Topology of mRNA Chain in Isolated Eukaryotic Double-Row Polyribosomes
Zh. A. Afonina1#, A. G. Myasnikov2#, N. F. Khabibullina1,3, A. Yu. Belorusova1,4, J.-F. Ménétret2, V. D. Vasiliev1, B. P. Klaholz2, V. A. Shirokov1, and A. S. Spirin1*
1Institute of Protein Research, Russian Academy of Sciences, 142290 Pushchino, Moscow Region, Russia; fax: +7 (495) 514-0218; E-mail: spirin@vega.protres.ru2Institute of Genetics and Molecular and Cellular Biology (IGBMC), Department of Integrated Structural Biology, Illkirch, F-67404 France; fax: +33-3886-5320; INSERM, U596, Illkirch, F-67400 France; CNRS, UMR7104, Illkirch, F-67400 France; Université de Strasbourg, Strasbourg, F-67000 France
3Current address: Research and Education Center “Bionanophysics”, Moscow Institute of Physics and Technology, 141700 Dolgoprudnyi, Russia
4Current address: Institute of Genetics and of Molecular and Cellular Biology (IGBMC), Illkirch, F-67404 France
# These authors contributed equally to this work.
* To whom correspondence should be addressed.
Received December 17, 2012; Revision received January 24, 2013
In the process of protein synthesis, the translating ribosomes of eukaryotic cells form polyribosomes that are found to be multiplex functional complexes possessing elements of ordered spatial organization. As revealed by a number of electron microscopy studies, the predominant visible configurations of the eukaryotic polyribosomes are circles (circular polyribosomes) and two-stranded formations (so-called double-row polyribosomes). The “long” (i.e. heavy loaded) polyribosomes are usually represented by double-row structures, which can be interpreted as either topologically circular (“collapsed rings”), or topologically linear (zigzags or helices). In the present work we have analyzed the mRNA path within the eukaryotic polyribosomes, isolated from a wheat germ cell-free translation system, by integrating two approaches: the visualization of mRNA ends in polyribosomes by marking them with gold nanoparticles (3′-end) and initiating 40S subunits (5′-end), as well as by the cryoelectron tomography. Examination of the location of the mRNA markers in polyribosomes and mutual orientation of ribosomes in them has shown that the double-row polyribosomes of the same sample can have both circular and linear arrangements of their mRNA.
KEY WORDS: eukaryotic polyribosomes, mRNA, circular translation, cryoelectron tomographyDOI: 10.1134/S0006297913050027
Abbreviations: CECF, continuous exchange cell-free system; CET, cryoelectron tomography; eIF, eukaryotic initiation factor; EM, electron microscopy; FITC, fluorescein isothiocyanate; FTSC, fluorescein-5-thiosemicarbazide; “MAFITC 10 nm gold”, anti-fluorescein isothiocyanate antibody conjugated with 10 nm gold particles; PABP, poly(A)-binding protein; UTR, untranslated region of mRNA.
The polyribosome is a group of ribosomes strung on an mRNA thread and
translating it sequentially in one direction, from the 5′
terminus to the 3′ terminus of the polyribonucleotide chain [1-6]. Polyribosomes are the major
form of translating ribosomes, universal for both pro- and eukaryotic
cells.
In pioneering electron microscopy (EM) studies of mammalian polyribosomes, it was observed that the spatial distribution of ribosomes in polysomes does not follow the simple “beads on a thread” model. Instead, polyribosomes usually reveal themselves as more or less ordered structures, most often circles, helices, and double rows [7-10]. On the other hand, functional assays demonstrated that polysomal ribosomes in living cells, as well as in cell-free systems, exchange slowly with free ribosomes and ribosomal subunits, and every next round of initiation/translation in a polysome pool is accomplished for the most part by terminating ribosomes [11-13]. From this the idea emerged that, when a eukaryotic polyribosome is “arranged in closed circle configuration”, its ribosomes “could move along the circular mRNA without being released” (cited from [11]). According to this model, “ribosomes that have completed one round of translation dissociate into RSU (ribosomal subunits) near the site where RSU will attach to mRNA and initiate a new round of translation”. “In this model, the 3′-end (termination site) of mRNA should be close to the 5′-end (initiation site) of the same mRNA” (cited from [13]). Later the circular configuration of eukaryotic polyribosomes was confirmed by EM analyses of membrane-bound and free cytoplasmic polyribosomes [14-16]. In the case of long mRNAs the double-row polyribosomes, which were also interpreted as circular polyribosomes (“collapsed circles”), represented the major fraction of membrane-bound polyribosomes [17].
In addition to the abovementioned structural and functional data, the synergistic effect of 5′ and 3′ untranslated regions (UTRs) on eukaryotic mRNA translation was shown, and then the idea of a noncovalent circularization was supported by discovery of direct interactions of cap-binding complex eIF4E-eIF4G with poly(A)-binding protein (PABP) on poly(A)-tail [18-23]. It was also demonstrated that free (isolated) 5′-capped mRNA with poly(A) sequence at the 3′-end acquires the circular form in the presence of initiation factor eIF4F and PABP [24]. The combination of all the cited structural and functional properties of eukaryotic mRNA and polyribosomes strongly substantiated the hypothesis of mRNA circularization and “circular translation” in polyribosomes, which became widely accepted [25, 26].
Polyribosomes visualized as circular structures and double rows were also shown to be formed during translation in eukaryotic cell-free systems [27, 28]. In a previous work, we demonstrated that efficient re-initiation of translation of the same mRNA by terminating ribosomes could occur in the double-row polyribosomes formed in a cell-free system; that made it possible to consider those polyribosomes as functionally circular [28]. In the circular model that we suggested for the double-row polyribosome, the mRNA termini should be located in proximity at the same polyribosome end.
