Destabilization of CH2 Domains in Intact IgG2 Is Accompanied by Reduced Ability to Inhibit Complement System Factor C1
M. A. Timchenko and V. M. Tischenko*
Institute of Theoretical and Experimental Biophysics, Russian Academy of Sciences, Institutskaya ul. 3, 142290 Pushchino, Moscow Region, Russia; fax: (4967) 33-0553; E-mail: tischen@vega.protres.ru* To whom correspondence should be addressed.
Received January 23, 2013; Revision received March 14, 2013
Fc fragments (hFc) of human myeloma IgG2 proteins LOM and SIN having core hinge (Cys-Cys-Val-Glu-Cys-Pro-Pro-Cys) were first obtained by a modified proteolytic procedure. The thermostability of CH2 domains inside of standard Fc, hFc fragments, and intact IgG2 LOM and SIN was studied by fluorescence spectroscopy. It was found that CH2 domains of intact IgG2 are destabilized. The destabilization is accompanied by reduced ability of IgG2 to inhibit the activation of complement system by classical pathway. This could be due to the decrease in the affinity of CH2 domains to factor C1q.
KEY WORDS: immunoglobulin IgG2, Fc fragment, CH2 domain, complement system factor C1q, myeloma, stabilityDOI: 10.1134/S0006297913060126
Abbreviations: DSS, disuccinimidyl suberate; FITC, fluorescein isothiocyanate; Fv-subfragment, structure formed by pair of variable domains VL-VH; ΔGst, Gibbs free energy of CH2 domain stabilization; H(L) chains, heavy (light) chains; IgG, immunoglobulins of G class; Tm, ΔHm, ΔSm, melting temperature, enthalpy, and entropy of CH2 domain, respectively; VH, variable domains of heavy chain; VL, variable domains of light chain.
IgG (H2L2) consists of two identical heavy and two
identical light chains. The N-terminal halves of H-chains and L-chains
form two antigen-binding Fab fragments, while the C-terminal halves of
H-chains make the one Fc fragment [1] responsible
for effector functions: interaction with the complement C1q protein and
with numerous cell receptors [2]. In the intact
molecule Fab and Fc are linked by the so-called “hinge”
region including the rigid core hinge formed by poly-L-proline double
helix stabilized by disulfide bridges, and flexible upper hinge and
lower hinge segments providing internal mobility in the molecule [3-6].
The Fc fragment obtained by restricted proteolysis contains CH2 and two CH3 domains as well as usually the hinge region (or its part) with interchain disulfide bonds. The close contacts between domains belonging to the neighboring chains are essential for IgG domains [7, 8]. The exception is CH2 domains, where inter-domain interactions come to a few contacts of their carbohydrate components, which are present at glycosylated conservative Asn297 residues [9, 10]. The observed type of contacts is in agreement with earlier obtained experimental data that the interacting domains form cooperative blocks [11-13] while CH2 domains are the same themselves [13, 14].
Human immunoglobulins IgG belonging to different subclasses differ significantly in their ability to perform different effector functions, in particular, to activate the complement system [15]. The complement factor C1q, which interacts with CH2 domains of IgG, plays the key role in activation of the complement system by the classical pathway [2]. It is known that IgG2 activates this process very weakly although its CH2 domains contain the sequence corresponding to the active center [16].
We previously showed that stabilities of the mentioned domains in the structure of an entire human IgG belonging to various subclasses differ considerably. The change in stability probably affects the ability of IgG of different subclasses to activate the complement system [17, 18]. If the above observation is not occasional, then one can suppose that it is true also for Fc fragments obtained from human IgG of all four subclasses. IgG2 with significantly shorter hinge region (especially upper hinge) containing four disulfide bridges between two identical 12-mer peptides is of special interest. These structural peculiarities make the IgG2 structure more rigid, causing some difficulties in Fc fragment isolation, and they apparently have a significant influence on the mentioned biological activity, which differs in intensity from that for IgG1 and IgG3 [2, 15, 19].
The goal of the present work was to study the thermodynamic stability of CH2 domains of human IgG2 LOM and SIN inside Fc fragments and the inhibitory activity of the latter towards complement factor C1.
