REVIEW: Biogenesis, Evolution, and Functions of Plant microRNAs
P. P. Pashkovskiy1 and S. S. Ryazansky2*
1Timiryazev Institute of Plant Physiology, Russian Academy of Sciences, ul. Botanicheskaya 35, 127276 Moscow, Russia; E-mail: pashkovskiy.pavel@gmail.com2Institute of Molecular Genetics, Russian Academy of Sciences, Kurchatov sq. 2, 123182 Moscow, Russia; E-mail: ryazansky@img.ras.ru
* To whom correspondence should be addressed.
Received January 21, 2013
This review focuses on the biological role of one class of plant small RNAs, ~22-nt microRNAs (miRNAs). The majority of plant miRNA targets are genes encoding the effector factors of cell signaling pathways. The regulation of their expression is necessary for both ontogenesis and rapid response of plants to biotic and abiotic stress factors. We also summarized current views on the biogenesis and evolution of plant miRNAs as well as the techniques used for their investigation.
KEY WORDS: microRNA, stress, plantDOI: 10.1134/S0006297913060084
BIOGENESIS AND MODE OF ACTION OF PLANT miRNAs
To date, several thousands of plant miRNAs related to more than 150 families are known (http://www.mirbase.org). The majority of miRNAs from model plants (e.g. Arabidopsis thaliana) are already described, and the extension of the miRNA list happens due to studies of new non-model plant species [1]. In contrast to metazoan miRNA genes, which are often organized into clusters and are located inside the introns of protein-coding genes, all plant miRNA genes are located in the intergenic spacer regions and rarely form clusters [2]. The majority of metazoan and plant miRNA genes are transcribed by RNA-polymerase II giving a primary miRNA precursor (pri-pre-miRNA or pri-miRNA) (Fig. 1) [3]. Pri-miRNA is recognized and further processed into precursor miRNA (pre-miRNA) by Dicer protein (Fig. 1) [4]. Dicer contains an RNA-binding PAZ (Piwi/Argonaute/Zwille) domain and two catalytic domains of RNase III. Plants have several paralogs of Dicer, but only Dicer1 (DCL1) catalyzes pre-miRNA processing, while other members of this family are involved in protection against viruses [5]. Pre-miRNA is able to adopt specific hairpin structure, which takes place in so-called nuclear D-bodies [6]. Excision of pre-miRNA from pri-miRNA involves DDL and HYL1 proteins, which bind DCL1 [7, 8], and SERRATE interacting with HYL1 [9, 10]; the functional role of these proteins remains to be studied. Further, DCL1 protein cleaves the hairpin pre-miRNA liberating a short (21-23 nt) miRNA duplex. Similar to other types of RNAs processed by the enzymes of the RNase III family, miRNA duplex bears two protruding nucleotides at the 3′-ends [11]. miRNA duplex is stabilized due to the methylation of 2′-OH groups of 3′-end protruding nucleotides by S-adenosyl-methionine-dependent methyltransferase HEN1 (HuaEnhancer1) [12]. In the absence of methylation, uridyl transferase adds uridyl residues to miRNA, inducing its degradation by specific nucleases [13]. Protein HASTY, a homolog of metazoan exportin-5, interacts specifically with protruding nucleotides and carries the duplex from the nucleus to the cytoplasm (Fig. 1) [14].
Fig. 1. Scheme of miRNA processing in plants. Transcript of the miRNA gene, pri-miRNA, binds to DDL, SE, and HYL1 proteins and is processed by DCL1 to miRNA/miRNA* duplex. The duplex is methylated on its 3′-ends by HEN1 and is actively transported to the cytoplasm by HASTY. There, one strand of the duplex binds AGO1 protein and is loaded into the RISC complex. Complementary binding of miRNA to the mRNA target leads to the digestion of the mRNA by AGO1 protein. In animals the RISC complex induces silencing even if the complementarity with 3′-UTR of the mRNA is partial. See text for further details.
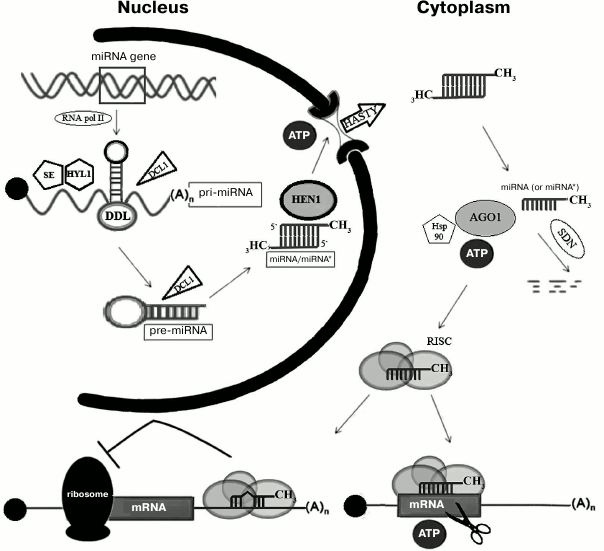
In the cytoplasm, one strand of miRNA duplex (mature, or “guide”, strand) is incorporated into miRNA–protein effector complex (RISC, RNA induced silencing complex) [15]. The second strand of the duplex (“passenger” strand), named miRNA*, is usually degraded (Fig. 1) [16, 17]. The choice of the guide strand is determined by the degree of stability of the ends of the duplex: the strand whose 5′-end is less stably paired will be selected as the guide strand [18, 19]. A key component of the effector complex RISC is a protein of the Argonaute (AGO) family. AGO proteins were first discovered in plants [20] and have been found in all eukaryotes, some archaea, and eubacteria (Aquifex aeolicus). It is believed that they evolved from translation initiation factors [21]. Usually, species encode several AGO paralogs, which participate in functioning of different types of small RNAs, including miRNAs. For example, there are 27 AGO paralogs in nematodes, 4 in mammals, and 10 in Arabidopsis [1]. AGO1 is involved in functioning of miRNAs in Arabidopsis [22], while in humans this role is played by the homologous hsAGO2 protein [23]. Metazoan AGO proteins are found predominantly in cytoplasmic P-bodies, where mRNAs are accumulated or degraded [24]. Plant RISC complex is recruited to the target mRNA, which is complementary to miRNA, and cleaves it due to the catalytic activity of AGO1 [22]. The interaction of a miRNA with AGO is an ATP-dependent process, which probably involves chaperone Hsp90, similar to animals [25].
Molecular mechanisms of AGO action and recognition of small RNAs appeared to be the same in plants and animals [26]. AGO proteins contain three domains with different functions – PAZ, MID, and PIWI domains. The MID (middle) domain binds the phosphate group on the 5′-end of the miRNA, which allows using the latter in several rounds of mRNA-target digestion. It should be noted that the MID domains of AGO paralogs from Arabidopsis have different affinities for different 5′-terminal nucleotides, thus binding different classes of small RNAs. AGO1 protein binds small RNAs (miRNA) if their first nucleotide is uridine, AGO2 and AGO4 prefer adenosine, while AGO5 prefers cytosine [27]. The PAZ domain binds the 3′-terminal nucleotide of the miRNA, in which ribose, as mentioned above, bears the 2′-hydroxymethyl modification. In animals, such modification of miRNAs has not been found [28]. Finally, the PIWI domain catalyzes the endonucleolytic cleavage of the phosphodiester bond between the tenth and eleventh nucleotide of the mRNA-target region, complemented to miRNA, thus generating products bearing 5′-phosphate and 3′-hydroxyl groups. The structure of the PIWI domain is similar to that of the ribonuclease catalytic domain of RNase H. The active center of PIWI is composed of three negatively charged amino acids, Asp-Asp-Glu, which bind Mg2+ ion [26].