At the same time, an alternative interpretation of the double-row polyribosome images is possible: it was reported that in such a polyribosome the mRNA path between ribosomes can have a zigzag or helical pattern [9, 29-31]. In this case the mRNA 5′ and 3′ termini appear on the opposite ends of the double-row polyribosome. In the recent papers of the Baumeister group [30, 31], structural organization of pro- and eukaryotic polyribosomes was studied by cryoelectron tomography. It was found that in the prokaryotic polyribosomes, which were visualized as double rows, ribosomes were arranged either in 3D helices with four ribosomes per turn, or in flat structures with zigzag path of mRNA [30]. Examination of eukaryotic polyribosomes in vivo in human glioma cells revealed long polyribosomes having helical configuration [31]. Polyribosome modeling based on the identified characteristic contacts between ribosomes also indicated that the helical type of polyribosome organization was most abundant. The authors pointed out that the hypothesis of mRNA circularization in eukaryotic polyribosomes was not supported by their data so far as all the analyzed polyribosomes had topologically linear path of the mRNA chain [31].
In this work, we have determined the topology of the mRNA chain in double-row polyribosomes formed in a wheat germ cell-free system using EM visualization of markers on mRNA 5′ and 3′ termini and by cryoelectron tomography (CET). The localization of the terminal markers in polyribosomes and the mutual orientation of polysomal ribosomes determined by CET suggested that the double-row polyribosomes can be of two coexisting types, possessing either topologically circular or topologically linear organization of their mRNA.
MATERIALS AND METHODS
Materials. Used in this study were: enzymes for DNA cloning and RNase inhibitor Ribolock (Fermentas, Lithuania); RNase inhibitor SUPERaseIn™ (Ambion, USA); yeast tRNA, creatine phosphate, and creatine phosphokinase (Roche Diagnostics, Germany); dithiothreitol, nucleoside triphosphates, and inorganic salts and buffers (Sigma, USA); cap-analog 3′-O-Me-m7G(5′)ppp(5′)G (New England Biolabs, USA); Sephacryl S-500 HR and MicroSpin G50 (GE Healthcare, USA); fluorescein-5-thiosemicarbazide (Invitrogen, USA); murine anti-fluorescein isothiocyanate antibodies conjugated to 10 nm colloidal gold (MAFITC-10 nm gold) (Aurion, The Netherlands).
Preparation of mRNA. Messenger RNA was prepared using the pTZβG-SBP-CBP-GFP-A30 plasmid containing under the T7 promoter the following sequences: 5′ UTR of β-globin mRNA (rabbit), coding sequence of GFP (cycle3 mutant) fused with N-end SBP-CBP tag (streptavidin- and calmodulin-binding peptides) and C-end His6-tag, and 3′-end A30 sequence. The GFPcycle3 sequence was amplified in PCR with the pBAD-GFP vector (Maxigen, USA) and primers 5′GATATACATATGGCTAGCAAAGGAGAAGAAC and 5′GACCATGGCCTTTGTAGAGCTCATCCATGCCATG; SBP-CBP sequence – in PCR with pCTAP-A vector (Stratagene, USA) and primers 5′CAGTGCTAGCATGGACGAGAAGACCACCGG and 5′CTGTCTAGAAAGTGCCCCGGAGGATGAG. The sequence of the 5′ UTR of the β-globin mRNA was obtained by extension of the complementary primers 5′ATAAGCTTCACACTTGCTTTTGACACAACTGTGTTTACTTGCA and 5′TTGGCCATTCTGTCTGTTTTGGGGGATTGCAAGTAAACACAGTTGT. All primers contained the restriction sites (underlined) for further cloning. The A30 sequence with flanking segments (including 50 nt spacer) was excised from pCITE-4a(+) (Novagen, USA) vector using XhoI and SpeI enzymes. The pTZβG-SBP-CBP-GFP-A30 plasmid was constructed in several steps using standard methods, the cloned sequence was inserted between HindIII and SalI of pTZ19R (Novagen) vector, and the insert was confirmed by sequencing of the cloning region. The capped mRNA (designated as cap-5′UTRβ-globin-GFP-3′UTR(A)30) was prepared by in vitro transcription of the pTZβG-SBP-CBP-GFP-A30 plasmid linearized at the unique XbaI site, as described previously [32], using the 3′-O-Me-m7G(5′)ppp(5′)G cap-analog. The total length of synthesized mRNA was 1144 nt with the length of coding sequence of 975 nt.
Preparation of ribosomal 40S subunits and ribosome-free extract. Ribosomal 40S subunits were isolated from wheat germ extract by centrifugation in 5-30% sucrose gradient in buffer 25 mM Hepes-KOH, pH 7.6, 500 mM KOAc, 5 mM Mg(OAc)2, 2 mM DTT (SW28, 24,000 rpm, 9 h). Fractions containing 40S subunits were concentrated on a Centricon 30 with simultaneous buffer change for buffer A (25 mM Hepes-KOH, pH 7.6, 100 mM KOAc, 5 mM Mg(OAc)2) containing 2 mM DTT. Ribosome-free extract (S100) was prepared by pelleting the ribosomes from the wheat germ extract through a 20% glycerol cushion in buffer A with 4 mM DTT (TLA-100.1, 67,000 rpm, 90 min); the resulting supernatant was dialyzed against the same buffer.
