REVIEW: Heparan Sulfate–Protein Binding Specificity
M. A. Nugent1,2,3*, J. Zaia1, and J. L. Spencer1
1Department of Biochemistry and 2Department of Ophthalmology, Boston University School of Medicine, 72 East Concord Street, Boston, MA 02118, USA; E-mail: mnugent@bu.edu3Department of Biomedical Engineering, Boston University, Boston, MA 02215, USA
* To whom correspondence should be addressed.
Received March 13, 2013
Heparan sulfate (HS) represents a large class of linear polysaccharides that are required for the function of all mammalian physiological systems. HS is characterized by a repeating disaccharide backbone that is subject to a wide range of modifications, making this class of macromolecules arguably the most information dense in all of biology. The majority of HS functions are associated with the ability to bind and regulate a wide range of proteins. Indeed, recent years have seen an explosion in the discovery of new activities for HS where it is now recognized that this class of glycans functions as co-receptors for growth factors and cytokines, modulates cellular uptake of lipoproteins, regulates protease activity, is critical to amyloid plaque formation, is used by opportunistic pathogens to enter cells, and may even participate in epigenetic regulation. This review will discuss the current state of understanding regarding the specificity of HS–protein binding and will describe the concept that protein binding to HS depends on the overall organization of domains within HS rather than fine structure.
KEY WORDS: heparin, heparan sulfate, glycosaminoglycans, proteoglycans, protein binding, bioinformaticsDOI: 10.1134/S0006297913070055
Abbreviations: ECM, extracellular matrix; FGF, fibroblast growth factor; FGFR, fibroblast growth factor receptor; HS, heparan sulfate; HSPGs, heparan sulfate proteoglycans.
Heparin and HS are linear polysaccharides characterized by repeating
disaccharide units of alternating N-substituted glucosamine and
hexuronic acid residues subject to selective modification including
sulfation of the N-position as well as the C-6 and C-3
O-positions of the glucosamine and the C-2 O-position of
the uronic acid [1-3] (Fig. 1). Thus, the 32 (or more) potential unique
disaccharide units and their grouping into structural motifs make this
class of compounds one of the most information dense in biology [4-6]. Unlike proteins, the
sequence and overall structure of these complex molecules are not
defined by a template. Instead, the specific structure is the result of
the action of at least 18 biosynthetic enzymes as well as the
postsynthetic processing by 6-O-sulfatases and heparanase [7] (Fig. 1). While the structure
of HS expressed by cells can rapidly change in response to specific
conditions, the specific mechanisms controlling HS biosynthesis and
postsynthetic modification remain to be defined.
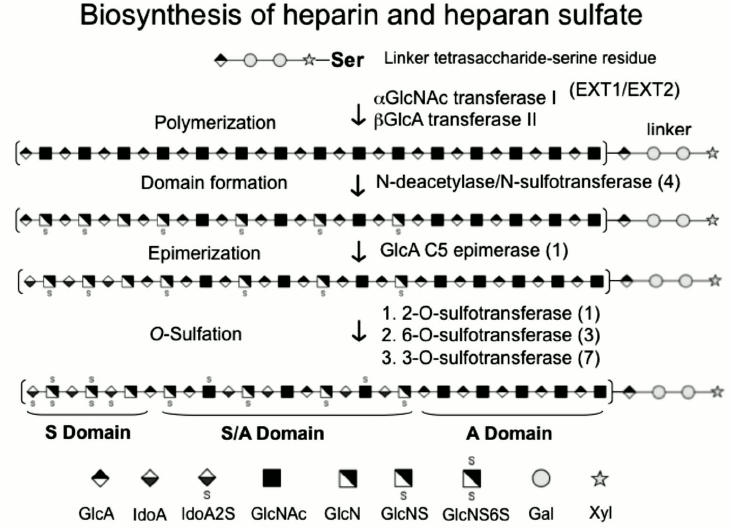
Fig. 1. Biosynthesis of heparin and heparan sulfate. The biosynthesis involves initial chain polymerization within the endoplasmic reticulum by Ext1 and 2. Deacetylation, epimerization, and sulfation of specific saccharide units occur in the Golgi apparatus through the action of four N-deacetylase/N-sulfotransferases, one C5-epimerase, one 2-O-sulfotransferase, three 6-O-sulfotransferases, and seven 3-O-sulfotransferases. S domain, sulfate-rich domain; A domain, under-sulfated domain.