Mechanisms of biogenesis and function of miRNAs in animals and plants are very similar. However, plant pre-miRNAs are longer (70-400 nt compared to 60-90 nt in animals) and have more complicated secondary structure [15, 29]. The terminal loop of plant pre-miRNA hairpin (20-75 nt compared to 7-12 nt in animals) is often branched [1, 30]. Plant pre-miRNAs, unlike metazoan ones, are conserved only in the region of the miRNA [19]. In animals, excision of pre-miRNA from pri-miRNA in the nucleus and of miRNA from pre-miRNA in the cytoplasm involves Drosha and Dicer proteins, respectively, while in plants in both processes in the nucleus only Dicer is involved. Metazoan mRNAs can contain several miRNA-binding sites, whereas plant mRNAs contain only one, with rare exceptions [29, 31, 32]. Silencing mechanisms are also significantly different (Fig. 1). In plants, miRNA is fully complementary to its mRNA target; binding of mRNA with miRNA-containing RISC complex leads to its cleavage by AGO. The majority of metazoan miRNAs are only partially complementary to the binding site, which is usually located in the 3′-UTR of mRNAs. Generally, the binding site contains several mismatches, including nucleotides 10 and 11, which prevents mRNA cleavage [33]. Perfect complementarity of the “seed” region (nucleotides 2-7) is indispensable for the recognition of the target [34]. In animals, miRNAs interfere with translation initiation, causing repression of gene expression [21]. It is assumed that AGO protein competes with translation initiation factor eIF-4E, which is consistent with the fact that AGO protein is able to bind the cap structure of mRNA [35]. Interestingly, in plants evolutionarily conserved miRNAs that are not fully complementary to mRNAs were found; in this case effective binding and cleavage of mRNA require the presence of 9-11 consecutive complementary base pairs [19, 36].
Regulation of microRNA expression. Mechanisms of regulation of miRNA processing and activity in plants are less studied than in animals. In the promoters of several plant miRNA genes, binding motifs of tissue-specific and stress-regulated transcription factors were found [37]. In A. thaliana a negative feedback regulation mechanism was discovered, in which genes coding for miRNA processing components are themselves miRNA targets. Thus, miR162 inhibits the expression of the DCL1 gene [38] and miR168 silence of the AGO1 gene [39]. It was also reported that DCL1 acts preferentially in vegetative organs, which may provide further tissue specificity for miRNA processing [5].
The timely degradation of miRNAs is also an important way of regulation of their activity. For example, in A. thaliana knockout of genes coding for SDN nucleases (small RNA dependent nucleases) cleaving miRNAs leads to their accumulation and appearance of numerous defects of plant development [40]. Similar nucleases in nematodes (XRN2/Rat1p) cleave small RNAs, including miRNAs, in the 5′→3′ direction, while in plants SDN nuclease acts in the opposite direction, from the 3′- to-5′-end [41, 42]. The presence of poly(U)-tail at the 3′-terminus of miRNAs inhibits their degradation by SDN nucleases [43].
An unusual mechanism of miRNA activity regulation, named “target mimicry”, which uses a long non-coding RNA (ncRNA), was demonstrated for miR399. Phosphate starvation induces the expression of the IPS1 pseudogene (Induced by Phosphate Starvation 1); the corresponding transcript contains several binding sites for miR399 [44]. Partial complementarity of IPS1 RNA with miR399 prevents its AGO-dependent cleavage, but it leads to a temporary decrease in free miR399 level. Therefore, another mRNA-target (PHO2) normally suppressed by miR399 becomes derepressed in case of phosphate starvation, which contributes to adaptation [44]. Later, metazoan ncRNAs with similar functions, called “competing endogenous RNAs” (ceRNAs), were also found [45].
miRNAs and ta-siRNAs. Study of miR173, miR828, and miR390 functions in Arabidopsis revealed several polyadenylated transcripts of non-coding genes as their targets, which are actually the precursors of another class of trans-acting small interfering RNAs (ta-siRNAs) [46]. It was found that ta-siRNA formation is triggered by miRNA: AGO1-dependent cleavage of RNA precursors initiates the binding of RNA-dependent RNA polymerase RDR6 with poly(A) tail of 3′-fragment and subsequent synthesis of the second RNA strand; the end of double-stranded RNA (dsRNA), which corresponds to the cleavage site, is recognized by the complex of two proteins, DRB4 and DCL4, and dsRNA is processed by DCL4 into 20-25-nt-long ta-siRNAs. Newly formed ta-siRNAs bind AGO proteins and suppress the expression of complementary genes through the cleavage of mRNA [46]. Since one round of precursor processing induces formation of multiple ta-siRNAs, one can assume that the latter amplify the effect of miRNAs. Thus, miR390 triggers the formation of many ta-siRNAs of TAS3 loci, which are able to regulate the expression of transcription factors ARF of the auxin hormone pathway. Interestingly, ta-siRNAs, unlike the initiating miRNAs, are capable of intercellular transport and participate in the formation of a gradient of regulated target expression within the tissues [46].
ORIGIN AND EVOLUTION OF miRNAs
It is proposed that the mechanism of gene silencing by complementary double-stranded RNAs (RNA interference, RNAi) is evolutionary ancient and was already present in the common eukaryotic ancestor. Loss of this mechanism in some eukaryotes (yeast) is considered to be as a recently acquired event [27]. Most probably, at the early stages of eukaryote evolution proteins similar to Dicer, Argonaute and RNA-dependent RNA polymerases being involved in some cell functions were recruited to the RNAi process by chance. Indeed, putative precursors of the Ago protein family played the role of translation initiation factors [47]. The most ancient function of the RNAi system is, obviously, the defense of a genome from penetration of exogenous genetic material, primarily from viruses. The acquisition by the RNAi mechanism of the ability to regulate the gene expression (by miRNA) can be considered as more recent evolutionary event [48]. This mechanism has opened for ancient eukaryotes wide opportunities for fine-tune regulation of cell activity in a constantly changing environment, which promoted its preservation in evolution.
Among thousands of miRNAs identified in plants, 100 families have been found in A. thaliana, and some of them have been found in mosses and Lycopsida. Out of 23 miRNAs families studied in A. thaliana, representatives of 11 families were also found in gymnosperm plants and eight families in ferns [49]. These conserved miRNAs, including miR156, miR160, miR319, and miR390, regulate expression of key transcription factors, which govern the transition of a plant into a new ontogenetic state, meristem formation, differentiation of cells and organs, and functioning of signaling pathways [50]. Over 400 million years, plant miRNAs (miR156, miR160, miR166, miR167, miR171, miR319, miR390, miR395, miR408, miR477, miR529, miR535, miR894) and their binding sites on mRNA-targets remain conserved [49, 51]. These facts clearly point to an ancient evolutionary origin of plant miRNAs and to the presence of conserved miRNAs that play a special role in regulation of ontogenesis. It should be noted that conserved miRNAs of multicellular plants were not found in the unicellular alga Chlamydomonas reinhardtii [52]. Moreover, only two miRNAs common to plants and animals (miR854 and miR855), which regulate the expression of genes from the same family of nuclear RNA-binding proteins hnRNP/UBP1, were found [53].