Cell-free translation in wheat germ system. Polyribosomes preparation was obtained by mRNA translation in the wheat germ continuous exchange cell-free system (CECF) as described in [28, 33]. The CECF system operates in dynamic regime providing replenishment of substrates and removal of low molecular weight products across a semi-permeable membrane, thus supporting steady conditions of translation reaction for many hours [34]. To do that, translation mixture is loaded into the dialysis reactor (see [28], Supplementary Data), and the outer chamber of the reactor contains the solution of substrates in a relevant buffer (“feeding solution”). The translation mixture contained 25 mM Hepes-KOH, pH 7.6, 3 mM Mg(OAc)2, 85 mM KOAc, 2 mM dithiothreitol, 0.25 mM spermidine, 2% glycerol, 0.01% NP40 detergent, 0.03% NaN3, 1 mM ATP, 0.4 mM GTP, 0.3 mM each of 20 amino acids, 0.05 mg/ml of total yeast tRNA, 16 mM creatine phosphate, 0.1 mg/ml of creatine phosphokinase, 500 U/ml of RNase inhibitor, 30% (v/v) of wheat germ extract, and 300 nM mRNA. The feeding solution contained the same components except tRNA, creatine phosphokinase, RNase inhibitor, wheat germ extract, and mRNA. Translation mixture was combined and preincubated for 2 min at 25°C prior the addition of the mRNA. A 100-µl aliquot of translation mixture was placed into the reaction chamber of the reactor; the outer chamber received 1 ml of feeding solution. The reactor was incubated at 25°C with agitation of the feeding solution. To monitor the translation reaction, 3-µl aliquots were taken at specific time points and quenched by addition of cycloheximide (0.01 mg/ml), and GFP fluorescence (excitation 395 nm, emission 510 nm) was measured after overnight incubation on ice. Translation reactions in the “polysome system” were performed under static condition in tubes (batch mode without dialysis). The composition of the polysome system was the same as in the CECF reaction; however, the wheat germ extract and mRNA were replaced with polyribosomes purified by gel chromatography (final concentration at least 5 OU260/ml) and S100 (30% v/v). Translation of modified mRNA was also performed in static mode (batch) using standard wheat germ extract.
Polyribosome isolation by gel chromatography. The translation reaction was terminated by addition of cycloheximide (0.01 mg/ml final concentration). A 300-µl portion of reaction mixture was clarified (12,000g, 10 min) and applied onto a 3-ml Sephacryl S-500 HR column (4 × 250 mm in 2 ml Costar Stripett; Sigma) equilibrated in buffer A containing 0.03% NaN3 and 0.01 mg/ml cycloheximide; when polyribosomes were prepared in the presence of edeine, the buffer was also supplemented with 1 µM edeine. Antibiotics were not added when polyribosomes were isolated for activity tests. Chromatography was performed in cold (4°C) at 10 µl/min flow rate collecting fractions of 35 µl volume; optical density in fractions was measured at 260 nm. Eluate fractions containing polyribosomes were pooled and used further for treatment or analysis. Sedimentation analysis in a sucrose gradient (15-45%) was performed as described earlier [28].
EM analysis of polyribosomes. Polyribosome suspension (3 µl, 1 OU260/ml in buffer A) was applied to an EM grid covered with a thin layer of carbon on a holey plastic film, and after 1 min the excess suspension was sucked up with a filter paper. The grid was washed twice with buffer A and negatively stained for 1 min with 2% uranyl acetate. The specimens were examined in Tecnai F20 (Philips, The Netherlands) or JEM 100C (JEOL, Japan) electron microscopes.
Chemical modification of 3′-end of mRNA. The 3′-ends of mRNA were modified with fluorescein-5-thiosemicarbazide (FTSC) after oxidative cleavage of the 3′-end ribose with sodium periodate. The cleavage was performed by 90-min incubation of cap-5′UTRβ-globin-GFP-3′UTR(A)30 mRNA in 66 mM NaOAc, pH 4.5, 5 mM NaIO4 buffer. After ethanol precipitation (2.5 volumes of ethanol), the mRNA precipitate was washed three times with 70% ethanol, after that the oxidized mRNA was incubated in 25 mM NaOAc, pH 6.1, 75% dimethyl sulfoxide buffer with 7.3 mM FTSC overnight at room temperature. The mRNA was precipitated and washed with ethanol as described above, and traces of FTSC were removed by gel filtration on a MicroSpin G50 column. The modification rate was found to be at least 70%, as calculated from the relative fluorescence of mRNA solution over the standard FTSC solution (excitation 492 nm, emission 519 nm).
Marking of the mRNA termini in polyribosomes. After 40 min translation of cap-5′UTRβ-globin-GFP-3′UTR(A)30 mRNA 3′-modified with FTSC, the translation mixture was supplemented with 40S subunits and, 1 min later, with edeine (3 OU260/ml and 2 µM final concentrations, respectively); 30 sec later translation was terminated by adding 0.1 mg/ml cycloheximide. The sample was clarified by centrifugation and subjected to gel chromatography on a Sephacryl S-500 HR column in the presence of 1 µM edeine. A 100-µl sample of polyribosome fraction (5-10 OU260/ml) was combined with 25 µl of colloidal gold conjugated with anti-FITC antibodies (MAFITC 10 nm gold) and 14 µl of SUPERaseIn™ RNase inhibitor (20 U/µl). The mixture was incubated for 30 min at room temperature, and then the unreacted conjugate was removed by one more chromatography on Sephacryl S-500 HR column. Chromatography fractions containing polyribosomes were pooled and analyzed by electron microscopy.