HS chains are found attached to core proteins in proteoglycans on cell surfaces and within the extracellular matrix (ECM) of nearly all mammalian cells and tissues [2, 6]. On cell surfaces, HS is mainly found attached to two classes of core proteins, the syndecans and glypicans. The syndecans (1 through 4) are characterized by a transmembrane core protein with HS and sometimes chondroitin sulfate chains attached to the region of the core protein extending from the cell surface into the pericellular matrix [8]. Glypicans (1 through 6), on the other hand, are anchored to the plasma membrane via glycosylphosphatidylinositol attached to a hydrophobic domain within the C-terminal region with the HS chains restricted to the last 50 amino acids such that they are held close to the membrane [9]. There are several heparan sulfate proteoglycans (HSPGs) found within the ECM including perlecan, agrin, and collagen XVIII [10, 11]. It is generally believed that the biological function of HSPGs are dependent on HS structure and localization of the HSPG, with cell surface HSPGs being implicated in controlling ligand–receptor interactions and ECM HSPGs being considered important modulators of intercellular molecular traffic [12, 13] (Fig. 2).
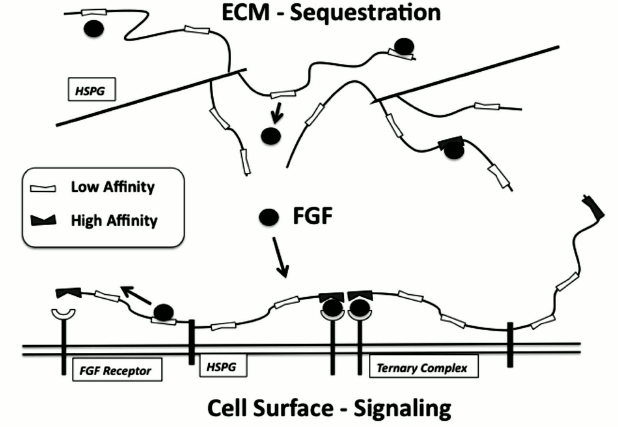
Fig. 2. Complex regulation of FGF by heparan sulfate. HS within the ECM and on the cell surface can participate in regulating FGF through binding to low-affinity non-selective sites and through interactions with high-affinity sites. Binding of FGF within the ECM can sequester or store FGF for pulse release at sites of injury. Binding of FGF by HS on the cell surface can provide a means for the cell to “capture” FGF and increase the likelihood of encounter with signaling receptors where it can form high-affinity ternary complexes.
In contrast to HS, the highly sulfated and more structurally uniform heparin is stored almost exclusively in the granules of cells of hematopoietic lineage, including connective tissue mast cells, as part of large heparin-proteoglycans that function to package inflammatory proteases [14-17]. In addition, a highly sulfated form of HS is present in leukocytes [18]. Heparin has been used clinically as an anti-coagulant for ~80 years and has also been shown to have anti-inflammatory activity and both anti- and pro-angiogenic activities [19-22]. The wide range of activities attributed to heparin and HS probably reflects the large number of proteins that these glycosaminoglycans bind and modulate [23-25]. While the majority of disaccharides within heparin contain 2-O, 6-O, and N-sulfate groups, HS is more structurally diverse with protein-binding sites more selectively expressed [23, 25]. Because of this structural diversity, it is thought that changes in HS as a function of environmental stimuli (e.g. tissue injury, ischemia) may lead to regulation of cellular responses to important extracellular proteins through alterations in HS–protein binding.
