REVIEW: New Functions of Small Nucleolar RNAs
J. A. Makarova1,2*, S. M. Ivanova3, A. G. Tonevitsky2,4,5, and A. I. Grigoriev6
1Engelhardt Institute of Molecular Biology, Russian Academy of Sciences, ul. Vavilova 32, 119991 Moscow, Russia; fax: (499) 135-1405; E-mail: j-makarova@yandex.ru2Science and Technology Center Bioclinicum, ul. Ugreshskaya 2/85, 115088 Moscow, Russia; E-mail: mail@bioclinicum.com
3Semashko Hospital at Lublino, ul. Stavropolskaya 23/1, 109386 Moscow, Russia; E-mail: ivanova.revm@yandex.ru
4Institute of General Pathology and Pathophysiology, Russian Academy of Medical Sciences, ul. Baltiiskaya 8, 125315 Moscow, Russia
5Lomonosov Moscow State University, 119991 Moscow, Russia; E-mail: tonevitsky@mail.ru
6State Research Center of the Russian Federation, Institute of Biomedical Problems, Russian Academy of Sciences, Khoroshevskoe Shosse 76A, 123007 Moscow, Russia; E-mail: grigoriev@imbp.ru
* To whom correspondence should be addressed.
Received January 30, 2013; Revision received February 21, 2013
Small nucleolar RNAs (snoRNAs) are one of the most abundant and well-studied groups of non-coding RNAs. snoRNAs are mostly engaged in processing of rRNA. However, recent data indicate that snoRNAs are also involved in other processes including regulation of alternative splicing, translation and oxidative stress. snoRNAs are also involved in pathogenesis of some hereditary diseases and cancer. Therefore, the range of snoRNAs’ functions is significantly wider than it has been assumed earlier.
KEY WORDS: snoRNA, scaRNA, sdRNA, miRNA, noncoding RNA, RNA silencing, oncogenesisDOI: 10.1134/S0006297913060096
Abbreviations: IRES, internal ribosome entry site; miRNAs, microRNAs; ncRNAs, noncoding RNAs; RNPs, ribonucleoproteins; scaRNAs, small Cajal body-specific RNAs; sdRNAs, sno-derived RNAs; snoRNAs, small nucleolar RNAs; snRNAs, small nuclear RNAs; vRNAs, vault RNAs; Ψ, pseudouridine.
Studies on RNAs that do not code proteins (ncRNAs) are among the most
intensively developing and interesting trends in modern biology. Since
the late 1990s, when functions of small nucleolar RNAs (snoRNAs) and
the phenomenon of RNA interference were described, the number of newly
detected ncRNAs is steadily increasing and becomes even higher than the
number of known proteins [1-3].
Recently introduced sequencing technologies have given new stimulus for
progress in this field [4, 5].
The cell was found to contain previously unknown families of ncRNAs,
and the well-studied ncRNAs were shown to have new, earlier unknown
functions [6]. Thus, snoRNAs, which represent a
well-studied group of ncRNAs, occur to be involved not only in
processing of rRNAs and small nuclear RNAs (snRNAs) [7]. snoRNAs also direct alternative splicing and are
involved in the cell response to stress, and dysregulation of their
expression can be associated with the appearance of some diseases.
Moreover, snoRNAs can give origin to shorter miRNA-like RNAs capable of
regulating translation due to complementary interactions with mRNA.
These functions of snoRNAs, as well as others known by now, are
discussed in the present review. It should be noted that new functions
and types of processing are also found in other cellular ncRNAs, namely
in RNA component of RNPs involved in development of drug resistance
(so-called vault particles) [8], Y RNA [9], 7SL RNA [10], and even in
tRNA whose functions have long been well known [11]. Thus, it has been shown that under stress
conditions, tRNAs produce shorter fragments responsible for repression
of translation [11]. Thus, the transcriptome
structure is more complicated and interesting than it was thought only
recently.
snoRNAs ARE INVOLVED IN PROCESSING OF rRNA
At present, the best-studied function of snoRNAs is their involvement in processing of rRNA. 18S, 5.8S, and 25/28S rRNAs are transcribed within a common precursor (pre-rRNA), which is cleaved yielding mature rRNA molecules. rRNA modifications occur cotranscriptionally, most frequently ribose being methylated at the 2′-O-position and uridine being converted to pseudouridine [12]. rRNA of vertebrates contains about 100 modifications of each type [13].
snoRNAs are involved in the cleavage of pre-rRNA and determine the modification sites. Based on the presence of characteristic elements of the nucleotide sequence, snoRNAs are divided into two families, C/D and H/ACA. Nearly all C/D RNAs direct 2′-O-methylation [14, 15], whereas the majority of H/ACA RNAs direct pseudouridylation of rRNA nucleotides [16].
snoRNAs of the C/D family are of ~70-90 nucleotides (nt) in length and contain conservative elements: boxes C (UGAUGA) and D (CUGA) located on the ends of the molecule and drawn in close proximity due to complementary interactions of its terminal regions (Fig. 1a). This results in formation of a so-called C/D motif, which includes the C and D boxes and the terminal double-stranded region. The C/D-motif acts as a scaffold for binding proteins that form C/D RNP: 15.5 kDa protein, NOP56, NOP58, and fibrillarin. This motif determines the stability and nucleolar localization of snoRNAs [17]. In the central region of a snoRNA molecule, there are C′ and D′ boxes that are copies of the C and D boxes (often imperfect) [18]. Towards the 5′-end from boxes D and/or D′, a so-called antisense element is located that is a sequence of 9-15 nt in length complementary to the rRNA region and capable of interacting with it. The rRNA nucleotide separated from box D/D′ by four nucleotides (Fig. 1a) is subjected to 2′-O-methylation. This modification is catalyzed by the protein fibrillarin [19]. Several C/D RNAs (SNORD3, SNORD14, SNORD22, SNORD118, and, probably SNORD13) are required for pre-rRNA cleavage [20-24]. They contain regions complementary to pre-rRNA and act as RNA-chaperones.
Fig. 1. snoRNAs of the C/D (a) and H/ACA (b) families. 2′-O-methylated nucleotides are indicated by asterisks; the pseudouridinylated nucleotide is shown with the sign Ψ.
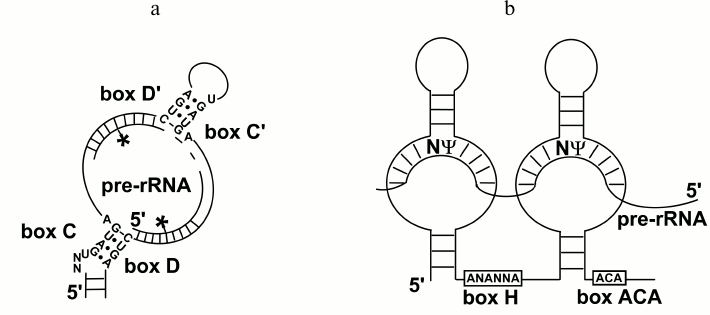
snoRNAs of the H/ACA family have length of about 150 nt. They are associated with four proteins (GAR1, NOP10, NHP2, and dyskerin), and contain conservative elements H (ANANNA) and ACA (ACA) located in the basement of two hairpins (Fig. 1b). In the middle part of the hairpins, antisense elements are located that capable of complementary interacting with rRNA fragments, as occurs in the case of C/D RNA. The rRNA nucleotide exposed in the produced one-stranded “window” is pseudouridinylated (Fig. 1b). The protein dyskerin is responsible for this modification [25]. The H/ACA RNA SNORA73, similarly to some C/D RNAs, does not direct modifications and is necessary for the cleavage of pre-rRNA [26]. The structure and functions of snoRNPs are discussed in reviews [19, 27, 28].
The role of rRNA modifications is a very interesting problem that is still far from its solution. The modifications are necessary for normal functioning of the ribosome and seem to be responsible for the correct packing of rRNA, stabilization of its structure, and for the correct interaction of rRNA with other participants of translation [29-32]. However, the corresponding mechanisms and functional role of each modification are still poorly studied.
Interestingly, the 3′-terminal part of the telomerase RNA forms a motif specific for H/ACA RNAs: two hairpins separated by single-stranded stretches containing the H and ACA boxes [33]. In humans this motif is formed by 240 of 451 nt of the telomerase RNA, and all four proteins of the H/ACA RNP are associated with it, but it does not direct pseudouridylation. This motif seems to be necessary for the stability, correct localization, and functioning of the telomerase [25].