Despite the fact that ancient plant miRNA genes are very conservative ones, some of them demonstrate polymorphism. For example, miRNA orthologs related to the conservative miR319 family show differences in pre-miRNA sequences and cis-regulatory elements of corresponding genes. This explains the species-specific particularities in regulation of their expression at transcriptional and post-transcriptional levels [54, 55]. In comparison to A. thaliana and pine tree, some miRNA genes of maize (miR319) that are involved in the mechanism of response to hypoxia contain several point mutations [56]. One can assume that the increase in plant adaptive capacity favored the stabilization of miRNA polymorphism in evolution.
Apart from conservative miRNAs, many non-conservative, species-specific miRNAs were found in plants. In early studies, it was not possible to identify evolutionarily young, poorly expressed miRNAs among highly expressed conservative ones. Only the development of new deep sequencing technologies for small RNAs overcame this methodological problem. It turned out that in plants there are much more species-specific miRNAs than conservative ones [2]. Systematic studies of mechanisms of cell specialization and plant metabolism under stress conditions also contribute to identification of novel non-conservative and species-specific miRNAs [57].
Conservative plant miRNA genes are represented by large families of homologous genes that have resulted from multiple duplications during evolution. The origin of new species-specific miRNA genes remains to be studied. Two possible mechanisms of their formation are being discussed. According to the first hypothesis, miRNA genes are derived from inverted duplications of protein-coding genes [58, 59]. Indeed, the miR163 gene has no orthologs or paralogs, and its pre-miRNA is homologous only to genes of S-adenosylmethionine-dependent methyltransferases [60]. It is believed that originally inverted repeats of protein-coding genes generated a population of heterogeneous siRNAs, similar to those, triggering the RNAi mechanism, formed by DCL4 and DCL3 from perfect hairpins [61]. If DCL1 is involved in processing, the hairpin is sliced in strictly defined positions, which leads to the generation of a set of discrete small RNAs. If the regulation of expression of the original protein-coding gene by newly formed discrete small RNAs turned out to be useful, for example, for reproductive isolation or survival under stress conditions, the corresponding region of the hairpin will undergo stabilizing evolutionary selection. At the same time, the sequences of the hairpin flanking the miRNA/miRNA* region will undergo directional evolutionary selection, which improves its stability and overall processing efficiency. Combination of stabilizing and directional selection results in the formation of a new miRNA gene, which will not be similar to the original protein-coding gene [58]. This model of the origin of miRNA genes is consistent with the low level of expression of young miRNA genes, which is essential for the reduction of energy costs during ATP-dependent formation of siRNA-like populations of small RNAs at the early stages of evolution [5]. This model is supported by the fact that DCL1 participates in processing of evolutionarily old pre-miRNAs, and DCL3 in processing of young ones [5]. The analysis of young miR824 gene polymorphism confirms the fact that the hairpin undergoes directional selection [62].
According to the second hypothesis of “spontaneous evolution”, miRNA genes derive from short- and medium-length partially self-complementary or inverted sequences, many of which are distributed randomly throughout the plant genome. Spontaneous transcription of such sequences will generate RNAs with hairpin-like secondary structure. If these sequences will be recognized by the pre-miRNA processing machinery and if the potential mRNA target with a partially complementary sequence will be present, subsequent stabilization of the functional link through the co-evolution of hairpin and target sequences may lead to fixation of new miRNAs [63]. Non-autonomous transposable elements, in particular small inverted repeats like MITE, could also be the source of new miRNAs [64]. Apparently, some miRNAs of rice emerged from mobile genetic elements [1]. A similar model of miRNA gene origin was also proposed for animals [65].
ROLE OF miRNAs IN STRESS RESPONSE
miRNAs are common regulators of gene expression and are involved in a large number of cellular processes [29]. It is believed that miRNAs form a regulatory network that helps in the spatial and temporal regulation of gene expression during development. Most of miRNA target genes encode transcription factors and other regulatory proteins associated with plant development. For example, miR393 and miR160 are involved in auxin homeostasis by regulating gene expression of auxin receptors and effector proteins of the auxin signaling pathway. Their absence in Arabidopsis results in abnormal plant development, serration or curling of leaf margins, formation of abnormal flowers, acceleration of flowering time, and reduced fertility [66]. The same miRNAs are involved in lateral root formation in gymnosperm and angiosperm plants [67]. miR164 is involved in petal and pistil organogenesis, regulating the expression of the Cup Shaped Cotyledon gene family (CUC1, 2, 3) [68]. RNAs miR165 and miR166 regulate genes of Phabulosa (PHB), Phavoluta (PHV), Revoluta (REV) and Kanadi (KAN1, 2, 3) families that are important for organ polar growth and correct orientation of stomata, xylem, and phloem [69]. The formation of flower primordia requires a complex work of several genes. One of these genes is APETALA2 (AP2) whose mRNA contains a binding site for miR172; overexpression of miR172 leads to the replacement of perianth generative tissues by somatic tissues [70].
Although the main function of plant miRNAs is associated with regulation of ontogenesis, to date an important set of experimental data has accumulated suggesting their possible role in stress response – neutralization of viral and bacterial infections, signal transduction under starvation or in presence of an excess of mineral elements, and regulation of genes of the antioxidant machinery in case of temperature changes and osmotic stress. Under stress conditions, plant growth slows significantly [71], which is, most likely, due to the perturbations in normal functioning of miRNAs and specific activity of certain miRNAs. The molecular mechanism of action under stress conditions is known only for some miRNAs, while for the other miRNAs only data concerning the alteration of their expression level is available. Although changes in expression of the majority of miRNAs are probably the result of nonspecific plant response to environmental conditions, some common tendencies reflecting the possible existence of groups of functionally similar miRNAs can be found.
First of all, one can discriminate a group of miRNAs whose expression pattern varies in almost all stress conditions (for example, miR393, miR160, and miR167) and a group of stress-specific miRNAs responding only to certain types of stress (miR395, miR399, etc.) (Fig. 2, a and b) [72]. The first group comprises miRNAs whose level of expression varies similarly or in opposite directions under stress conditions. For example, it was found that drought or salt stress causes the upregulation of miR393, miR160, and miR167 in most plant species (Fig. 2, a and b) accompanied by a slowing of plant growth and development rates [72]. As described below, these miRNAs are involved in regulation of auxin signaling pathway that is important for all aspects of plant physiology. As an example of miRNA whose expression changes in different directions in species-specific manner, one can take miR169, which is involved in drought and salt stress response in A. thaliana, Medicago sativa [73], and Triticum aestivum. Under stress conditions this RNA is downregulated, while its target, transcription factor NFYA5, is activated [74]. Artificial overexpression of NFYA5 increases the resistance of A. thaliana to drought, while miR169 overexpression reduces it [75]. In rice, on the contrary, drought induces upregulation of miR169g [75] (Fig. 2a), indicating that miR169 orthologs have different functions in these species. Cold provokes the upregulation of miR168 in poplar and A. thaliana and, in contrast, downregulation in rice; the opposite effects were observed for miR171 (Fig. 2, a and b) [76].