HS is essential for embryonic development [26] and required for the function of all adult physiological systems. HS structure and expression can change rapidly during development [26-28], indicating that alterations in HS might be a key signal of functional changes in cells and tissues. While the complete mechanisms remain unknown, it is generally believed that HS function is mediated by the ability of HS to bind and regulate proteins. A recent bioinformatics analysis of the HS interactome identified 435 human proteins that interact with HS or the structurally related heparin [29]. Network analysis of HS-binding proteins revealed enrichment in processes such as cell–cell communication, wound healing, immune response, defense response, and regulation of cell proliferation. Not surprisingly, HS has been implicated in many human diseases including cardiovascular disease, chronic obstructive pulmonary disease, cancer, infectious disease, amyloidosis, and HIV/AIDS [30-41]. As such, there are a number of drugs currently in development that aim to either interfere with or replicate the protein binding/regulating function of HS. The motivation for the development of HS-based drugs is driven in part by the long-standing clinical success of unfractionated and low-molecular-weight heparin as well as anecdotal evidence of secondary benefits of heparin unrelated to antithrombotic activities [32-34, 42]. While recent research has produced considerable insight, the chemical mechanisms underlying HS–protein binding affinity and specificity remain to be fully defined. Thus, the aim of this review is to evaluate the current state of understanding of the structural specificity that underlies HS–protein binding by discussing a few specific examples in detail.
HEPARAN SULFATE–PROTEIN BINDING
Based on the finding that antithrombin III binds specifically to a rare pentasaccharide sequence present in heparin, early models proposed the existence of protein-specific binding-site sequences encoded within the primary structure of HS [43]. However, recent findings indicate that the formal binding-site sequence specificity of antithrombin III is the exception and not the rule. Instead, a model has been proposed where proteins bind to localized sulfate-rich domains (NS domains) that are distributed throughout the HS chain separated by intervening under-sulfated regions (NA domains) [23, 24]. While short sulfated oligosaccharides (degree of polymerization (dp) 4-8) have been isolated to support this concept, full biological activity generally requires longer HS chains with more complex structure [5, 6].
Consistent with the concept that positioning of sulfate residues is critical for protein binding, most heparin-binding proteins have clusters of basic amino acid residues that have been shown to be required for binding heparin [43]. However, an energetic analysis of heparin–protein binding using isothermal calorimetry indicates that ~70% of the binding energy involves non-ionic interactions such as hydrogen bonds and van der Waals forces [44]. This analysis, like most previous studies on HS–protein binding, used highly sulfated heparin as a model for HS, which may have biased the data toward identifying important ionic interactions because of the nearly uniform highly sulfated structure of heparin. Thus, it is likely that intervening NA domains function as more than simple spacer regions and instead might participate directly in protein binding.
Work by our group has also noted that a range of heparin-binding growth factors can compete for one another’s binding to large-capacity but low-affinity sites on HS while also showing specific high-affinity binding to low-density binding sites [45, 46]. These findings are beginning to indicate that protein binding to HS is neither purely discrete and specific nor general and nonspecific. Instead a concept has been emerging whereby HS activity reflects the composite of its ability to bind a range of proteins in a variety of ways with some binding events leading to specific tight complexes while others providing a means to modulate protein availability and movement within the peri- and extracellular space (Fig. 2).
In a recent elegant study, FGF2 (fibroblast growth factor 2) movement within the pericellular matrix was tracked using single molecular imaging to produce compelling evidence that binding sites on HS chains are arranged in a nonrandom heterogeneous network [47]. As such, FGF2 movement appears to involve translocation from one HS-binding site to another where the rate of movement apparently relates to the distribution and the selectivity of these binding sites. Indeed, previous studies from our group concluded that a binding and diffusion model effectively describes the transport of growth factors within isolated basement membrane [48].
HS CONTROL OF FIBROBLAST GROWTH FACTOR 2
Fibroblast growth factors (FGFs) represent a large family of related proteins that bind to heparin and HS and show a wide range of important biological activities including regulation of cell proliferation, migration, differentiation, and tissue morphogenesis [49, 50]. The involvement of HS in the FGF system has been studied extensively and now represents a classic mechanism by which HS can modulate protein–protein interactions [51]. Although the majority of these studies focused on the role of HS in modulating FGF1 and FGF2, the fundamental mechanism has now been extended to understand other FGF family members as well as other heparin-binding growth factors. HS has been shown to play a key role as a co-receptor that stabilizes FGF–FGF receptor (FGFR) complex formation through interactions with both proteins. While studies have shown that short (dp 6-8) heparin- or HS-derived oligosaccharides are sufficient to bind and promote FGF2 activity, longer HS chains containing significantly reduced overall sulfate density are more active in mole/mole comparisons with high-affinity FGF2-binding oligosaccharides [52, 53]. These observations indicate that longer chains may contain structural functionality that is required for activity and that NA domains comprise a portion of the protein binding sites. One possibility is that spacer regions act to properly position multiple sulfate-rich domains for binding to FGF and its receptor proteins to enhance binding avidity. It has also been suggested that longer HS chains are required for the formation of higher-order multimeric complexes of FGF–FGFRs on the cell surface [53].