MATERIALS AND METHODS
Isolation and typing of myeloma IgG1 MUR and IgG2 LOM and SIN and preparation of Fc fragments and CH2 and CH3 domains of these proteins. IgG1 MUR and IgG2 LOM and SIN were isolated from blood serum of multiple myeloma patients by a standard procedure [17]. Further we used proteins during their storage time (2 days at 0°C in solution). Ig subclass was checked by monospecific antisera in a double agar gel immunodiffusion test [20]. Fc fragments of IgG1 MUR were obtained by a standard procedure [13, 17]. Fc fragments from IgG2 LOM and SIN were obtained by two approaches: standard restricted proteolysis using papain [17] and trypsin [21] and our modified procedure of trypsinolysis. In the last case the proteolysis was carried out for 2 h in the presence of double excess of Fv subfragments obtained from rabbit antibodies against CH2 domains of IgG2 (see below). The reaction was carried out in 1 mM phosphate buffer, pH 6.0, containing 2 mM cysteine with enzyme to substrate ratio of 1 : 20 at 35°C. Formed Fc and Fab fragments of LOM and SIN were separated from Fv subfragments and remaining intact IgG by gel filtration at pH 4.0 (10 mM glycine buffer). Then Fc and Fab fragments were separated by ion-exchange chromatography. All experiments were carried out according to standard procedures [17, 22]. The Fc and Fab fragments were identified by immunoelectrophoresis with antiserum to light and heavy chains of IgG as well as to all proteins of blood serum [23].
CH2 domains were obtained from papain-digested Fc fragments of IgG2 by trypsinolysis as described earlier [24]. However, first, the incubation before the proteolysis was at pH 2.8 instead of pH 2.5 [13]. Second, the reaction was carried out in the presence of Fab fragments from rabbit antibodies against CH2 domains at CH2 to Fab ratio of 1 : 1 for the stabilization of labile structure of these domains [25, 26]. After the completion of reaction, this complex was dissociated at pH 4.0 (10 mM glycine buffer), and the two proteins were separated by gel filtration under the mentioned conditions. CH3 domains (pFc′-fragments) were obtained by pepsinolysis at enzyme to substrate ratio of 1 : 100 in 100 mM acetate buffer, pH 4.5, according to a standard procedure [27]. The homogeneity of studied fragments was checked by SDS-PAGE [28].
Preparation of antibodies, their Fab fragments, and Fv subfragments against CH2 and CH3 domains. Both ordinary and fluorescently labeled antibodies against CH2 and CH3 domains of IgG were obtained by immunization of a rabbit with labeled and unlabeled CH2 and CH3 domains, correspondingly [13]. Fab fragments of these antibodies were obtained by standard papain digestion [11, 17, 22]. Fv subfragments were obtained by low-temperature pepsinolysis as described in our paper [29]. Its VH and VL domains were cross-linked by homobifunctional reagent (DSS) by a procedure described previously [30, 31].
Preparation of Fc fragments with selectively fluorescently labeled CH2 and CH3 domains. The Fc fragments were modified by the fluorescent dye FITC in 200 mM borate buffer, pH 9.1, containing 150 mM NaCl for 12 h at 4°C at protein to dye ratio of 2 : 1 [13]. The only deviation from the above-mentioned procedure is a slight decrease in pH of the solution (from 9.1 to 8.8). The quantity of dye incorporated into the Fc fragment was calculated from the ratio of the absorbance at 495 nm to that at 470 nm [32]. Fc fragments with selectively fluorescence labeled CH2 or CH3 domains were isolated by affinity chromatography using rabbit antibodies as described in [13, 14]. This approach is based on the fact that for some antibodies the epitope is sensitive not only to dye but also to adjacent regions of the protein matrix. Therefore Fc fragments with the corresponding labeled domains were eluted from the column by immobilized rabbit antibodies or by FITC-CH2 domains or FITC-CH3 domains.
