REVIEW: piRNA Clusters as a Main Source of Small RNAs in the Animal Germline
I. A. Olovnikov and A. I. Kalmykova*
Institute of Molecular Genetics, Russian Academy of Sciences, pl. Akademika Kurchatova 2, 123182 Moscow, Russia; fax: (499) 196-0221; E-mail: allakalm@img.ras.ru* To whom correspondence should be addressed.
Received February 4, 2013
PIWI subfamily Argonaute proteins and small RNAs bound to them (PIWI interacting RNA, piRNA) control mobilization of transposable elements (TE) in the animal germline. piRNAs are generated by distinct genomic regions termed piRNA clusters. piRNA clusters are often extensive loci enriched in damaged fragments of TEs. New TE integration into piRNA clusters causes production of TE-specific piRNAs and repression of cognate sequences. piRNAs are thought to be generated from long single-stranded precursors encoded by piRNA clusters. Special chromatin structures might be essential to distinguish these genomic loci as a source for piRNAs. In this review, we present recent findings on the structural organization of piRNA clusters and piRNA biogenesis in Drosophila and other organisms, which are important for understanding a key epigenetic mechanism that provides defense against TE expansion.
KEY WORDS: piRNA, PIWI, transposable elements, germline, chromatin, DrosophilaDOI: 10.1134/S0006297913060035
Small RNAs associated with Argonaute (Ago) family proteins play an important role in regulation of gene expression at all stages of development in the most studied eukaryotes. Analysis of small RNAs from animal germlines revealed, in addition to siRNA (small interfering RNA) and miRNA (micro RNA), a distinct class of small RNAs that were termed piRNAs (Piwi interacting RNA) [1, 2]. As it is seen from their name, piRNA biology is tightly linked to Piwi subfamily proteins of Ago family. The major function of piRNAs is to protect the genome from activity of transposable elements (TE), over-expression of which in germ cells may lead to high frequency of transpositions and transmission of harmful insertion mutations to the next generations. piRNAs recognize TE mRNAs and cause their degradation due to endonuclease activity of Piwi proteins.
piRNA biogenesis differs from that of siRNA and miRNA by some peculiarities. piRNAs are longer (~24-28 nt versus ~21 nt for siRNAs and ~22 nt for miRNAs in fruitfly) and are characterized by preference for definite nucleotides at some positions. For piRNA, similarly to siRNA, 2′-O-methylation is characteristic [3-8]. piRNA production does not depend on type III RNA endonucleases (Dicer) that slice double-stranded RNAs and are necessary for maturation of siRNAs and miRNAs [3, 9]. This circumstance along with other data evidences formation of piRNAs from single-stranded precursors. The mechanism of piRNAs excision from a long precursor transcript, the process termed as primary processing of piRNAs, is still enigmatic. Presence of uridine at the first position (1U) is characteristic for the majority of primary piRNAs [6, 10, 11]. Finally, sequencing and mapping of piRNAs on the genome show that piRNAs mostly originate from discrete loci lacking genes but enriched in fragments of degraded TEs [6]. These loci, termed piRNA clusters, are numerous (for example, ~150 clusters in Drosophila) and may be very long (more than 200,000 base pairs) [6]. In Drosophila piRNA clusters are localized, as a rule, in pericentromeric or subtelomeric regions of chromosomes, and compose about 3.5% of the genome.
Like many others members of the Argonaute family, Piwi subfamily proteins possess slicing activity, i.e. an ability to cut mRNAs that are complementary to small RNAs [4, 5]. This process is termed post-transcriptional gene silencing (PTGS). For the piRNA-dependent PTGS compartmentalization is a necessary condition: for example, majority of known factors of piRNA pathway in germ cells are localized in perinuclear granules that form a particular structure – “nuage” (for cloud in French) [12, 13]. However, in many species Ago proteins may function in the nucleus, where they cause transcriptional silencing of sequences that are complementary to small RNA (TGS – transcriptional gene silencing). TGS was first described in yeast and plants [14, 15] and, presently, its role in RNA silencing is established in animals. In mammals piRNAs induce methylation of promoters of homologous loci, and this leads to transcriptional silencing of retrotransposons [16]. It has been shown in Drosophila that transcriptional activity of RNA-polymerase on sequences of TEs increase in the absence of piRNA [17]. Genome-wide analysis has demonstrated that disturbance in piRNA-dependent TGS leads to increase in level of RNA-polymerase II on TE promoters and, as consequence, activation of transcription in Drosophila ovarian somatic cells [18].
piRNA clusters represent a key component of the piRNA pathway, because their transcripts give rise to small RNAs that participate in two critical processes – transcriptional and post-transcriptional repression of TEs. Moreover, piRNA clusters provide adaptive piRNA-response during invasion of new TEs, because integration of TEs into these loci provokes synthesis of new piRNAs. This review discusses data accumulated on piRNA clusters biology, primarily in Drosophila.
ORIGIN OF piRNAs IN Drosophila GERMLINE
The role of piRNAs is best studied in Drosophila ovaries. The Drosophila ovary is composed of ovarioles, each of which represents a number of egg chambers at consecutive stages of development (Fig. 1). The egg chamber contains germ cells (oocyte along with 15 nurse cells connected by channels) and somatic follicular cells surrounding the oocyte. Follicular and nurse cells necrotize to the moment of egg laying. Each ovariole contains a germarium at its apical end. Each germarium contains somatic and germ stem cells which give rise to egg chambers. The main branch of piRNA pathway functions in ovarian germ cells, because all three Piwi subfamily proteins – nuclear protein Piwi and cytoplasmic factors – Aubergine (Aub) and Ago3 – are expressed there. Only one protein from the Piwi subfamily, Piwi itself, is expressed in follicular cells. Studies of piRNA populations in ovaries of Aub and Ago3 mutants allowed dividing of TEs into three classes depending on in which tissue they are regulated – in germ or follicular cells or with mixed regulation. This classification is based on sites of TE expression and on inhibition of their activity by piRNA. Below structural and functional peculiarities of piRNA clusters in follicular and germ cells in ovaries and testes of Drosophila melanogaster are considered.
Fig. 1. piRNA clusters in Drosophila melanogaster ovary. Top: schematic representation of Drosophila ovary. On the left – depiction of the entire ovary; on the right – structure of ovariole. Bottom: organization of piRNA clusters in somatic and germ cells [6]. Density of unique piRNA mappers is shown for a particular chromosome in each box. piRNA clusters are located predominantly in pericentromeric regions of chromosomes. Below each chromosome are blowups of major piRNA clusters (germ cells: locus 42AB on chromosome 2; follicular cells: locus 20A on X-chromosome, also known as flamenco). Orientations of TE fragment are indicated by arrows and density of piRNAs on genomic + and – strands is plotted. piRNAs originate from both strands of a germ line cluster, but only from one strand in the case of a somatic cluster.
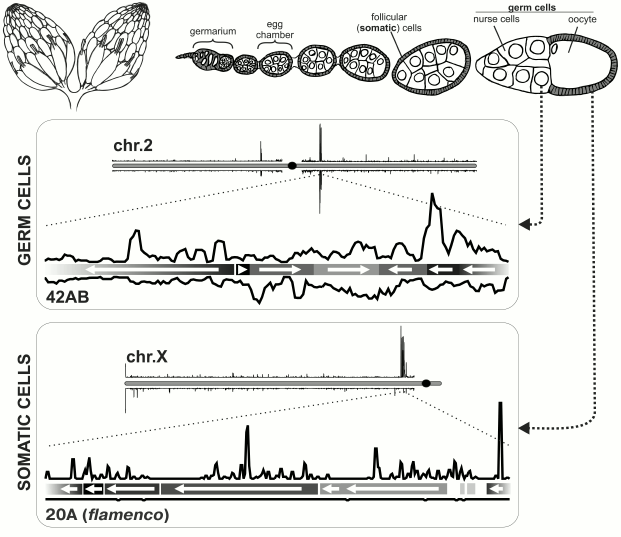
piRNA clusters in follicular cells of Drosophila. Follicular cells of Drosophila represent the simplest model of the piRNA pathway. Most of the factors required for antisense TE-specific piRNA amplification in germ cells (see below) are not expressed here [11, 19]. Also, in these cells there is no perinuclear nuage that contains numerous components of the piRNA pathway in germ cells. Only primary piRNA processing that consists of slicing of transcripts encoded by piRNA clusters, their binding to Piwi protein and subsequent shortening from 3′-end (3′-5′ trimming and 2′-O-methylation) takes place in follicular cells [11, 20]. Comparison of piRNA populations from the ovaries and laid eggs that have no follicular cells showed that somatic piRNAs mainly originate from pericentromeric locus on X-chromosome that was previously called flamenco. This locus is required for control of activity of retrovirus-like retrotransposons gypsy, ZAM, and Idefix, expressed in follicular cells and able to infect germ cells [11, 21-23]. This observation was supported by sequencing of piRNAs from ovaries with disturbed germ line piRNA pathway [10], and also from cultured Drosophila ovarian somatic cells (OSC) [19, 24]. The vast majority of unique piRNAs from flamenco locus map only to one genomic strand (Fig. 1). Importantly, TE fragments are located in reverse orientation to the piRNA-producing strand in this locus [6]. Thus, enrichment in piRNAs that are complementary to active transposon mRNAs and therefore effective in TE suppression is achieved [25].