There remains considerable debate over the nature and stoichiometry of the active complexes, with data supporting various models of FGF–FGFR–HS complexes (i.e. 2 : 2 : 1, 2 : 2 : 2, 4 : 4 : 2) [54-58]. We propose that there are multiple competent complexes whose signal intensity and duration may depend on the nature of the HS chain and the specific complex type that forms. For instance, our previous data indicate that proteoglycans containing multiple HS chains that localize to lipid raft domains may be able to capture and retain FGF2 to enhance prolonged FGFR signaling [59, 60]. In this regard, we have also shown that FGF2 binds to multiple classes of binding sites within HS, with some being non-selective and low-affinity sites but present at very high levels, and others being more specific high-affinity sites but rare [45]. Recently we isolated FGF2-binding hexasaccharides from a library generated from HS using SEC (size-exclusion chromatography) followed by hydrophobic trapping and MS, and characterized low- and high-affinity binders [52]. The low-affinity sites were mostly derived from internal regions of the HS chain, whereas the high-affinity sites were enriched in non-reducing-end NS domains. We also noted that the high-affinity binders were selectively more active at promoting FGF2-induced cell proliferation using a BaF32 cell system. These results support a 1 : 1 ratio model for HS chains in the FGF system, where high-affinity NS sites at the non-reducing end might selectively participate in the formation of high-affinity FGF ternary complexes (Fig. 2).
Longer chains may also be required to enhance ligand–receptor interactions through nonspecific low-affinity interactions with under-sulfated regions that may increase FGF–FGFR encounter frequency [46, 60, 61]. While the structural requirements for heparin–FGF binding have been described and receptor specificity has been defined for the FGF system’s 23 ligands and seven receptor variants [62, 63], there remains little known about the HS structural requirements for interaction with FGF receptors or how distinct domain organization within HS can modulate FGF–FGFR complex formation. Similar gaps in knowledge exist for a wide range of heparin-binding protein systems. Thus, the ability of HS to participate in other growth factor–receptor systems, modify protease activity, alter protein structure, and mediate enzyme–substrate association is likely to be dependent on higher-order HS structure, yet to date there have been frustratingly few methods available to allow for the study of HS function at this level.
HS-MEDIATED PROTEASE INHIBITION IN EMPHYSEMA
Chronic obstructive pulmonary disease (COPD), which includes emphysema, is the fourth leading cause of death in the United States, accounting for more than 120,000 deaths in 2007 [64]. COPD comprises multiple disease types including chronic bronchitis and emphysema. Emphysema, in particular, is characterized by progressive destruction of elastin within the lung leading to airspace enlargement, decreased tissue compliance, and reduced gas exchange. Seminal findings, first reported in the 1960s, identified a strong link between emphysema and a deficiency in the natural protease inhibitor, α-1 anti-trypsin (AAT), leading to the hypothesis that this disease is the result of an imbalance between proteases and antiproteases [65, 66]. Since these initial observations, a preponderance of evidence has implicated leukocyte-derived elastases as major culprits in emphysema. For example, mice deficient in neutrophil elastase show ~60% reduced airspace enlargement compared with wild-type counterparts in response to cigarette smoke [67]. Hence, there is strong interest in understanding how to best regulate the levels and activities of proteolytic enzymes such as neutrophil elastase within the lung. Consequently, considerable effort has been directed toward producing effective elastase inhibitors to treat emphysema [68, 69], yet to date, none have proven to be clinically effective.