Determination of molecular mass. The molecular masses of proteins were determined by high-speed (weight average molecular mass Mw) and low-speed (Z-average molar mass Mz) equilibrium centrifugation by the methods of Yphantis [33] and of Van Holde-Baldwin [34] using Beckman Spinco model E (Beckman Coulter, USA) and MOM (MOM, Hungary) analytical ultracentrifuges at 20°C. The equilibrium centrifugation was carried out with incomplete filling of cells to reach equilibrium in a short time during the experiment. It is well known that the time needed to reach the equilibrium in a cell is proportional to the square of liquid layer height [35].
Study of conformational changes in Fc fragments by fluorescence spectroscopy. The temperature-induced conformational transitions in proteins were studied by measuring fluorescence intensity of the solution in the 460-600-nm range with 450-nm excitation using a Shimadzu RF-5301 PC spectrofluorimeter (Shimadzu, Japan) with temperature-controlled cell. Protein concentrations were in the range 0.05-0.25 mg/ml. The warming rate was varied in the range 0.25-2°C/min. The samples were gel filtered before any experiments using Ultragels AcA-34 or AcA-44 (LKB, Sweden) equilibrated with corresponding buffers. Experimental temperature dependences of fluorescence were analyzed by described methods [13, 36, 37].
Determination of inhibitory activity of Ig and Fc fragments. The ability of both Ig and Fc fragments to interact with complement system proteins was estimated by a procedure described in [38, 39] and already used by us previously [30]. This procedure is based on the determination of residual activity of complement C1 factor. The activity of C1 is due to the presence of C1q molecules not bound to Ig or Fc and causing erythrocyte lysis [38, 39]. The C1q protein was isolated by a procedure described in [40] with modification proposed in our paper [41].
RESULTS AND DISCUSSION
In most cases, the restricted proteolysis of human IgG2 immunoglobulins by various proteases results in hydrolysis of a peptide bond in the lower hinge region with generation of Fc fragment [21, 42-46], where two newly formed N-terminal regions of heavy chains are not bound by covalent bonds and dissociate under denaturing conditions. It should be noted that for Fc fragments generated by proteolysis of IgG of other subclasses, core hinge and, therefore, interchain disulfide bonds are preserved [4, 47-50].
We used a new approach for the isolation of Fc fragments of IgG2 that preserves the interchain disulfide bonds. On one hand, it was made by destabilization of core hinge by partial reducing of disulfide bonds by cysteine and increasing temperature up to 35°C. On the other hand, the structure of CH2 domains exposed to the proteases was stabilized by Fv subfragments from rabbit antibodies against these domains.
The following trypsinolysis results in the cleavage of the Lys218–Cys219 peptide bond [18] instead of Pro230–Ala231, which occurs in the standard proteolysis [43]. As shown in Fig. 1, the molecular mass of hFc fragments of IgG2 LOM obtained by this approach is greater than the same of Fc fragments (see lanes 1 and 2). The analysis of lanes 1 and 6 indicates that there are disulfide bonds among hFc chains supporting the quaternary structure of the fragment together with intensive noncovalent interactions between CH3 domains [2, 4-6, 8-11, 13]. Importantly, to avoid protein degradation the labile structure of CH2 domains should be stabilized by Fv subfragments obtained from rabbit antibodies against these domains (see lanes 3-5).
Fig. 1. SDS-PAGE of proteolytic products of IgG2 LOM: 1) hFc fragment with reducing agent, the fragment was obtained by trypsinolysis at the IgG2 to rabbit Fv IgG against CH2 domains ratio of 1 : 2; 2) Fc fragment without reducing agent, the fragment was obtained by standard papain digestion [18, 44]; 3-5) Fc fragments obtained by trypsinolysis at the IgG2 LOM to Fv subfragments ratio of 1 : 1, 1 : 0.5, 1 : 0.2, correspondingly; 6) hFc fragment without reducing agent, the fragment was obtained by trypsinolysis at the IgG2 to rabbit Fv IgG against CH2 domains ratio of 1 : 2; 7) molecular weight markers. The results for IgG2 SIN are similar.