Previously, it was shown that integration of transgenes into distal region of flamenco impairs functioning of the locus and activates the gypsy retrotransposon, indicating the presence of a discrete promoter that initiated transcription of a precursor [26]. Indeed, amount of piRNAs from flamenco in these mutants is strongly decreased [6]. Recently, the presence of the discrete promoter of RNA polymerase II in the distal region of flamenco was demonstrated on OSC by chromatin immunoprecipitation followed by deep sequencing (ChIP-seq) [18]. Dependence of expression of germ line piRNA clusters on RNA polymerase II activity has also been shown in silkworm [27, 28]. RNA polymerase II seems to be responsible for piRNA cluster transcription in different organisms.
Drosophila ovarian somatic cells also produce piRNAs from mRNAs of genes [24, 29]. The main source of genic piRNAs is a gene that encodes transcription factor Traffic Jam (tj). The prominent feature of genic piRNAs is that they are produced, predominantly, from 3′ untranslated gene region (UTR). The reason for production of piRNAs from 3′UTR of the tj gene has not been elucidated, but it was found that any random sequence being integrated in tj 3′UTR, starts to generate piRNA [30]. Genic piRNAs were also described for germ cells of Drosophila, silkworms, and mice [6, 31, 32]. The predominant production of piRNAs from 3′UTR is also observed in mouse genes [29]. Presently, it is not clear if genic piRNAs play a biological role in Drosophila or they are a result of erroneous processing. The latter is evidenced by low abundance of genic piRNAs in comparison with transposon piRNAs along with the presence of some basal amount of piRNAs from practically all mRNAs. Seemingly, one exception is the hsp70 gene that produces numerous piRNAs in Drosophila germ cells [33]; however, the role of these piRNAs remains unclear.
Follicular cells and ovarian somatic cell culture may be used in studies of many processes linked with piRNA biology. Certain progress in understanding of primary processing has been achieved [24, 34-36]. Primary processing is beleived to take place in cytoplasmic granules termed Yb-bodies and containing factors Yb and Armitage (Armi) [34]. It was suggested that protein Zuc is responsible for slicing of long piRNA precursors; however, the role of its endonuclease activity in this process has not yet been proven [37-40].
As mentioned above, Piwi is the only nuclear representative of Piwi proteins in Drosophila. Therefore, it has been suggested that, unlike Aub and Ago3 localized in nuage and utilizing their catalytic centers to slice mRNAs that are complementary to piRNAs, Piwi is involved in suppression of TE expression at the level of TE chromatin alterations. Indeed, its nuclear localization, but not catalytic “slicing” activity, is important for TE suppressing ability [18, 34, 41]. Ovarian somatic cells were used to demonstrate that Piwi participates in transcriptional TE silencing [18] (Fig. 2). It was shown that Piwi loaded with piRNAs induces accumulation of histone H3 tri-methylated on lysine 9 (H3K9me3) on transcribed TE sequences, which decreases RNA-polymerase II occupancy and inhibits transcription. A factor Maelstrom (Mael) that is necessary for silencing, but does not influence piRNA biogenesis, is also involved in this process [18]. Thus, only the nuclear branch of piRNA pathway silences active transposons in follicular cells.
Fig. 2. piRNAs biogenesis in Drosophila ovaries. a) Maternally inherited piRNAs or piRNAs produced as a result of the primary processing of cluster transcripts are utilized in the secondary processing − amplification of piRNAs complementary to TEs (“ping-pong”). b) Piwi–piRNA complex in follicular cells is able recognize a homologous locus (probably, co-transcriptionally) that leads to accumultaion of H3K9me3 chromatin mark that, in turn, induces transcriptional silencing with involvement of Maelstrom [18]. This mechanism also operates in germ cell nuclei. c) piRNAs produced by one genomic locus allow recognizing another homologous locus, which leads to its transformation into de novo piRNAs cluster [52]. Processes for which the molecular mechanism is unknown are marked by question marks.
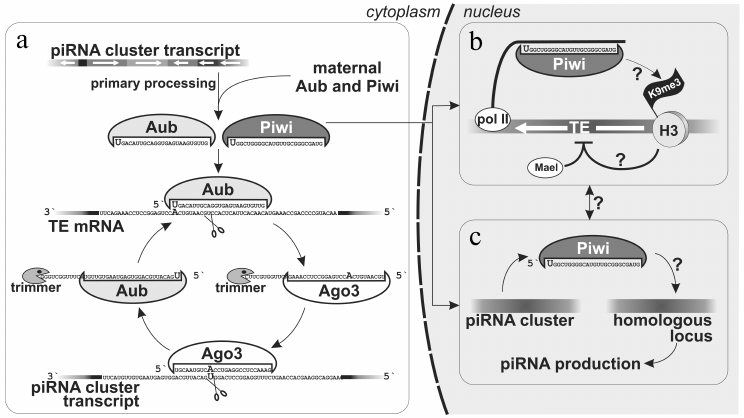
piRNA clusters in Drosophila ovarian germ cells. The main branch of the piRNA pathway functions in germ cells, and study of its principles is critical for understanding piRNA biology in general. Analysis of piRNAs from Drosophila germ cells showed that the majority of piRNAs is complementary to TEs [3, 10, 11]. piRNA clusters are predominantly localized in pericentromeric and subtelomeric regions and generate the bulk of piRNAs. However, in contrast to flamenco, piRNAs originate from both strands of piRNA clusters, and TE orientation in them is random (Fig. 1). Such clusters are termed “double-stranded” piRNA clusters. A question arises: how is the bias towards anti-sense transposon-specific piRNAs achieved in the total piRNA population? To date the answer is not completely clear, but a “ping-pong” model based on the analysis of piRNAs bound to each of the three proteins of the Piwi subfamily explains the main stages of piRNA biogenesis in germ cells [4, 6] (Fig. 2). Piwi and Aub bind to piRNAs that predominantly have 1U and are complementary to TE, whereas piRNAs bound to Ago3 have adenine at position 10 and mainly originate from sense TE transcripts. According to the “ping-pong” model: 1) primary piRNAs originating directly from cluster transcripts (equally from plus and minus strands) bind to Aub and Piwi proteins; 2) Aub loaded with anti-sense piRNAs slices TE mRNAs and this is the major goal of the entire piRNA pathway; 3) Ago3 protein binds to piRNAs that are processed from TE mRNAs as a result of this slicing; 4) completing this cycle, Ago3 loaded with sense piRNA induces excision of complementary piRNAs that are anti-sense to TEs and are present among transcripts of piRNA clusters giving rise to new anti-sense effector piRNAs in complexes with Aub. Multiple repeats of this cycles lead to amplification of anti-sense piRNA processed from transcripts of piRNA clusters. Possibly, an active expression of clusters and, consequently, anti-sense TE transcripts in combination with high level of Aub expression allows accumulation of a large amount of anti-sense piRNAs. However, the reason why different proteins have strand preferences remains unexplained. Ping-pong probably occurs in nuage (role of some nuage proteins is studied in [12, 13, 36, 42-45]). Notably, the oocyte remains in transcriptionally inert state (at meiotic pachytene stage) over all the developmental stage of the egg chamber. Germ cells, in which the above mentioned events take place, are called trophocytes (nurse cells) – the terminally differentiated cells with polytenized genome that play a major role in oocyte development. Lack of Aub, Ago3 and many other factors of the piRNA pathway in follicular cells leads to absence of ping-pong (and nuage) and necessity to accumulate TEs in orientation opposite to cluster transcription. Thus, double-stranded germ line clusters are more flexible but require numerous factors which role remains largely enigmatic.