In addition to natural protein-based inhibitors of neutrophil elastase, heparin and HS have been shown to bind and inhibit this enzyme [70, 71]. In the lung, HS has been shown to play critical roles in development [28, 72, 73], to be targets of elastase, and to participate in ECM assembly [74-82]. Indeed, heparin and HS inhibit elastase-mediated airspace enlargement in mice [83, 84]. While it appears that HS is involved in the lung response to elastase at many levels, there remain considerable gaps in our understanding of the specific mechanisms of these processes. More generally it is interesting to note that HS has been implicated as a modulator of multiple mediators of lung injury that underlie the pathogenesis of emphysema. For example, HS has been suggested to provide protection against oxidative stress directly [85] and through interactions with extracellular superoxide dismutase (ecSOD) [86-88], store and protect vascular endothelial growth factor and facilitate its receptor activation [89-93], inhibit elastase activity [70, 83, 84, 94], control cell–ECM adhesion [95-98] and contribute to the mechanical properties and stability of the alveolar wall [99], and potentially influence chromatin structure through alterations in histone acetylation [100].
Work by our group has focused on the inhibition of elastase by heparin and HS [70], with the goal of understanding how natural feedback mechanisms involving HS within the lung might normally be involved in limiting elastase activity. Moreover, the ability of elastase to release HS has a number of consequences with regard to the storage and release of critical regulatory growth factors such as FGF2, transforming growth factor β, and heparin-binding epidermal growth factor [77, 78, 101-105].
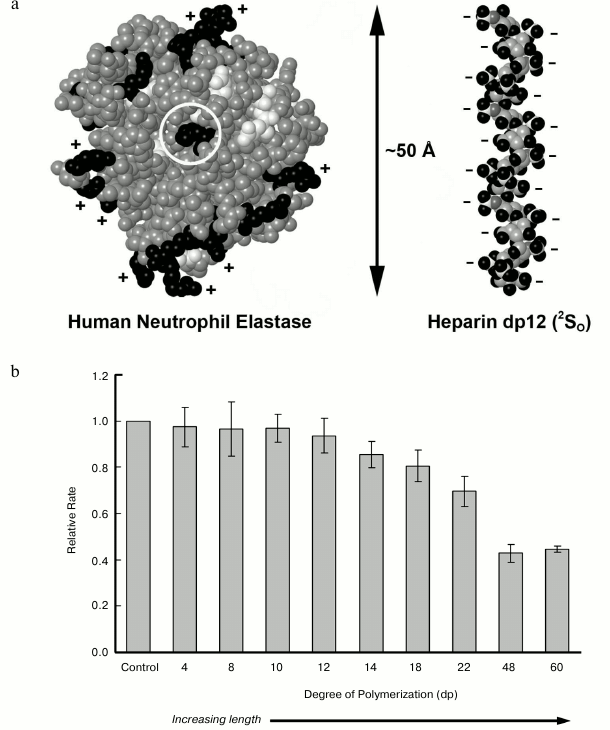
Our analysis of heparin/HS inhibition of elastase revealed that heparin acts as a hyperbolic, tight-binding competitive inhibitor of elastase as indicated by an increase in Km with no change in Vm and incomplete inhibition of activity even in the presence of high heparin concentrations [70]. We further established that the chemical structure of heparin is critical for inhibition as a minimum length of 12 monosaccharide residues was required for activity, and selectively de-sulfated heparin preparations were less active (Fig. 3). These length requirements were consistent with molecular docking studies that indicated that relatively long HS chains would be required to bridge the entire elastase molecule [70]. Interestingly, we and others have shown that HS can inhibit elastase, yet it is unlikely to ever have such long stretches of highly sulfated heparin-like domains, suggesting that contiguous NS domains are not required for inhibition. Instead, we have proposed, based on structural analysis of elastase-inhibitory HS, that optimal inhibition may require two separate NS domains separated and properly spaced by an under-sulfated region [106]. Because an extended NS domain has been found as a general feature of HS chains regardless of organ source [107], there may be implications of such domains for elastase regulation.