In addition to rRNA, snoRNAs have other targets. Thus, some snoRNAs are involved in modification of the snRNA U6, which contains 2′-O-methylated ribose and pseudouridine [34]. Modifications of the snRNA U6 occur in the nucleolus. Other snRNAs are also modified by C/D and H/ACA RNPs, but these modifications occur not in the nucleolus but in so-called Cajal bodies, which are spherical formations inside the nucleus [35] (Fig. 2). The main function of these bodies is likely the assemblage of snRNP complexes, which later are transported into other compartments of the nucleus. C/D and H/ACA RNAs from the Cajal bodies were called scaRNAs (small Cajal body-specific RNAs) [36]. Targets of about 20 snoRNAs are unknown [37]. These snoRNAs are especially interesting because their functions are still unknown, and their elucidation would shed new light on the involvement of snoRNAs in cellular processes.
Fig. 2. Involvement of snoRNAs in various cellular processes.
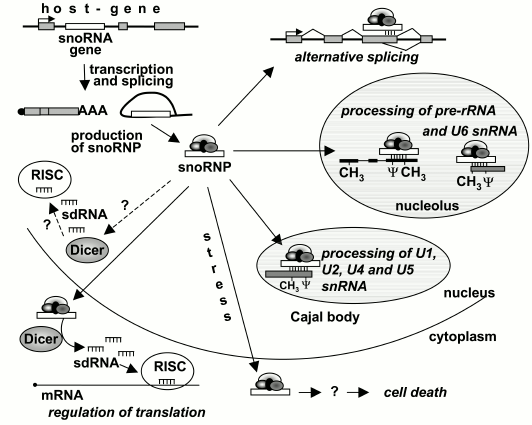
snoRNAs of vertebrates are encoded unusually: nearly all their genes are located within introns of other genes, one gene per intron, and snoRNAs are processed on splicing of the “host-gene” mRNA (Fig. 2). Many of host genes encode proteins involved in translation and its regulation, including translation factors, ribosomal proteins, and nucleolar proteins. In addition, more than dozen host genes are known whose exons do not encode any polypeptide [38-40].
Retroposons Alu located in introns were recently shown to encode H/ACA RNAs. RNAs produced upon splicing of Alu are associated with all four proteins of the H/ACA RNP, but they are located not in the nucleolus or Cajal bodies but in the nucleoplasm. Several hundreds of such AluACA RNAs are already described, and they seem to form a large new group of H/ACA RNAs with yet unknown functions [41].
A polyribonucleotide component of RNase MRP (265 nt in humans) is also assigned to nucleolar RNAs. This endonuclease found in all eukaryotes is located in the nucleolus and mitochondria. During replication of DNA, the RNase MRP forms in mitochondria the 3′-end of the RNA-primer, and this has determined its name (mitochondrial RNA processing) [42]. However, the major part of the RNase MRP is located in the nucleolus, where it cleaves pre-rRNA in site A3 of the first internal transcribed spacer (ITS1) releasing 5.8S rRNA.
The RNase MRP also has other substrates, in particular, mRNA of cyclin B2, and thus it is involved in the control of the cell cycle [43, 44].
snoRNAs CAN SERVE AS PRECURSORS OF miRNAs
During deep sequencing of small RNAs (19-40 nt), it was revealed in 2008 that snoRNAs as well as other small RNAs can yield shorter products [7]. Similar data were obtained later by other researchers [45-48]. Bioinformatic analysis of more than twenty libraries of small RNAs from various organisms has shown that more than half of the snoRNAs from vertebrates, arabidopsis, and yeast give rise to short fragments called sno-derived RNAs (sdRNAs) [49]. sdRNAs were also found in the parasitic protozoon Giardia lamblia [50]. Many sdRNAs originating from the same snoRNA were found to have identical nucleotide sequences. In some cases, these sdRNAs similarly to miRNAs [51] differ in several terminal nucleotides. In a number of cases a particular sdRNA was found in several organisms [45]. These data suggest that many of sdRNAs could be produced due to specific processing and not to degradation of snoRNAs [48, 52].
sdRNAs from different families of snoRNAs have some specific features. In humans, the majority of sdRNAs originating from C/D snoRNAs form two classes, with 17-19 nt and about 30 nt size, and the shorter RNAs are mainly produced from the 5′-terminal part of the molecule, whereas the longer RNAs are produced from the middle part [49]. Human H/ACA sdRNAs are usually produced from the 3′-terminal hairpin of snoRNAs, and the size of the majority of them is 20-24 nt, which corresponds to the size of miRNAs [45, 49]. In fact, similarly to miRNAs, H/ACA sdRNAs bound with immunoprecipitated proteins Ago (Ago1-4) were found in human cells [45, 46]. These proteins are the main components of the RNA-induced silencing complex (RISC), which is responsible for gene silencing: miRNA is associated with the protein Ago and the complementary interaction of miRNA with an mRNA-target results in suppression of translation [53].
Using reporter constructions that contain sequences complementary to sdRNA in the 3′-UTR of the luciferase gene, both H/ACA and C/D sdRNAs were shown to suppress the expression of the gene [45, 47]. However, it is still unclear whether sdRNAs have targets among cellular RNAs. Up to now, such a target has been found only for one sdRNA produced from SCARNA15 [45]. Nevertheless, it is reasonable to expect that cellular targets will be detected and that sdRNAs are really capable of functioning as miRNAs, because a region of miRNA complementary to mRNA is usually very short and, therefore, has to occur in various cellular mRNAs. Moreover, some miRNAs found in independent studies correspond to fragments of snoRNAs (table). Genome browsers allow to clearly demonstrate these findings (Fig. 3).
Human snoRNAs fragments of which are annotated as miRNAs
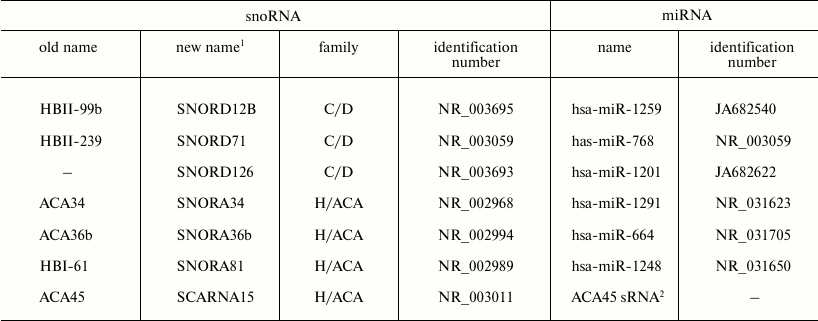
1 According to the new nomenclature adopted in 2006 by the
International HUGO Gene Nomenclature Committee, names of snoRNAs of the
C/D family are represented as SNORDn, where “n” is the
snoRNA number, names of the H/ACA family are presented as SNORAn, and
names of scaRNAs – as SCARNAn. Nevertheless, names of
snoRNAs given by their first discoverers before introduction of the new
nomenclature are still widely used.
2 The ability of ACA45 sRNA to silence the cellular CDC2L6
mRNA has been demonstrated in [45]. In mice, the
ACA45 sRNA homolog is annotated as miR-1839-5p miRNA (NR_035501).
Fig. 3. An example of a snoRNA gene independently annotated as the gene of miRNA. The gene of the human SNORA36B snoRNA is simultaneously annotated as the gene of miR-664 miRNA. The mature miRNA (miR-664-3p), its precursor (miR-664), and SNORA36B snoRNA are shown. Below small RNAs are shown detected during deep sequencing and corresponding to fragments of SNORA36B snoRNA. The image was obtained using the genome browser of the University of California at Santa Cruz (UCSC, http://genome.ucsc.edu).

sdRNAs are mainly located in the nucleus, and only some H/ACA sdRNAs are found in the cytoplasm [54]. Mechanisms of formation of sdRNAs are not yet clear, although involvement of some proteins of the RNA-silencing system in their processing has been shown [49]. It seems that some other yet unidentified proteins can also contribute to this processing because knockout of the main proteins of the RNA-silencing pathway only slightly decreases the number of sdRNAs [49]. The processing pathways seem to be different for different families of snoRNAs and even for different snoRNAs.