The phenomenon of hybrid dysgenesis demonstrates efficiency of germ line double-strand clusters in acquisition of “immunity” against new TEs [46]. In hybrid dysgenesis, uncontrolled amplification of a certain class of TEs, which is brought by paternal chromosomes and absent in the maternal genome, causes sterility of the next generation [47, 48]. This phenomenon is not observed in reciprocal cross, when new TEs are inherited from mother, due to inheritance of maternal piRNAs through the oocyte. Thus, maternally deposited piRNA-complexes are critical for fertility of progeny. Importantly, hybrid dysgenesis is not absolute in many cases; with aging, dysgenic females begin to lay increasing number of viable eggs. This is a result of piRNA cluster activation. Data on hybrid dysgenesis linked with transposon P-element evidence accumulation with time of a sufficient number of piRNAs that are complementary to P-element and are originated from transcripts of paternal immune clusters [49]. Interestingly, in case of P-element dysgenesis in ovaries of disgenetic females, for unknown reasons, activation of some other transposons, which were present and repressed in the maternal genome, takes place in addition to that of the P-element. In the case of dysgenesis induced by retrotransposon I, accumulation of piRNAs from degraded ancestor I-element copies that are present in maternal piRNA clusters is observed with aging, so the progeny of older females is less sensitive to introduction of active I-element than progeny of young females [50]. Interestingly, this process is additive: sensitivity to introduction of I-element decreases with number of generations. Thus, piRNA clusters keep information regarding TE infections during the genome evolution and transmit it through the maternal line in the form of piRNAs.
What are the mechanisms of piRNA-dependent repression in germ cells? The role of piRNAs is not restricted to direct cleavage of TE transcripts due to endonuclease activity of Piwi subfamily proteins. It has been shown that impairment of germ line piRNA pathway leads to transcriptional activation of retrotransposons HeT-A, TART, and I-element that are typical representatives of TEs expressed in germ cells [17]. Moreover, it has been shown in our laboratory that a small population of piRNAs homologous to I-element is able to induce formation of de novo active piRNA clusters essentially from any sequences that have homology to these piRNAs and are expressed in germ cells [33]. This phenomenon was revealed as the lack of hybrid dysgenesis in descendants of females with initial high sensitivity to I-element, but carrying transgene with transcribed fragment of I-element [51]. It may be suggested that there is a co-transcriptional process in the nucleus, and this process depends on Piwi–piRNA complexes (since Piwi is the only nuclear protein in Piwi subfamily) and stimulates additional production of piRNAs to increase further TE suppression. Thus, suppression of TEs in germ cells with involvement of piRNAs is performed at post-transcriptional and transcriptional levels and, possibly, by formation of new piRNA clusters. However, this suggestion requires additional confirmation.
The role of piRNAs in establishing new piRNA clusters has also been demonstrated in the elegant work [52] that showed piRNA clusters to be paramutagenic. In this case, paramutagenesis means transfer of ability to produce piRNAs from one allele to another homologous allele. Copies of the transgene arranged in tandem and containing lacZ gene and, due to unknown reason, producing piRNAs in ovaries (and, as consequence, not expressing lacZ) were able to activate production of piRNAs by other transgenic loci that initially expressed lacZ and did not generate piRNAs (Fig. 2c). This effect was retained in many generations and, moreover, the para-mutagenized allele acquired properties of a para-mutagene.
The phenomenon of telomeric trans-silencing is a prominent example of trans-effects associated with activity of germline piRNA clusters [53]. Telomerase activity necessary for prevention of terminal incomplete replication of chromosomes in most eukaryotes is absent in the Drosophila genome. Drosophila telomeres have a very different structure from mammalian ones: they contain tandem LINE-like retrotransposons HeT-A, TART, and TAHRE, which are attached by their 3′ end to ends of chromosomes [54, 55]. So-called telomere-associated sequences (TAS) – tandem repeats of satellite-like nature – immediately follow terminal retroelements. The telomeric trans-silencing occurs if transgene integrated in TAS and containing lacZ gene is able to inhibit expression of lacZ from another transgene located in euchromatin. It is known that TAS is an active piRNA cluster [6]. A suggestion that trans-silencing is achieved by production of lacZ-specific piRNAs from transgene in TAS was confirmed by Northern-blot analysis and deep sequencing of small RNAs [30, 56]. Here it is necessary to note that telomeric retrotransposons in Drosophila produce multiple piRNAs in germ cells; however, it is still unclear whether all copies of telomeric retroelements participate in piRNA production. Telomeres consist of active and ordered copies of retroelements unlike double-stranded germ line piRNA clusters composed of randomly arranged inactive TE fragments. A question arises: why telomeres, which are arranged differently from regular germ line clusters, are able to produce piRNAs (see next section)? It may be suggested that telomeres were para-mutagenized a long time ago with involvement of piRNAs that were homologous to telomeric retrotransposons and remain to be in this state due to stability of paramutagenized alleles.
piRNA clusters in Drosophila testis. All described effects associated with expression of piRNAs in Drosophila were observed in the female germ line. How is the piRNA system organized in testis of Drosophila? It has been shown that Aub and Ago3 are expressed in male germ cells [8, 57]. Piwi is present, predominantly, in apical somatic germ cells of testis [5], but there are also indications that it is expressed in germ stem cells [58]. Processing of piRNAs in germ cells of Drosophila testis takes place in nuage and nuage-associated structures – “pING bodies” [57, 59]. The structure of piRNA clusters in somatic cells of Drosophila testis was not studied. The main source of piRNAs in testis is the Su(Ste) locus localized on the Y-chromosome and necessary for suppression of homologous Stellate (Ste) gene locus [8, 60]. Su(Ste)-specific piRNAs were the first discovered piRNAs [61]. piRNAs originate almost exclusively from one strand of Su(Ste) that is complementary to Ste transcripts; their generation along the sequence Su(Ste) is very irregular: ~65% of all Su(Ste)-derived piRNAs bound to Aub protein are identical to each other and have practically no ping-pong pairs [57]. Mutations in genes of the piRNA pathway or deletions in the Su(Ste) locus lead to sterilization of males due to accumulation of crystals of a protein encoded by genes of Ste locus in spermatocytes [8, 61, 62]. Interestingly, the Su(Ste) locus is found in D. melanogaster only, so Su(Ste) may be considered as species-specific piRNA cluster. The second major source of piRNAs in testis is the At-chX locus that, probably, regulates expression of vasa gene, the classic marker of germ cells, which is necessary for piRNA biogenesis in these cells [57]. piRNAs homologous to satellite-like repeats of the Responder locus are necessary for chromatin compaction during the process of spermatogenesis [63]. The role of piRNAs in suppression of TEs in testis is still unclear, because mutations of Aub and Ago3 have practically no influence on their expression [57]. It has been shown that some TE families, namely mdg1 and copia retrotransposons, are activated in piwi mutants in testis [64]. Seemingly, piRNAs do not play key role in suppression of TE activity in Drosophila testis, in contrast to mouse, where the important function of the piRNA pathway is the suppression of TEs at a pre-meiotic stage of spermatogenesis [16].
FACTORS NECESSARY FOR piRNA CLUSTER ACTIVITY. STRUCTURE OF
CHROMATIN IN piRNA CLUSTERS
What properties make piRNA-generating loci different from all others? One possible explanation is the existence of special factors or chromatin modifications that mark clusters and are necessary for targeting their transcripts for degradation by piRNA machinery. As mentioned above, piRNA clusters in Drosophila are localized in pericentromeric and subtelomeric regions of chromosomes. Arrangement of these regions in special chromatin compartments suggests that such localization is a determining factor for piRNAs production. However, being transferred to euchromatin within a transgene, a part of a pericentromeric piRNA cluster efficiently generates piRNAs [30]. Mouse pachytene piRNA clusters are also located outside of heterochromatin regions of chromosomes (see below), but they generate very abundant piRNAs. Finally, it is known that transcription of long piRNA precursors is possible due to ignoring of termination and splicing signals [30]. These data show that piRNA clusters possess distinct properties, which also may be applied to any sequences they incorporate, and these properties are not dependent on the genomic environment.
Recently, data were obtained (see below) that provide evidence in favor of a linkage between chromatin status of clusters and their piRNA-producing ability. Mutations of factors that are necessary for functioning of the clusters may lead to dramatic disruptions of nuage and the piRNA pathway, while, vice versa, mutations of cytoplasmic components of the piRNA pathway do not influence correct localization of cluster-specific chromatin factors [65, 66]. Thus, specific structure of chromatin of piRNA clusters is critical for functioning of the entire piRNA pathway.
The mechanisms underlying link between structure of chromatin and processing of small RNAs were studied in detail in the yeast Schizosaccharomyces pombe [67]. Despite the fact that yeast is a unicellular eukaryote lacking piRNA system, there are some similarities between the processes of chromatin formation in pericentromeric loci producing small RNAs in yeast S. pombe and those in Drosophila piRNA clusters. Tandem repeats dg/dh localized in pericentromeres of yeast produce double-stranded RNAs that undergo processing with involvement of Dicer, followed by loading of single-stranded 21-nt RNAs into Ago1. In the nucleus, Ago1, as a part of RITS (RNAi-induced initiation of transcriptional silencing) complex, recognizes transcripts of a pericentromeric locus due to complementary interaction with small RNAs and induces its silencing by recruiting of H3K9-histone methyltransferase Clr4 [68, 69]. Histone tag H3K9me is recognized by Swi6 protein, the ortholog of a classical heterochromatin factor HP1 (Heterochromatin Protein 1). Because of the presence of Chp1 protein possessing chromo-domain that recognizes H3K9me, RITS is able to bind pericentromeric locus modified by its own activity. Silencing is increased due to association of Clr4 with RNA-dependent RNA polymerase (RdRP), which allows generating additional double-stranded RNAs from this locus [70, 71]. Paradoxically, Swi6 induces not only silencing by recruiting of histone deacetylase Clr3, but also activation of transcription due to interaction with factor Epe1 [72]. Therefore, a balance between two antagonistic yet interdependent processes – transcription and silencing – is achieved.