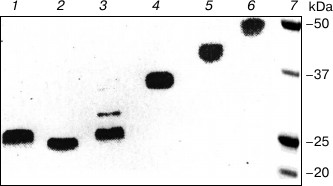
The new Fc fragment of IgG2 obtained by the modified procedure of proteolysis we called hFc fragment emphasizing that this fragment keeps both lower hinge and core hinge in contrast to the ordinary Fc fragment of IgG2 obtained by standard papain digestion [17, 43] and having only the lower hinge. Thermodynamic properties and functional activity of the studied hFc fragments of LOM and SIN proteins are much different from those for both ordinary Fc fragments and Fc subunits inside intact IgG2.
It should be emphasized that according to analytical centrifugation data the molecular masses of both intact IgG2s are equal: the weight average molecular masses Mw determined by high-speed centrifugation were 151 ± 6 kDa for LOM protein and 149 ± 6 kDa for SIN protein, Z average molar masses Mz defined by the low-speed method were 153 ± 9 kDa for LOM protein and 152 ± 9 kDa for SIN protein. The weight average and Z average molecular masses were determined for hFc fragments and their values are Mw 52 ± 3 kDa for hFc LOM and 51 ± 3 kDa for hFc SIN, Mz 53 ± 3 kDa for hFc LOM and 50 ± 3 kDa for hFc SIN. Mw and Mz are sensitive to the presence of aggregates, and coincidence of molecular masses points out that the percentage of aggregates in studied samples is less than 3% [35]. Besides, the results of melting of proteins given below do not depend upon the concentrations of the studied samples and, therefore, reflect the intramolecular processes1.
1 Despite of the absence of interchain disulfide bonds in the Fc fragment obtained by standard procedures, the thermodynamic parameters of the CH2 domain melting of this fragment are practically independent of protein concentration. This can be explained in two ways: first, the mentioned domain melts the first and, second, the CH3 domain melting temperature is rather higher that for the CH2 domain and these two transitions do not overlap. The results for temperature-induced conformational transitions both for hFc and ordinary Fc fragments of LOM protein with selectively fluorescently labeled CH2 domains and for intact IgG2 LOM are presented in Fig. 2. The corresponding melting curve for Fc IgG1 MUR is also shown for comparison. One can see from Fig. 2 that transition temperatures depend on both the environment of the domains and the type of subclass. Also, it is seen that the stability of the CH2 domains in hFc structure is higher than their stability inside ordinary Fc fragment. This is not surprising because the presence of disulfide bonds in the N-terminal part of Fc fragment stabilizes CH2 domains there and decreases the entropy of their transitions between native and denatured states. Such a picture is usually observed for both globular [51-53] and fibrillar proteins [41, 54, 55].
Fig. 2. Dependence of fraction of denatured state on temperature for CH2 domains inside IgG2 LOM (closed triangles) and hFc (closed circles), Fc fragments (open triangles), and Fc IgG1 (open circles) calculated from fluorescence data. The same results were also obtained for CH2 domains of SIN. Experiments were carried out in 10 mM acetate buffer, pH 5.5.
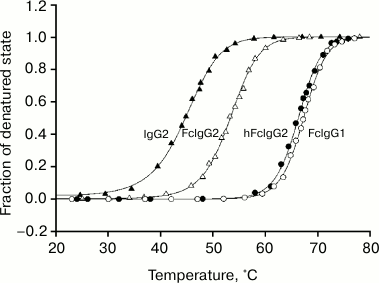
The most notable fact is that the stability of the CH2 domains in the hFc fragments is higher than their stability inside intact IgG2. This means that the indicated domains are destabilized by existed interactions between Fab and Fc subunits in intact IgG2 LOM and SIN molecules. As follows from the data represented in Fig. 2 and in the table, the effective values of enthalpy calculated from equilibrium melting curves for the mentioned domains using the known formula
ΔHm = RT2dlnK/dT
(where K is an equilibrium constant) are also higher for CH2 domains belonging to the hFc fragment. It is necessary to note that all experimental results are independent of warming rate. This fact along with the full reversibility of the melting process means that the data characterize an equilibrium process and, therefore, are truly thermodynamic.