Fig. 2. Venn diagrams showing the repertoire of miRNAs with changing level of expression under abiotic stress conditions in dicotyledonous (A. thaliana) (a) and monocotyledonous (O. sativa) (b) plants.
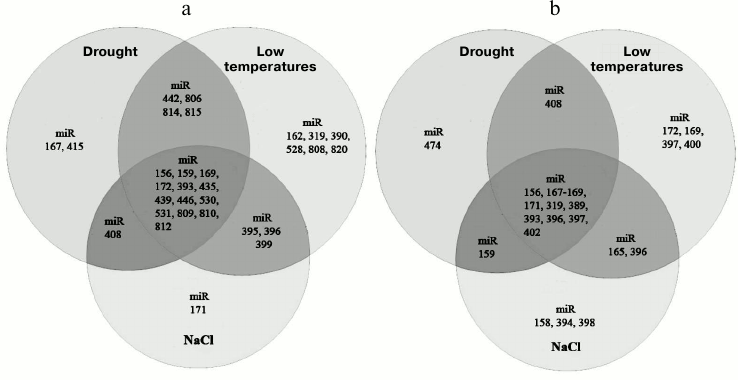
In different varieties with different degree of stress tolerance, the levels of miRNA expression under stress conditions may vary to different extents. Thus, drought induces upregulation of miR166, miR169 and miR1513 in a drought-sensitive variety of soybean, while in drought-tolerant varieties these miRNAs are downregulated [72]. A similar effect is observed in maize varieties with different degree of salt sensitivity [77]. Heating of wheat seedlings to 40°C revealed differences in miRNA expression profiles of heat-sensitive and heat-tolerant varieties of wheat [78]. These miRNAs are probably involved in determination of the degree of stress-adaptation in different varieties.
Finally, miRNA expression levels may change under stress conditions in different directions in different organs. For example, in barley dehydration induces the upregulation of miR166, miR156, miR171, and miR408 only in leaves, while in roots, to the contrary, these miRNAs are downregulated [79]. Similarly, under osmotic stress in Thellungiella salsuginea (halophila) miR398 is downregulated in roots, but upregulated in leaves [80]. This effect is probably determined by the different degree of susceptibility of different organs to the stress and the necessity to generate a biological response especially in those tissues that are more exposed to the stress; it may also indicate the existence of miRNA transport between different organs.
Role of miRNAs in auxin homeostasis under stress. Common strategies of plant survival under stress conditions include slowing of growth and the reduction of metabolic rate, which are needed for the redistribution of energy and other resources for better adaptation. The auxin signaling pathway is the main regulator of plant growth and development; it is involved in the development of adaptation to various types of stresses [81]. In this pathway the binding of auxin to the auxin receptor TiR1 results in the degradation of Aux/IAA proteins and activation of multiple transcription factors ARF (auxin response factor), which are normally suppressed by Aux/IAA proteins. ARF factors regulate the transcription of the Aux/IAA and GH3 genes and short auxin-dependent RNA (SAUR) [82]. It was demonstrated that in Arabidopsis eight ARF transcription factors out of 23 are targets for two miRNAs and ta-siRNAs: ARF10, ARF16, and ARF17 are targets for miR160; ARF6 and ARF8 are the targets for miR167; ARF2, ARF3, and ARF4 are the targets for miR390-dependent ta-siRNAs of the TAS3 locus [72]. Among them, ARF6 and ARF8 are transcription activators, and the others are repressors. Moreover, mRNA of the auxin receptor TiR1 itself is a target of miR393. Drought, salinity, abscisic acid, ultraviolet (UV-B, 280-315 nm), high and low temperatures, as well as bacterial infections cause the upregulation of miR160, miR167, and miR393 in different plant species [72]. The upregulation of miR160 and miR167 provokes the upregulation of their targets ARF6 and ARF8 and, conversely, the downregulation of ARF10, ARF16, and ARF17. On the other hand, the upregulation of miR393 entails the downregulation of TiR1, which ultimately leads to the inactivation of certain ARF transcription factors. Under different stress conditions miR390 is downregulated, stimulating the expression of ARF2, ARF3, and ARF4 [72]. Although the precise role of each ARF transcription factor in stress response is not fully understood, it is clear that differential regulation of ARF gene expression, mediated by miRNAs, can play an important role in adaptation and slowing of plant growth rates. It is interesting to note that miR160 activation under stress conditions leads not only to the downregulation of its target, the transcriptional repressor ARF17, but also stimulates the transcription of the GH3 gene regulated by ARF17. The GH3 gene codes for an auxin-binding protein; therefore, its upregulation under stress conditions probably leads to decrease in the intracellular level of free auxin [66]. Thus, another potential function of miR160 is the reduction of the amount of auxin to the normal level, which can help in restoration of the ability of the plant to respond to the new stress conditions. Indeed, miR160a null mutants of A. thaliana demonstrated decreased response to auxin [72].
Role of miRNAs in macronutrient homeostasis. Plants developed different mechanisms of adaptation to nitrogen starvation. In A. thaliana, miR393 is involved in activation of growth and formation of the root system induced by the absence of nitrogen; the upregulation of this miRNA is detected already two hours after the reduction of the amount of available nitrogen [83]. Activation of miR393 in roots leads to downregulation of its target AFB3 coding for a component of the auxin signaling pathway and inhibiting the growth of primary and secondary roots. A similar mode of action was shown for miR167, which induces the downregulation of transcription factor ARF8 that stimulates the formation of secondary roots. During nitrogen starvation, a decrease in miR167 amount in the pericycle and the root cap of A. thaliana was observed, accompanied by the upregulation of ARF8 [84].
When phosphate, a key player of cell energy-dependent processes (in the form of ATP), is lacking in the medium a rapid upregulation of miR399, miR827, miR2111, miR156, miR778, miR828, miR169, miR395, miR398, and miR399* is detected, which indicates the important role of miRNA in maintenance of the phosphate level in plants [85]. Among all these miRNAs, the function of only one miR399 in A. thaliana is studied. Upregulation of miR399 leads to the downregulation of its target – mRNA of ubiquitin ligase PHO2/UBC24, which is involved in their transport from old to young leaves [86]. Interestingly, miR827 and miR2111, also upregulated by phosphate starvation, downregulate genes coding for ubiquitin ligases E3 [87]. This might signify that phosphate starvation can induce the inhibition of ATP-dependent processes of protein degradation in proteasomes, which reduces in turn the need for energy-consuming new protein synthesis.
Sulfur, available for plants in the soil in the form of sulfates, is incorporated in cysteine, methionine, polypeptides (e.g. glutathione), phenolic compounds phytoalexins, and glucosinolates, which play an important role in plant development in optimal and stress conditions. During sulfate deprivation, sulfur uptake and metabolism are regulated at different levels, and miRNAs also play a role in these processes. Thus in Arabidopsis, miR395 induced during sulfate deprivation regulates the expression of genes coding for the membrane sulfate transporters SULTR2, APS1, APS3, and APS4 and is involved in sulfate redistribution between leaves [88]. It is also known that in colza that miR395, miR156, miR160, miR164, miR167, miR168, and miR394 are also upregulated during sulfate deprivation, suggesting that these miRNAs might potentially regulate the expression of genes coding for ARF transcription factors of the auxin-signaling pathway [88].