A number of works that will be reviewed below demonstrate similarity in functioning of pericentromeric loci in yeast and piRNA clusters in Drosophila. It is considered that the process in yeast is initiated by products of degradation (Dicer-independent) of both strands of the pericentromeric locus that are loaded in Ago1 and initiate the described silencing/transcription cycle [73]. In the case of the piRNA pathway, processing of small RNAs also occurs from single-stranded precursors independently on Dicer; however, details of this process remain to be determined. Like in yeast, nuclear Piwi protein loaded with piRNAs in Drosophila is able to induce processes of heterochromatization of sequences homologous to the piRNAs [18]. Finally, similarly to pericentromeres in yeast, piRNA clusters must also be recognized by Ago family protein (in this case – Piwi) and undergo silencing. However, because piRNA clusters must be actively transcribed to produce piRNA precursors, then, seemingly, there is a mechanism that prevents their inactivation. To date no activity of RdRP that amplifies substrate for production of small RNAs in yeast has been demonstrated for Drosophila, but a number of factors are revealed that are necessary for expression of clusters and, probably, for their protection from the above-mentioned silencing. Thus, it seems that “heterochromatization” of piRNA clusters causes an opposite effect – activation of their transcription. Below we consider factors mutations of which impair functioning of piRNA clusters and, consequently, oogenesis as a whole.
Some H3K9-histone methyltransferases, including dSETDB1, SU(VAR)3-9, and dG9a, are expressed in Drosophila ovary. Described SU(VAR)3-9 and dG9a mutations do not cause sterility, whereas dSETDB1 is necessary for oogenesis [74-76]. Interestingly, in the case of SU(VAR)3-9 mutation, trimethylation of lysine 9 in histone H3 (H3K9me3) is disrupted in the whole ovary, excluding the germarium, but this does not influence oogenesis in general [75]. Like SU(VAR)3-9, dSETDB1 leads to H3K9 trimethylation that is important for early oogenesis. dSETDB1 mutations lead to disappearance of H3K9me3 in germ and somatic cells of the germarium, reduction of piRNA amount, accumulation of double-stranded breaks, and activation of TEs [77]. Despite the fact that H3K9me3 is a typical marker of pericentromeric heterochromatin in many tissues, the specific pattern of dSETDB1 expression and characteristic effects of the mutation suggest its special role in the ovary that is associated, namely, with functioning of piRNA clusters, but not with pericentromeric regions of chromosomes on the whole. Immunoprecipitation and deep sequencing of DNA (ChIP-seq) bound with H3K9me3 revealed accumulation of this chromatin modification on piRNA clusters in ovaries. Comparison of piRNAs from dSETDB1 mutants with control showed reduction of levels of piRNAs originated from germline as well as somatic clusters. There is also a moderate decrease of transcription level of somatic and germ clusters in the dSETDB1 mutant. Thus, heterochromatin mark H3K9me3 is able to stimulate transcription of piRNA clusters, which is reminiscent of events taking place in pericentromeric heterochromatin in S. pombe. It may not be excluded that marking of clusters with H3K9me3 has temporary character and it is substituted by another chromatin modification at later stages of oogenesis, because presence of H3K9me3 is apparently not critical for activity of piRNA clusters here. dSETDB1 is also active in testis, and it is able to mono- and tri-methylate H3K9 [78], although the link between this process and the piRNA pathway was not studied.
It is not clear what protein binds with H3K9me3 deposited in the germarium by dSETDB1, but it was shown that localization of HP1 (HP1A) here coincides with H3K9me3, and it is disrupted in the case of dSETDB1 mutation [75]. The role of HP1 in determining activity of clusters is not clear, but it is known that binding of HP1 with piRNA clusters in germ cells and in cultured OSC does not depend on Piwi [79]. It is also known that HP1 binds telomeres, which are active piRNA clusters, although the main role for HP1 in association with a telomere is formation of protective complex [80]. Rhino (HP1D), another representative of the HP1 subfamily, is necessary for functioning of double-stranded clusters [65], but modification of chromatin, to which this protein binds, remains unknown. Rhino is expressed in female germ cells; its mutations lead to sterility and demonstrate a phenotype of the piRNA pathway mutations: disruption of oogenesis (defects of dorsoventral polarity) increased frequency of double-stranded breaks caused by activation of transposons [65, 81]. Nuage is also disrupted in rhino mutants, the amount of piRNAs is decreased by 80%, and the ping-pong is impaired. Rhino is necessary for expression of double-stranded germ cell clusters; its mutation does not affect production of piRNAs by the follicular cluster flamenco. Thus, despite the fact that Rhino represents the HP1 family, it seems that this factor is necessary for transcription activation, which reminds of the dual (silencing and activating) function of Swi6 in S. pombe pericentromeres. It has been suggested that the role of Rhino (and Cutoff, see below) consists in protection of piRNA clusters from heterochromatinization typical for pericentromeres, and, hence, in providing their effective transcription [82]. However, it is known that Rhino is not specific to pericentromeres in general, but only to piRNA clusters. Thus, it may be suggested that Rhino is a particular paralog of HP1 with transcription-activating function, probably protecting piRNA clusters from silencing induced by Piwi.
Protein Cutoff, an ortholog of the yeast transcription termination factor Rai1, plays an important role in functioning of germ line piRNA clusters [82, 83]. Physical interaction of Cutoff with double-stranded clusters was proven by ChIP-qPCR. Interestingly, the specific nuclear localization characteristic for Cutoff is impaired in rhino mutants, and cutoff mutants demonstrate delocalization of Rhino, which indicates interdependent co-localization of these proteins at germ line double-stranded clusters in Drosophila. Cutoff mutants demonstrate decreased transcription of double-stranded piRNA clusters. Different levels of decrease at different sites within the same cluster (42AB) suggested that germinal clusters produce multiple transcripts instead of a discrete transcript; however, this hypothesis requires confirmation. Reduction of piRNAs amount by 75%, disappearance of ping-pong and TE activation are observed in cutoff mutants. The certain role of Cutoff in piRNA clusters remains to be elucidated.
It has been recently shown that UAP56, an RNA splicing and nuclear export factor, also co-localizes with Rhino in Drosophila germ cells [66]. Mutation of rhino induced delocalization of UAP56 from nuclear granules, while UAP56 is not essential for correct localization of Rhino at early stages of oogenesis. It is known that the pericentromeric chromatin, in which piRNA clusters are localized, is located mainly at the nuclear periphery. It was shown that there is a strong correlation between localization of chromatin associated with Rhino and UAP56 and Vasa, the central component of nuage [66]. The authors suggest that UAP56 is a factor necessary for targeted delivery of transcripts of piRNA clusters to nuage granules, in which subsequent events of piRNA processing take place. Activation of transposons caused by strong decrease of piRNA derived from double-stranded clusters is observed in UAP56 mutants, although the level of expression of these clusters is not reduced. This indicates to participation of UAP56 in the export of transcripts of piRNA clusters, but not in their transcription. Although UAP56 is expressed in all tissues, the studied UAP56 mutations do not influence the somatic branch of the piRNA pathway, which suggests distinct function for this protein in germ cells.
The structure of chromatin in piRNA clusters in Drosophila testis has not been analyzed yet, but lack of Piwi, Rhino, and Cutoff in testicular germ cells suggests that the structure of chromatin in piRNA clusters here differs from clusters in ovaries.
As mentioned in the beginning of this chapter, piRNA clusters must possess special properties to give rise to long transcripts and target them for piRNA processing. Presently, there is no certain picture of events in regulation of transcription of clusters. Seemingly, factors that are necessary for functioning of piRNA clusters allow interpreting trimethylation of H3K9 as the activation signal that initiates their transcription and processing with production of piRNAs.