Thermodynamic parameters obtained by melting of intact IgG and their hFc
and Fc fragments
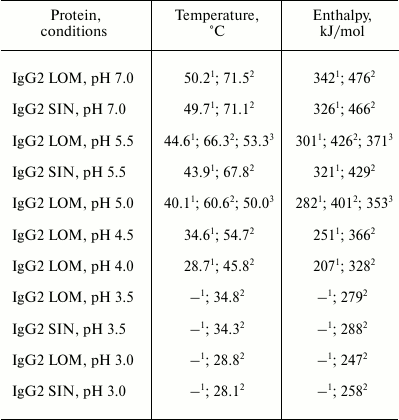
Note: Measurements were carried out in 10 mM glycine buffer in the
range of pH 3.0-3.5, 10 mM acetate buffer in the range of
pH 4.0-5.5, and 1 mM phosphate buffer at pH 7.0. The
accuracy of denaturation temperature determination (midpoint of
transition) is ± 0.7ºC, and the accuracy of determination
of effective enthalpy of denaturation (melting) is ± 12%.
1 Data obtained by melting of CH2 domains inside
the intact protein.
2 Data obtained by melting CH2 domains inside the
hFc fragment.
3 Data obtained by melting CH2 domains inside
ordinary Fc fragments.
It is important that the changes in stability of these domains are accompanied by changes in their biological activity.
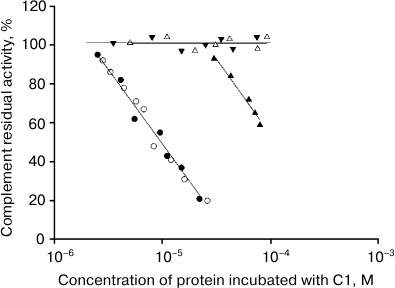
The dependence of C1 complement factor residual activity upon the concentration of intact IgG2 protein and its fragments is presented in Fig. 3. It is evident that hFc fragments inhibit the sheep erythrocyte lysis reaction more effective than corresponding intact IgG2, most probably due to their higher affinity to C1q factor [39].
Fig. 3. Dependence of C1 complement factor residual activity upon the concentration of IgG2 (closed triangles), hFc IgG2 (closed circles), Fc IgG1 (open circles), Fab IgG2 (open triangles), and pFc′ IgG2 (closed inverted triangles). The residual activity of C1 factor of the complement system in the erythrocyte lysis reaction was determined after its preincubation with the studied protein. Each point was defined from not less than three experiments.
Efforts to obtain Fc fragments with core hinge by restricted proteolysis failed for a long time. Only in 2012 data on the melting of this fragment isolated as a recombinant protein from a eukaryotic cell were published [56]. Our results are in good agreement with the above-mentioned data, though the accuracy of determination of thermodynamic parameters including transition temperature is higher in the case of differential methods (scanning microcalorimetry) than integral methods (optical methods, in our case, fluorescence spectroscopy). However, it should be noted that the set of methods used in our work reliably indicates that the melting curves observed by fluorescence spectroscopy are valid and reflect the intramolecular processes.
Indeed, in our previous work [13] the thermodynamic properties of a number of Fc fragments from IgG1 were studied with both scanning microcalorimetry and fluorescence spectroscopy, and it was found that the difference in melting temperatures of labeled CH2 and CH3 domains detected by the two methods is less than 0.7ºC. At the same time, the control calorimetric measurements for hFc of IgG2 LOM at pH 7.0 and 4.0 revealed, that the stability of labeled CH2 domains also changes in the indicated pH range (data are not shown), so the use of fluorescent dye had no serious effect on the stability of the studied domain. It should be noted that the protein oligomeric state was carefully controlled in all experiments. The protein was in the monomeric form and the stability of CH2 domains and their enthalpy of melting did not depend on the concentration, whereas in paper [56] the profile of melting curves was concentration dependent. The latter could be explained by the presence of protein fraction with aggregative properties (so-called A-form [57-59]). The possible involvement of CH2 domains in formation of this form was first shown in [60-63]. All these arguments definitely indicate that our results are truly thermodynamic and characterize exclusively intramolecular conformational transformations.
