL lipase, also contains a long (320 amino acid residue) N-terminal domain participating in protein–protein interactions [28]. To examine the structural and functional significance of the N-terminal region in Lip1Pc, we constructed five deletion forms of this protein and compared their properties with the wild-type enzyme. All of the mutant proteins were produced in E. coli cells and purified according to the same protocol as Lip1Pc.
Complete loss of activity observed as a result of deletion of the first 110 and 127 amino acid residues of Lip1Pc in ND3 and ND4 and internal deletion of 50 residues in ND5 suggested the indispensable role of the N-terminal domain of the protein activity. Albeit ND1 and ND2 deletion forms with truncations of 42 and 70 residues correspondingly preserved their catalytic activity, it was reduced by 30-35% against p-NPB (C4). Deletions slightly narrowed the substrate specificity of the mutants; the preference for C10 and C12 substrates against C16 became more expressed in the activity profiles of ND1 and ND2. The optimum temperature of ND2 activity decreased by 5°C (from 25 to 20°C), and the temperature ranges of ND1 and ND2 activities were narrowed. Unexpectedly, in comparison with Lip1Pc, the stability of both mutants after incubation at 30°C slightly increased by 13 and 17% for ND1 and ND2, respectively. Thermostabilization was even more pronounced in the case of ND2: incubation at 40°C led to 9% activity decrease, while the full-length Lip1Pc lost 53% of its activity (Fig. 6).
The stabilizing effect of the deletions presumably reflects more compact conformation of the deleted forms in comparison with the full-length protein. Cold-active enzymes are known to possess flexible structure, provided by increased exposure of hydrophobic residues, limited number of stabilizing interactions, extended loops, and other factors [29]. As a result, thermostability of these proteins could be increased by mutagenesis without influencing catalytic properties [30]. Structural changes in the Lip1Pc mutant variants as a consequence of deletions presumably resulted in altered conformation of a substrate tunnel and associated decreased ability to utilize long-chain p-nitrophenyl esters.
Initial analysis of the available structures of the enzymes belonging to the HSL family suggested a role for the N-terminus in the formation of a substrate tunnel and limiting access to the catalytic site of the protein [31]. Studies of the N-terminal subdomain of EST2 esterase from the thermophilic microorganism A. acidocaldarius also demonstrated its importance for determining thermostability and temperature optimum of enzyme activity [27]. Deletions of this region completely inactivated EST2 esterase and cold-adapted lipase from Psychrobacter sp. TA144 [25]. Results of the study of the deletion forms of Lip1Pc confirm the impact of the N-terminal domain of the protein on the activity and thermostability of HSL family lipases and demonstrate the perspectives of protein engineering applications to modify the catalytic properties of a recombinant enzyme.
In conclusion, we have for the first time cloned and characterized a cold-active lipase from P. cryohalolentis K5T with high activity at low temperatures and broad substrate specificity. These properties make Lip1Pc an attractive candidate for various biotechnological applications and prove that the microbial community of cryopegs may be an important resource for prospecting novel cold-active lipases and other enzymes. It should be noted that up to now no three-dimensional structure has been solved for the members of the HSL family of lipolytic enzymes from psychrotrophic or psychrophilic organisms. Construction of the expression system and preliminary study of Lip1Pc properties in the course of the current work constitute the basis for consecutive structural investigation of this protein that will contribute to understanding of enzymatic activities at low temperatures and their optimization for biotechnological applications.
The authors thank Dr. V. A. Shcherbakova for providing the strain Psychrobacter cryohalolentis K5T.
This work was supported by the Russian Academy of Sciences programs “The Origin of the Biosphere and the Evolution of Geo-biological Systems. The Microbial Biosphere” and “Molecular and Cellular Biology” and Russian Foundation Basic Research grant No. 13-04-00366.
REFERENCES
1.Jaeger, K. E., Dijkstra, B. W., and Reetz, M. T.
(1999) Ann. Rev. Microbiol., 53, 315-351.
2.Reetz, M. T. (2002) Curr. Opin. Chem. Biol.,
6, 145-150.
3.Ollis, D. L., Cheah, E., Cygler, M., Dijkstra, B.,
Frolow, F., Franken, S. M., Harel, M., Remington, S. J., Silman, I.,
and Schrag, J. (1992) Protein Eng., 5, 197-211.
4.Arpigny, J., and Jaeger, K. (1999) Biochem.
J., 343, 177-183.
5.Gerday, C., Aittaleb, M., Bentahir, M., Chessa, J.
P., Claverie, P., Collins, T., D’Amico, S., Dumont, J., Garsoux,
G., Georlette, D., Hoyoux, A., Lonhienne, T., Meuwis, M. A., and
Feller, G. (2000) Trends Biotechnol., 18, 103-107.
6.Joseph, B., Ramteke, P. W., and Thomas, G. (2008)
Biotechnol. Adv., 26, 457-470.
