REVIEW: Heterochromatin Formation and Transcription in Relation to trans-Inactivation of Genes and Their Spatial Organization in the Nucleus
A. S. Shatskikh and V. A. Gvozdev*
Institute of Molecular Genetics, Russian Academy of Sciences, pl. Kurchatova 2, 123182 Moscow, Russia; E-mail: gvozdev@img.ras.ru* To whom correspondence should be addressed.
Received March 19, 2013
The role of transcription in heterochromatin formation in the nuclei of eukaryotes, originally shown for the pericentromeric heterochromatin assembly in fission yeasts, has now become an accepted paradigm extended to multicellular eukaryotes. It has been shown that small RNAs involved in the RNA interference system in its broadest sense can play an important role in this multi-step process — they are recognized by complementary interactions with the newly formed nuclear transcripts and recruit protein complexes to the local genomic sites for heterochromatinization. The role of transcription as a trigger of this process at the sites of genomic repeats will be considered in this review using various examples of heterochromatin formation, with an emphasis on discussion of its role in trans-chromosomal interactions causing gene inactivation.
KEY WORDS: heterochromatin, transcription, epigenetics, trans-inactivation, paramutation, variegated position effectDOI: 10.1134/S0006297913060060
The term heterochromatin was dubbed in the beginning of the last century for the chromosome regions of eukaryotes that remain condensed at the stage of interphase [1]. Heterochromatin regions occur in all eukaryotes and, in accordance with classical notions, have been considered as transcriptionally inactive regions of the genome. The question about the functional role of these genomic regions is still quite unresolved in spite of impressive progress, e.g. elucidation of their role in immune defense against transposons [2, 3]. The influence of heterochromatin on gene expression was demonstrated in Drosophila after the discovery of phenomenon of variegated position effect that has been the object of research in many reviews [4-7]. The position effect manifests itself in cis-inactivation (inherited in a number of cell generations) of euchromatic genes transposed to heterochromatin or when euchromatin is incorporated into heterochromatin. This phenomenon, demonstrating the possibility of reversible gene inactivation, is a classical example of epigenetic inheritance. The molecular mechanisms responsible for the emergence of epigenetic modifications of the genes, altering their expression, are now intensively studied and extensively used in practice: it would be enough to mention the role of these researches in the study of the causes of cancer [8, 9]. Heterochromatin is characterized by the presence of repeated nucleotide sequences of different nature; the functional role of many of them is still unclear. The role of heterochromatinization may be to maintain the stability of these genomic regions, since the formation of compact heterochromatin prevents the recombination of repeats and the loss of repeating copies, including the ribosomal RNA (rDNA) genes [10]. Some other functionally important genomic regions represented by repeats are also localized in heterochromatin. For instance, the defective transposon copies grouped in heterochromatin perform the repression of transposition of their active copies in the genome [2, 3]. Heterochromatic regions are involved in chromosomal pairing, providing strict segregation of the chromosomes in mitosis and meiosis [10, 12], and they determine spatial localization of genes in the nucleus, which is associated with the level of their expression [13-15].
This review is devoted to modern concepts on the role of transcription in the formation of heterochromatic regions of the genome. The heterochromatin formation mechanisms coupled with transcription of these regions may involve the systems responsible for the generation of small RNA (siRNA and piRNA) complementary to the nascent transcripts. The results of rapidly developing multi-aspect studies of this problem are discussed in other articles in this issue as well [3, 16, 17]. Here we will consider a number of other examples of silencing, demonstrating the inactivation of genomic repeats coupled with their transcription. Special attention will be given to discussion of the phenomenon of gene trans-inactivation. Trans-inactivation consists in appearance in the chromosome of the repressive structure of chromatin that can be extended to the homologous regions of the other chromosome. The cases of trans-inactivation related to gene heterochromatinization have been described in animals and plants. The results of some studies suggest that trans-inactivation may be associated with the pairing of homologous regions of the chromosomes, and inactivation may be determined by the shift of inactivated target in the space of the nucleus. In this context, we will consider individual examples of “spatial kissing” of local chromosome regions, which is accompanied by heterochromatin formation, and discuss the notions of the role of transcription in trans-inactivation.
The modern concepts of heterochromatin formation suggest that this process is triggered, paradoxically, by transcription coupled with inactivation [18]. As regards the classical gene position effects accompanied by cis-inactivation and its extension to the neighboring euchromatin genes, this idea has not yet been sufficiently proved experimentally, though there are some indications of the weakening of silencing in the case of mutations that disturb the formation and function of piRNA-dependent repression [19]. The role of transcription in piRNA dependent silencing followed by inactivation of a target and expansion of inactive chromatin to the neighboring genes has been recently shown in Drosophila by the example of inactivation of the regions adjacent to transposon insertions [20]. These results suggest a similar mechanism of heterochromatin formation and distribution in the case of position effects.
FORMATION OF HETEROCHROMATIN AREAS IN THE CENTROMERE AND
NUCLEOLUS ORGANIZER REGIONS
Let us first consider the role of transcription in the formation of constitutive heterochromatic areas [16] in centromeric regions. The role of transcription of genomic regions acquiring the properties of heterochromatin was proved for the first time as a result of detailed genetic and molecular studies of the formation of centromeric heterochromatin in the chromosomes of fission yeasts (for review, see [16, 21, 22]). The transcripts of these repeat-rich noncoding regions attract, with the involvement of complementary siRNA, chromatin modification complexes responsible for heterochromatinization [23, 24]. This results in formation of centromeric heterochromatin regions responsible for correct chromosome disjunction during cell division. Note that the silencing of the euchromatic loci of fission yeasts may not involve siRNA, but target transcription is necessary for its inactivation. For instance, it occurs during the inactivation of meiotic genes in vegetatively propagating cells of fission yeasts [25]. This is the case of formation of facultative heterochromatin (see review [16] concerning constitutive and facultative heterochromatin) in fission yeasts. The dynamics of pericentromeric heterochromatin formation in multicellular eukaryotes has only begun to be studied, but recently obtained results demonstrate the role of transcription of the satellite DNA of pericentromeric regions in heterochromatin formation at the early stages of mouse embryogenesis.