Fig. 3. Inhibition of neutrophil elastase by heparan sulfate. a) Space-filling structures for human neutrophil elastase based on the solved X-ray crystal structure 1HNE [108], and for a heparin-derived dodecasaccharide based on NMR analysis (1HPN) [109]. b) The relative rate of human neutrophil elastase digestion of a pseudosubstrate N-Suc-(Ala)3-pNA in the presence of the indicated heparin-derived oligosaccharide. Degree of polymerization is indicated on the x-axis. Reactions were carried out for 1 h at 27°C. Experimental details of elastase enzyme assay conditions are described in [70].
DETERMINING HS DOMAIN STRUCTURE
Although it was initially thought that specific sequences within HS would prove to be responsible for the ability to bind and modulate the wide array of heparin-binding proteins, it is now thought that the particular arrangement and spacing of structural domains may underlie HS activity [25]. While it is clear that the complex structure of HS underlies the ability to bind and modulate protein function, technology for determining how HS structure defines function remains lacking. Despite the common analytical procedures that exist to sequence nucleic acids and proteins, the ability to fully characterize HS remains elusive. We suggest that this is due to the unique level of complexity and non-template-driven biosynthesis that makes HS resistant to traditional approaches used to define biological polymers. Thus, fundamental understanding of HS biochemistry will require a different conceptual model. Whereas the function of a given protein can be evaluated based on its unique folded structure, it appears that the function of HS emerges from the arrangement and density of particular domain patterns (i.e. local arrangement of various sequence clusters).
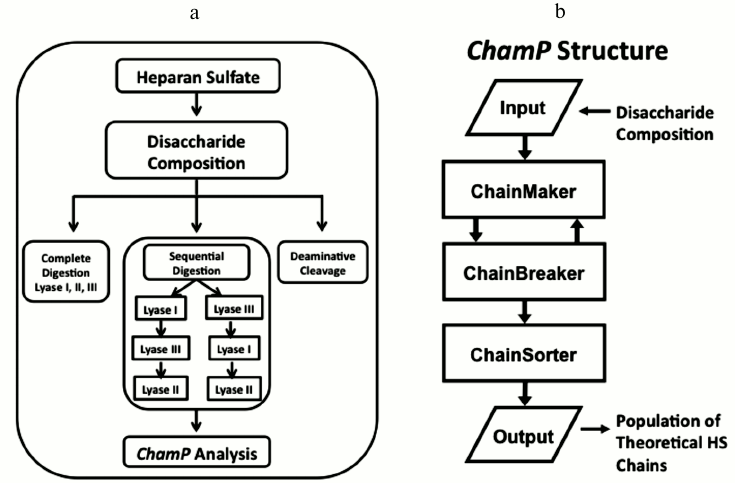
As analytical methods have advanced to the point that complete HS compositional information can be produced in high throughput, the need for new bioinformatics algorithms to model HS structure has become critical. To meet this need, our lab recently developed the foundation of a computational method, the ChainMaker Program (ChamP), for deciphering the domain pattern and sequences of complex mixtures of HS chains based on disaccharide compositional analysis [106] (Fig. 4). ChamP uses disaccharide composition data in conjunction with known biosynthetic rules to model the overall structures of a population of HS chains. This process begins with complete digestion of HS with heparin lyases I, II, and III to obtain the overall composition of the major disaccharides. HS samples are also subjected to selective and sequential digestion with these enzymes, and the release of each disaccharide is measured as a relative fraction of the total. Then, based on defined substrate cleavage specificities for the various heparin lyases, ChamP produces populations of theoretical HS chains with structures that match the compositional data and meet biosynthetic rules. Each chain is then subjected to a chain breaker routine where it is digested with the various heparinase enzymes alone and in sequence in silico, and theoretical disaccharides released are compared with those measured experimentally. Chains that do not produce degradation profiles matching those measured experimentally are discarded, and the process is repeated until a sufficient number of unique chains are produced (generally 100-200 chains). The final population of HS chains provides a representation of the mixed biological population that can be evaluated and sorted for any property of interest. For instance, the output chains can be searched for a specific sequence or general characteristic that may impart function. Measured values for glucuronic versus iduronic acid residues from deaminative cleavage as well as chain length distribution can also be included as part of the ChamP input to improve the accuracy of the output chains.
Fig. 4. Heparan sulfate analytical work-up for ChamP analysis. a) Schema of HS workflow to obtain disaccharide data for ChamP input. b) Structure of ChamP analysis.