The nuclear endonuclease Drosha and the RNA-binding protein DGCR8 are not involved in formation of the majority of H/ACA sdRNAs [45]. These two proteins involved in RNA silencing are the main components of a complex named “microprocessor”. The “microprocessor” processes primary transcripts of miRNA genes: it recognizes within them a hairpin with length of about 70 nt (i.e. a pre-miRNA), which contains the miRNA sequence and cuts this hairpin from the primary transcript [55]. H/ACA snoRNAs are similar to pre-miRNA in size and secondary structure, but the “microprocessor” role in this case is played by a spliceosome, which cuts the snoRNAs from the primary transcript and exonucleases, which destroy intronic sequences flanking the snoRNAs. Note that in addition to miRNAs produced from snoRNAs, other miRNAs are also detected that mature in a noncanonical pathway, i.e. without processing by Drosha/DGCR8. These are, for example, miRNAs processed from the so-called mirtrons – short introns that upon slicing produce secondary structures similar to secondary structures of pre-miRNAs [56]. As in the case of H/ACA RNAs, a spliceosome and in some cases exonucleases act as “microprocessor” [57, 58]. Endonuclease Dicer, which is one of the main enzymes of the RNA-silencing pathway, is involved in the production of H/ACA sdRNAs [45]. During processing of “canonic” miRNAs, Dicer in a complex with the RNA-binding protein TRBP cuts from pre-miRNA a double-stranded fragment with length of about 22 nt. Then one of the strands degrades, whereas the mature miRNA becomes a RISC component [55]. H/ACA snoRNAs contain two hairpins reminiscent of the usual substrate of Dicer. This seems to explain the ability of Dicer to processes H/ACA snoRNAs with generation of sdRNAs with 20-24 nt in length, which later associate with Ago proteins and can be involved in RNA silencing.
Endogenous substrates of Dicer are very diverse. In addition to snoRNAs, many other ncRNAs can serve as such substrates: tRNAs [11], vRNAs [8], 7SL RNA [10], and even transcripts of the Alu retroposon [59]. This can be explained by the small size and pronounced secondary structures of all these ncRNAs that makes them similar to pre-miRNAs. Note that according to numerous observations, Dicer is located in the cytoplasm of mammalian cells [55, 60], whereas C/D sdRNAs and the majority of H/ACA sdRNAs are located in the nucleus [54, 61]. Therefore, the sdRNA processing by Dicer still needs further studies. Note that in addition to other components of the RNA-silencing system, Dicer has been recently found in the nucleus of mammalian cells [62-65], and its knockdown leads to defects in rRNA processing and changes in nucleolus structure [63].
Processing of C/D sdRNAs is much less studied. The predicted secondary structures of C/D RNAs are much more diverse than those of H/ACA RNAs, and C/D sdRNAs are located in the hairpin rather seldom. Processing of the majority of C/D sdRNAs seems not to depend on Drosha/DGCR8 and on Dicer [49]. Note that in many cases sequences of C/D sdRNAs include the box C or C′. It seems that their processing can occur due to recognizing the conservative elements of the nucleotide sequence rather than the characteristic secondary structures [47].
sdRNAs apparently represent a heterogeneous population that includes products of both degradation and specific processing. Several processing pathways are likely to exist, resulting in production of sdRNAs with different length and functions. So far, a pathway resulting in production of miRNA-like sdRNAs involved in the gene silencing is best studied. However, these data do not exhaust the diversity and functions of sdRNAs. Thus, the protein DGCR8, which is a component of the “microprocessor”, is able to associate with full-size snoRNAs and destabilize them, probably due to activity of a yet unknown endonuclease that associates with DGCR8. This results in generation of sdRNAs that are different from those involved in the silencing [66]. Their functions still need elucidation.
snoRNAs DIRECT ALTERNATIVE SPLICING
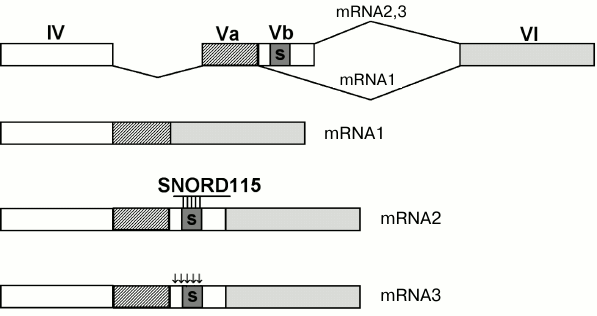
In 2006, snoRNAs were shown to direct alternative splicing [67] (Fig. 2). The SNORD115 snoRNA whose target was unknown earlier was found to have a long (18 nt) anti-sense element complementary to the exon Vb region of the serotonin receptor 5-HT2C mRNA (Fig. 4). The exon Vb contains a silencer of splicing that prevents the inclusion of Vb into the mRNA that results in formation of a short mRNA form encoding a shortened nonfunctional protein (Fig. 4, mRNA1) [68]. The SNORD115 snoRNA inhibits the action of the silencer and provides the inclusion of exon Vb into the mRNA. This results in a normal receptor with high sensitivity to serotonin (Fig. 4, mRNA2) [67, 69]. There is also another pathway of inhibiting the silencer: the exon Vb contains five nucleotides that undergo editing – adenosine is deaminated producing inosine. The silencer stops functioning, and exon Vb is incorporated into the mRNA. However, during translation inosine is read as guanosine; therefore, the receptor contains some amino acid substitutions that results in its decreased sensitivity to serotonin (Fig. 4, mRNA3) [70]. Normally, the cell contains both the edited and non-edited forms of the receptor. This seems to be a putative pathway for the fine regulation of brain cell receptivity for serotonin. At present, there is more evidence of involvement of snoRNAs in the regulation of alternative splicing [48, 71].
Fig. 4. Regulation of alternative splicing of the serotonin receptor mRNA. Exons are shown as rectangles and numerated with Roman numerals. Black arrows show sites of editing; the silencer sequence is indicated by the letter “s”.
The alternative splicing mechanisms with involvement of snoRNAs are still unclear. By now, two putative schemes have been proposed. According to the first, snoRNAs direct 2′-O-methylation of the branchpoint adenosine. This inhibits splicing, and the exon flanking the intron from the 3′-end is excluded from the mature mRNA. Although up to now such a mechanism has not been described for cellular snoRNAs, artificial snoRNAs are shown to direct 2′-O-methylation of the branchpoint adenosine of cellular and viral pre-mRNAs. This leads to generation of shortened mRNAs [72-75]. According to the other scheme, splicing is regulated not by full-size snoRNAs but by sdRNAs produced as a result of their processing. The available data are contradictory [71, 76, 77], but some of these sdRNAs are shown to be associated with nuclear hnRNPs involved in the splice site selection [71]. It seems that similarly to antisense oligonucleotides, sdRNAs complementarily interact with pre-mRNA and prevent it from interaction with splicing factors, or, in other cases, they may associate with splicing factors and bring them to mRNA. And just such a mode of regulation has been proposed for the SNORD115 snoRNA [71]. Note that snoRNAs not only regulate alternative splicing, but they can also decrease the level of mRNA targets and proteins encoded by them, and the stretch complementary to mRNA can be removed from D/D′ boxes, and thus the observed effect is not associated with 2′-O-methylation of the RNA target [78]. This knockdown is likely performed by sdRNAs generated as a result of snoRNA processing and located not only in the nucleolus, but also in the nucleoplasm [48, 79].
Targets are unknown for more than 20 of snoRNAs [37]. Probably these targets will be identified as mRNAs, and snoRNAs will function as regulators of their splicing, stability, and translation. Note in this connection that pseudouridylation of mRNA stop codons promotes their specific recognition by tRNA, and instead of terminating the translation an amino acid is incorporated and the protein synthesis is continued. Thus, pseudouridylation of the codon UGA (ΨGA) resulted in incorporation of tyrosine or phenylalanine [80].
Unfortunately, the search for targets for such snoRNAs is difficult because every snoRNA, due to small size of the antisense element, has hundreds of potential targets. However, on searching for targets for the SNORD116 snoRNA the majority of targets were found inside genes subjected to alternative splicing [81]. In general, it is very interesting to know how many targets every snoRNA can have. It is especially interesting now when nucleolar RNAs have been shown to reach pre-mRNAs generally believed to be located in the nucleoplasm. It has also been found that one to three mismatches in the antisense element/RNA-target duplex do not prevent the regulation of splicing through snoRNAs [71]. It seems that the traditional view that the number of targets should be equal to the number of antisense elements needs to be changed. Thus, the search for additional targets for the SNORD115 snoRNA revealed that it could regulate the splicing of at least five mRNAs and provide both inclusion and exclusion of exons [71]. Probably, the situation will be similar to that observed for the U1 snRNA that is involved in splicing and usually is imperfectly complementary to its numerous targets and also to miRNAs, each of which can also have several targets and be imperfectly complementary to them.
snoRNAs ARE INVOLVED IN CELL RESPONSE TO STRESS
The nucleolus is one of the key participants of the cell response to stress. Various stress conditions can cause changes in the nucleolus and even its destruction. Many nucleolar proteins are moved to other compartments of the cell and contribute to the cell response. Thus, under stress conditions some ribosomal proteins of mammalian cells are moved into the cytoplasm and interact there with ubiquitin ligase MDM2. Under normal conditions, MDM2 ubiquitinates the transcriptional factor p53 that leads to its degradation. Interaction with the ribosomal proteins inhibits MDM2 that stabilizes p53. As a result, the cell stops dividing or enters apoptosis [82].