Cryoelectron tomography of polyribosomes. Samples of polyribosome fractions after gel chromatography were diluted to 1.5 OU260/ml with buffer A. Colloidal gold markers (10 nm gold–protein A conjugate) were added to give a final distribution of 10 to 20 beads per EM field. A 3-μl aliquot of the sample was applied to 300 mesh holey carbon grids in the Vitrobot apparatus (FEI, The Netherlands) at 20°C, 95% humidity. Excess sample volume was blotted automatically with filter paper from both sides of the grid, and it was immediately plunged into liquid ethane (–196°C). Cryoelectron tomography was recorded on Polara F30 FEG instrument (FEI) at 150 kV acceleration voltage and nominal underfocus of Δz = 3 μm. Tilt series were recorded between −66° and +66° with tilt angle increment of 1°-3° and with cumulative electron dose not exceeding 60 eˉ/Å2. Data were acquired using a 4096 × 4096 CCD Eagle (FEI) camera. The image alignment and tomogram reconstruction were performed using the ETOMO (IMOD software, Boulder Laboratory for 3-D EM, University of Colorado, USA) and inspect3D (FEI) software. Analysis of ribosome positioning and orientation in polyribosomes was conducted using software packages XMIPP (Spanish National Center of Biotechnology, Madrid) [35], IMAGIC (Image Science Software GmbH, Germany), and Chimera [36]. In this way, maximum-likelihood analysis of a set of sub-tomograms (polysomal ribosomes) of the reconstructed tomogram accompanied by an orientation survey for each sub-tomogram yielded the averaged ribosome model as well as rotation and shift parameters for each polysomal ribosome. The tomogram was interpreted by fitting the averaged ribosome model into the initial electron density map according to the rotation and shift parameters found for each ribosome. As a result, we obtained the reconstructed models of individual polyribosomes.
RESULTS
Formation and isolation of polyribosomes. As demonstrated earlier, the polyribosomes formed during translation in the wheat germ CECF system were structurally and functionally similar to cellular polyribosomes [27, 28]. In this work, formation of polyribosomes on the capped mRNA (cap-5′UTRβ-globin-GFP-3′UTR(A)30) occurred during 1 h of translation in such a system, followed by isolation of polyribosomes from the translation mixture. The polyribosomes were purified to remove the system components, primarily free 40S subunits and free mRNA, to facilitate the subsequent formation and analysis of the complex between polyribosomes containing 3′-modified mRNA, MAFITC 10 nm gold conjugate particles, and initiating 40S subunits.
The commonly used method of polyribosome purification by pelleting in an ultracentrifuge is too harsh in our case, as conglutination in a pellet can alter the structural organization of polyribosomes. An alternative and gentler method for isolation of polyribosomes is gel chromatography [37]. For separation of polyribosome complexes (molecular mass n·3.5·106 Da, where n is a number of ribosomes in the polyribosome) from monoribosomes (3.5·106 Da) and other cell-free components, fractionation on Sephacryl S500 HR was found most reliable (fractionation range 4·104 -2·107 Da).
The gel chromatography was performed on a Sephacryl S500 HR column in buffer A with salt composition close to that of the translation mixture. In the chromatography profile shown in Fig. 1a, the first peak corresponds to polyribosomes eluted in the void volume, the second peak contains monoribosomes, and the last one contains mRNA, tRNA, proteins, and other components of the reaction mixture. Separation of monoribosomes and polyribosomes, and also the integrity of the latter, were confirmed by EM-analysis of fractions I-III (Fig. 1b). The major part of the polyribosomes appeared as characteristic double rows of ribosomal particles. The isolated polyribosomes were also analyzed by sedimentation in a sucrose gradient. Figure 1c shows the sedimentation profile of the total polyribosome fraction after gel chromatography in comparison with the sedimentation profile of the original translation system, the source of the isolated polyribosomes. At equivalent loading, the two profiles coincided in the polyribosome zone, while monosomes and disomes comprised only a minor fraction in the case of the isolated polyribosome sample.
Fig. 1. Isolation and characterization of polyribosomes from translation reaction mixture. a) UV profile of polyribosome chromatography on Sephacryl S-500 HR column (3 ml); sample – 300 µl of the reaction mixture after 1 h of CECF translation. Fractions containing the “long” polyribosomes heavy-loaded with ribosomes (fraction I), “short” – low-loaded – polyribosomes (fraction II), and monoribosomes (80S) (fraction III) are indicated. b) EM images (negative contrast) of respective fractions of polyribosomes and ribosomes. Scale bar, 200 nm. c) Sedimentation profiles of the translation reaction mixture (dotted line) and the combined polyribosome fraction (fractions I + II) isolated by gel chromatography (solid line). Arabic numerals indicate the polyribosome “size” – number of ribosomes per polysomal mRNA. The gradients were loaded with 25 µl of reaction mixture or 65 µl of isolated polyribosomes (proportionally to dilution during chromatography). d) Comparison of translational activity of initial (1) and “polysomal” (2) translation systems; the polyribosome concentrations in the translation mixtures were 21 and 4.8 OU260/ml, respectively.