COMPARISON OF piRNA CLUSTERS FROM DIFFERENT ORGANISMS
The existing data regarding the piRNA pathway in different organisms suggest that piRNA clusters differ in their structure. The piRNA pathway in the silkworm Bombyx mori is the best studied in insects, with the exception of Drosophila [20, 27, 28, 32, 84-86]. Silkworm germ cells have nuage, and orthologs of many proteins of the Drosophila piRNA pathway are encoded in the genome. There are piRNAs complementary to TEs, ping-pong is observed and other properties specific for Drosophila piRNAs are also characteristic for silkworm piRNAs [28, 32]. It is important to note that only two proteins of the Piwi subfamily are encoded in the silkworm genome, Siwi and BmAgo3, which are orthologs of Drosophila cytoplasmic proteins Aub and Ago3, respectively. Seemingly, there is no nuclear branch of the piRNA pathway in silkworm, which is likely the reason why piRNA clusters here differ from Drosophila: they have small size, present in euchromatin, often overlap with genes and are enriched by chromatin modifications typical for actively transcribed regions (H3K4me2, H3K4me3, H3K9ac) [28]. There is no accumulation of H3K9me3 observed for piRNA clusters in silkworm. Moreover, there are no orthologs of Rhino and Cutoff encoded in the silkworm genome. These data highlight the role of nuclear Piwi subfamily proteins in the local modification of piRNA-producing regions. An interesting feature of silkworm piRNA clusters was revealed with the use of cultivated germ cells BmN4: insertion of a transgene often occurs in the same piRNA cluster Torimochi, which leads to effective piRNA production by transgene sequences and silencing of the reporter gene [27]. Such homing of TEs to piRNA clusters has not been described for other organisms.
A similar pattern of piRNA distribution is observed in the mosquito Aedes aegypti: piRNAs originate here from euchromatin regions, including many genes, and a fraction of piRNAs specific to TEs constitutes only 19% [87]. This is surprising when accounting for the fact that TEs constitute 47% of A. aegypti genome against 16% in Drosophila. In total, piRNA clusters cover here one fifth of the genome, and the density of TEs in clusters does not exceed the average density for the rest of genome. The most active clusters, like flamenco in D. melanogaster, are single-stranded ones, and they contain TEs and genes in orientation opposite to transcription, although double-stranded clusters are also present here. Most piRNAs specific to TEs are anti-sense, and this is also similar to follicular piRNAs in Drosophila. These data were obtained for small RNAs libraries made from total RNA of mosquitoes [87]. Most probably, the majority of piRNAs in these libraries does not originate from gonads, because in A. aegypti, in contrast to Drosophila, small RNAs with properties typical for piRNA are detected in somatic cells [88]. Seven Piwi subfamily proteins are encoded in the A. aegypti genome, and some of them are likely to be expressed outside gonads [88, 89]. Moreover, ping-pong amplification of piRNAs in somatic tissues was first described in this species. It is also interesting that viral infections induce occurrence of virus-specific piRNAs in somatic cells in A. aegypti and A. albopictus, which indicates a possible role of piRNAs in the anti-virus response.
Mammalian piRNA pathway is best studied in mouse testis. Three proteins of the Piwi subfamily – Mili, Miwi, and Miwi2 – are expressed at different stages of mouse spermatogenesis and bind piRNAs of different origin (Fig. 3).
Fig. 3. piRNAs biogenesis in mouse spermatogenesis. a) Mili, Miwi2, and Miwi proteins are expressed at different stages of mouse spermatogenesis. Mili and Miwi2 participate in TE DNA methylation. Mili is also expressed at later stages including meiosis. During the pachytene stage, Mili and Miwi bind to piRNAs derived from discrete loci that are not enriched in TEs. b) Density of pachytene Miwi-bound piRNAs along mouse chromosome 17 is plotted on the top. Several piRNA clusters generate piRNAs from both strands, but others are single-stranded. Bottom: the biggest pachytene piRNAs cluster that possesses characteristic “divergent” structure, i.e. represents two adjacent clusters, which produce piRNAs from opposite strands [30].
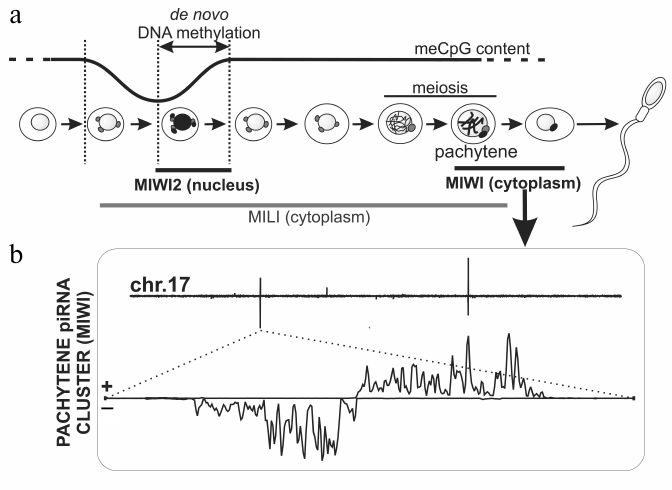
In mouse embryogenesis, removal of DNA methylation and its subsequent restoration (13.5-18.5 days after fertilization) in germ cells take place immediately after their segregation from soma [90] (see review by A. Bortvin in this issue). Methylation of DNA mainly occurs at TE sequences. Miwi2 is the only nuclear protein of the Piwi subfamily in mice, its expression is restricted to the beginning of DNA methylation in precursor cells of spermatogonia (prospermatogonia), and Miwi2 mutations lead to disruption of this process, activation of TEs, and infertility [16, 91]. Accordingly, ~80% of piRNAs bound with Miwi2 are derived from TE sequences. The majority of these piRNAs are anti-sense to TEs. Seemingly, primary processing happens here predominantly on sense mRNAs of individual TEs, which are therefore piRNA clusters themselves that produce piRNAs, which bind with Mili, another Piwi protein. Double-stranded and single-stranded clusters expressed in prospermatogonia are reminiscent of germinal and somatic Drosophila piRNA clusters; however, they give rise to smaller amount of piRNA than individual TE copies. In total, they occupy ~0.2% of the mouse genome. A similar picture is observed for Danio rerio, where primarily active TE copies, but not piRNA clusters, contribute to production of piRNAs bound with Ziwi, the Piwi subfamily protein [9]. Among Drosophila species, production of primary piRNAs by individual copies of transposon (Ulysses) was described for Drosophila virilis only [92]. Biochemical details of TE methylation in mouse and the role of Miwi2 in the process remain to be studied; however, it is suggested that methylation of histone (H3K9) is a primary modification, which initiates DNA methylation [93].
Mili protein is expressed throughout mouse spermatogenesis, and it is necessary for TE methylation in spermatogonia as well as at subsequent stages until formation of the round spermatids [94-97]. In addition to piRNAs homologous to TEs and TE-containing piRNA clusters, a fraction of piRNAs of genic origin (29%) binds with Mili. Interestingly, they originate mostly from 3′UTR like in Drosophila follicular cells. Finally, a significant fraction of pre-meiotic piRNAs bound with Mili (28%) maps to non-annotated genome regions. The function of these piRNAs is still unknown.
Like Miwi2, expression of a third mouse Piwi protein, Miwi, is restricted to a distinct stage of spermatogenesis – the pachytene stage of meiosis [98]. Miwi along with Mili bind to the most abundant mouse piRNAs, 95.5% of which originate from discrete loci containing no annotated sequences uniformly scattered on all autosomes and often having a particular “divergent” arrangement (Fig. 3) [30, 94, 95, 99]. It is suggested that divergent transcription of piRNA precursors from a common central promoter leads to appearance of two neighboring clusters, in which piRNAs map to opposite strands of genomic DNA [94]. The function of pachytene piRNA clusters is enigmatic; however, taking into account the time of their expression and presence on all chromosomes with exception of X and Y, it has been suggested that they are necessary for conjugation of homologous chromosomes in meiosis. Interestingly, piRNA clusters syntenic to those in mouse, but different in sequences, are detected in human and rat. This probably means that the fact of transcription of a particular chromosome region is important, but a sequence of piRNA precursor or piRNAs themselves is not [94, 99]. The role of chromatin in expression of mouse piRNA clusters has not yet been studied. Notably, the role of the piRNA pathway in mouse oogenesis ramains unexplored; it should be noted, however, that female Mili mutants are viable and fertile [97].