Role of miRNAs in micronutrient homeostasis. Copper is a cofactor of many enzymes, such as plastocyanin, necessary for photosynthesis; cytochrome c, a component of the mitochondrial redox chain; and ethylene response proteins. Under stress conditions, copper participates in the neutralization of reactive oxygen species. In plants, conservative miR397, miR398, miR408, and miR857 are involved in regulation of copper homeostasis [89]. These miRNAs can regulate the expression of genes coding for copper-containing superoxide dismutases (Cu/Zn-SOD), plastocyanin, and some laccases (laccases 2-4, 7, 12, 13, and 17) [72]. The functions of miR398 are the most studied. During copper deprivation, miR398 is activated and leads to the downregulation of copper-containing Cu/Zn-SODs with different cellular localization (CSD1, CSD2) and the CCS1 gene, coding for SOD chaperone, in order to preserve the activity of copper-containing proteins involved in the most important processes of photosynthesis and respiration, plastocyanin and cytochrome c oxidase [57, 90]. It is interesting to mention that during oxidative stress miR398, in contrast, is downregulated. This is due to the fact that under oxidative stress the concentration of reactive oxygen species increases dangerously, and for their urgent detoxification elevated activity of superoxide dismutases CSD1 and CSD2 is needed [91]. Thus, miR398 is an interesting example of a universal miRNA whose expression changes depending upon the physiological state of the plant, and it is involved in the development of cell response to different types of stress.
Under certain conditions, miRNAs can selectively regulate the expression of target genes. Thus, abiotic stress-induced downregulation of miR398 leads to the upregulation of both CSD1 and CSD2, while under biotic stress the downregulation of miR398 leads to the upregulation of only CSD1, but not CSD2 [92]. Similarly, activation of miR393 in the absence of nitrogen leads only to the downregulation of AFB3, having no effect on three other AFB genes with complementary binding sites [83]. The mechanism of this selective regulation remains unclear. Perhaps miRNA expression and expression of a target gene are spatially and temporally separated, as was shown for miR395 and AST68 [88]. It is also possible that RISC complexes do not recognize certain mRNA targets because of their interaction with other RNA-binding proteins or because of the secondary structure, as shown in animals [93].
Iron, copper, zinc, and molybdenum micronutrients are also involved in biological processes, and plants need them in certain amounts. On the other hand, compounds containing these elements can accumulate in tissues, causing a number of diseases. Lead, mercury, cadmium, and aluminum have no beneficial role in biological processes and are toxic [94]. They cause a cytostatic effect in roots first of all, and the accumulation of these metals causes oxidative stress, lipid peroxidation, and DNA damage. Although miRNAs specifically responsible for the heavy metal response have not been identified, expression of several miRNAs regulating plant growth rate under stress conditions is altered in presence of heavy metals [95]. These miRNAs include miR319, which regulates transcription factor TCP, miR393, regulating the auxin receptor gene TIR1, and miR390, which directly regulates the genes of transcription factors of the auxin signaling pathway ARF [96]. Treatment of dicotyledonous plants with cadmium changes the expression of conserved miR160 and miR167 (target – ARF), and miR164 (target – CUC2, necessary for meristem formation). In monocotyledonous plants, cadmium alters the expression of miR602 and miR604. miR602 regulates the gene of xyloglucan endotransglucosylase/hydrolase – an enzyme that catalyzes the temporal reorganization of the cell wall polysaccharides and thus promotes cell growth [68, 97]. miR604 regulates the expression of the lipid transporter gene (LTP), which is involved in hormone regulation, cutin and wax formation, as well as in plant response to pathogens [98].
Role of miRNAs in immune response to pathogens. Plants, which are affected by bacterial or viral pathogens, are able to detect the latter at the molecular level and to protect organs and tissues by changing the activity of protective and signaling genes by changing the levels of hormones and other metabolites [99]. Although siRNAs are the main type of small RNAs that is involved in plant protection against bacterial and especially viral infections [99], there are some data about miRNAs participating in these processes. Thus, leaf infection of A. thaliana with bacterial pathogen P. syringae leads to the upregulation of miR393, miR319, miR158-160, and miR165-167 and downregulation of miR390, miR408, and miR398 [100]. Upregulation of miR393 was also observed after treatment of plants with bacterial protein flagellin-22 [79]. At the same time, as mentioned before, miR390, miR393 miR160, and miR167 regulate the auxin signaling pathway necessary for increasing host plant resistance to infection.
Role of miRNA* passenger strand in stress tolerance. Initially, miRNAs* generated from pre-miRNAs and complementary to functional mature miRNAs were thought to be quickly degraded and accumulate only in tiny quantities. However, it was shown for Drosophila and mammals that miRNAs* may also play an important role in regulation of gene expression [101]. Moreover, so-called effect of “arm switching” is often observed during pre-miRNA processing, when miRNAs* are accumulated in larger amounts than corresponding miRNAs [101]. Similarly, in plants miRNAs* can be accumulated to a significant physiological level under certain conditions. For example, miR393* is highly expressed in leaves of A. thaliana infected with bacterial pathogen P. syringae. Upregulation of miR393* promotes plant resistance to bacterial infection by regulating expression of the vacuole-localized product of MEMB12 (SNARE) gene that is able to influence the process of cell vesicular transport [100]. Interestingly, miR393 and miR393* regulate the expression of TiR1 and SNARE, respectively, which are involved in promoting plant resistance to pathogens. miR399* accumulates in leaves of A. thaliana under phosphate deprivation [85, 86]. miR395* is constitutively expressed at high levels in sorghum (Sorghum moench) grown under optimal nutrient conditions. Probably, functions of miR395*, in contrast to miR395, might not be related to sulfur metabolism [72]. It become obvious that in order to list all possible functions of miRNAs under normal and stress conditions, targets of both guide and passenger strands of miRNA should be considered.
METHODS OF STUDYING miRNAs
Methods of studying miRNAs include different versions of nucleic acid hybridization (in situ, Northern-blot hybridization, microarrays, etc.), different variants of PCR, and next-generation deep-sequencing technology. With the help of bioinformatics, these approaches provide a possibility to characterize the special features of miRNAs and can help to avoid ambiguous interpretations of data.
Nucleic acid hybridization using radioactive probes (Northern-blot analysis), used to determine level and profile of miRNA expression, is highly sensitivity and allows detection of low quantities of small RNAs. In many cases this method is sufficient. However, the specificity of this method is not enough for a differential analysis of closely related miRNAs from the same family that differ only in several nucleotides. This problem can be partially overcome by using probes based on LNA (locked nucleic acid), in which 2′- and 4′-OH groups of ribose are bound with a methylene bridge. DNA probe containing LNA-nucleotides recognizes miRNA more specifically due to the increase in hybridization temperature [102]. Another disadvantage of hybridization is the need for a significant amount of the material (up to 100 µg of total RNA), which in some cases is not possible. In situ hybridization combined with confocal microscopy allows determining the spatial and temporal miRNA expression patterns. However, this approach is semi-quantitative and is used only in combination with other methods.