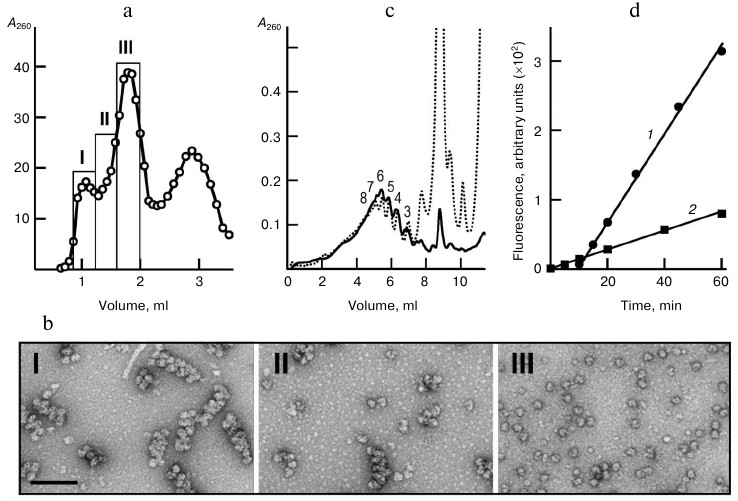
Functional properties of the isolated polyribosomes were analyzed in a cell-free translation system with ribosome-free wheat germ extract. Such a system containing ribosomes and mRNA only as the components of polyribosomes (“polyribosome translation system”) performed protein synthesis with a constant rate throughout the recording time (Fig. 1d). The activity of isolated polyribosomes (per OU of polyribosomes) in this system was found to be 80% of the activity of polyribosomes in the original system, as calculated from comparison of GFP synthesis rates in both systems at concentrations of polyribosomes 4.8 OU260/ml in the first and 21 OU260/ml in the second system. It is noteworthy that in the original translation system the accumulation of fluorescent (full-size) GFP started only after some lag-period dictated by the time when the early ribosomes read out the full mRNA sequence (about 10 min in this system) and release the full-size product. In the polyribosome system, the fluorescent protein accumulated from the very start of incubation due to completion of polypeptides that were partially pre-synthesized in the original system. All this proved the functionality of the isolated polyribosomes once again.
Thus, the isolation procedure did not affect the functional activity and integrity of the polyribosomes, and therefore the isolated samples of polyribosomes were considered suitable for structural studies, including the cryoelectron tomography. Besides, the efficient polysome translation system based on polyribosomes isolated by gel chromatography may be offered as a useful instrument for the analysis of the translation processes in polyribosomes.
Visualization of 5′ and 3′ mRNA termini in polyribosomes. To directly localize the mRNA 5′ and 3′ termini in polyribosomes, we used specific markers for labeling mRNA termini with subsequent visualization of the markers by conventional EM. When both termini bear the markers, their adjacent location at the same end of the polyribosome should testify to circular organization of mRNA in it, while the two markers separated between the ends should indicate the topologically linear organization of mRNA.
The mRNA 3′-end was oxidized with periodate and then modified with fluorescein-thiosemicarbazide (FTSC). The modified 3′-end was subsequently marked in polyribosomes by 10 nm gold particles conjugated with antibodies to fluorescein isothiocyanate (MAFITC 10 nm gold). Since in the co-transcriptional capping of mRNA we used cap-analog with blocked 3′-hydroxylic group at m7G, the cap-side of mRNA was immune to oxidation and modification. The FTSC-modified mRNA preserved translational activity and ability to form polyribosomes.
For marking the 5′-terminus of the polyribosomal mRNA, we made use of 40S subunits arrested at the start codon in the presence of edeine, the antibiotic that blocks interaction of 60S subunit with initiating 40S subunit; as a result, the 40S subunit remains non-coupled with the 60S subunit and bound at the 5′-terminus of the polyribosome.
In the experiment on marking the polysome termini, the translation system containing polyribosomes formed on the modified mRNA was supplemented with 40S subunits and edeine; after a short incubation, the translation was stopped by the addition of cycloheximide. Polyribosomes were isolated on the Sephacryl S500 HR column and incubated with MAFITC 10 nm gold conjugate. The polyribosomes were chromatographically purified again to remove unbound gold conjugate particles. In a control reaction, polyribosomes were formed by the translation of non-modified mRNA, and 40S subunits and edeine were not added.
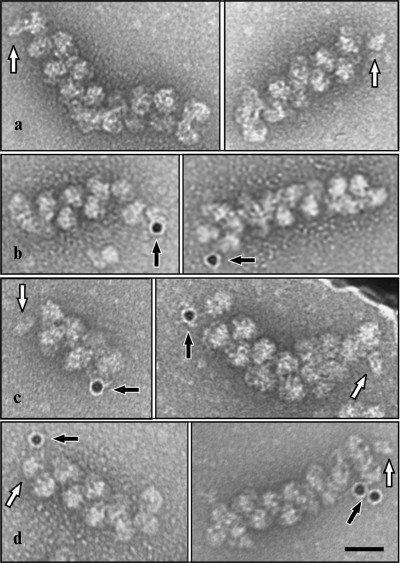
EM analysis of the polyribosomes marked in this way showed 20% yield of polyribosome complexes with 10 nm gold particles (Fig. 2), while free gold particles were absent in the EM field; in the control sample complexes were not found. The initiating 40S subunits associated with polyribosomes were observed more rarely. It should be noted that in the original reaction mixture, initiating 40S subunits were found in more than 20% of the polyribosomes; it is probable that the repeated chromatography purification procedures resulted in significant loss of the 40S particles from polyribosomes. In polyribosomes containing a single marker (either 40S subunit or 10 nm gold particle), the marker was always found at one of the ends of the polyribosome (Fig. 3, a and b). In polyribosomes bearing the two markers simultaneously (5′-40S subunit and 3′-10 nm gold particle) they were found either on the opposite ends (Fig. 3c), or on the same end of the polyribosome (Fig. 3d).