Numerous Ago family proteins, among which PRG-1 and PRG-2 are the closest orthologs of Drosophila Piwi proteins, are expressed in Caenorhabditis elegans germ cells [100, 101]. PRG-1 binds with so-called 21U-RNAs (RNA of 21 nucleotide length that have uridine in the first position), which are C. elegans piRNAs. In contrast to Drosophila piRNAs, 21U-RNAs are able to repress only one class of TEs (Tc3), and their main function appears to be to recognize self from nonself and silence new genomic insertions [100, 102-105]. The majority of 21U-RNAs are encoded by two genomic clusters that differ from the above-described piRNA clusters by individual transcription of each piRNA rather than processing from long precursors [106-109]. Another interesting property of 21U-RNAs is the absence of perfectly complementary targets in the genome for most of them, similar to some classes of mouse piRNAs. It is thought that 21U-RNAs, after processing and loading in PRG-1, are utilized for scanning the transcriptome in search for foreign sequences [102]. Host genes expressed in germ cells are protected from silencing by complementary small RNAs loaded in another Argonaute, CSR-1. Transcripts that are homologous to 21U-RNA, but are not protected by CSR-1, undergo processing by RdRP and Argonaute WAGO (worm-specific AGOs) subfamily proteins, which leads to formation of secondary small RNAs termed 22G-RNAs (RNA 22 nucleotides in length with preference for guanidine in the first position). WAGO induce silencing of foreign sequences at the chromatin level [105]. Details of the mechanism of this genomic surveillance remain to be elucidated.
Although key data on function of piRNAs and Piwi proteins were obtained in studies of multicellular organisms, it is necessary to note that Piwi subfamily proteins have been studied in ciliates, unicellular eukaryotic organisms, where they participate in global remodeling of the genome and gene silencing [110, 111]. This indicates to an evolutionarily conserved role of Piwi proteins and small RNAs bound to them in regulation of activity of genes.
UNSOLVED QUESTIONS OF BIOLOGY OF piRNA CLUSTERS
Despite significant progress in piRNA biology, there are still gaps in understanding of fundamental mechanisms underlying this pathway. It is obvious that Piwi subfamily proteins and special small RNAs, piRNAs, are the most conservative components of the piRNA system. Functions of piRNAs, their genomic origins, and processing vary in many aspects, even in relatively close organisms. Nevertheless, piRNA clusters, regions producing numerous piRNAs, exist in practically all studied multicellular organisms. The question how these regions are recognized and their transcripts are processed with production of piRNAs remains the main unsolved problem in piRNA biology.
It is possible that in the case of piRNA clusters in Drosophila germ cells, bidirectional transcription plays an important role. Indeed, this feature is not characteristic for other genomic regions, it is known to terminate transcription and may probably provide a signal for processing [112]. Some animal TEs have internal bidirectional promoter, for example, human LINE1 and Drosophila telomere retrotransposons [113-116]. Perhaps this property stimulates production of piRNAs by telomeres in Drosophila and individual mammalian TEs. However, this model does not explain how single-stranded clusters are recognized. In particular, why do some genes produce piRNAs?
It is still not clear whether inherited piRNAs can play role in activation of piRNA clusters in progeny. As described above, it seems that the activation of parental piRNA clusters suppresses P-element hybrid dysgenesis with age. The mechanism of this activation is not clear: are existing maternal piRNAs that are homologous to paternal clusters important for this process? Is chromatin altered on paternal clusters of aged flies, or do all changes take place only at the level of secondary amplification of distinct piRNAs? In other words, what roles do transcriptional and post-transcriptional processes play in initiation of piRNA processing? Also, the question why a number of TEs, including those not unique to paternal clusters, are activated in disgenesis and why the nuage and piRNA pathway in general are disordered is still enigmatic. Despite attempts to explain this fact by systemic disorders in gametogenesis caused by numerous TE transpositions and appearance of double-stranded breaks [83, 117], the nature of such deep interrelationship is still unclear. In conclusion, it has to be noted that artificial clusters that produce abundant piRNAs are also known [52]. To date, it is still unclear what triggered these transgenes to produce small RNA and how their chromatin is organized. Perhaps studies of such “model” piRNA clusters will provide answers to many of existing questions in the field.
The authors are grateful to A. A. Aravin for fruitful discussion and critical remarks.
This work was supported by grants from the Russian Foundation for Basic Research (grant 12-04-00797a) and the program “Wildlife: Current State and Development" of the Presidium of RAS.
REFERENCES
1.Siomi, M. C., Sato, K., Pezic, D., and Aravin, A.
A. (2011) Nat. Rev. Mol. Cell Biol., 12, 246-258.
2.Senti, K. A., and Brennecke, J. (2010) Trends
Genet., 26, 499-509.
3.Vagin, V. V., Sigova, A., Li, C., Seitz, H.,
Gvozdev, V., and Zamore, P. D. (2006) Science, 313,
320-324.
4.Gunawardane, L. S., Saito, K., Nishida, K. M.,
Miyoshi, K., Kawamura, Y., Nagami, T., Siomi, H., and Siomi, M. C.
(2007) Science, 315, 1587-1590.
5.Saito, K., Nishida, K. M., Mori, T., Kawamura, Y.,
Miyoshi, K., Nagami, T., Siomi, H., and Siomi, M. C. (2006) Genes
Dev., 20, 2214-2222.
6.Brennecke, J., Aravin, A. A., Stark, A., Dus, M.,
Kellis, M., Sachidanandam, R., and Hannon, G. J. (2007) Cell,
128, 1089-1103.
7.Horwich, M. D., Li, C., Matranga, C., Vagin, V.,
Farley, G., Wang, P., and Zamore, P. D. (2007) Curr. Biol.,
17, 1265-1272.
8.Aravin, A. A., Klenov, M. S., Vagin, V. V.,
Bantignies, F., Cavalli, G., and Gvozdev, V. A. (2004) Mol. Cell
Biol., 24, 6742-6750.
9.Houwing, S., Kamminga, L. M., Berezikov, E.,
Cronembold, D., Girard, A., van den Elst, H., Filippov, D. V., Blaser,
H., Raz, E., Moens, C. B., Plasterk, R. H., Hannon, G. J., Draper, B.
W., and Ketting, R. F. (2007) Cell, 129, 69-82.
10.Li, C., Vagin, V. V., Lee, S., Xu, J., Ma, S.,
Xi, H., Seitz, H., Horwich, M. D., Syrzycka, M., Honda, B. M., Kittler,
E. L., Zapp, M. L., Klattenhoff, C., Schulz, N., Theurkauf, W. E.,
Weng, Z., and Zamore, P. D. (2009) Cell, 137,
509-521.
11.Malone, C. D., Brennecke, J., Dus, M., Stark, A.,
McCombie, W. R., Sachidanandam, R., and Hannon, G. J. (2009)
Cell, 137, 522-535.
12.Lim, A. K., and Kai, T. (2007) Proc. Natl.
Acad. Sci. USA, 104, 6714-6719.
13.Findley, S. D., Tamanaha, M., Clegg, N. J., and
Ruohola-Baker, H. (2003) Development, 130, 859-871.
14.Hall, I. M., Shankaranarayana, G. D., Noma, K.,
Ayoub, N., Cohen, A., and Grewal, S. I. (2002) Science,
297, 2232-2237.
15.Onodera, Y., Haag, J. R., Ream, T., Costa Nunes,
P., Pontes, O., and Pikaard, C. S. (2005) Cell, 120,
613-622.
16.Aravin, A. A., Sachidanandam, R.,
Bourc’his, D., Schaefer, C., Pezic, D., Toth, K. F., Bestor, T.,
and Hannon, G. J. (2008) Mol. Cell, 31, 785-799.
17.Shpiz, S., Olovnikov, I., Sergeeva, A., Lavrov,
S., Abramov, Y., Savitsky, M., and Kalmykova, A. (2011) Nucleic
Acids Res., 39, 8703-8711.
18.Sienski, G., Donertas, D., and Brennecke, J.
(2012) Cell, 151, 964-980.
19.Lau, N. C., Robine, N., Martin, R., Chung, W. J.,
Niki, Y., Berezikov, E., and Lai, E. C. (2009) Genome Res.,
19, 1776-1785.
20.Kawaoka, S., Izumi, N., Katsuma, S., and Tomari,
Y. (2011) Mol. Cell, 43, 1015-1022.
21.Pelisson, A., Song, S. U., Prud’homme, N.,
Smith, P. A., Bucheton, A., and Corces, V. G. (1994) EMBO J.,
13, 4401-4411.
22.Desset, S., Meignin, C., Dastugue, B., and Vaury,
C. (2003) Genetics, 164, 501-509.
23.Prud’homme, N., Gans, M., Masson, M.,
Terzian, C., and Bucheton, A. (1995) Genetics, 139,
697-711.
24.Saito, K., Inagaki, S., Mituyama, T., Kawamura,
Y., Ono, Y., Sakota, E., Kotani, H., Asai, K., Siomi, H., and Siomi, M.
C. (2009) Nature, 461, 1296-1299.
25.Pelisson, A., Sarot, E., Payen-Groschene, G., and
Bucheton, A. (2007) J. Virol., 81, 1951-1960.
26.Robert, V., Prud’homme, N., Kim, A.,
Bucheton, A., and Pelisson, A. (2001) Genetics, 158,
701-713.