Microarray technology allows analysis of the level of expression of many miRNA genes simultaneously. This method has a number of obvious advantages compared to standard methods of nucleic acid hybridization: this high-throughput technology allows making the analysis rather quickly using a very well developed system of positive and negative controls. However, this method is also semi-quantitative, and results obtained by this method require further verification [102].
Another method to measure miRNA expression level is PCR. First, one can use long (up to 2 kb) polyadenylated miRNA precursors (pri-miRNAs) as a template for reverse transcription followed by quantitative PCR. However, in this case the estimated number of mature miRNAs will be not correct, while pri-miRNA processing is a multistep regulated process. Currently, a wide range of PCR variations are being developed involving the ligation of oligonucleotide adapter molecules to mature miRNAs with subsequent amplification with primers designed for adapter and miRNA. These methods can be used for most miRNAs; however, in some cases methylation of 3′-end of miRNA (in plants) makes it difficult to use it [103].
One of the fast developing methods of studying miRNA functions is the method of next-generation deep-sequencing (NGS) [104]. This method provides the means for quick and precise identification of miRNA and pre-miRNA sequences, and it allows analysis of the number of copies of a miRNA in one individual sample. However, this method requires expensive equipment and sophisticated technique for sample preparation. It includes mandatory computer processing that requires high computational performance [105].
Another powerful technique is the directed mutagenesis of miRNA pathway components and miRNAs themselves. This method allows rather accurate study of specific features of biogenesis, regulation, and functioning of miRNA in cells and certain physiological processes. However, this method is time-consuming and can produce false-positive results.
Bioinformatics remains an integral part of all methods aimed at studying of miRNAs [106]. Computer modeling and modern software have made possible experiments in silico, providing the possibility to work with one or many sequences to predict possible miRNA targets, visualize models of hairpin precursors, simulate global regulatory miRNA–mRNA networks, and analyze the results of sequencing. Bioinformatics is an essential part of any experiment aimed at studying small RNAs (particularly miRNAs), which makes them efficient. However, this method always requires experimental verification [1].
Study of miRNAs developed into an independent field rather recently. Nowadays commercial antibodies and oligonucleotides are available, facilitating fast and budget-friendly analysis of miRNAs for a wide range of researchers. Study of specific features of miRNAs and expression of their target genes, miRNA processing regulation, and functioning of RISC complex requires a complex approach with a combination of methods including indispensable bioinformatic analysis [1].
A wide range of methods developed over the past ten years has enriched our knowledge about miRNA-mediated regulation of gene expression and allowed us to study in detail the process of miRNA biogenesis, their mode of action, and their biological functions. The field of plant miRNAs is still at the stage of data accumulation. It became obvious that regulation of most biological processes in plants is somehow linked to miRNA activity. Some of the main plant miRNA targets are the genes of universal transcription factors involved in the control of growth and development as effector components of signaling pathways, including, first of all, ARF genes of the auxin signaling pathway. Salt stress and drought- and micronutrient depletion-induced changes in expression of these miRNAs lead to slowing of growth and contribute to development of adaptation. However, functions of many other miRNAs whose expression change under stress conditions are still unknown, but they are intensively studied.
In conclusion, the practical value of studying miRNA functions on agronomic crops should be mentioned. On one hand, cultivated plants are exposed to a range of biotic and abiotic stresses, compared to the model plants, and therefore represent a suitable system for studying miRNAs functions in nature. In depth study of these functions opens perspectives to create next-generation transgenic crops (for example, with altered level of expression of individual miRNAs), which are able to better realize their adaptive capacity to maintain productivity under stress conditions. Another prospective will be to use cultivated plants to produce active substances for medical purposes. It was shown recently that plant miR168a, consumed by mammals, is found in their serum and tissues; in liver this miRNA is capable to downregulate the LDLRAP1 gene (low-density lipoprotein receptor adapter protein 1), thus adjusting the low-density lipoprotein content in plasma [107]. A significant level of miR168a expression in rice – one of the most popular agronomic crops – opens the possibility to use this effect for treatment and prevention of atherosclerosis in humans [107]. If preservation of activity of consumed miRNA is a common phenomenon, it may be possible to use transgenic plants with altered expression of miRNAs for therapeutic purposes.
The authors are grateful to V. V. Kuznetsov and V. A. Gvozdev for precious advice in writing this review.
This work was partially supported by grants from the federal program “Scientific and Pedagogical Personnel of Innovative Russia for 2009-2013” (No. 14.740.11.1484) and Russian Foundation for Basic Research (No. 12-04-31691 and 11-04-01305a) as well as by the program of fundamental research of the Russian Academy of Sciences “Molecular and Cellular Biology”.
REFERENCES
1.Voinnet, O. (2009) Cell, 136,
669-687.
2.Zhang, B., Pan, X., Cannon, C. H., Cobb, G. P., and
Anderson, T. A. (2006) Plant J., 46, 243-259.
3.Lee, Y., Kim, M., Han, J., Yeom, K. H., Lee, S.,
Baek, S. H., and Kim, V. N. (2004) EMBO J., 23,
4051-4060.
4.Kim, V. N. (2005) Nat. Rev. Mol. Cell Biol.,
6, 376-385.
5.Vazquez, F., Blevins, T., Ailhas, J., Boller, T.,
and Meins, F., Jr. (2008) Nucleic Acids Res., 36,
6429-6438.
6.Fang, Y., and Spector, D. L. (2007) Curr.
Biol., 17, 818-823.
7.Yu, B., Bi, L., Zheng, B., Ji, L., Chevalier, D.,
Agarwal, M., Ramachandran, V., Li, W., Lagrange, T., Walker, J. C., and
Chen, X. (2008) Proc. Natl. Acad. Sci. USA, 105,
10073-10078.
8.Kurihara, Y., Takashi, Y., and Watanabe, Y. (2006)
RNA, 12, 206-212.
9.Yang, L., Liu, Z., Lu, F., Dong, A., and Huang, H.
(2006) Plant J., 47, 841-850.
10.Lobbes, D., Rallapalli, G., Schmidt, D. D.,
Martin, C., and Clarke, J. (2006) EMBO Rep., 7,
1052-1058.
11.Xie, Z., Allen, E., Fahlgren, N., Calamar, A.,
Givan, S. A., and Carrington, J. C. (2005) Plant Physiol.,
138, 2145-2154.
12.Yu, B., Yang, Z., Li, J., Minakhina, S., Yang,
M., Padgett, R. W., Steward, R., and Chen, X. (2005) Science,
307, 932-935.
13.Li, J., Yang, Z., Yu, B., Liu, J., and Chen, X.
(2005) Curr. Biol., 15, 1501-1507.
14.Park, M. Y., Wu, G., Gonzalez-Sulser, A.,
Vaucheret, H., and Poethig, R. S. (2005) Proc. Natl. Acad. Sci.
USA, 102, 3691-3696.
15.Reinhart, B. J., Weinstein, E. G., Rhoades, M.
W., Bartel, B., and Bartel, D. P. (2002) Genes Dev., 16,
1616-1626.
16.Matranga, C., Tomari, Y., Shin, C., Bartel, D.