7.Zhang, A., Gao, R., Diao, N., Xie, G., Gao, G., and
Cao, S. (2009) J. Mol. Catal. B: Enzymatic, 56,
78-84.
8.Zhang, J., Lin, S., and Zeng, R. (2007) J.
Microbiol. Biotechnol., 17, 604-610.
9.Chen, R., Guo, L., and Dang, H. (2011) World J.
Microbiol. Biotechnol., 27, 431-441.
10.Tolstikhin, N. I., and Tolstikhin, O. N. (1976)
in Groundwater and Surface Water in the Permafrost Region, Chap.
IX. General Permafrost Studies, Environment Canada, Inland
Waters Directorate, Ottawa, Technical Bulletin No. 97, p. 25.
11.Gilichinsky, D., Rivkina, E., Bakermans, C.,
Shcherbakova, V., Petrovskaya, L., Ozerskaya, S., Ivanushkina, N.,
Kochkina, G., Laurinavichuis, K., and Pecheritsina, S. (2005) FEMS
Microbiol. Ecol., 53, 117-128.
12.Petrovskaya, L., Novototskaya-Vlasova, K.,
Spirina, E., Khokhlova, G., Rivkina, E., Dolgikh, D., and Kirpichnikov,
M. (2012) DAN, 445, 102-105.
13.Bakermans, C., Ayala-del-Rio, H. L., Ponder, M.
A., Vishnivetskaya, T., Gilichinsky, D., Thomashov, M. F., and Tiedje,
J. M. (2006) Int. J. Syst. Evol. Microbiol., 56,
1285-1291.
14.Novototskaya-Vlasova, K., Petrovskaya, L.,
Yakimov, S., and Gilichinsky, D. (2012) FEMS, 82,
367-375.
15.Altschul, S. F., Gish, W., Miller, W., Myers, E.
W., and Lipman, D. J. (1990) J. Mol. Biol., 215,
403-410.
16.Sambrook, J., and Russell, D. W. (2001)
Molecular Cloning: a Laboratory Manual, 3rd Edn., Cold Spring
Harbor Laboratory Press, Cold Spring Harbor, NY, Vol. 1, p. 152.
17.Horton, R. M., Hunt, H. D., Ho, S. N., Pullen, J.
K., and Pease, L. R. (1989) Gene, 77, 61-68.
18.Kulakova, L., Galkin, A., Nakayama, T., Nishino,
T., and Esaki, N. (2004) Biochim. Biophys. Acta,
1696, 59-65.
19.Xuezheng, L., Shuoshuo, C., Guoying, X., Shuai,
W., Ning, D., and Jihong, S. (2010) Polar Res., 29,
421-429.
20.Parra, L., Reyes, F., Acevedo, J. P., Salazar,
O., Andrews, B. A., and Asenjo, J. A. (2008) Enzyme Microb.
Technol., 42, 371-377.
21.Feller, G., Thiry, M., Arpigny, J. L., and
Gerday, C. (1991) Gene, 102, 111-115.
22.Mandrich, L., Pezzullo, M., Del Vecchio, P.,
Barone, G., Rossi, M., and Manco, G. (2004) J. Mol. Biol.,
335, 357-369.
23.Langin, D., Laurell, H., Holst, L. S., Belfrage,
P., and Holm, C. (1993) Proc. Natl. Acad. Sci. USA,
90, 4897.
24.Arpigny, J. L., Lamotte, J., and Gerday, C.
(1997) J. Mol. Catal. B Enzym., 3, 29-35.
25.De Santi, C., Tutino, M. L., Mandrich, L.,
Giuliani, M., Parrilli, E., Del Vecchio, P., and De Pascale, D. (2010)
Biochimie, 92, 949-957.
26.Nardini, M., and Dijkstra, B. W. (1999) Curr.
Opin. Struct. Biol., 9, 732-737.
27.Mandrich, L., Merone, L., Pezzullo, M., Cipolla,
L., Nicotra, F., Rossi, M., and Manco, G. (2005) J. Mol. Biol.,
345, 501-512.
28.Shen, W. J., Sridhar, K., Bernlohr, D. A., and
Kraemer, F. B. (1999) Proc. Natl. Acad. Sci. USA, 96,
5528.
29.Feller, G., and Gerday, C. (1997) Cell. Mol.
Life Sci., 53, 830-841.
30.Gatti-Lafranconi, P., Caldarazzo, S. M., Villa,
A., Alberghina, L., and Lotti, M. (2008) FEBS Lett., 582,
2313-2318.
31.Wei, Y., Contreras, J. A., Sheffield, P.,
Osterlund, T., Derewenda, U., Kneusel, R. E., Matern, U., Holm, C., and
Derewenda, Z. S. (1999) Nat. Struct. Biol., 6,
340-345.