Fig. 2. EM image (negative contrast) of polyribosomes formed on a 3′FTSC-modified mRNA and then incubated with MAFITC 10 nm gold conjugate (for details see “Results”). All the polyribosomes associated with gold particles in this EM field are shown enlarged in inserts.
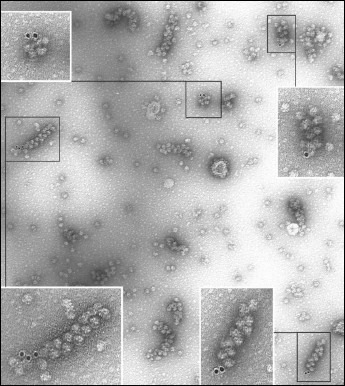
Fig. 3. Marking of the mRNA termini in polyribosomes formed on 3′FTSC-modified mRNA, incubated with edeine and then with MAFITC 10 nm gold (see “Results”). The panels show EM images of polyribosomes containing the initiating 40S subunit (a), polyribosomes associated with 10 nm gold particles (anti-FITC conjugated) (b), as well as polyribosomes bearing both markers simultaneously (initiating 40S subunit on 5′-terminus and 10 nm gold particle on 3′-terminus of mRNA) either separately at distant ends of polyribosome (c) or alongside at the same end of the polyribosome (d). Arrows indicate the initiating 40S subunits (white arrow) and 10 nm gold particles (black arrow). Scale bar 50 nm.
Cryoelectron tomography of polyribosomes. The application of cryoelectron tomography (CET) for ascertaining the 3D structure of polyribosomes started recently in several groups [30, 31, 38, 39]. The CET technique makes possible the analysis of polyribosomes under conditions most favorable for preservation of their native structure. The method includes flash-freezing of the object under investigation at ultra low temperature, resulting in conservation of the object in an amorphous ice layer, with subsequent taking of EM photos at different angles in the range of –70° to +70° and using the resulting projections for reconstruction of the 3D structure of the object [40]. The CET methodology accompanied with sub-tomogram averaging allows determination of the arrangement and mutual orientation of ribosomes in the polyribosome under investigation. As the mRNA entrance and exit sites in ribosome are well known [41, 42], the direction of the mRNA path in each ribosome within the polyribosome can also be unambiguously determined. Using the CET technique and the data obtained on mutual orientation of all ribosomes in each polyribosome studied, we determined the plausible path of the mRNA chain in every individual polyribosome, provided that the mRNA chain connects neighboring ribosomes and does not form knots.
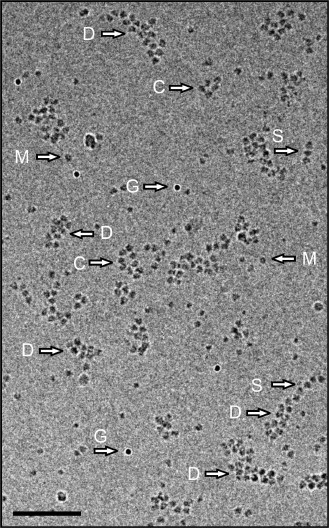
Figure 4 shows the cryo-EM field of a preparation in which polyribosomes of different shapes are displayed. Single-row, circular, and double-row polyribosomes containing from 3-4 to 12-14 ribosomes per polyribosome are seen. It should be mentioned that in such a 2D image the 3D helical polyribosomes may also look like double-row formations; they can be distinguished from the flat double-row polyribosomes only in the reconstructed tomogram. The content of monoribosomes in the sample was low, this making unlikely their occasional appearance in the vicinity of polyribosomes.
Fig. 4. Cryo-EM image of polyribosomes isolated by gel chromatography. Arrows point to single-row (S), double-row (D), and morphologically circular (ring-like) (C) polyribosomes, monoribosomes (M), and 10 nm gold particles (G). Scale bar 200 nm. The cumulative dose for this image was 20 eˉ/Å2.
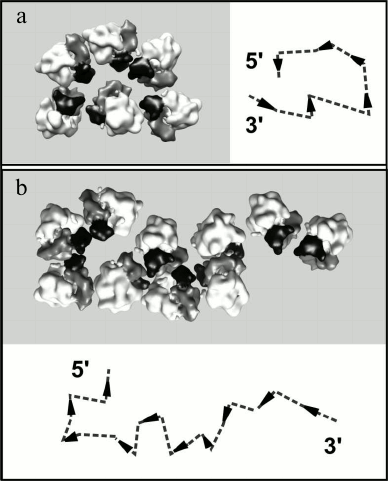
The analysis of the reconstructed polyribosome models showed that in the double-row polyribosomes both topologically linear and circular paths of mRNA could be observed. Figure 5 demonstrates the reconstructed models of the characteristic types of double-row polyribosomes and the plausible path of mRNA in them. Arrowheads indicate the direction of the mRNA chain in each ribosome (towards the 5′ terminus), while dotted lines connect the entry and exit sites of mRNA in the neighboring ribosomes according to the abovementioned criteria. It is seen that the ribosomes of the first type of polyribosomes (Fig. 5a) are arranged in the way that directs the mRNA chain to the right in the lower row and to the left in the upper row, thus implying a circular path and adjacent termini of mRNA in the polyribosome. In the scheme the 5′ and 3′ termini are depicted at the end of the double-row, where they have been typically observed in EM localization of the labeled mRNA termini. In the polyribosomes of the other type (Fig. 5b), the ribosomes are oriented in such a way that the mRNA chain aims in the same direction in both rows; on the whole, the mRNA path exhibits the zigzag (topologically linear) character, with 5′ and 3′ termini at the two opposite ends of the polyribosome. The double-row polyribosomes comprised one fourth of the total polyribosome population of the samples analyzed, and the circular topology of mRNA was found in about 10% of the double-row polyribosomes.