P., and Zamore, P. D. (2005) Cell, 123, 607-620.
17.Chapman, E. J., and Carrington, J. C. (2007)
Nat. Rev. Genet., 8, 884-896.
18.Schwarz, D. S., Hutvagner, G., Du, T., Xu, Z.,
Aronin, N., and Zamore, P. D. (2003) Cell, 115,
199-208.
19.Jones-Rhoades, M. W., and Bartel, D. P. (2004)
Mol. Cell, 14, 787-799.
20.Bohmert, K., Camus, I., Bellini, C., Bouchez, D.,
Caboche, M., and Benning, C. (1998) EMBO J., 17,
170-180.
21.Humphreys, D. T., Westman, B. J., Martin, D. I.,
and Preiss, T. (2005) Proc. Natl. Acad. Sci. USA, 102,
16961-16966.
22.Baumberger, N., and Baulcombe, D. C. (2005)
Proc. Natl. Acad. Sci. USA, 102, 11928-11933.
23.Beitzinger, M., Peters, L., Zhu, J. Y., Kremmer,
E., and Meister, G. (2007) RNA Biol., 4, 76-84.
24.Wang, Y., Juranek, S., Li, H., Sheng, G., Wardle,
G. S., Tuschl, T., and Patel, D. J. (2009) Nature, 461,
754-761.
25.Iki, T., Yoshikawa, M., Nishikiori, M., Jaudal,
M. C., Matsumoto-Yokoyama, E., Mitsuhara, I., Meshi, T., and Ishikawa,
M. (2010) Mol. Cell, 39, 282-291.
26.Yuan, Y. R., Pei, Y., Ma, J. B., Kuryavyi, V.,
Zhadina, M., Meister, G., Chen, H. Y., Dauter, Z., Tuschl, T., and
Patel, D. J. (2005) Mol. Cell, 19, 405-419.
27.Cenik, E. S., and Zamore, P. D. (2011) Curr.
Biol., 21, R446-R449.
28.Ghildiyal, M., and Zamore, P. D. (2009) Nat.
Rev. Genet., 10, 94-108.
29.Bartel, D. P. (2004) Cell, 116,
281-297.
30.Iwai, N., and Naraba, H. (2005) Biochem.
Biophys. Res. Commun., 331, 1439-1444.
31.Pillai, R. S., Artus, C. G., and Filipowicz, W.
(2004) RNA, 10, 1518-1525.
32.Aleman, L. M., Doench, J., and Sharp, P. A.
(2007) RNA, 13, 385-395.
33.Haley, B., and Zamore, P. D. (2004) Nat.
Struct. Mol. Biol., 11, 599-606.
34.Lewis, B. P., Burge, C. B., and Bartel, D. P.
(2005) Cell, 120, 15-20.
35.Kiriakidou, M., Tan, G. S., Lamprinaki, S., De
Planell-Saguer, M., Nelson, P. T., and Mourelatos, Z. (2007)
Cell, 129, 1141-1151.
36.Dunoyer, P., Lecellier, C. H., Parizotto, E. A.,
Himber, C., and Voinnet, O. (2004) Plant Cell, 16,
1235-1250.
37.Kawashima, C. G., Yoshimoto, N.,
Maruyama-Nakashita, A., Tsuchiya, Y. N., Saito, K., Takahashi, H., and
Dalmay, T. (2009) Plant J., 57, 313-321.
38.Xie, Z., Kasschau, K. D., and Carrington, J. C.
(2003) Curr. Biol., 13, 784-789.
39.Vaucheret, H., Vazquez, F., Crete, P., and
Bartel, D. P. (2004) Genes Dev., 18, 1187-1197.
40.Ramachandran, V., and Chen, X. (2008)
Science, 321, 1490-1492.
41.Kennedy, S., Wang, D., and Ruvkun, G. (2004)
Nature, 427, 645-649.
42.Chatterjee, S., and Grosshans, H. (2009)
Nature, 461, 546-549.
43.Kai, Z. S., and Pasquinelli, A. E. (2010) Nat.
Struct. Mol. Biol., 17, 5-10.
44.Franco-Zorrilla, J. M., Valli, A., Todesco, M.,
Mateos, I., Puga, M. I., Rubio-Somoza, I., Leyva, A., Weigel, D.,
Garcia, J. A., and Paz-Ares, J. (2007) Nat. Genet., 39,
1033-1037.
45.Salmena, L., Poliseno, L., Tay, Y., Kats, L., and
Pandolfi, P. P. (2011) Cell, 146, 353-358.
46.Allen, E., and Howell, M. D. (2010) Semin.
Cell Dev. Biol., 21, 798-804.
47.Anantharaman, V., Koonin, E. V., and Aravind, L.
(2002) Nucleic Acids Res., 30, 1427-1464.
48.Buchon, N., and Vaury, C. (2006) Heredity
(Edinb.), 96, 195-202.
49.Axtell, M. J., and Bowman, J. L. (2008) Trends
Plant Sci., 13, 343-349.
50.Martin, R. C., Liu, P. P., Goloviznina, N. A.,
and Nonogaki, H. (2010) J. Exp. Bot., 61, 2229-2234.
51.Arazi, T. (2012) Plant Mol. Biol.,
80, 55-65.
52.Molnar, A., Schwach, F., Studholme, D. J.,
Thuenemann, E. C., and Baulcombe, D. C. (2007) Nature,
447, 1126-1129.
53.Arteaga-Vazquez, M., Caballero-Perez, J., and
Vielle-Calzada, J. P. (2006) Plant Cell, 18,
3355-3369.
54.Palatnik, J. F., Wollmann, H., Schommer, C.,
Schwab, R., Boisbouvier, J., Rodriguez, R., Warthmann, N., Allen, E.,
Dezulian, T., Huson, D., Carrington, J. C., and Weigel, D. (2007)
Dev. Cell, 13, 115-125.
55.Warthmann, N., Das, S., Lanz, C., and Weigel, D.
(2008) Mol. Biol. Evol., 25, 892-902.
56.Moldovan, D., Spriggs, A., Yang, J., Pogson, B.
J., Dennis, E. S., and Wilson, I. W. (2010) J. Exp. Bot.,
61, 165-177.
57.Beauclair, L., Yu, A., and Bouche, N. (2010)
Plant J., 62, 454-462.
58.Fahlgren, N., Howell, M. D., Kasschau, K. D.,
Chapman, E. J., Sullivan, C. M., Cumbie, J. S., Givan, S. A., Law, T.
F., Grant, S. R., Dangl, J. L., and Carrington, J. C. (2007) PLoS
One, 2, e219.
59.Omelyantchuk, N. A., Kuznetsova, T. N., and
Katokhin, A. V. (2005) in Informatsionnyi Vestnik VOGiS, pp.
440-450.
60.Allen, E., Xie, Z., Gustafson, A. M., Sung, G.
H., Spatafora, J. W., and Carrington, J. C. (2004) Nat. Genet.,
36, 1282-1290.
61.Dunoyer, P., Himber, C., Ruiz-Ferrer, V., Alioua,
A., and Voinnet, O. (2007) Nat. Genet., 39, 848-856.