27.Kawaoka, S., Mitsutake, H., Kiuchi, T.,
Kobayashi, M., Yoshikawa, M., Suzuki, Y., Sugano, S., Shimada, T.,
Kobayashi, J., Tomari, Y., and Katsuma, S. (2012) RNA,
18, 265-273.
28.Kawaoka, S., Hara, K., Shoji, K., Kobayashi, M.,
Shimada, T., Sugano, S., Tomari, Y., Suzuki, Y., and Katsuma, S. (2013)
Nucleic Acids Res., 41, 1581-1590.
29.Robine, N., Lau, N. C., Balla, S., Jin, Z.,
Okamura, K., Kuramochi-Miyagawa, S., Blower, M. D., and Lai, E. C.
(2009) Curr. Biol., 19, 2066-2076.
30.Muerdter, F., Olovnikov, I., Molaro, A., Rozhkov,
N. V., Czech, B., Gordon, A., Hannon, G. J., and Aravin, A. A. (2012)
RNA, 18, 42-52.
31.Aravin, A. A., Hannon, G. J., and Brennecke, J.
(2007) Science, 318, 761-764.
32.Kawaoka, S., Hayashi, N., Suzuki, Y., Abe, H.,
Sugano, S., Tomari, Y., Shimada, T., and Katsuma, S. (2009) RNA,
15, 1258-1264.
33.Olovnikov, I., Ryazansky, S., Shpiz, S., Lavrov,
S., Abramov, Y., Vaury, C., Jensen, S., and Kalmykova, A. (2013)
Nucleic Acids Res., PMID: 23620285; doi: 10.1093/nar/gkt310.
34.Saito, K., Ishizu, H., Komai, M., Kotani, H.,
Kawamura, Y., Nishida, K. M., Siomi, H., and Siomi, M. C. (2010)
Genes Dev., 24, 2493-2498.
35.Olivieri, D., Sykora, M. M., Sachidanandam, R.,
Mechtler, K., and Brennecke, J. (2010) EMBO J., 29,
3301-3317.
36.Handler, D., Olivieri, D., Novatchkova, M.,
Gruber, F. S., Meixner, K., Mechtler, K., Stark, A., Sachidanandam, R.,
and Brennecke, J. (2011) EMBO J., 30, 3977-3993.
37.Ipsaro, J. J., Haase, A. D., Knott, S. R.,
Joshua-Tor, L., and Hannon, G. J. (2012) Nature, 491,
279-283.
38.Nishimasu, H., Ishizu, H., Saito, K., Fukuhara,
S., Kamatani, M. K., Bonnefond, L., Matsumoto, N., Nishizawa, T.,
Nakanaga, K., Aoki, J., Ishitani, R., Siomi, H., Siomi, M. C., and
Nureki, O. (2012) Nature, 491, 284-287.
39.Voigt, F., Reuter, M., Kasaruho, A., Schulz, E.
C., Pillai, R. S., and Barabas, O. (2012) RNA, 18,
2128-2134.
40.Pane, A., Wehr, K., and Schupbach, T. (2007)
Dev. Cell, 12, 851-862.
41.Klenov, M. S., Sokolova, O. A., Yakushev, E. Y.,
Stolyarenko, A. D., Mikhaleva, E. A., Lavrov, S. A., and Gvozdev, V. A.
(2011) Proc. Natl. Acad. Sci. USA, 108, 18760-18765.
42.Anand, A., and Kai, T. (2012) EMBO J.,
31, 870-882.
43.Zhang, Z., Xu, J., Koppetsch, B. S., Wang, J.,
Tipping, C., Ma, S., Weng, Z., Theurkauf, W. E., and Zamore, P. D.
(2011) Mol. Cell, 44, 572-584.
44.Patil, V. S., and Kai, T. (2010) Curr.
Biol., 20, 724-730.
45.Siomi, M. C., Mannen, T., and Siomi, H. (2010)
Genes Dev., 24, 636-646.
46.Brennecke, J., Malone, C. D., Aravin, A. A.,
Sachidanandam, R., Stark, A., and Hannon, G. J. (2008) Science,
322, 1387-1392.
47.Bucheton, A. (1990) Trends Genet.,
6, 16-21.
48.Kidwell, M. G., Kidwell, J. F., and Sved, J. A.
(1977) Genetics, 86, 813-833.
49.Khurana, J. S., Wang, J., Xu, J., Koppetsch, B.
S., Thomson, T. C., Nowosielska, A., Li, C., Zamore, P. D., Weng, Z.,
and Theurkauf, W. E. (2011) Cell, 147, 1551-1563.
50.Grentzinger, T., Armenise, C., Brun, C., Mugat,
B., Serrano, V., Pelisson, A., and Chambeyron, S. (2012) Genome
Res., 22, 1877-1888.
51.Jensen, S., Gassama, M. P., and Heidmann, T.
(1999) Nat. Genet., 21, 209-212.
52.De Vanssay, A., Bouge, A. L., Boivin, A.,
Hermant, C., Teysset, L., Delmarre, V., Antoniewski, C., and Ronsseray,
S. (2012) Nature, 490, 112-115.
53.Ronsseray, S., Josse, T., Boivin, A., and
Anxolabehere, D. (2003) Genetica, 117, 327-335.
54.Pardue, M. L., and DeBaryshe, P. G. (2003)
Annu. Rev. Genet., 37, 485-511.
55.Shpiz, S., and Kalmykova, A. (2012) in Reviews
on Selected Topics of Telomere Biology (Li, B., ed.) Intech,
Rijeka, pp. 31-56.
56.Todeschini, A. L., Teysset, L., Delmarre, V., and
Ronsseray, S. (2010) PLoS One, 5, e11032.
57.Nagao, A., Mituyama, T., Huang, H., Chen, D.,
Siomi, M. C., and Siomi, H. (2010) RNA, 16,
2503-2515.
58.Cox, D. N., Chao, A., and Lin, H. (2000)
Development, 127, 503-514.
59.Kibanov, M. V., Egorova, K. S., Ryazansky, S. S.,
Sokolova, O. A., Kotov, A. A., Olenkina, O. M., Stolyarenko, A. D.,
Gvozdev, V. A., and Olenina, L. V. (2011) Mol. Biol. Cell,
22, 3410-3419.
60.Nishida, K. M., Saito, K., Mori, T., Kawamura,
Y., Nagami-Okada, T., Inagaki, S., Siomi, H., and Siomi, M. C. (2007)
RNA, 13, 1911-1922.
61.Aravin, A. A., Naumova, N. M., Tulin, A. V.,
Vagin, V. V., Rozovsky, Y. M., and Gvozdev, V. A. (2001) Curr.
Biol., 11, 1017-1027.
62.Bozzetti, M. P., Massari, S., Finelli, P.,
Meggio, F., Pinna, L. A., Boldyreff, B., Issinger, O. G., Palumbo, G.,
Ciriaco, C., Bonaccorsi, S., et al. (1995) Proc. Natl. Acad. Sci.
USA, 92, 6067-6071.
63.Gell, S. L., and Reenan, R. A. (2012)
Genetics, 193, 771-784.
64.Kalmykova, A. I., Klenov, M. S., and Gvozdev, V.
A. (2005) Nucleic Acids Res., 33, 2052-2059.
65.Klattenhoff, C., Xi, H., Li, C., Lee, S., Xu, J.,
Khurana, J. S., Zhang, F., Schultz, N., Koppetsch, B. S., Nowosielska,
A., Seitz, H., Zamore, P. D., Weng, Z., and Theurkauf, W. E. (2009)
Cell, 138, 1137-1149.
66.Zhang, F., Wang, J., Xu, J., Zhang, Z.,
Koppetsch, B. S., Schultz, N., Vreven, T., Meignin, C., Davis, I.,
Zamore, P. D., Weng, Z., and Theurkauf, W. E. (2012) Cell,
151, 871-884.
67.Zofall, M., and Grewal, S. I. (2006) Cold
Spring Harb. Symp. Quant. Biol., 71, 487-496.
68.Volpe, T. A., Kidner, C., Hall, I. M., Teng, G.,
Grewal, S. I., and Martienssen, R. A. (2002) Science,
297, 1833-1837.
69.Verdel, A., Jia, S., Gerber, S., Sugiyama, T.,
Gygi, S., Grewal, S. I., and Moazed, D. (2004) Science,
303, 672-676.
70.Motamedi, M. R., Verdel, A., Colmenares, S. U.,
Gerber, S. A., Gygi, S. P., and Moazed, D. (2004) Cell,
119, 789-802.
71.Sugiyama, T., Cam, H., Verdel, A., Moazed, D.,
and Grewal, S. I. (2005) Proc. Natl. Acad. Sci. USA, 102,
152-157.
72.Zofall, M., and Grewal, S. I. (2006) Mol.