Fig. 5. Reconstructed models of topologically circular (a) and topologically linear (b) double-row polyribosomes. Polysomal ribosomes are represented by the averaged ribosome model in orientations determined for each ribosome during the averaging procedure. The head of the 40S subunit is shown in black, the body of the 40S subunit in gray, and the 60S subunit in white. In the schemes the orientation of the mRNA chain in each ribosome is symbolized by an arrowhead directed from the mRNA entry to mRNA exit sites of the ribosome. The deduced path of the mRNA chain through the ribosome is shown by a dashed line.
DISCUSSION
Information on the structural organization of polyribosomes is important for understanding the processes of polyribosome formation and growth, termination and re-initiation of translation, ribosome recycling in polyribosomes, as well as the regulation of protein synthesis at the polyribosome level. As mentioned in the introduction, the idea of circular translation was suggested and met some confirming data already in the 1960s [11-13]. Later it was revived and expanded by several groups on the ground of discovering the functional synergism and the direct interaction between capped 5′-terminus and polyadenylated 3′-terminus of mRNA mediated by eIF4E/eIF4G initiation factors and PABP (see references in the introductory part). On the other hand, structural data has been obtained recently favoring an alternative model, where the eukaryotic polyribosome, visualized as a double row in flat EM images, was reported to have topologically linear mRNA path with the ribosome particles arranged in zig-zag or 3D helices [31]. Earlier this type of polyribosome organization was found in prokaryotic systems [30].
We analyzed the character of the mRNA path in eukaryotic polyribosomes obtained by cell-free translation of capped polyadenylated mRNA. We integrated two methodological approaches – EM visualization of marked mRNA termini in polyribosomes and cryoelectron tomography (CET). The major focus was to determine the mRNA path in the double-row polyribosomes, especially in regard to the existing alternative models. The double-row polyribosomes used to be interpreted as circular structures with antiparallel arrangement of the opposite sides of the collapsed ring [17, 27, 28]. However, no direct structural evidence for this interpretation was published.
Our results of EM visualization of the markers at the 5′ and 3′ termini of mRNA in the [5′-40S]–polyribosomes–[3′-10 nm gold] complex demonstrated that in polyribosomes bearing both markers simultaneously, they were displayed more frequently at the opposite ends of polyribosomes. Yet, we also observed them occurring simultaneously at the same end of the polyribosome (Fig. 3d). This implies that polyribosomes possessing double-row morphology may be topologically different, with either linear or circular path of the mRNA. The existence of two types of mRNA topology in the double-row polyribosomes was also confirmed by our CET analysis.
Thus, the main fact we revealed is that among double-row polyribosomes of the same specimen, both circular and linear topology of mRNA may occur. One may assume that the circular organization of mRNA is preserved in the double-row polyribosomes, which have arisen by collapsing the circular (ring-shaped) polyribosomes. At the same time, a major part of the double-row polyribosomes was found possessing the zigzag-like, topologically linear mRNA path. The latter may be an intermediate stage in the process of further transformations of the polyribosome structure.
The authors are grateful to K. S. Vasilenko, V. A. Kolb and A. V. Efimov for valuable discussions and suggestions. We thank E. A. Arutuinian, M. V. Dontsova, E. A. Chernousova and M. A. Stoilov for skillful technical assistance.
This work was supported by the Program on Molecular and Cell Biology of the Russian Academy of Sciences and by the Russian Foundation for Basic Research (grant Nos. 06-04-49152 and 09-04-01726), as well as by the Agence Nationale pour la Recherche (ANR), the Centre Nationale pour la Recherche Scientifique (CNRS), the European Molecular Biology Organization (EMBO) Young Investigator Programme (YIP), the European Commission as SPINE2-complexes (contract n_ LSHG-CT-2006-031220), the European Research Council (ERC Starting Grant) and by Award Number R03TW008217 from the Fogarty International Center. Z. A. A. was supported by the ARCUS (Alsace/Russia-Ukraine) program and by an EMBO short-term fellowship (ASTF 388.00-2009), A. G. M. by a fellowship from the Fondation pour la Recherche Medicale (FRM) and by the ANR, J-F. M. by the FRM. The electron microscope facility is supported by the Alsace Region, the FRM, IBiSA platform program, INSERM, CNRS and the Association pour la Recherche sur le Cancer (ARC).
REFERENCES
1.Warner, J. R., Rich, A., and Hall, C. (1962)
Science, 138, 1399-1403.
2.Warner, J. R., Knopf, P. M., and Rich, A. (1963)
Proc. Natl. Acad. Sci. USA, 49, 122-129.
3.Rich, A., Warner, J. R., and Goodman, H. M. (1963)
Cold Spring Harbor Symp. Quant. Biol., 28, 269-285.
4.Gierer, A. (1963) J. Mol. Biol., 6,
148-157.
5.Wettstein, F. O., Staehelin, T., and Noll, H.
(1963) Nature, 197, 430-435.
6.Penman, S., Scherrer, K., Becker, Y., and Darnell,
J. E. (1963) Proc. Natl. Acad. Sci. USA, 49, 654-662.
7.Palade, G. E. (1955) J. Biophys. Biochem.
Cytol., 1, 59-68.
8.Mathias, A. P., Williamson, R., Huxley, H. E., and
Page, S. (1964) J. Mol. Biol., 9, 154-167.
9.Shelton, E., and Kuff, E. L. (1966) J. Mol.