In 2011, the RNA component of the nucleolus, a snoRNA, was shown to also contribute to response to stress. Under conditions of hypoxia, expression of the SNORD14A and SNORD83B snoRNAs significantly increased in neuronal stem cells [83]. SNORD14A is required for normal translation because it is involved in the pre-rRNA cleavage, whereas the target of SNORD83B is unknown. In the same year, expression of three snoRNAs (SNORD32A, SNORD33, and SNORD35A) was shown to significantly increase under conditions of oxidative stress and on treatment of cells with excess fatty acids (palmitate) [84]. All these snoRNAs are encoded by introns of the ribosomal protein RPL13A gene, and rRNA nucleotides serve as their targets, although involvement of these snoRNAs in rRNA methylation was not shown directly [85]. The loss of these snoRNAs causes cell resistance to metabolic stress, i.e. they are key participants of the stress response pathways, including induction of cell death. Under stress conditions, full-length forms of these snoRNAs are accumulated not in the nucleus but in the cytoplasm. This is not accompanied by an increase in the extent of methylation of rRNA; therefore, their effect seems to be not associated with the modification of rRNA [84]. The presence of snoRNAs in the cytoplasm, and only during the stage of processing, was found earlier only for independently transcribing snoRNAs (SNORD3, SNORD13, and SNORD118) [19], whereas the presence of intronic snoRNAs was shown for the first time. Probably, in addition to rRNA, these snoRNAs also have additional targets among mRNAs and, after entering the cytoplasm under conditions of metabolic stress complementarily interact with them and regulate their translation. The pathways of snoRNA involvement in the regulation of cellular processes can be even more diverse. Thus, there are some indications that the H/ACA snoRNA ACA11 inhibits the cell response under conditions of oxidative stress [86].
CHANGES IN EXPRESSION OF snoRNAs AND ASSOCIATED PROTEINS CAN
CONTRIBUTE TO THE DEVELOPMENT OF SOME DISEASES
Recent data indicates that changes in expression of individual snoRNAs can be significant not only for intracellular processes, but also for the whole organism. This can be exemplified by the results of medical studies presented below. For example, changes in expression of some snoRNAs can lead to severe diseases. Besides, expression of some snoRNAs is changed during various infections, acute exercise, and in some other situations.
It was thought for a long time that the loss of individual snoRNAs and of the corresponding modifications of rRNAs, with some exceptions, have no serious consequences for the cell [87-91]. Later studies with more sensitive methods have shown that changes in the level of modifications of the majority of rRNA sites does really influence translation, and the absence of modifications in certain sites is more pronounced phenotypically than in some other sites [92-97]. However, the majority of such works were performed on yeast or on prokaryotic systems and not on multicellular organisms. Recent studies described below demonstrate that changes in the general level of modifications of rRNA nucleotides in vertebrates as well as changes in the modification degree of individual rRNA nucleotides (associated with changes in the expression of individual snoRNAs) are significant for functioning of the organism. Changes in expression of snoRNAs with unknown targets are also shown to induce pronounced phenotypical manifestations. Note that sometimes it is difficult to interpret the data because the observed effects can be caused by other, “non-canonical” functions of snoRNAs that have been discussed above and elucidation of which is only at the beginning.
snoRNAs are involved in pathogenesis of some hereditary and autoimmune diseases. For some hereditary diseases, snoRNAs and proteins of snoRNPs are found that contribute to their pathogenesis. Thus, the absence in human cells of the C/D snoRNA SNORD116 and, probably, of the SNORD115 causes development of Prader–Willi syndrome (PWS), which is a severe hereditary disease associated with obesity, mental retardation, and some other symptoms [98]. SNORD115 and SNORD116 are rather unusual human snoRNAs because they are encoded not by individual genes, but by two clusters containing, respectively, 48 and 29 genes [37] and are mainly expressed in the brain [99]. Moreover, these clusters are located within introns of the gene of the giant (~460,000 nt) dicistron SNURF-SNRPN-UBE3A AS RNA (one intron – one gene) and undergo imprinting: these snoRNAs are expressed from the paternal allele [100, 101]. A putative involvement of these genes in the development of PWS has been under discussion for about ten years, up to the finding in 2008 of a patient with a microdeletion resulting in complete loss only of the SNORD116 snoRNA genes [102]. The patient had all the main diagnostic manifestations of PWS. This was the first direct evidence that the loss of a snoRNA can be the cause of a human disease. The SNORD116 target is unknown. Its discovery seems to elucidate not only mechanisms leading to PWS, but seems promising for discovery of new pathways of snoRNA involvement in the regulation of cellular processes.
The above-mentioned patient had some unusual phenotypic features [102]. Therefore, other genes are likely to contribute also, although to lesser degree, to development of the PWS-characteristic phenotype [102]. Such genes can be SNORD115 snoRNA genes located adjacent to SNORD116 genes and the serotonin receptor 5-HT2C gene regulated by SNORD115 (see above). Although the data are somewhat contradictory, changes in the processing of 5-HT2C mRNA associated with the absence of SNORD115 have been shown to cause the PWS-specific phenotypic features [67, 103-106]. Thus, dysregulated snoRNAs expression definitely contributes to the development of PWS.
Mutations in the genes encoding the H/ACA RNP proteins, dyskerin, and significantly less frequently NOP10 and NHP2 cause dyskeratosis congenita – a hereditary disease associated with bone marrow failure, dystrophic changes in the skin, and other manifestations. Tissues with a high level of proliferative activity are damaged first. Patients with this disease have shortened telomeres and display a tendency for malignancies [107-109]. Dyskeratosis can also be caused by mutations in the H/ACA RNA-like domain of the telomerase RNA [110]. And it is unclear whether the disease is caused by defects in the synthesis of telomeres, disorders in rRNA processing, or is due to both mechanisms. Mutations in the gene of dyskerin are found to cause a decrease in the amount of telomerase RNA. And cells with a decreased level of telomerase RNA seem to be incapable of maintaining telomere length [108]. On the other hand, a decrease in pseudouridylation of rRNA in mice with a hypomorphic mutation of the dyskerin gene is observed in the first generation, concurrently with the first dyskeratosis manifestations, whereas the shortening of telomeres is observed only beginning from the fourth generation simultaneously with an increase in the number of diskeratosis symptoms [111]. Deregulation of ribosome biogenesis probably initiates the appearance of dyskeratosis, and defects in synthesis of telomeres can determine the subsequent development of the disease [111]. Similar data were obtained for Danio rerio: a decrease in the expression of two other H/ACA RNP proteins, NOP10 [112] and Gar1p [113], induces defects in the synthesis of ribosomes that results in stabilization of p53, p53-dependent death of hematopoietic stem cells [112], and in a decrease in the blood cell counts specific for dyskeratosis [113]. Thus, the pathogenesis of this disease is contributed to by both defects in the processing of ribosomes and defects in synthesis of telomeres, and both effects are caused by impaired functioning of H/ACA snoRNPs.
Mutations in the gene encoding another snoRNA, MRP, lead to some diseases, in particular, to the cartilage-hair hypoplasia and some others. Patients with these diseases also have an increased tendency to malignancies [114].
Antibodies to snoRNPs are found in various autoimmune diseases. Thus, antibodies to the MRP RNase (anti-Th/T0 antibodies) and fibrillarin (a C/D snoRNP component) have been found in patients with systemic sclerosis, systemic lupus erythematosus, and some other diseases [43, 115, 116]. Interestingly, salts of mercury, and also of silver and gold, induce production of antibodies to fibrillarin [117, 118]. Antibodies to H/ACA RNPs can be found very seldom [116]. Pathogenic effect of antibodies to snoRNPs is unknown, but they are very useful for diagnosis of autoimmune diseases and prediction of their development [43, 119, 120].
snoRNAs are involved in oncogenesis. During recent years, data have been published about disturbances in the expression of some snoRNAs in various kinds of cancer, and the amount of such data is steadily increasing [121, 122]. Such data indicate that snoRNAs can contribute to oncogenesis. Thus, genes encoding some snoRNAs have been found within loci that are often amplified in non-small cell lung cancer, and expression of the encoded snoRNAs is increased in the tumors [123]. An increased expression of one of these snoRNAs (H/ACA RNA SNORA42) in tissues correlates with unfavorable outcome of the disease and can be used for predicting the disease course. A putative mechanism of the SNORA42 oncogenic action can be realized through suppression by its hyperexpression of p53-dependent apoptosis [124]. Increased levels of the C/D snoRNAs SNORD33, SNORD66, and SNORD76 are detected not only in tumors, but also in the plasma, and this can be used for the early and non-invasive diagnosis of non-small cell lung cancer [123].