Cell, 22, 681-692.
73.Halic, M., and Moazed, D. (2010) Cell,
140, 504-516.
74.Clough, E., Moon, W., Wang, S., Smith, K., and
Hazelrigg, T. (2007) Development, 134, 157-165.
75.Yoon, J., Lee, K. S., Park, J. S., Yu, K., Paik,
S. G., and Kang, Y. K. (2008) PLoS One, 3, e2234.
76.Tschiersch, B., Hofmann, A., Krauss, V., Dorn,
R., Korge, G., and Reuter, G. (1994) EMBO J., 13,
3822-3831.
77.Rangan, P., Malone, C. D., Navarro, C., Newbold,
S. P., Hayes, P. S., Sachidanandam, R., Hannon, G. J., and Lehmann, R.
(2011) Curr. Biol., 21, 1373-1379.
78.Ushijima, Y., Inoue, Y. H., Konishi, T.,
Kitazawa, D., Yoshida, H., Shimaji, K., Kimura, H., and Yamaguchi, M.
(2012) Chromosome Res., 20, 319-331.
79.Moshkovich, N., and Lei, E. P. (2010) PLoS
Genet., 6, e1000880.
80.Fanti, L., Giovinazzo, G., Berloco, M., and
Pimpinelli, S. (1998) Mol. Cell, 2, 527-538.
81.Volpe, A. M., Horowitz, H., Grafer, C. M.,
Jackson, S. M., and Berg, C. A. (2001) Genetics, 159,
1117-1134.
82.Pane, A., Jiang, P., Zhao, D. Y., Singh, M., and
Schupbach, T. (2011) EMBO J., 30, 4601-4615.
83.Chen, Y., Pane, A., and Schupbach, T. (2007)
Curr. Biol., 17, 637-642.
84.Kawaoka, S., Arai, Y., Kadota, K., Suzuki, Y.,
Hara, K., Sugano, S., Shimizu, K., Tomari, Y., Shimada, T., and
Katsuma, S. (2011) RNA, 17, 1401-1407.
85.Kawaoka, S., Hayashi, N., Katsuma, S., Kishino,
H., Kohara, Y., Mita, K., and Shimada, T. (2008) Insect Biochem.
Mol. Biol., 38, 1058-1065.
86.Kawaoka, S., Minami, K., Katsuma, S., Mita, K.,
and Shimada, T. (2008) Biochem. Biophys. Res. Commun.,
367, 755-760.
87.Arensburger, P., Hice, R. H., Wright, J. A.,
Craig, N. L., and Atkinson, P. W. (2011) BMC Genomics,
12, 606.
88.Morazzani, E. M., Wiley, M. R., Murreddu, M. G.,
Adelman, Z. N., and Myles, K. M. (2012) PLoS Pathog., 8,
e1002470.
89.Campbell, C. L., Black, W. C. T., Hess, A. M.,
and Foy, B. D. (2008) BMC Genomics, 9, 425.
90.Van der Heijden, G. W., and Bortvin, A. (2009)
Epigenetics, 4, 76-79.
91.Carmell, M. A., Girard, A., van de Kant, H. J.,
Bourc’his, D., Bestor, T. H., de Rooij, D. G., and Hannon, G. J.
(2007) Dev. Cell, 12, 503-514.
92.Rozhkov, N. V., Aravin, A. A., Zelentsova, E. S.,
Schostak, N. G., Sachidanandam, R., McCombie, W. R., Hannon, G. J., and
Evgen’ev, M. B. (2010) RNA, 16, 1634-1645.
93.Aravin, A. A., and Bourc’his, D. (2008)
Genes Dev., 22, 970-975.
94.Aravin, A., Gaidatzis, D., Pfeffer, S.,
Lagos-Quintana, M., Landgraf, P., Iovino, N., Morris, P., Brownstein,
M. J., Kuramochi-Miyagawa, S., Nakano, T., Chien, M., Russo, J. J., Ju,
J., Sheridan, R., Sander, C., Zavolan, M., and Tuschl, T. (2006)
Nature, 442, 203-207.
95.Klenov, M. S., Lavrov, S. A., Stolyarenko, A. D.,
Ryazansky, S. S., Aravin, A. A., Tuschl, T., and Gvozdev, V. A. (2007)
Nucleic Acids Res., 35, 5430-5438.
96.Kuramochi-Miyagawa, S., Kimura, T., Yomogida, K.,
Kuroiwa, A., Tadokoro, Y., Fujita, Y., Sato, M., Matsuda, Y., and
Nakano, T. (2001) Mech. Dev., 108, 121-133.
97.Kuramochi-Miyagawa, S., Kimura, T., Ijiri, T. W.,
Isobe, T., Asada, N., Fujita, Y., Ikawa, M., Iwai, N., Okabe, M., Deng,
W., Lin, H., Matsuda, Y., and Nakano, T. (2004) Development,
131, 839-849.
98.Deng, W., and Lin, H. (2002) Dev. Cell,
2, 819-830.
99.Girard, A., Sachidanandam, R., Hannon, G. J., and
Carmell, M. A. (2006) Nature, 442, 199-202.
100.Das, P. P., Bagijn, M. P., Goldstein, L. D.,
Woolford, J. R., Lehrbach, N. J., Sapetschnig, A., Buhecha, H. R.,
Gilchrist, M. J., Howe, K. L., Stark, R., Matthews, N., Berezikov, E.,
Ketting, R. F., Tavare, S., and Miska, E. A. (2008) Mol. Cell,
31, 79-90.
101.Batista, P. J., Ruby, J. G., Claycomb, J. M.,
Chiang, R., Fahlgren, N., Kasschau, K. D., Chaves, D. A., Gu, W.,
Vasale, J. J., Duan, S., Conte, D., Jr., Luo, S., Schroth, G. P.,
Carrington, J. C., Bartel, D. P., and Mello, C. C. (2008) Mol.
Cell, 31, 67-78.
102.Lee, H. C., Gu, W., Shirayama, M., Youngman,
E., Conte, D., Jr., and Mello, C. C. (2012) Cell, 150,
78-87.
103.Shirayama, M., Seth, M., Lee, H. C., Gu, W.,
Ishidate, T., Conte, D., Jr., and Mello, C. C. (2012) Cell,
150, 65-77.
104.Bagijn, M. P., Goldstein, L. D., Sapetschnig,
A., Weick, E. M., Bouasker, S., Lehrbach, N. J., Simard, M. J., and
Miska, E. A. (2012) Science, 337, 574-578.
105.Ashe, A., Sapetschnig, A., Weick, E. M.,
Mitchell, J., Bagijn, M. P., Cording, A. C., Doebley, A. L., Goldstein,
L. D., Lehrbach, N. J., Le Pen, J., Pintacuda, G., Sakaguchi, A.,
Sarkies, P., Ahmed, S., and Miska, E. A. (2012) Cell,
150, 88-99.
106.Ruby, J. G., Jan, C., Player, C., Axtell, M.
J., Lee, W., Nusbaum, C., Ge, H., and Bartel, D. P. (2006) Cell,
127, 1193-1207.
107.Cecere, G., Zheng, G. X., Mansisidor, A. R.,
Klymko, K. E., and Grishok, A. (2012) Mol. Cell, 47,
734-745.
108.Gu, W., Lee, H. C., Chaves, D., Youngman, E.
M., Pazour, G. J., Conte, D., Jr., and Mello, C. C. (2012) Cell,
151, 1488-1500.
109.Bushati, N., Stark, A., Brennecke, J., and
Cohen, S. M. (2008) Curr. Biol., 18, 501-506.
110.Mochizuki, K., Fine, N. A., Fujisawa, T., and
Gorovsky, M. A. (2002) Cell, 110, 689-699.
111.Bouhouche, K., Gout, J. F., Kapusta, A.,
Betermier, M., and Meyer, E. (2011) Nucleic Acids Res.,
39, 4249-4264.
112.Hobson, D. J., Wei, W., Steinmetz, L. M., and
Svejstrup, J. Q. (2012) Mol. Cell, 48, 365-374.
113.Maxwell, P. H., Belote, J. M., and Levis, R. W.
(2006) Nucleic Acids Res., 34, 5498-5507.
114.Minchiotti, G., Contursi, C., and Di Nocera, P.
P. (1997) J. Mol. Biol., 267, 37-46.
115.Shpiz, S., Kwon, D., Rozovsky, Y., and
Kalmykova, A. (2009) Nucleic Acids Res., 37, 268-278.
116.Speek, M. (2001) Mol. Cell. Biol.,
21, 1973-1985.
117.Klattenhoff, C., Bratu, D. P.,
McGinnis-Schultz, N., Koppetsch, B. S., Cook, H. A., and Theurkauf, W.
E. (2007) Dev. Cell, 12, 45-55.