Some works show transcription of the regions of mammalian constitutive heterochromatin represented by the repeated sequences of the pericentomeric satellite DNA [26-28]. There is indirect evidence of potential involvement of siRNA in heterochromatinization of centromeric regions in mammals because the assembly of heterochromatin and the function of the centromeres are disturbed in the absence of Dicer protein responsible for siRNA formation [29, 30]. However, as yet there are no direct data on the involvement of an RNAi mechanism in formation of pericentromeric heterochromatin in mammals. Transcription of pericentromeric satellite repeats in mice is regulated during early embryonic development, and its beginning temporally coincides with the organization of chromocenters formed as a result of spatial aggregation of the centromeric regions of chromosomes [31]. The chromocenter (heterochromatic compartment) starts to be formed during embryo development at the stage of two cells [32, 33], when there appears a peak of bidirectional transcription (direct and opposite) of pericentromeric satellites followed by rapid diminution in their transcription already at the stage of four cells. Directed suppression of satellite transcription by anti-sense oligonucleotides resulted in arrested development and the impairment of chromocenter formation. It is supposed that the transcripts can participate in the assembly of heterochromatin proteins on satellite DNA. The chromatin protein HP1a, which is often considered as a repressor protein, begins to be found in the chromocenter also at the stage of two cells. The mutation substituting arginine for lysine in position 27 in histone H3.3 and preventing the emergence of the H3.3K27me3 heterochromatin modification impairs the binding of HP1b to the pericentromeric regions. At the same time, chromosome segregation is impaired as well [32]. Note that the protein HP1 and its orthologs are active RNA-binding proteins [34, 35]. The direct transcripts of the satellites showed partial colocalization with HP1a. It seems that the binding of HP1 to the pericentromeric loci mediated by noncoding RNA attracts the repressor complexes of histone modification. The embryos expressing the mutant HP1 with impaired “hinge domain”, which is responsible for the RNA-binding activity of this protein, demonstrate impaired chromosome disjunction [31]. It is interesting that the impaired chromosome disjunction may be compensated by introduction of a double-stranded RNA with the nucleotide sequence corresponding to the satellites [32]. This result may be an indirect evidence of potential involvement of the RNAi and siRNA mechanisms in the formation of pericentromeric heterochromatin in mammals. However, different temporal profiles of transcription of both strands, as well as non-overlapping of the intracellular localization of direct and reverse transcripts, may be indicative of their different functions not depending on RNAi.
One more example, demonstrating the dependence of heterochromatin formation on transcription, concerns the silencing of the ribosomal RNA (rDNA) genes adjacent to constitutive heterochromatic regions. Heterochromatin formation on the repeated copies of ribosomal DNA is not only considered as a mean of preventing intrachromosomal recombination between the homologous copies of these repeats, which would result in their loss [36], but also as a mean of silencing of some part of rDNA clusters that are probably excessive for a cell and an organism. The rDNA gene copies are separated by spacers, which have been considered previously as untranscribed regions. The characteristic negative modifications are introduced into the histones of mammalian rDNA chromatin, followed by rDNA methylation, only during the transcription of spacer regions (promoters of the rDNA genes). These transcripts showing specific features of secondary structure attract the silencing protein complexes (histone modifiers), DNA methyltransferase, and subunits of the complexes of chromatin remodeling, all together providing the establishment of repressor epigenetic modifications in the rDNA gene clusters [37, 38]. These results supplement the existing notions of the role of nascent RNA as a factor attracting mammalian proteins to certain regulatory regions of the DNA. To date, there is no direct evidence of involvement of the RNAi (siRNA) mechanism in the silencing of rDNA genes.
Trans-INACTIVATION INDUCED BY THE CLUSTERS OF
HETEROCHROMATINIZED REPEATS IN Drosophila
It is particularly interesting to consider the functioning of artificial clusters represented by the tandem copies of transgenes in Drosophila, because these examples can demonstrate not only the case of trans-inactivation in connection with the gene position effect, but also the role of transcription of inactivated regions in the transfer of silencing to the homologous chromosome. Transgenes flanked by terminal repeats of a transposon can move in the presence of active transposase. The authors of [39], when trying to move them, observed the formation in euchromatin of tandemly organized transgenic clusters, which included the reporter genes mini-white (determining eye color) and lacZ (encoding β-galactosidase). The clusters demonstrated mosaic inactivation of these reporters clonally inherited in somatic cells that is typical for position-effect variegation [40, 41]. Not only the inactivation of transgenes within a cluster but also extension of inactivation to the adjacent genes was observed. The typical modifiers of position effect (mutations in the gene encoding the heterochromatin protein HP1 and heterochromatic Y-chromosome) suppressed the inactivation. The silencing was intensified due to shortening of a distance from the cluster to the block of constitutive heterochromatin on the chromosome map, that is also typical for classical position effects. The character of expression of the reporter genes within artificial tandem clusters and the influence of modifiers on these genes shows that the formed artificial heterochromatin behaves like a native one and can simulate the characteristics of classical position effect induced by chromosomal rearrangements causing gene transposition to heterochromatin. In all likelihood, the repeated sequences within the cluster are recognized by the mechanisms inducing directed heterochromatinization. The intensity of inactivation in the cluster was proportional to the number of repeats in the latter [41].