Hyperexpression of four other snoRNAs (C/D RNA SNORD25, SNORD27, SNORD30, and SNORD31) correlates with a rapid transition of smoldering multiple myeloma to active multiple myeloma [125]. Remarkably, all these snoRNAs are encoded by introns of the same host gene. This host gene contains in its introns four other snoRNAs, and its own product is ncRNA (UHG) [40].
snoRNAs can act not only as oncogenes, but also as oncosuppressors. Thus, the C/D snoRNAs SNORD43, SNORD44, and SNORD48 seem to be putative suppressors of breast cancer: low level of their expression is associated with low survival [126]. The SNORD44 snoRNA is encoded by an intron of the non-protein-coding gas5 gene. This gene was initially discovered in a screen for potential tumor suppressor genes [127], and later its introns were shown to encode ten different snoRNAs [128]. ncRNA gas5 induces apoptosis, and its expression is decreased in breast cancer [129]. Interestingly, some isoforms of gas5 regulating apoptosis contain unspliced introns with sequences of snoRNAs [129]. Moreover, a case of B-cellular lymphoma has been described with a translocation resulting in joining of the gas5 gene with the gene BCL6, the expression of which is often disturbed in this disease: the chimerical gene contained the gas5 promoter and its first three exons, two genes of snoRNAs located within introns of gas5, and also a full-size gene of BCL6 [130]. Together these data suggest that not only transcripts of the gas5 but also snoRNAs from its introns are involved in oncogenesis.
Another snoRNA, C/D RNA SNORD50, encoded by introns of the noncoding host gene (U50HG) seems to be a suppressor of breast and prostate cancer [131, 132]. U50HG is localized at the break point of chromosomal translocation observed in B-cellular lymphoma [133]. Mechanisms of the involvement of SNORD50 in oncogenesis are still unclear, but its expression is often decreased in prostate and breast cancer [131, 132]. In some cases, SNORD50 contains a two-nucleotide deletion, which does not affect the antisense element. However, in the homozygous state this deletion has a statistically significant correlation with prostate cancer [132].
Recent data indicates that in tumor tissues dozens of snoRNA genes have aberrant expression. Thus, in the cell lines K562 and HL60 isolated from patients with leukemia there were multiple changes in the expression of snoRNA genes in comparison with normal lymphocytes [134]. Studies on genes differentially expressed in tumors sensitive and resistant to tamoxifen used for treatment of breast cancer revealed among them genes encoding snoRNAs [135]. Expression of dozens of snoRNA genes is deregulated in leukemias, peripheral T-cellular lymphomas, and prostate cancer [136-138], and in many cases their expression level was decreased [136, 137]. In contrast, in testicular germ cell tumor the expression level of some snoRNAs was increased [139]. A change in the methylation status of host gene promoters is one of the possible mechanisms for changing the expression level of snoRNAs [139, 140].
Protein components of snoRNPs are also involved in oncogenesis. Thus, in prostate cancer the level of dyskerin expression is increased, and this correlates with tumor progression [141], and in hepatocellular carcinoma hyperexpression of dyskerin is an unfavorable prognostic factor [142].
Mechanisms of involvement of snoRNAs and snoRNP proteins in oncogenesis are now under intensive study. In particular, deregulation of snoRNA expression changes the pattern of rRNA modification, and this leads to the impairment of translation. Thus, in aggressive forms of breast cancer rRNA methylation is increased and the synthesis of ribosomes becomes more intensive [143], whereas in dyskeratosis (i.e. with decreased pseudouridylation of rRNA) the translation of IRES-containing mRNAs is impaired, including the translation of mRNA of the tumor suppressor protein p27 [144]. This seems to be an explanation of the increased tendency for cancer in patients with dyskeratosis. The translation of IRES-containing mRNAs, in particular those of p53 and p27, is also deregulated because of a decrease in the 2′-O-methylation degree of rRNA [32]. Expression of the majority of snoRNAs and, possibly, the modification degree of the corresponding rRNA sites are decreased in the studied tumors [136, 137]. Other mechanisms of involvement of snoRNA in oncogenesis can be based on their ability to direct alternative splicing and give rise to miRNA-like small RNAs. Simultaneously with studies on molecular mechanisms of the involvement of snoRNAs in oncogenesis, the possibility is actively studied of using data on the expression level of snoRNAs for early diagnosis of cancer, prediction of disease outcome, and for choice of therapy.
Expression level of snoRNAs changes in different status of the organism. The expression level of snoRNAs can change during embryogenesis. Corresponding changes are revealed in mice during differentiation of the embryonic stem cells into neurons [145, 146] and also on comparing myocardial tissues from infants and fetus [147].
Suppressed expression of three C/D snoRNAs (SNORD26, SNORD44, and SNORD78) in Danio rerio leads to a decrease in the methylation level of the corresponding rRNA sites and to developmental disorders [148]. But it is still unclear whether these disorders are associated with changes in the pattern of rRNA modification or with the loss of miRNAs possibly produced from two of the studied snoRNAs (SNORD44 and SNORD78) [47].
The expression level of snoRNAs is different in different tissues. Moreover, different snoRNAs are presented differently in the same tissue [149, 150]. This finding was rather unexpected because the majority of snoRNAs is involved in rRNA modification, and it was reasonable to expect that they all should be expressed to a similar extent.
Expression of several dozens of snoRNAs is increased in athletes during vigorous exercises, and these changes arise as early as 15 min after the beginning of the exercise. snoRNAs are probably involved in the organism’s adaptation to stress. Interestingly, expression of the majority of correspondent host gene does not change [151].
Expression of many snoRNAs changes during viral infections [152, 153] and also during surgical interventions [154]. Changes in the expression profile of some snoRNAs have been detected in the myocardium of infants with congenital heart defect (Tetralogy of Fallot) [147] and in leukocytes of American soldiers with craniocerebral injury obtained during recent military operations [155]. In both cases, expression of the majority of snoRNAs was lower than in healthy humans.
The physiological significance and molecular mechanisms of these changes still require elucidation.
In conclusion, within recent years numerous and unexpected data were obtained on the role of snoRNAs in the organism. On one hand, new molecular mechanisms of the involvement of snoRNAs in cellular processes are described and, on the other hand, changes in the expression level of snoRNAs are shown to be significant for clinical practice. At present, there is a new splash of interest in this field; the data at hand seem to be only the top of an iceberg, whereas the world of snoRNAs still conceals many enigmas.
We are grateful to D. A. Kramerov and N. S. Vasetsky for their help.
This work was supported by the Russian Foundation for Basic Research (project No. 11-04-00439), by the Molecular and Cellular Biology Program of the Russian Academy of Sciences, and by the Russian Ministry for Science (project Nos. 16.522.11.2004 and 16.552.11.7057).
REFERENCES
1.Rinn, J. L., and Chang, H. Y. (2012) Annu. Rev.
Biochem., 81, 145-166.
2.Tuck, A. C., and Tollervey, D. (2011) Trends
Genet., 27, 422-432.
3.Siomi, M. C., Sato, K., Pezic, D., and Aravin, A.
A. (2011) Nat. Rev. Mol. Cell. Biol., 12, 246-258.
4.Wang, Z., Gerstein, M., and Snyder, M. (2009)
Nat. Rev. Genet., 10, 57-63.
5.Metzker, M. L. (2010) Nat. Rev. Genet.,
11, 31-46.
6.Rother, S., and Meister, G. (2011)
Biochimie, 93, 1905-1915.
7.Kawaji, H., Nakamura, M., Takahashi, Y., Sandelin,
A., Katayama, S., Fukuda, S., Daub, C. O., Kai, C., Kawai, J., Yasuda,
J., Carninci, P., and Hayashizaki, Y. (2008) BMC Genomics,
9, 157.
8.Persson, H., Kvist, A., Vallon-Christersson, J.,
Medstrand, P., Borg, A., and Rovira, C. (2009) Nat. Cell Biol.,
11, 1268-1271.
9.Nicolas, F. E., Hall, A. E., Csorba, T., Turnbull,
C., and Dalmay, T. (2012) FEBS Lett., 586, 1226-1230.
10.Ren, Y. F., Li, G., Wu, J., Xue, Y. F., Song, Y.
J., Lv, L., Zhang, X. J., and Tang, K. F. (2012) PLoS One,
7, e40705.
11.Sobala, A., and Hutvagner, G. (2011) Wiley
Interdiscip. Rev. RNA, 2, 853-862.
12.Panse, V. G., and Johnson, A. W. (2010) Trends
Biochem. Sci., 35, 260-266.
13.Maden, B. E. (1990) Prog. Nucleic Acids Res.
Mol. Biol., 39, 241-303.
14.Kiss-Laszlo, Z., Henry, Y., Bachellerie, J. P.,
Caizergues-Ferrer, M., and Kiss, T. (1996) Cell, 85,
1077-1088.
15.Cavaille, J., Nicoloso, M., and Bachellerie, J.