Biol., 22, 23-31.
10.Dallner, G., Siekevitz, P., and Palade, G. E.
(1966) J. Cell Biol., 30, 73-96.
11.Philipps, G. R. (1965) Nature, 205,
53-56.
12.Adamson, S. D., Howard, G. A., and Herbert, E.
(1969) Cold Spring Harbor Symp. Quant. Biol., 34,
547-554.
13.Baglioni, C., Vesco, C., and Jacobs-Lorena, M.
(1969) Cold Spring Harbor Symp. Quant. Biol., 34,
555-565.
14.Christensen, A. K., Kahn, L. E., and Bourne, C.
M. (1987) Amer. J. Anat., 178, 1-10.
15.Yoshida, T., Wakiyama, M., Yazaki, K., and Miura,
K.-I. (1997) J. Electron Microscopy (Japan), 46,
503-506.
16.Yasaki, K., Yoshida, T., Wakiyama, M., and Miura,
K.-I. (2000) J. Electron Microscopy (Japan), 49,
663-668.
17.Christensen, A. K., and Bourne, C. M. (1999)
Anat. Record, 255, 116-129.
18.Gallie, D. R., and Walbot, V. (1990) Genes
Dev., 4, 1149-1157.
19.Gallie, D. R. (1991) Genes Dev., 5,
2108-2116.
20.Le, H., Tanguay, R. L., Balasta, M. L., Wei, C.
C., Browning, K. S., Metz, A. M., Goss, D. J., and Gallie, D. R. (1997)
J. Biol. Chem., 272, 16247-16255.
21.Tarun, S. Z. J., and Sachs, A. B. (1996) EMBO
J., 15, 7168-7177.
22.Imataka, H., Gradi, A., and Sonenberg, N. (1998)
EMBO J., 17, 7480-7489.
23.Borman, A. M., Michel, Y. M., Malnou, C. E., and
Kean, K. M. (2002) J. Biol. Chem., 277, 36818-36824.
24.Wells, S., Hillner, P., Vale, R., and Sachs, A.
(1998) Mol. Cell, 2, 135-140.
25.Jacobson, A. (1996) in Translational Control
(Hershey, J. W. B., Mathews, M. B., and Sonenberg, N., eds.) Cold
Spring Harbor Laboratory Press, Cold Spring Harbor, N. Y., pp.
451-480.
26.Preiss, T., and Hentze, M. W. (1999) Curr.
Opin. Genet. Dev., 9, 515-521.
27.Madin, K., Sawasaki, T., Kamura, N., Takai, K.,
Ogasawara, T., Yazaki, K., Takei, T., Miura, K.-I., and Endo, Y. (2004)
FEBS Lett., 562, 155-159.
28.Kopeina, G. S., Afonina, Zh. A., Gromova, K. V.,
Shirokov, V. A., Vasiliev, V. D., and Spirin, A. S. (2008) Nucleic
Acids Res., 36, 2476-2488.
29.Martin, K. A., and Miller, O. L., Jr. (1983)
Dev. Biol., 98, 338-348.
30.Brandt, F., Etchells, S. A., Ortiz, J.
O. M., Elcock, A. H., Hartl, F. U., and Baumeister, W. (2009)
Cell, 136, 261-271.
31.Brandt, F., Carlson, L. A., Hartl, F. U.,
Baumeister, W., and Grunewald, K. (2010) Mol. Cell, 39,
560-569.
32.Vassilenko, K. S., Alekhina, O. M., Dmitriev, S.
E., Shatsky, I. N., and Spirin, A. S. (2011) Nucleic Acids Res.,
39, 5555-5567.
33.Shirokov, V. A., Kommer, A., Kolb, V. A., and
Spirin, A. S. (2007) in Methods in Molecular Biology, Vol. 375
(Grandi, G., ed.) Humana Press Inc., Totowa, NJ, pp. 19-55.
34.Spirin, A. S. (2004) Trends Biotech.,
22, 538-545.
35.Sorzano, C. O. S., Marabini, R.,
Velazquez-Muriel, J., Bilbao-Castro, J. R., Scheres, S. H. W., Carazo,
J. M., and Pascual-Montano, A. (2004) J. Struct. Biol.,
148, 194-204.
36.Pettersen, E. F., Goddard, T.
D., Huang, C. C., Couch, G. S., Greenblatt,
D. M., Meng, E. C., and Ferrin, T. E. (2004) J.
Comput. Chem., 25, 1605-1612.
37.Eschenfeldt, W. H., and Patterson, R. J. (1975)
Prep. Biochem., 5, 247-255.
38.Simonetti, A., Marzi, S.,
Myasnikov, A. G., Menetret, J.-F., and Klaholz, B. P. (2011) in
Ribosomes. Structure, Function and Dynamics (Rodnina, M.,
Wintermeyer, W., and Green, R., eds.) Springer, Wien-N. Y., pp.
113-128.
39.Pfeffer, S., Brandt, F., Hrabe, T., Lang, S.,
Eibauer, M., Zimmermann, R., and Förster, F. (2012)
Structure, 20, 1508-1518.
40.Lucić, V., Förster, F., and
Baumeister, W. (2005) Ann. Rev. Biochem., 74,
833-865.
41.Evstafieva, A. G., Shatsky, I. N., Bogdanov,
A. A., Semenkov, Y. P., and Vasiliev, V. D. (1983) EMBO
J., 2, 799-804.
42.Yusupova, G. Zh., Yusupov, M. M., Cate, J. H. D.,
and Noller, H. F. (2001) Cell, 106, 233-241.