It is still unclear, whether heterochromatinization of artificial clusters depends on their transcription. The same question concerns cis-inactivation due to position-effect. However, recently obtained data make it possible to consider the transcriptional activity of artificial clusters as a factor of epigenetic inheritance of their inactivation. Transcriptional activity of the clusters influences their ability not only to cause mosaic inactivation of genes within a cluster, but also to have a trans-inactivating effect on such a cluster in the homologous chromosome. The revealed effect of trans-interaction between the clusters resembles the phenomenon of classical epigenetic gene inactivation – paramutation in plants (see below).
Artificial clusters may differ in the degree of cis-inactivation of the reporter transgenes within a cluster. As a result of irradiation of the BX-2 cluster with a weak level of inactivation, the T-1 cluster with strong inactivation was obtained with no change in the number of repeats. The molecular mechanism responsible for intensification of inactivation has not been identified yet. In somatic cells, the tandem cluster proved to be able to induce trans-inactivation of the mini-white reporters in the homologous chromosome if they were located in the same region according to the chromosome map [41]. This suggests the necessity of pairing of the homologous chromosome regions for the establishment of trans-inactivation. In germline cells, the T-1 cluster induced trans-silencing of the homologous lacZ reporter [42]; however, in germinal cells the lacZ transgene as a target of silencing could be localized at random sites on the homologous chromosome or even on the other chromosomes. The absence of HP1 protein, which had a suppressing effect on the silencing in somatic tissue, did not affect the efficiency of inactivation in germinal tissues. The mechanisms of inactivation induced by the cluster seem to be different in somatic and germinal tissues. The inactivation of lacZ was inherited by a progeny of females but not males, demonstrating the phenomenon of cytoplasmic inheritance of silencing. It was shown that transgene trans-inactivation induced by the cluster of repeats in the germline was associated with the production of small RNA formed during transcription of the reporter genes within the cluster [43]. Two subpopulations of small RNA (21 and 23-28 nt) were found, the former seems to be similar to siRNA and the latter to piRNA. Trans-silencing of the reporter gene in the germline did not occur due to the disturbance of the piRNA silencing system in the presence of the aubergine mutation, while the absence of the Dicer-2 protein involved in siRNA biogenesis had no effect on trans-silencing. The Aub protein is known to participate in the cycle of piRNA amplification – ping-pong cycle [2, 3, 16]. The ability of the cluster for trans-inactivation did not depend on the number of primary transcripts but was determined by their processing with the formation of small RNA. This is indicative of the role of post-transcriptional processes in establishment of the silencing. The maternal effect in trans-inactivation inheritance seems to be based on the cytoplasmic transfer of piRNA inducing the biogenesis of new piRNA during the processing of the transcripts of homologous transgenes.
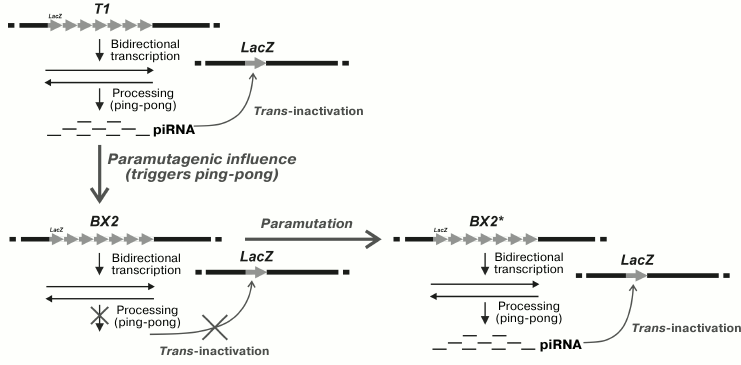
It has been revealed that the ability of the T1 cluster to induce trans-silencing of the reporter transgene in the germline cells can be transmitted to the BX-2 cluster lacking this property [43]. This new property of the BX-2 cluster appears if both clusters are presented in a single genome. The process of trans-interaction between the clusters is schematically shown in Fig. 1. The new ability of the BX-2 cluster for trans-inactivation was epigenetically inherited in the successive generations. The cluster with the acquired ability to trans-inactivate the reporter transgene becomes capable of transmitting this property to another cluster that did not possess it previously. It seems that the mechanism of such transition is associated with the induction of piRNA biogenesis as a result of activation of the processing of primary transcripts of the cluster. Phenomenologically, such inter-allelic transition of the state of cluster expression resembles the epigenetic phenomenon established previously: paramutation, which demonstrates one of the interesting cases of trans-inactivation and its epigenetic inheritance.
Fig. 1. Trans-interaction in the germ cells of D. melanogaster of transgene clusters with different epigenetic states is accompanied by acquisition by one of the clusters of the ability to inactivate single homologous transgenes in different genomic sites. T1, the cluster capable of trans-inactivation of the reporter transgene; BX2, the cluster incapable of trans-inactivation of the reporter transgene; BX2*, BX2 cluster with the acquired ability for trans-inactivation of the reporter transgene; LacZ, the reporter transgene.