P. (1996) Nature, 383, 732-735.
16.Ganot, P., Bortolin, M. L., and Kiss, T. (1997)
Cell, 89, 799-809.
17.Samarsky, D. A., Fournier, M. J., Singer, R. H.,
and Bertrand, E. (1998) EMBO J., 17, 3747-3757.
18.Kiss-Laszlo, Z., Henry, Y., and Kiss, T. (1998)
EMBO J., 17, 797-807.
19.Watkins, N. J., and Bohnsack, M. T. (2012)
Wiley Interdiscip. Rev. RNA, 3, 397-414.
20.Borovjagin, A. V., and Gerbi, S. A. (1999) J.
Mol. Biol., 286, 1347-1363.
21.Enright, C. A., Maxwell, E. S., Eliceiri, G. L.,
and Sollner-Webb, B. (1996) RNA, 2, 1094-1099.
22.Tycowski, K. T., Shu, M. D., and Steitz, J. A.
(1994) Science, 266, 1558-1561.
23.Peculis, B. A., and Steitz, J. A. (1993)
Cell, 73, 1233-1245.
24.Cavaille, J., Hadjiolov, A. A., and Bachellerie,
J. P. (1996) Eur. J. Biochem., 242, 206-213.
25.Kiss, T., Fayet-Lebaron, E., and Jady, B. E.
(2010) Mol. Cell, 37, 597-606.
26.Morrissey, J. P., and Tollervey, D. (1993)
Mol. Cell Biol., 13, 2469-2477.
27.Makarova, J. A., and Kramerov, D. A. (2007)
Mol. Biol. (Moscow), 41, 246-259.
28.Hamma, T., and Ferre-D’Amare, A. R. (2010)
J. Biol. Chem., 285, 805-809.
29.Baxter-Roshek, J. L., Petrov, A. N., and Dinman,
J. D. (2007) PLoS One, 2, e174.
30.Blanchard, S. C., and Puglisi, J. D. (2001)
Proc. Natl. Acad. Sci. USA, 98, 3720-3725.
31.Liu, B., Liang, X. H., Piekna-Przybylska, D.,
Liu, Q., and Fournier, M. J. (2008) RNA Biol., 5,
249-254.
32.Basu, A., Das, P., Chaudhuri, S., Bevilacqua, E.,
Andrews, J., Barik, S., Hatzoglou, M., Komar, A. A., and Mazumder, B.
(2011) Mol. Cell Biol., 31, 4482-4499.
33.Mitchell, J. R., Cheng, J., and Collins, K.
(1999) Mol. Cell Biol., 19, 567-576.
34.Tycowski, K. T., You, Z. H., Graham, P. J., and
Steitz, J. A. (1998) Mol. Cell, 2, 629-638.
35.Nizami, Z., Deryusheva, S., and Gall, J. G.
(2010) Cold Spring Harb. Perspect. Biol., 2, a000653.
36.Darzacq, X., Jady, B. E., Verheggen, C., Kiss, A.
M., Bertrand, E., and Kiss, T. (2002) EMBO J., 21,
2746-2756.
37.Lestrade, L., and Weber, M. J. (2006) Nucleic
Acids Res., 34, D158-162.
38.Makarova, J. A., and Kramerov, D. A. (2007)
Genetika, 43, 149-158.
39.Makarova, J. A., and Kramerov, D. A. (2005)
Gene, 363, 51-60.
40.Tycowski, K. T., Shu, M. D., and Steitz, J. A.
(1996) Nature, 379, 464-466.
41.Jady, B. E., Ketele, A., and Kiss, T. (2012)
Genes Dev., 26, 1897-1910.
42.Shadel, G. S., and Clayton, D. A. (1997) Annu.
Rev. Biochem., 66, 409-435.
43.Mattijssen, S., Welting, T. J., and Pruijn, G. J.
(2010) Wiley Interdiscip. Rev. RNA, 1, 102-116.
44.Martin, A. N., and Li, Y. (2007) Cell
Res., 17, 219-226.
45.Ender, C., Krek, A., Friedlander, M. R.,
Beitzinger, M., Weinmann, L., Chen, W., Pfeffer, S., Rajewsky, N., and
Meister, G. (2008) Mol. Cell, 32, 519-528.
46.Burroughs, A. M., Ando, Y., de Hoon, M. J.,
Tomaru, Y., Suzuki, H., Hayashizaki, Y., and Daub, C. O. (2011) RNA
Biol., 8, 158-177.
47.Brameier, M., Herwig, A., Reinhardt, R., Walter,
L., and Gruber, J. (2011) Nucleic Acids Res., 39,
675-686.
48.Scott, M. S., Ono, M., Yamada, K., Endo, A.,
Barton, G. J., and Lamond, A. I. (2012) Nucleic Acids Res.,
40, 3676-3688.
49.Taft, R. J., Glazov, E. A., Lassmann, T.,
Hayashizaki, Y., Carninci, P., and Mattick, J. S. (2009) RNA,
15, 1233-1240.
50.Saraiya, A. A., and Wang, C. C. (2008) PLoS
Pathog., 4, e1000224.
51.Starega-Roslan, J., Krol, J., Koscianska, E.,
Kozlowski, P., Szlachcic, W. J., Sobczak, K., and Krzyzosiak, W. J.
(2011) Nucleic Acids Res., 39, 257-268.
52.Langenberger, D., Bermudez-Santana, C. I.,
Stadler, P. F., and Hoffmann, S. (2010) Pac. Symp. Biocomput.,
80-87.
53.Fabian, M. R., and Sonenberg, N. (2012) Nat.
Struct. Mol. Biol., 19, 586-593.
54.Taft, R. J., Simons, C., Nahkuri, S., Oey, H.,
Korbie, D. J., Mercer, T. R., Holst, J., Ritchie, W., Wong, J. J.,
Rasko, J. E., Rokhsar, D. S., Degnan, B. M., and Mattick, J. S. (2010)
Nat. Struct. Mol. Biol., 17, 1030-1034.
55.Okamura, K. (2012) Wiley Interdiscip. Rev.
RNA, 3, 351-368.
56.Ruby, J. G., Jan, C. H., and Bartel, D. P. (2007)
Nature, 448, 83-86.
57.Westholm, J. O., and Lai, E. C. (2011)
Biochimie, 93, 1897-1904.
58.Babiarz, J. E., Ruby, J. G., Wang, Y., Bartel, D.
P., and Blelloch, R. (2008) Genes Dev., 22,
2773-2785.
59.Kaneko, H., Dridi, S., Tarallo, V., Gelfand, B.
D., Fowler, B. J., Cho, W. G., Kleinman, M. E., Ponicsan, S. L.,
Hauswirth, W. W., Chiodo, V. A., Kariko, K., Yoo, J. W., Lee, D. K.,
Hadziahmetovic, M., Song, Y., Misra, S., Chaudhuri, G., Buaas, F. W.,
Braun, R. E., Hinton, D. R., Zhang, Q., Grossniklaus, H. E., Provis, J.
M., Madigan, M. C., Milam, A. H., Justice, N. L., Albuquerque, R. J.,
Blandford, A. D., Bogdanovich, S., Hirano, Y., Witta, J., Fuchs, E.,
Littman, D. R., Ambati, B. K., Rudin, C. M., Chong, M. M., Provost, P.,
Kugel, J. F., Goodrich, J. A., Dunaief, J. L., Baffi, J. Z., and
Ambati, J. (2011) Nature, 471, 325-330.
60.Smalheiser, N. R. (2008) Biochim. Biophys.
Acta, 1779, 678-681.
61.Liao, J. Y., Ma, L. M., Guo, Y. H., Zhang, Y. C.,
Zhou, H., Shao, P., Chen, Y. Q., and Qu, L. H. (2010) PLoS One,
5, e10563.
62.Ando, Y., Tomaru, Y., Morinaga, A., Burroughs, A.
M., Kawaji, H., Kubosaki, A., Kimura, R., Tagata, M., Ino, Y., Hirano,
H., Chiba, J., Suzuki, H., Carninci, P., and Hayashizaki, Y. (2011)
PLoS One, 6, e23385.
63.Liang, X. H., and Crooke, S. T. (2011) Nucleic
Acids Res., 39, 4875-4889.
64.Sinkkonen, L., Hugenschmidt, T., Filipowicz, W.,
and Svoboda, P. (2010) PLoS One, 5, e12175.
65.Ohrt, T., Muetze, J., Svoboda, P., and Schwille,
P. (2012) Curr. Top. Med. Chem., 12, 79-88.
66.Macias, S., Plass, M., Stajuda, A., Michlewski,
G., Eyras, E., and Caceres, J. F. (2012) Nat. Struct. Mol.
Biol., 19, 760-766.