To date, the problem of different activity of subclasses of human IgG is far from solution despite its importance and the great number of works in this field [1, 15, 19, 64]. The only hypothesis concerning the low activity of IgG2 consists in steric hindrance for interaction of C1q factor with CH2 domains from the side of Fab subunits due to the short length of the upper hinge [1]. The proposed approach in our work is based on the study of stability of CH2 domains inside intact protein and hFc fragment with the hinge. The decrease in melting temperature Tm = ΔHm/ΔSm of the mentioned domains inside of intact protein indicates the increasing role of entropy in the Gibbs free energy equation ΔGst = ΔHm – TΔSm. It can reflect the increase in intramolecular mobility of CH2 domains in the IgG2 structure. The significantly faster kinetics of hydrogen exchange for CH2 domains inside IgG2 in comparison with those inside hFc fragment confirms this suggestion [18].
It is known that the measurement of enthalpy at different temperatures by scanning microcalorimetry permits the direct calculation of all necessary thermodynamic functions with high accuracy [65]. However, here we could use only integral curves for analysis. This prevented analysis with high accuracy, and it was restricted mainly by the above-mentioned qualitative considerations.
Summarizing our data and that of the literature, we conclude that the state and activity of human IgG depend on both the changes in the primary structure of CH2 domains and their interaction with Fab subunits. The latter modulate this state, decreasing their stability and increasing the entropy. This entropy increase results in decrease in Gibbs energy of interaction between IgG and C1q (or association constants). In contrast, the fixation of these domains will be accompanied by loss of entropy, which is dependent on the mobility of the protein structure.
The interaction of subunits in IgG molecules is realized only at their close distance to each other. According to electron microscopy data, the individual IgG2 molecules represent compact structures consisting of three well-expressed formations, and in some molecules one formation contacts with two others [66]. These formations could be interpreted as Fc and Fab subunits, correspondingly. The small sizes of hinge in IgG2 provide just this situation. The closer contact between subunits could proceed by the mechanism demonstrated for hog IgG [67] and human IgG1 [68] by electron microscopy. The authors found that Ig molecules can have pyramidal form instead of planar. The mobility of subunits in IgG2 molecules was confirmed both by fluorescence depolarization in solution [69] and by X-ray structural analysis [70]. Thus, the data on IgG2 structure point to possible interactions between Fab subunits and the Fc subunit.
The authors are grateful to A. A. Timchenko for comments and helpful suggestions.
This work was supported by the Russian Foundation for Basic Research (grant 11-04-00064-a).
REFERENCES
1.Porter, R. R. (1959) Biochem. J., 73,
119-126.
2.Burton, D. R., and Woof, J. M. (1992) Adv.
Immunol., 51, 1-84.
3.Marquart, M., Deisenhofer, J., Huber, R., and Palm,
W. (1980) J. Mol. Biol., 141, 369-391.
4.Harris, L. J., Larson, S. B., Hasel, K. W., Day.
J., Greenwood, A., and McPherson, A. (1992) Nature, 360,
369-372.
5.Harris, L. J., Skaletsky, E., and McPherson, A.
(1998) J. Mol. Biol., 275, 861-872.
6.Saphire, E. O., Stanfield, R. L., Crispin, M. D.,
Parren, P. W., Rudd, P. M., Dwek, R. A., Burton, D. R., and Wilson, I.
A. (2002) J. Mol. Biol., 319, 9-18.
7.Saul, F. A., Amzel, L. M., and Poljak, R. J. (1978)
J. Biol. Chem., 253, 585-597.
8.Deisenhofer, J. (1981) Biochemistry,
20, 2361-2370.
9.Sutton, B. J., and Phillips, D. C. (1983)
Biochem. Soc. Trans., 11, 130-132.
10.Padlan, E. F. (1994) Mol. Immunol.,
31, 169-217.
11.Tischenko, V. M., Zav’yalov, V. P.,
Medgyesi, G. A., Potekhin, S. A., and Privalov, P. L. (1982) Eur. J.
Biochem., 126, 517-521.
12.Zav’yalov, V. P., and Tishchenko, V. M.
(1991) Scand. J. Immunol., 33, 755-762.
13.Tischenko, V. M., Abramov, V. M., and
Zav’yalov, V. P. (1998) Biochemistry, 37,
5576-5581.
14.Tischenko, V. M. (2000) J. Therm. Anal.
Cal., 62, 63-68.
15.Burton, D. R. (1985) Mol. Immunol.,
22, 161-206.