PARAMUTATION IN PLANTS
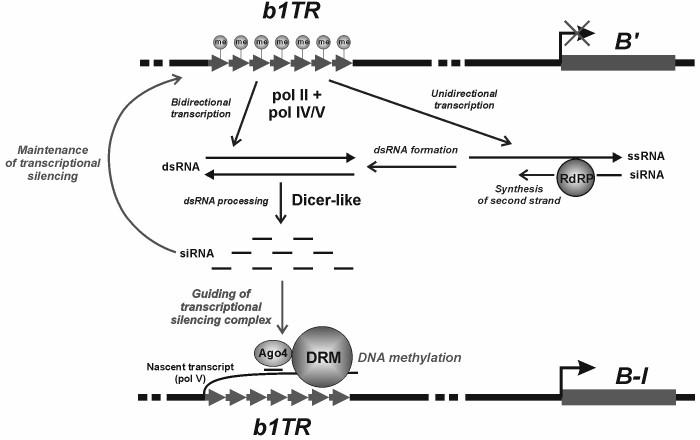
The phenomenon of paramutation was described for the first time in plants. Now we will consider the classic examples of paramutation and its potential underlying mechanisms. In case of paramutation, trans-inactivation is also based on the transcription of interacting alleles; however, the system of silencing with the involvement of piRNA has not been found in plants, though the role of another small RNA (siRNA) in chromatin silencing has been well studied [44, 45]. Initially, paramutation was found in maize [46] in the locus regulating pigment formation. Paramutation occurs during trans-interaction of two homologous loci: active and inactivated. As a result, the active locus is inactivated with no change in its nucleotide sequence. The inactive state is inherited epigenetically in the successive generations [47, 48]. The interacting alleles of the loci are called epialleles. The epiallele with inactivating effect is designated as paramutagenic, while the inactivated one is designated as paramutable. As a rule, the inactivated epiallele becomes capable, in its turn, of exerting the paramutagenic effect. Paramutation may emerge spontaneously, in the absence of the paramutagenic allele, whereas spontaneous reversion of inactive state into active one is observed in exceptional cases [49]. The classic example of paramutation was revealed in maize for the b1 locus encoding anthocyanins’ biogenesis and production of plant color. The regulation of b1 expression is controlled by the b1TR enhancer located at a distance of 100 kb from the protein-encoding gene [50]. The enhancer consists of tandem repeats. The repressed paramutagenic epiallele B′ can inactivate the active paramutable B-I (B-Intense) with its level of expression exceeding 10-30 times the expression of B′ [51]. The DNA of the b1TR enhancer of the epiallele B′ is heavily methylated, and chromatin of the epiallele B′, in contrast to B-I, shows a repressive histone modification (H3K27me2) [52, 53]. Paramutation leads to a drastic increase in the level of DNA methylation of the B-I enhancer, as well as repressive chromatin modifications (H3K9me2 and H3K27me2) in this region [53]. Methylation of the enhancer repeats is supposed first of all to determine the emergence of paramutation, while histone modifications mainly affect the tissue-specific regulation of expression, though the coordinated participation of both processes in paramutation must not be ruled out.
The molecular mechanism of paramutation is being intensively studied. It seems that the transcription of interacting loci with the involvement of specialized plant RNA polymerases (pol IV and pol V) plays the key role in inter-allelic interaction [54]. The paramutated state is induced with participation of RNA-dependent RNA polymerase [55] supplying the double-stranded RNA, which in turn is a siRNA precursor most likely processed with the involvement of the Dicer protein. The special protein of the Argonaute family (AGO4), which is known to participate in RNA-dependent DNA silencing in plants [56], is supposed to bind siRNA and provide complementary interactions between siRNA and the transcript of the paramutable locus. In its turn, it attracts the proteins of chromatin remodeling and DNA methyltransferase. Many details of molecular interactions between proteins and RNAs during the establishment of paramutated state are still undiscovered. It seems that the mechanisms of emergence of paramutated states may appreciably vary for different loci, but the cumulative experimental result obtained to date allows their consideration in accordance with the presented scheme (Fig. 2). The role of spatial pairing of homologous loci that facilitates the paramutagenic effect is suggested as well. One should not rule out the role of physical interaction between the alleles, which may either result in the direct transition of repressor epigenetic labels from the inactive locus to the homologous paramutable locus or provide the displacements of the paramutable epiallele, together with the paramutagenic epiallele, to the transcriptionally inactive compartment of the nucleus [57].
Fig. 2. Hypothetical mechanism of paramutation induction. b1TR, the b1 gene enhancer consisting of the repeats whose methylation is accompanied by suppression of the gene expression; B′ and B-I, paramutagenic and paramutable epialleles of the b1 gene, respectively; ssRNA, single-stranded RNA; dsRNA, double-stranded RNA; RdRP, RNA-dependent RNA polymerase; Ago4, the protein of the Argonaute family involved in RNA-dependent DNA methylation in plants; DRM, DNA methyltransferase of plants.
Trans-INTERACTIONS AND HETEROCHROMATIN FORMATION DURING
THE INACTIVATION OF MAMMALIAN X-CHROMOSOME AND IN THE PHENOMENON OF
NUCLEOLAR DOMINANCE IN Drosophila
Inactivation of one of the two X-chromosomes in mammals, accompanied by the formation of facultative heterochromatin (compact Barr body), as is known, calls for coordinated transcription of a number of noncoding RNAs on the inactivated X-chromosome [58]. The transcripts attract a number of protein complexes providing the introduction of negative epigenetic labels of histones and DNA methylation to the inactivated X-chromosome. Without considering the successive steps of inactivation, we will note only the stage of pairing of two X-chromosomes as a necessary event for “asymmetric inactivation” of one of the two X-chromosomes. Effective pairing of the chromosomes becomes possible only due to transcription of noncoding Tsix RNA in the regions of “inactivation centers” [58]. Such pairing is a basis for asymmetrical redistribution of definite ribonucleoprotein complexes between the X-chromosomes and for random selection of only one of the X-chromosomes as inactive. As a result, the transcription of noncoding Xist RNA begins on the inactivated X-chromosome and extends along the whole X-chromosome, attracting protein complexes to establish and consolidate the inactivation. Here, we should emphasize not so much the role of transcription and extension along the chromosome of the noncoding Xist RNA, which attracts the hierarchy of silencing complexes, as the role of Tsix transcription in spatial orientation of the chromosomes. An analogous situation is observed also during the analysis of the phenomenon of nucleolar dominance in Drosophila [59] described for the rRNA gene clusters and consisting in the dominance of cluster expression in the Y-chromosome under repression of the homologous cluster in the X-chromosome [58]. Note that heterochromatinization and repression of the rDNA cluster in the X-chromosome occur after somatic pairing of the X and Y chromosomes in the region of rDNA localization.