62.De Meaux, J., Hu, J. Y., Tartler, U., and Goebel,
U. (2008) Proc. Natl. Acad. Sci. USA, 105, 8994-8999.
63.Felippes, F. F., Schneeberger, K., Dezulian, T.,
Huson, D. H., and Weigel, D. (2008) RNA, 14,
2455-2459.
64.Piriyapongsa, J., and Jordan, I. K. (2008)
RNA, 14, 814-821.
65.Niwa, R., and Slack, F. J. (2007) Curr. Opin.
Genet. Dev., 17, 145-150.
66.Mallory, A. C., Bartel, D. P., and Bartel, B.
(2005) Plant Cell, 17, 1360-1375.
67.Guo, H. S., Xie, Q., Fei, J. F., and Chua, N. H.
(2005) Plant Cell, 17, 1376-1386.
68.Mallory, A. C., Dugas, D. V., Bartel, D. P., and
Bartel, B. (2004) Curr. Biol., 14, 1035-1046.
69.McHale, N. A., and Koning, R. E. (2004) Plant
Cell, 16, 1730-1740.
70.Chen, X. (2004) Science, 303,
2022-2025.
71.Baranenko, V. V. (2006) Tsitologiya,
48, 465-474.
72.Sunkar, R., Li, Y. F., and Jagadeeswaran, G.
(2012) Trends Plant Sci., 17, 196-203.
73.Trindade, I., Capitao, C., Dalmay, T., Fevereiro,
M. P., and Santos, D. M. (2010) Planta, 231, 705-716.
74.Li, W. X., Oono, Y., Zhu, J., He, X. J., Wu, J.
M., Iida, K., Lu, X. Y., Cui, X., Jin, H., and Zhu, J. K. (2008)
Plant Cell, 20, 2238-2251.
75.Zhao, B., Liang, R., Ge, L., Li, W., Xiao, H.,
Lin, H., Ruan, K., and Jin, Y. (2007) Biochem. Biophys. Res.
Commun., 354, 585-590.
76.Lv, D. K., Bai, X., Li, Y., Ding, X. D., Ge, Y.,
Cai, H., Ji, W., Wu, N., and Zhu, Y. M. (2010) Gene, 459,
39-47.
77.Ding, D., Zhang, L., Wang, H., Liu, Z., Zhang,
Z., and Zheng, Y. (2009) Ann. Bot., 103, 29-38.
78.Xin, M., Wang, Y., Yao, Y., Song, N., Hu, Z.,
Qin, D., Xie, C., Peng, H., Ni, Z., and Sun, Q. (2011) BMC Plant
Biol., 11, 61.
79.Kantar, M., Unver, T., and Budak, H. (2010)
Funct. Integr. Genom., 10, 493-507.
80.Pashkovskii, P. P., Ryazanskii, S. S., Radyukina,
N. L., Gvozdev, V. A., and Kuznetsov, Vl. V. (2010) Rus. J. Plant
Physiol., 57, 707-714.
81.Navarro, L., Dunoyer, P., Jay, F., Arnold, B.,
Dharmasiri, N., Estelle, M., Voinnet, O., and Jones, J. D. (2006)
Science, 312, 436-439.
82.Chapman, E. J., and Estelle, M. (2009) Annu.
Rev. Genet., 43, 265-285.
83.Vidal, E. A., Araus, V., Lu, C., Parry, G.,
Green, P. J., Coruzzi, G. M., and Gutierrez, R. A. (2010) Proc.
Natl. Acad. Sci. USA, 107, 4477-4482.
84.Gifford, M. L., Dean, A., Gutierrez, R. A.,
Coruzzi, G. M., and Birnbaum, K. D. (2008) Proc. Natl. Acad. Sci.
USA, 105, 803-808.
85.Hsieh, L. C., Lin, S. I., Shih, A. C., Chen, J.
W., Lin, W. Y., Tseng, C. Y., Li, W. H., and Chiou, T. J. (2009)
Plant Physiol., 151, 2120-2132.
86.Pant, B. D., Buhtz, A., Kehr, J., and Scheible,
W. R. (2008) Plant J., 53, 731-738.
87.Pant, B. D., Musialak-Lange, M., Nuc, P., May,
P., Buhtz, A., Kehr, J., Walther, D., and Scheible, W. R. (2009)
Plant Physiol., 150, 1541-1555.
88.Liang, G., and Yu, D. (2010) Plant Signal.
Behav., 5, 1257-1259.
89.Burkhead, J. L., Reynolds, K. A., Abdel-Ghany, S.
E., Cohu, C. M., and Pilon, M. (2009) New Phytol., 182,
799-816.
90.Pashkovsky, P. P., and Radyukina, N. L. (2011)
Vestnik Tomsk. Univer. Biol., 3, 147-151.
91.Abdel-Ghany, S. E., and Pilon, M. (2008) J.
Biol. Chem., 283, 15932-15945.
92.Jagadeeswaran, G., Saini, A., and Sunkar, R.
(2009) Planta, 229, 1009-1014.
93.Ameres, S. L., Martinez, J., and Schroeder, R.
(2007) Cell, 130, 101-112.
94.Maksymiec, W. (2007) Acta Physiol. Plant.,
29, 177-187.
95.Ding, Y., Chen, Z., and Zhu, C. (2011) J. Exp.
Bot., 62, 3563-3573.
96.Chen, L., Wang, T., Zhao, M., Tian, Q., and
Zhang, W. H. (2012) Planta, 235, 375-386.
97.Huang, S. Q., Peng, J., Qiu, C. X., and Yang, Z.
M. (2009) J. Inorg. Biochem., 103, 282-287.
98.Kim, T. H., Park, J. H., Kim, M. C., and Cho, S.
H. (2008) J. Plant Physiol., 165, 345-349.
99.Ruiz-Ferrer, V., and Voinnet, O. (2009) Annu.
Rev. Plant Biol., 60, 485-510.
100.Zhang, W., Gao, S., Zhou, X., Chellappan, P.,
Chen, Z., Zhou, X., Zhang, X., Fromuth, N., Coutino, G., Coffey, M.,
and Jin, H. (2011) Plant Mol. Biol., 75, 93-105.
101.Berezikov, E. (2011) Nat. Rev.
Genet., 12, 846-860.
102.Castoldi, M., Schmidt, S., Benes, V., Noerholm,
M., Kulozik, A. E., Hentze, M. W., and Muckenthaler, M. U. (2006)
RNA, 12, 913-920.
103.Shi, R., and Chiang, V. L. (2005)
Biotechniques, 39, 519-525.
104.Brautigam, A., and Gowik, U. (2010) Plant
Biol. (Stuttg.), 12, 831-841.
105.Zhang, Y., Jiang, W. K., and Gao, L. Z. (2011)
PLoS One, 6, e28073.
106.Studholme, D. J. (2012) Brief Funct.
Genom., 11, 71-85.
107.Zhang, L., Hou, D., Chen, X., Li, D., Zhu, L.,
Zhang, Y., Li, J., Bian, Z., Liang, X., Cai, X., Yin, Y., Wang, C.,
Zhang, T., Zhu, D., Zhang, D., Xu, J., Chen, Q., Ba, Y., Liu, J., Wang,
Q., Chen, J., Wang, J., Wang, M., Zhang, Q., Zhang, J., Zen, K., and
Zhang, C. Y. (2012) Cell Res., 22, 107-126.