16.Duncan, A. R., and Winter, G. (1988)
Nature, 332, 738-740.
17.Denesyuk, A. I., Tishchenko, V. M., Abramov, V.
M., and Zav’yalov, V. P. (1983) Mol. Biol. (Moscow),
17, 1262-1271.
18.Tishchenko, V. M. (2013) Mol. Biol.
(Moscow), in press.
19.Van Loghem, E. (1986) Monographs Allergy,
19, 40-51.
20.Ouchterlony, O. (1965) in Immunochemie,
15. Colloquim Ges. Physl. Chem., Springer-Verlag,
Berlin-Heidelberg-New York, pp. 13-35.
21.Vogt, R. A., and Michaelsen, T. E. (1987)
Scand. J. Immunol., 26, 59-69.
22.Frangione, B., Franklin, E. C., Fudenberg, H. H.,
and Koshland, M. E. (1966) J. Exp. Med., 124,
715-732.
23.Grabar, P., and Williams, C. A. (1953)
Biochim. Biophys. Acta, 10, 193-194.
24.Ellerson, J. R., Yasmeen, D., Painter, R. H., and
Dorrington, K. J. (1972) FEBS Lett., 24, 318-322.
25.Tischenko, V. M., Khristoforov, V. S., and
Blizniukov, O. P. (2009) Mol. Biol. (Moscow), 43,
148-156.
26.Tishchenko, V. M. (2011) Mol. Biol.
(Moscow), 45, 1055-1064.
27.Turner, M. W., Bennich, H. H., and Natvig, J. B.
(1970) Clin. Exp. Immunol., 7, 603-625.
28.Laemmli, U. K. (1970) Nature, 227,
680-685.
29.Zav’yalov, V. P., and Tishchenko, V. M.
(1991) Scand. J. Immunol., 33, 755-762.
30.Tischenko, V. M. (2001) Biochemistry
(Moscow), 66, 1671-1675.
31.Bliznyukov, O. P., Kozmin, L. D., Vysotskaya, L.
L., Golenkov, A. K., Tischenko, V. M., Samoylovich, M. P., and
Klimovich, V. B. (2005) Biochemistry (Moscow), 70,
556-567.
32.Mercola, D. A., Morris, J. W., and Arquilla, E.
R. (1972) Biochemistry, 11, 3860-3874.
33.Yphantis, D. A. (1964) Biochemistry,
3, 297-317.
34.Van Holde, K. E., and Baldwin, R. L. (1958) J.
Phys. Chem., 62, 734-743.
35.Bowen, T. (1971) in An Introduction to
Ultracentrifugation (Degly, S., ed.), Wiley-Interscience,
London-N.-Y.-Sydney-Toronto, pp. 107-108.
36.Tischenko, V. M., and Zav’yalov, V. P.
(2002) Immunol. Lett., 84, 241-245.
37.Tishchenko, V. M. (2000) Mol. Biol.
(Moscow), 34, 116-122.
38.Augener, W., Grey, H. M., Cooper, N. R., and
Muller-Eberhard, H. J. (1971) Immunochemistry, 8,
1011-1019.
39.Yasmeen, D., Ellerson, J. R., Dorrington, K. J.,
and Painter, R. H. (1976) J. Immunol., 116, 518-526.
40.Assimeh, S. N., Bing, D. H., and Painter, R. H.
(1974) J. Immunol., 113, 225-234.
41.Tischenko, V. M., Ichtchenko, A. M., Andreyev, C.
V., and Kajava, A. V. (1993) J. Mol. Biol., 234,
654-660.
42.Medgyesi, G. A., Jakab, M., Nagy, M. C., and
Gergely, J. (1971) Acta Biochim. Biophys. Acad. Sci. Hung.,
6, 405-414.
43.Wang, A. C., and Fudenberg, H. H. (1972)
Nature New Biol., 240, 24-26.
44.Solomon, A., Gramse, M., and Havemann, K. (1978)
Eur. J. Immunol., 8, 782-785.
45.Baici, A., Knopfel, M., Fehr, K., Skvaril, F.,
and Boni, A. (1980) Scand. J. Immunol., 12, 41-50.