Trans-INACTIVATION DETERMINED BY HETEROCHROMATINIZATION OF
THE HOMOLOGOUS CHROMOSOME
Trans-interactions between two X-chromosomes in mammals proved to be a trigger of random inactivation of one of them in the case of transcription of noncoding RNA in the inactivation center. It seems attractive to emphasize the putative role of transcription when considering the phenomena of trans-inactivation in Drosophila caused by cis-heterochromatinization of the homologous region of opposite chromosome as a result of its eu-heterochromatic chromosome rearrangement (gene position effect). These are rare phenomena, and only two genetic systems demonstrating this type of silencing have been described so far. Trans-inactivation of this type was shown for the first time for the brown-Dominant (bwD) allele inactivating in the homologous chromosome the normal allele of the brown (bw+) gene determining eye color [60]. The bwD mutation was caused by insertion of the block of satellite DNA with the AAGAG repeat into the coding region of the brown gene [61]. This phenomenon has been studied in quite a number of works [62-65]. The second case of such trans-silencing was revealed in the analysis of interaction between the normal chromosome and the eu-heterochromatic rearrangement characterized by the typical effect of cis-inactivation of the genes adjacent to heterochromatin that is usually observed in case of position effects [66]. The rearrangement was an inversion (In(2)A4) formed as a result of one break in the satellite DNA in heterochromatin of chromosome 2 and the other break in euchromatin of the left arm of this chromosome (Fig. 3). Trans-inactivation of the mini-white reporter transgenes was observed in heterozygotes carrying the normal and homologous rearranged chromosomes. The transgenes located on the chromosome map in the regions close to the sites of inversion break underwent mosaic trans-inactivation (Fig. 3).
Fig. 3. In(2)A4 inversion with breaks in euchromatin and in satellite DNA of the pericentromeric region induces mosaic trans-inactivation of the reporter transgene in euchromatin of the non-rearranged homologous chromosome. Hc, heterochromatin; C, centromere; A, B, C, D are endogenous genes in the region of inversion.
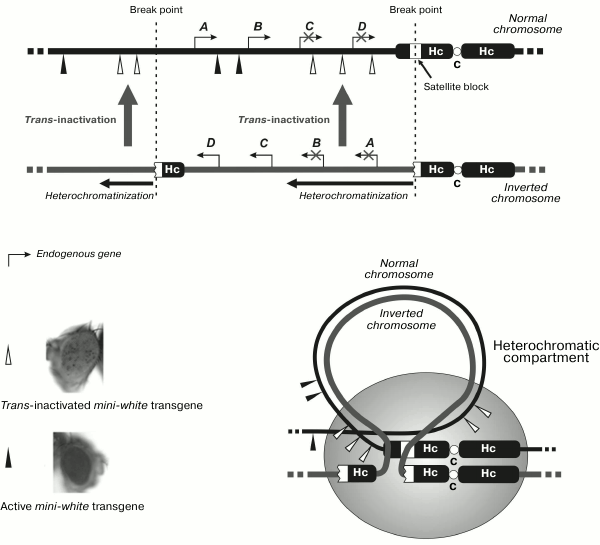
Study of trans-inactivation caused by the bwD allele has revealed that satellite block inducing inactivation drags the paired homologous locus of the normal chromosome into the heterochromatic compartment, resulting in heterochromatinization of the latter [67]. Hybridization in situ with fluorescent probes demonstrated [66, 68, 69] the movement of inactivated chromosome regions to the heterochromatic compartment of the nucleus in the cells of imaginal discs and neuroblasts. In some cells, dragging of the euchromatic gene into heterochromatin was not revealed, being in agreement with the observation of mosaic and stochastic inactivation. Simultaneous analysis of the activity and spatial localization of the gene in separate cells showed the correlation between the silencing and the presence of inactivated region in the heterochromatic compartment [70]; the indications of such correlation between inactivation and gene shift to heterochromatin were also obtained for trans-interactions between the normal and inverted chromosomes [71]. Molecular analysis of the chromatin structure of transgenes trans-inactivated by the bwD [72] showed that the silencing was accompanied by compaction of the nucleosomal structure and accumulation of the HP1 protein. The trans-effect was not accompanied by increase of heterochromatic repressive histone modification H3K9me2 or decrease in the level of euchromatic ones (H3K9Ac2 and H3K14Ac2) that may be indicative of their insignificant role in the establishment of trans-activation.