67.Kishore, S., and Stamm, S. (2006) Science,
311, 230-232.
68.Canton, H., Emeson, R. B., Barker, E. L.,
Backstrom, J. R., Lu, J. T., Chang, M. S., and Sanders-Bush, E. (1996)
Mol. Pharmacol., 50, 799-807.
69.Khanna, A., and Stamm, S. (2010) RNA
Biol., 7, 480-485.
70.Burns, C. M., Chu, H., Rueter, S. M., Hutchinson,
L. K., Canton, H., Sanders-Bush, E., and Emeson, R. B. (1997)
Nature, 387, 303-308.
71.Kishore, S., Khanna, A., Zhang, Z., Hui, J.,
Balwierz, P. J., Stefan, M., Beach, C., Nicholls, R. D., Zavolan, M.,
and Stamm, S. (2010) Hum. Mol. Genet., 19, 1153-1164.
72.Ge, J., Liu, H., and Yu, Y. T. (2010) RNA,
16, 1078-1085.
73.Semenov, D. V., Vratskih, O. V., Kuligina, E. V.,
and Richter, V. A. (2008) Ann. N. Y. Acad. Sci., 1137,
119-124.
74.Stepanov, G. A., Semenov, D. V., Kuligina, E. V.,
Koval, O. A., Rabinov, I. V., Kit, Y. Y., and Richter, V. A. (2012)
Acta Naturae, 4, 32-41.
75.Zhao, X., and Yu, Y. T. (2008) Nat.
Methods, 5, 95-100.
76.Shen, M., Eyras, E., Wu, J., Khanna, A., Josiah,
S., Rederstorff, M., Zhang, M. Q., and Stamm, S. (2011) Nucleic
Acids Res., 39, 9720-9730.
77.Bortolin-Cavaille, M. L., and Cavaille, J. (2012)
Nucleic Acids Res., 40, 6800-6807.
78.Ono, M., Yamada, K., Avolio, F., Scott, M. S.,
van Koningsbruggen, S., Barton, G. J., and Lamond, A. I. (2010) Mol.
Biol. Cell, 21, 1569-1584.
79.Ono, M., Scott, M. S., Yamada, K., Avolio, F.,
Barton, G. J., and Lamond, A. I. (2011) Nucleic Acids Res.,
39, 3879-3891.
80.Karijolich, J., and Yu, Y. T. (2011)
Nature, 474, 395-398.
81.Bazeley, P. S., Shepelev, V., Talebizadeh, Z.,
Butler, M. G., Fedorova, L., Filatov, V., and Fedorov, A. (2008)
Gene, 408, 172-179.
82.Suzuki, A., Kogo, R., Kawahara, K., Sasaki, M.,
Nishio, M., Maehama, T., Sasaki, T., Mimori, K., and Mori, M. (2012)
Cancer Sci., 103, 632-637.
83.Liu, Z. H., Yang, G., Zhao, T., Cao, G. J.,
Xiong, L., Xia, W., Huang, X., Wu, L. Y., Wu, K., Fan, M., Shao, N. S.,
and Zhu, L. L. (2011) Cell. Mol. Neurobiol., 31, 1-5.
84.Michel, C. I., Holley, C. L., Scruggs, B. S.,
Sidhu, R., Brookheart, R. T., Listenberger, L. L., Behlke, M. A., Ory,
D. S., and Schaffer, J. E. (2011) Cell Metab., 14,
33-44.
85.Nicoloso, M., Qu, L. H., Michot, B., and
Bachellerie, J. P. (1996) J. Mol. Biol., 260,
178-195.
86.Chu, L., Su, M. Y., Maggi, L. B., Jr., Lu, L.,
Mullins, C., Crosby, S., Huang, G., Chng, W. J., Vij, R., and Tomasson,
M. H. (2012) J. Clin. Invest., 122, 2793-2806.
87.Parker, R., Simmons, T., Shuster, E. O.,
Siliciano, P. G., and Guthrie, C. (1988) Mol. Cell Biol.,
8, 3150-3159.
88.Li, S. G., Zhou, H., Luo, Y. P., Zhang, P., and
Qu, L. H. (2005) J. Biol. Chem., 280, 16446-16455.
89.Lowe, T. M., and Eddy, S. R. (1999)
Science, 283, 1168-1171.
90.Qu, L. H., Henras, A., Lu, Y. J., Zhou, H., Zhou,
W. X., Zhu, Y. Q., Zhao, J., Henry, Y., Caizergues-Ferrer, M., and
Bachellerie, J. P. (1999) Mol. Cell Biol., 19,
1144-1158.
91.Piekna-Przybylska, D., Decatur, W. A., and
Fournier, M. J. (2007) RNA, 13, 305-312.
92.Liang, X. H., Liu, Q., and Fournier, M. J. (2007)
Mol. Cell, 28, 965-977.
93.Esguerra, J., Warringer, J., and Blomberg, A.
(2008) RNA, 14, 649-656.
94.Liang, X. H., Liu, Q., and Fournier, M. J. (2009)
RNA, 15, 1716-1728.
95.King, T. H., Liu, B., McCully, R. R., and
Fournier, M. J. (2003) Mol. Cell, 11, 425-435.
96.Badis, G., Fromont-Racine, M., and Jacquier, A.
(2003) RNA, 9, 771-779.
97.Baudin-Baillieu, A., Fabret, C., Liang, X. H.,
Piekna-Przybylska, D., Fournier, M. J., and Rousset, J. P. (2009)
Nucleic Acids Res., 37, 7665-7677.
98.Cassidy, S. B., Schwartz, S., Miller, J. L., and
Driscoll, D. J. (2012) Genet. Med., 14, 10-26.
99.Cavaille, J., Buiting, K., Kiefmann, M., Lalande,
M., Brannan, C. I., Horsthemke, B., Bachellerie, J. P., Brosius, J.,
and Huttenhofer, A. (2000) Proc. Natl. Acad. Sci. USA,
97, 14311-14316.
100.Gray, T. A., Saitoh, S., and Nicholls, R. D.
(1999) Proc. Natl. Acad. Sci. USA, 96, 5616-5621.
101.Runte, M., Huttenhofer, A., Gross, S.,
Kiefmann, M., Horsthemke, B., and Buiting, K. (2001) Hum. Mol.
Genet., 10, 2687-2700.
102.Sahoo, T., del Gaudio, D., German, J. R.,
Shinawi, M., Peters, S. U., Person, R. E., Garnica, A., Cheung, S. W.,
and Beaudet, A. L. (2008) Nat. Genet., 40, 719-721.
103.Runte, M., Varon, R., Horn, D., Horsthemke, B.,
and Buiting, K. (2005) Hum. Genet., 116, 228-230.
104.Burger, J., Horn, D., Tonnies, H., Neitzel, H.,
and Reis, A. (2002) Am. J. Med. Genet., 111, 233-237.
105.Doe, C. M., Relkovic, D., Garfield, A. S.,
Dalley, J. W., Theobald, D. E., Humby, T., Wilkinson, L. S., and Isles,
A. R. (2009) Hum. Mol. Genet., 18, 2140-2148.
106.Morabito, M. V., Abbas, A. I., Hood, J. L.,
Kesterson, R. A., Jacobs, M. M., Kump, D. S., Hachey, D. L., Roth, B.
L., and Emeson, R. B. (2010) Neurobiol. Dis., 39,
169-180.
107.Nelson, N. D., and Bertuch, A. A. (2012)
Mutat. Res., 730, 43-51.
108.Mason, P. J., and Bessler, M. (2011) Cancer
Genet., 204, 635-645.
109.Vulliamy, T., Beswick, R., Kirwan, M., Marrone,
A., Digweed, M., Walne, A., and Dokal, I. (2008) Proc. Natl. Acad.
Sci. USA, 105, 8073-8078.
110.Mason, P. J., Wilson, D. B., and Bessler, M.
(2005) Curr. Mol. Med., 5, 159-170.
111.Ruggero, D., Grisendi, S., Piazza, F., Rego,
E., Mari, F., Rao, P. H., Cordon-Cardo, C., and Pandolfi, P. P. (2003)
Science, 299, 259-262.
112.Pereboom, T. C., van Weele, L. J., Bondt, A.,
and MacInnes, A. W. (2011) Blood, 118, 5458-5465.
113.Zhang, Y., Morimoto, K., Danilova, N., Zhang,
B., and Lin, S. (2012) PLoS One, 7, e30188.
114.Narla, A., and Ebert, B. L. (2010)
Blood, 115, 3196-3205.
115.Chung, L., and Utz, P. J. (2004) Curr.
Rheumatol. Rep., 6, 156-163.
116.Van Eenennaam, H., Vogelzangs, J. H.,
Bisschops, L., Te Boome, L. C., Seelig, H. P., Renz, M., De Rooij, D.