46.Baici, A., Knopfel, M., and Fehr, K. (1982)
Scand. J. Immunol., 16, 487-498.
47.Michaelsen, T. E., Frangione, B., and Franklin,
E. C. (1977) J. Biol. Chem., 252, 883-889.
48.Isenman, D. E., Dorrington, K. J., and Painter,
R. H. (1975) J. Immunol., 114, 1726-1729.
49.Ryazantsev, S., Tishchenko, V., Vasiliev, V.,
Zav’yalov, V., and Abramov, V. (1990) Eur. J. Biochem.,
190, 393-399.
50.Tischenko, V. M., and Zav’yalov, V. P.
(2003) Immunol. Lett., 86, 281-285.
51.Gong, R., Vu, B. K., Feng, Y., Prieto, D. A.,
Dyba, M. A., Walsh, J. D., Prabakaran, P., Veenstra, T. D., Tarasov, S.
G., Ishima, R., and Dimitrov, D. S. (2009) J. Biol. Chem.,
284, 14203-14210.
52.Piatek, R., Bruzdziak, P., Wojciechowski, M.,
Zalewska-Piatek, B., and Kur, J. (2010) Biochemistry, 49,
1460-1468.
53.Gong, R., Wang, Y., Feng, Y., Zhao, Q., and
Dimitrov, D. S. (2011) J. Biol. Chem., 286,
27288-27293.
54.McBride, O., and Harrington, W. F. (1967)
Biochemistry, 6, 1484-1498.
55.Privalov, P. L., Tiktopulo, E. I., and Tischenko,
V. M. (1979) J. Mol. Biol., 127, 203-216.
56.Latypov, R. F., Hogan, S., Lau, H., Gadgil, H.,
and Liu, D. (2012) J. Biol. Chem., 287, 1381-1396.
57.Buchner, J., Renner, M., Lilie, H., Hinz, H. J.,
Jaenicke, R., Kiefhabel, T., and Rudolph, R. (1991)
Biochemistry, 30, 6922-6929.
58.Lilie, H., and Buchner, J. (1995) FEBS
Lett., 362, 43-46.
59.Thies, M. J., Kammermeier, R., Richter, K., and
Buchner, J. (2001) J. Mol. Biol., 309, 1077-1085.
60.Kravchuk, Z. I., Vlasov, A. P., Lyakhnovich, G.
V., and Martsev, S. P. (1994) Biochemistry (Moscow), 59,
1079-1092.
61.Martsev, S. P., Kravchuk, Z. I., and Vlasov, A.
P. (1994) Immunol. Lett., 43, 149-152.
62.Martsev, S. P., Kravchuk, Z. I., Vlasov, A. P.,
and Lyakhnovich, G. V. (1995) FEBS Lett., 361,
173-175.
63.Vlasov, A. P., Kravchuk, Z. I., and Martsev, S.
P. (1996) Biochemistry (Moscow), 61, 155-172.
64.Michaelsen, T. E., Sandlie, I., Bratlie, D. B.,
Sandin, R. H., and Ihle, O. (2009) Scand. J. Immunol.,
70, 553-564.
65.Privalov, P. L. (1979) Adv. Prot. Chem.,
33, 167-241.
66.Tischenko, V. M. (1999) Biophysical
Meeting, Abstract book, Moscow, Russia, P. 81.
67.Ryazantsev, S. N., Vasiliev, V. D., Abramov, V.
M., Franek, F., and Zav’yalov, V. P. (1989) FEBS Lett.,
244, 291-295.
68.Ryazantsev, S. N., Abramov, V. M.,
Zav’yalov, V. P., and Vasiliev, V. D. (1990) FEBS Lett.,
275, 221-225.
69.Dangl, J. L., Wensel, T. G., Morrison, S. L.,
Stryer, L., Herzenberg, L. A., and Oi, V. T. (1988) EMBO J.,
7, 1989-1994.
70.Ely, K. R., Colman, P. M., Abola, E. E., Hess, A.
C., Peabody, D. S., Parr, D. M., Connell, G. E., Laschinger, C. A., and
Edmundson, A. B. (1978) Biochemistry, 17, 820-823.