Are transcription processes involved in the described effects of trans-inactivation with heterochromatin formation, as it has been shown for paramutation in plants or for mammalian X-chromosome inactivation? Such possibility may be supported by the above-mentioned data on the temporal correlation between the transcription of satellite pericentromeric DNA and the formation of chromocenters in the early development in mammals. Indications of the existence of bidirectional transcription of satellite repeats in Drosophila were obtained as well [73]. The number of these transcripts increased in the presence of mutation in the spn-E and aub genes involved in the piRNA-dependent system of silencing. This effect of the spn-E mutation correlated with the characteristics of active chromatin of the satellites: the content of positive H3K9ac modification and the amount of the TAF1 protein (a component of the RNA polymerase II transcription complex) increased. Note that trans-inactivation caused by the In(2)A4 inversion was completely eliminated in the presence of mutation inactivating the histone methyltransferase SetDB/EGG responsible for the H3K9me3 modification. This modification is considered as a repressive one; however, it has been shown that SetDB/EGG is necessary for the transcription of heterochromatic regions participating in production of piRNA precursors [74]. Therefore, it may be supposed that the described variant of trans-inactivation calls for the transcription of interacting heterochromatic region with its target in euchromatin for the establishment of trans-inactivation. In addition, the SAYP protein being a subunit of chromatin remodeling complex [75], simultaneously found in the Drosophila heterochromatin, proved to be necessary for the induction of trans-inactivation of reporter genes in the normal chromosome being in heterozygous state with the In(2)A4 inversion. Hypothetical concepts of the role of chromatin binding of various transcription factors as a prerequisite for transcription of satellite DNA repeats intended for silencing now begin to be confirmed [76]. Undoubtedly, further studies must be performed to directly demonstrate the role of transcription in the considered cases of trans-inactivation in Drosophila.
In addition to the above effects of trans-interactions in Drosophila, we will consider the results of work indicating the role of transcription in heterochromatin formation in Drosophila and the possibility of non-Mendelian paramutation like inheritance of heterochromatic state. Recently it has been shown that the activating transcription factor dATF-2 participates in heterochromatin formation, and its absence suppresses cis-inactivation of the euchromatic gene in the case of classical position effect in Drosophila [77]. Mutations in the respective orthologous genes in fission yeasts also prevented the organization of constitutive chromatin and transcription of the target of heterochromatinization (the mating loci). However, in this case heterochromatin formation did not require the functioning of the RNA-interference system [78]. This result of the assessment of the negative effect of the d-ATF-2PB mutation on the ability of adjacent heterochromatin to extend the silencing to the adjacent genes once again indicates the role of transcription in the establishment of cis-inactivation as a result of classical position-effect variegation. Colocalization of the dATF-2 factor with the DNA of constitutive heterochromatin demonstrated by chromatin immunoprecipitation was impaired under stress conditions and accompanied by the disturbance of silencing caused by position effect. However, the most interesting result of the work [77] is a demonstration of the fact that the state of heterochromatin damaged by stress can be inherited by the homologous chromosome and maintained in descendants. The authors fairly compare the revealed phenomenon with the paramutagenic effect and assume the involvement of RNA in this process. When discussing the phenomenon of gene trans-inactivation determined by heterochromatinization in one of the chromosomes, it was noted that it was accompanied by the dragging of inactivated targets into the heterochromatic compartment of the nucleus. It is considered that gene inactivation coupled with the movement of gene loci within the nuclear space can be performed with the involvement of Polycomb repressor complexes forming intranuclear bodies (Polycomb bodies) [79], which carry the genes of not one but different chromosomes, demonstrating trans-interactions. It is known that noncoding nascent transcripts may serve as platforms for recruitment of Polycomb complexes [80]. Noncoding RNAs also participate in reversible gene movement from these bodies to inter-chromatin granules (speckles), where these genes are actively transcribed [81]. Cook’s notion [82] of nuclear factories of transcription gradually wins recognition from researchers, demonstrating the role of transcription factors and mysterious insulators in their formation [83, 84]. At present, RNA is considered as an architectural component forming intranuclear bodies [85]. Originally, the concepts of the role of transcription in the formation of “silent” heterochromatin sounded like an oxymoron. We will dare to suggest that the specialized transcription factories can be spatial intranuclear compartments for implementation of the silencing of genomic regions. Future studies will prove or disprove these concepts.
This work was supported by the Russian Foundation for Basic Research (project No. 11-04-00017-a) and the Program of the Presidium of the Russian Academy of Sciences “Molecular and Cell Biology”.
REFERENCES
1.Heitz, E. (1929) Ber. Dt. Bot. Ges.,
47, 274-284.
2.Brennecke, J., Aravin, A. A., Stark, A., Dus, M.,
Kellis, M., Sachidanandam, R., and Hannon, G. J. (2007) Cell,
128, 1089-1103.
3.Olovnikov, I. A., and Kalmykova, A. I. (2013)
Biochemistry (Moscow), 78, 572-584.
4.Eissenberg, J. C., and Reuter, G. (2009) Int.
Rev. Cell Mol. Biol., 273, 1-47.
5.Henikoff, S. (1994) Genetics, 138,
1-5.
6.Henikoff, S. (1990) Trends. Genet.,
6, 422-426.
7.Elgin, S. C. R., and Reuter, G. (2007) in
Epigenetics (Allis, D., Jenuwein, T., and Reinberg, D., eds.)
CSHL Press, pp. 81-100.
8.Berdasco, M., and Esteller, M. (2013) Hum.
Genet., 132, 359-383.
9.Zhu, J., Adli, M., Zou, J. Y., Verstappen, G.,
Coyne, M., Zhang, X., Durham, T., Miri, M., Deshpande, V., De Jager, P.
L., Bennett, D. A., Houmard, J. A., Muoio, D. M., Onder, T. T.,
Camahort, R., Cowan, C. A., Meissner, A., Epstein, C. B., Shoresh, N.,
and Bernstein, B. E. (2013) Cell, 152, 642-654.
10.Peng, J. C., and Karpen, G. H. (2007) Nat.
Cell Biol., 9, 25-35.
11.Karpen, G. H., Le, M. H., and Le, H. (1996)
Science, 273, 118-122.
12.Dernburg, A. F., Sedat, J. W., and Hawley, R. S.
(1996) Cell, 86, 135-146.
13.Kosak, S. T., Scalzo, D., Alworth, S. V., Li, F.,
Palmer, S., Enver, T., Lee, J. S., and Groudine, M. (2007) PLoS
Biol., 5, 2602-2613.