J., Brouwer, R., Pluk, H., Pruijn, G. J., Van Venrooij, W. J., and Van
Den Hoogen, F. H. (2002) Clin. Exp. Immunol., 130,
532-540.
117.Pollard, K. M., Hultman, P., and Kono, D. H.
(2010) Chem. Res. Toxicol., 23, 455-466.
118.Yang, J. M., Baserga, S. J., Turley, S. J., and
Pollard, K. M. (2001) Clin. Immunol., 101, 38-50.
119.Welting, T. J., Raijmakers, R., and Pruijn, G.
J. (2003) Autoimmun. Rev., 2, 313-321.
120.Aggarwal, R., Lucas, M., Fertig, N., Oddis, C.
V., and Medsger, T. A., Jr. (2009) Arthritis. Rheum., 60,
1112-1118.
121.Mannoor, K., Liao, J., and Jiang, F. (2012)
Biochim. Biophys. Acta, 1826, 121-128.
122.Williams, G. T., and Farzaneh, F. (2012)
Nat. Rev. Cancer, 12, 84-88.
123.Liao, J., Yu, L., Mei, Y., Guarnera, M., Shen,
J., Li, R., Liu, Z., and Jiang, F. (2010) Mol. Cancer, 9,
198.
124.Mei, Y. P., Liao, J. P., Shen, J., Yu, L., Liu,
B. L., Liu, L., Li, R. Y., Ji, L., Dorsey, S. G., Jiang, Z. R., Katz,
R. L., Wang, J. Y., and Jiang, F. (2012) Oncogene, 31,
2794-2804.
125.Lopez-Corral, L., Mateos, M. V., Corchete, L.
A., Sarasquete, M. E., de la Rubia, J., de Arriba, F., Lahuerta, J. J.,
Garcia-Sanz, R., San Miguel, J. F., and Gutierrez, N. C. (2012)
Haematologica, 97, 1439-1443.
126.Gee, H. E., Buffa, F. M., Camps, C.,
Ramachandran, A., Leek, R., Taylor, M., Patil, M., Sheldon, H., Betts,
G., Homer, J., West, C., Ragoussis, J., and Harris, A. L. (2011) Br.
J. Cancer, 104, 1168-1177.
127.Schneider, C., King, R. M., and Philipson, L.
(1988) Cell, 54, 787-793.
128.Smith, C. M., and Steitz, J. A. (1998) Mol.
Cell Biol., 18, 6897-6909.
129.Mourtada-Maarabouni, M., Pickard, M. R., Hedge,
V. L., Farzaneh, F., and Williams, G. T. (2009) Oncogene,
28, 195-208.
130.Nakamura, Y., Takahashi, N., Kakegawa, E.,
Yoshida, K., Ito, Y., Kayano, H., Niitsu, N., Jinnai, I., and Bessho,
M. (2008) Cancer Genet. Cytogenet., 182, 144-149.
131.Dong, X. Y., Guo, P., Boyd, J., Sun, X., Li,
Q., Zhou, W., and Dong, J. T. (2009) J. Genet. Genomics,
36, 447-454.
132.Dong, X. Y., Rodriguez, C., Guo, P., Sun, X.,
Talbot, J. T., Zhou, W., Petros, J., Li, Q., Vessella, R. L., Kibel, A.
S., Stevens, V. L., Calle, E. E., and Dong, J. T. (2008) Hum. Mol.
Genet., 17, 1031-1042.
133.Tanaka, R., Satoh, H., Moriyama, M., Satoh, K.,
Morishita, Y., Yoshida, S., Watanabe, T., Nakamura, Y., and Mori, S.
(2000) Genes Cells, 5, 277-287.
134.Vaz, C., Ahmad, H. M., Sharma, P., Gupta, R.,
Kumar, L., Kulshreshtha, R., and Bhattacharya, A. (2010) BMC
Genomics, 11, 288.
135.Huber-Keener, K. J., Liu, X., Wang, Z., Wang,
Y., Freeman, W., Wu, S., Planas-Silva, M. D., Ren, X., Cheng, Y.,
Zhang, Y., Vrana, K., Liu, C. G., Yang, J. M., and Wu, R. (2012)
PLoS One, 7, e41333.
136.Valleron, W., Laprevotte, E., Gautier, E. F.,
Quelen, C., Demur, C., Delabesse, E., Agirre, X., Prosper, F., Kiss,
T., and Brousset, P. (2012) Leukemia, 26, 2052-2060.
137.Valleron, W., Ysebaert, L., Berquet, L.,
Fataccioli, V., Quelen, C., Martin, A., Parrens, M., Lamant, L., de
Leval, L., Gisselbrecht, C., Gaulard, P., and Brousset, P. (2012)
Blood, 120, 3997-4005.
138.Martens-Uzunova, E. S., Jalava, S. E., Dits, N.
F., van Leenders, G. J., Moller, S., Trapman, J., Bangma, C. H.,
Litman, T., Visakorpi, T., and Jenster, G. (2012) Oncogene,
31, 978-991.
139.Cheung, H. H., Lee, T. L., Davis, A. J., Taft,
D. H., Rennert, O. M., and Chan, W. Y. (2010) Br. J. Cancer,
102, 419-427.
140.Ferreira, H. J., Heyn, H., Moutinho, C., and
Esteller, M. (2012) RNA Biol., 9, 881-890.
141.Sieron, P., Hader, C., Hatina, J., Engers, R.,
Wlazlinski, A., Muller, M., and Schulz, W. A. (2009) Br. J.
Cancer, 101, 1410-1416.
142.Liu, B., Zhang, J., Huang, C., and Liu, H.
(2012) PLoS One, 7, e43147.
143.Belin, S., Beghin, A., Solano-Gonzalez, E.,
Bezin, L., Brunet-Manquat, S., Textoris, J., Prats, A. C., Mertani, H.
C., Dumontet, C., and Diaz, J. J. (2009) PLoS One, 4,
e7147.
144.Yoon, A., Peng, G., Brandenburger, Y., Zollo,
O., Xu, W., Rego, E., and Ruggero, D. (2006) Science,
312, 902-906.
145.Skreka, K., Schafferer, S., Nat, I. R.,
Zywicki, M., Salti, A., Apostolova, G., Griehl, M., Rederstorff, M.,
Dechant, G., and Huttenhofer, A. (2012) Nucleic Acids Res.,
40, 6001-6015.
146.Skreka, K., Zywicki, M., Karbiener, M.,
Huttenhofer, A., Scheideler, M., and Rederstorff, M. (2012) J.
Nucleic Acids, 2012, 283560.
147.O’Brien, J. E., Jr., Kibiryeva, N., Zhou,
X. G., Marshall, J. A., Lofland, G. K., Artman, M., Chen, J., and
Bittel, D. C. (2012) Circ. Cardiovasc. Genet., 5,
279-286.
148.Higa-Nakamine, S., Suzuki, T., Uechi, T.,
Chakraborty, A., Nakajima, Y., Nakamura, M., Hirano, N., and Kenmochi,
N. (2012) Nucleic Acids Res., 40, 391-398.
149.Castle, J. C., Armour, C. D., Lower, M.,
Haynor, D., Biery, M., Bouzek, H., Chen, R., Jackson, S., Johnson, J.
M., Rohl, C. A., and Raymond, C. K. (2010) PLoS One, 5,
e11779.
150.Yan, D., He, D., He, S., Chen, X., Fan, Z., and
Chen, R. (2011) PLoS One, 6, e21652.
151.Sakharov, D. A., Maltseva, D. V., Riabenko, E.
A., Shkurnikov, M. U., Northoff, H., Tonevitsky, A. G., and Grigoriev,
A. I. (2012) Eur. J. Appl. Physiol., 112, 963-972.
152.Hutzinger, R., Mrazek, J., Vorwerk, S., and
Huttenhofer, A. (2010) RNA Biol., 7, 586-595.
153.Peng, X., Gralinski, L., Ferris, M. T.,
Frieman, M. B., Thomas, M. J., Proll, S., Korth, M. J., Tisoncik, J.
R., Heise, M., Luo, S., Schroth, G. P., Tumpey, T. M., Li, C., Kawaoka,
Y., Baric, R. S., and Katze, M. G. (2011) MBio, 2.
154.Tamboli, R. A., Hajri, T., Jiang, A.,
Marks-Shulman, P. A., Williams, D. B., Clements, R. H., Melvin, W.,
Bowen, B. P., Shyr, Y., Abumrad, N. N., and Flynn, C. R. (2011) PLoS
One, 6, e28577.
155.Pasinetti, G. M., Ho, L., Dooley, C., Abbi, B.,
and Lange, G. (2012) Am. J. Neurodegener. Dis., 1,
88-98.