14.Pecinka, A., Kato, N., Meister, A., Probst, A.
V., Schubert, I., and Lam, E. (2005) J. Cell Sci., 118,
3751-3758.
15.Fransz, P., Soppe, W., and Schubert, I. (2003)
Chromosome Res., 11, 227-240.
16.Sentmanat, M., Wang, S. H., and Elgin, S. C. R.
(2013) Biochemistry (Moscow), 78, 562-571.
17.Bortvin, A. (2013) Biochemistry (Moscow),
78, 592-602.
18.Grewal, S. I., and Elgin, S. C. (2007)
Nature, 447, 399-406.
19.Pal-Bhadra, M., Leibovitch, B. A., Gandhi, S. G.,
Rao, M., Bhadra, U., Birchler, J. A., and Elgin, S. C. (2004)
Science, 303, 669-672.
20.Sienski, G., Donertas, D., and Brennecke, J.
(2012) Cell, 151, 964-980.
21.Reyes-Turcu, F. E., and Grewal, S. I. (2012)
Curr. Opin. Genet. Dev., 22, 156-163.
22.Grewal, S. I. (2010) Curr. Opin. Genet.
Dev., 20, 134-141.
23.Aygun, O., and Grewal, S. I. (2010) Cold
Spring Harb. Symp. Quant. Biol., 75, 259-267.
24.Woolcock, K. J., and Buhler, M. (2013) Curr.
Opin. Cell Biol., 25, 1-6.
25.Yamanaka, S., Mehta, S., Reyes-Turcu, F. E.,
Zhuang, F., Fuchs, R. T., Rong, Y., Robb, G. B., and Grewal, S. I.
(2013) Nature, 493, 557-560.
26.Ting, D. T., Lipson, D., Paul, S., Brannigan, B.
W., Akhavanfard, S., Coffman, E. J., Contino, G., Deshpande, V.,
Iafrate, A. J., Letovsky, S., Rivera, M. N., Bardeesy, N., Maheswaran,
S., and Haber, D. A. (2011) Science, 331, 593-596.
27.Chan, F. L., Marshall, O. J., Saffery, R., Kim,
B. W., Earle, E., Choo, K. H., and Wong, L. H. (2012) Proc. Natl.
Acad. Sci. USA, 109, 1979-1984.
28.Bouzinba-Segard, H., Guais, A., and Francastel,
C. (2006) Proc. Natl. Acad. Sci. USA, 103, 8709-8714.
29.Fukagawa, T., Nogami, M., Yoshikawa M., Ikeno,
M., Okazaki, T., Takami, Y., Nakayama, T., and Oshimura, M. (2004)
Nat. Cell Biol., 8, 784-791.
30.Kanellopoulou, C., Muljo, S. A., Kung, A. L.,
Ganesan, S., Drapkin, R., Jenuwein, T., Livingston, D. M., and
Rajewsky, K. (2005) Genes Dev., 19, 489-501.
31.Probst, A. V., and Almouzni, G. (2011) Trends
Genet., 27, 177-185.
32.Santenard, A., Ziegler-Birling, C., Koch, M.,
Tora, L., Bannister, A. J., and Torres-Padilla, M. E. (2010) Nat.
Cell Biol., 12, 853-862.
33.Probst, A. V., Okamoto, I., Casanova, M., El
Marjou, F., Le Baccon, P., and Almouzni, G. (2010) Dev. Cell,
19, 625-638.
34.Buhler, M., and Hiller, S. (2012) Cell
Cycle, 11, 3907-3908.
35.Keller, C., Adaixo, R., Stunnenberg, R.,
Woolcock, K. J., Hiller, S., and Buhler, M. (2012) Mol. Cell,
47, 215-227.
36.Larson, K., Yan, S. J., Tsurumi, A., Liu, J.,
Zhou, J., Gaur, K., Guo, D., Eickbush, T. H., and Li, W. X. (2012)
PLoS Genet., 8, 1-10.
37.Schmitz, K. M., Mayer, C., Postepska, A., and
Grummt, I. (2010) Genes Dev., 24, 2264-2269.
38.Santoro, R., Schmitz, K. M., Sandoval, J., and
Grummt, I. (2010) EMBO Rep., 11, 52-58.
39.Dorer, D. R., and Henikoff, S. (1994)
Cell, 77, 993-1002.
40.Martin-Morris, L. E., Csink, A. K., Dorer, D. R.,
Talbert, P. B., and Henikoff, S. (1997) Genetics, 147,
671-677.
41.Dorer, D. R., and Henikoff, S. (1997)
Genetics, 147, 1181-1190.
42.Ronsseray, S., Boivin, A., and Anxolabehere, D.
(2001) Genetics, 159, 1631-1642.
43.De Vanssay, A., Bouge, A. L., Boivin, A.,
Hermant, C., Teysset, L., Delmarre, V., Antoniewski, C., and Ronsseray,
S. (2012) Nature, 490, 112-115.
44.Castel, S. E., and Martienssen, R. A. (2013)
Nat. Rev. Genet., 14, 100-112.
45.Van, Ex. F., Jacob, Y., and Martienssen, R. A.
(2011) Curr. Opin. Plant Biol., 14, 588-593.
46.Brink, R. A. (1956) Genetics, 41,
872-889.
47.Chandler, V. L., and Stam, M. (2004) Nat. Rev.
Genet., 5, 532-544.
48.Stam, M. (2009) Mol. Plant., 2,
578-588.
49.Chandler, V. L., Eggleston, W. B., and Dorweiler,
J. E. (2000) Plant Mol. Biol., 43, 121-145.
50.Stam, M., Belele, C., Ramakrishna, W., Dorweiler,
J. E., Bennetzen, J. L., and Chandler, V. L. (2002) Genetics,
162, 917-930.
51.Patterson, G. I., Thorpe, C. J., and Chandler, V.
L. (1993) Genetics, 135, 881-894.
52.Stam, M., Belele, C., Dorweiler, J. E., and
Chandler, V. L. (2002) Genes Dev., 16, 1906-1918.
53.Haring, M., Bader, R., Louwers, M., Schwabe, A.,
van Driel, R., and Stam, M. (2010) Plant J., 63,
366-378.
54.Wierzbicki, A. T. (2012) Curr. Opin. Plant
Biol., 15, 517-522.
55.Alleman, M., Sidorenko, L., McGinnis, K.,
Seshadri, V., Dorweiler, J. E., White, J., Sikkink, K., and Chandler,
V. L. (2006) Nature, 442, 295-298.
56.Matzke, M., Kanno, T., Daxinger, L., Huettel, B.,
and Matzke, A. J. (2009) Curr. Opin. Cell Biol., 21,
367-376.
57.Brzeski, J., and Brzeska, K. (2011) Wiley
Interdiscip. Rev. RNA, 2, 863-874.
58.Lee, J. (2011) Nat. Rev. Mol. Cell Biol.,
12, 815-826.
59.Greil, F., and Ahmad, K. (2012) Genetics,
191, 1119-1128.
60.Henikoff, S., and Dreesen, T. D. (1989) Proc.
Natl. Acad. Sci. USA, 86, 6704-6708.
61.Platero, J. S., Csink, A. K., Quintanilla, A.,
and Henikoff, S. (1998) J. Cell Biol., 140,
1297-1306.
62.Henikoff, S., Jackson, J. M., and Talbert, P. B.
(1995) Genetics, 140, 1007-1017.
63.Csink, A. K., Bounoutas, A., Griffith, M. L.,
Sabl, J. F., and Sage, B. T. (2002) Genetics, 160,
257-269.
64.Sage, B. T., Wu, M. D., and Csink, A. K. (2008)
Genetics, 178, 749-759.
65.Schneiderman, J. I., Goldstein, S., and Ahmad, K.
(2010) PLoS Genet., 6, 1-12.
66.Abramov, Yu. A., Kibanov, M. V., Gvozdev, V. A.,
and Lavrov, S. A. (2011) Dokl. Akad. Nauk, 437,
261-265.
67.Sass, G. L., and Henikoff, S. (1999)
Genetics, 152, 595-604.
68.Dernburg, A. F., Broman, K. W., Fung, J. C.,
Marshall, W. F., Philips, J., Agard, D. A., and Sedat, J. W. (1996)
Cell, 85, 745-759.
69.Csink, A. K., and Henikoff, S. (1996)
Nature, 381, 529-531.
70.Harmon, B., and Sedat, J. (2005) PLoS
Biol., 3, 450-462.
71.Lavrov, S. A., Shatskikh, A. S., Kibanov, M. V.,
and Gvozdev, V. A. (2013) Mol. Biol. (Moscow), 47,
286-291.
72.Nisha, P., Plank, J. L., and Csink, A. K. (2008)
Genetics, 179, 359-373.
73.Usakin, L., Abad, J., Vagin, V. V., de Pablos,
B., Villasante, A., and Gvozdev, V. A. (2007) Genetics,
176, 1343-1349.
74.Rangan, P., Malone, C. D., Navarro, C., Newbold,
S. P., Hayes, P. S., Sachidanandam, R., Hannon, G. J., and Lehmann, R.
(2011) Curr. Biol., 21, 1373-1379.
75.Panov, V. V., Kuzmina, J. L., Doronin, S. A.,
Kopantseva, M. R., Nabirochkina, E. N., Georgieva, S. G., Vorobyeva, N.
E., and Shidlovskii, Y. V. (2012) Nucleic Acids Res., 40,
2445-2453.
76.Bulut-Karslioglu, A., Perrera, V., Scaranaro, M.,
de la Rosa-Velazquez, I. A., van de Nobelen, S., Shukeir, N., Popow,
J., Gerle, B., Opravil, S., Pagani, M., Meidhof, S., Brabletz, T.,
Manke, T., Lachner, M., and Jenuwein, T. (2012) Nat. Struct. Mol.
Biol., 19, 1023-1030.
77.Seong, K. H., Li, D., Shimizu, H., Nakamura, R.,
and Ishii, S. (2011) Cell, 145, 1049-1061.
78.Jia, S., Noma, K., and Grewal, S. I. (2004)
Science, 304, 1971-1976.
79.Pirrotta, V., and Li, H. B. (2012) Curr. Opin.
Genet. Dev., 22, 101-109.
80.Aguilo, F., Zhou, M. M., and Walsh, M. J. (2011)
Cancer Res., 71, 5365-5369.
81.Yang, L., Lin, C., Liu, W., Zhang, J., Ohgi, K.
A., Grinstein, J. D., Dorrestein, P. C., and Rosenfeld, M. G. (2011)
Cell, 147, 773-788.
82.Iborra, F. J., Pombo, A., Jackson, D. A., and
Cook, P. R. (1996) J. Cell Sci., 109, 1427-1436.
83.Rasin, S. V., Gavrilov, A. A., and Yarovaya, O.
V. (2010) Biochemistry (Moscow), 75, 1307-1315.
84.Edelman, L. B., and Fraser, P. (2012) Curr.
Opin. Genet. Dev., 22, 110-114.
85.Caudron-Herger, M., and Rippr, K. (2012) Curr.
Opin. Genet. Dev., 22, 179-187.