HS is involved in nearly every cellular process at some level where it appears to function as a critical tuning factor that can enhance or attenuate the activity and/or dynamics of a wide range of bioactive proteins. It is generally accepted that this wide range of functions is encoded within HS structure, yet how HS structure is controlled by cells as a means to modulate protein interactions and activity remains poorly understood. Moreover, the standard paradigm in biochemical research of isolating an active factor from a complex mixture of molecules may not translate well to HS where the density and specific arrangement of “active” motifs within a given chain and within a population of chains may ultimately dictate biological function. Therefore, expansion of new and emerging technologies is needed to allow populations of HS chains to be defined based on their domain structure in order to gain insight into how HS is used by cells and tissues to maintain homeostasis and to respond to environmental challenges.
This work was supported in part by NIH grants R01 HL088572, R01 HL098950, and P41 GM104603, grant M2012014 from the Bright Focus Foundation, and a Departmental grant from the Massachusetts Lions Eye Research Fund, Inc.
REFERENCES
1.Casu, B., and Lindahl, U. (2001) Adv.
Carbohydrate Chem. Biochem., 57, 159-206.
2.Bishop, J. R., Schuksz, M., and Esko, J. D. (2007)
Nature, 446, 1030-1037.
3.Sasisekharan, R., and Venkataraman, G. (2000)
Curr. Opin. Chem. Biol., 4, 626-631.
4.Nugent, M. A. (2000) Proc. Natl. Acad. Sci.
USA, 97, 10301-10303.
5.Gallagher, J. T. (2001) J. Clin. Invest.,
108, 357-361.
6.Esko, J. D., and Selleck, S. B. (2002) Annu.
Rev. Biochem., 71, 435-471.
7.Dhoot, G. K., Gustafsson, M. K., Ai, X., Sun, W.,
Standiford, D. M., and Emerson, C. P., Jr. (2001) Science,
293, 1663-1666.
8.Alexopoulou, A. N., Multhaupt, H. A., and Couchman,
J. R. (2007) Int. J. Biochem. Cell Biol., 39,
505-528.
9.Saunders, S., Paine-Saunders, S., and Lander, A. D.
(1997) Devel. Biol., 190, 78-93.
10.Iozzo, R. V. (1998) Annu. Rev. Biochem.,
67, 609-652.
11.Iozzo, R. V., Zoeller, J. J., and Nystrom, A.
(2009) Mol. Cells, 27, 503-513.
12.Nugent, M. A., and Iozzo, R. V. (2000) Int. J.
Biochem. Cell Biol., 32, 115-120.
13.Nugent, M. A., Forsten-Williams, K., Karnovsky,
M. J., and Edelman, E. R. (2005) in Chemistry and Biology of Heparin
and Heparan Sulfate (Garg, H. G., ed.) Elsevier Ltd., pp.
533-570.
14.Metcalfe, D. D., Baram, D., and Mekori, Y. A.
(1997) Physiol. Rev., 77, 1033-1079.
15.Forsberg, E., Pejler, G., Ringvall, M.,
Lunderius, C., Tomasini-Johansson, B., Kusche-Gullberg, M., Eriksson,
I., Ledin, J., Hellman, L., and Kjellen, L. (1999) Nature,
400, 773-776.
16.Humphries, D. E., Wong, G. W., Friend, D. S.,
Gurish, M. F., Qiu, W. T., Huang, C., Sharpe, A. H., and Stevens, R. L.
(1999) Nature, 400, 769-772.
17.Humphries, D. E., Wong, G. W., Friend, D. S.,
Gurish, M. F., and Stevens, R. L. (1999) J. Histochem.
Cytochem., 47, 1645D-1646.
18.Shao, C., Shi, X., White, M., Huang, Y.,
Hartshorn, K., and Zaia, J. (2013) Febs J., in press.
19.Folkman, J., and Shing, Y. (1992) in Heparin
and Related Polysaccharides (Lane, D. A., et al., eds.) Plenum
Press, New York, pp. 355-364.
20.Presta, M., Leali, D., Stabile, H., Ronca, R.,
Camozzi, M., Coco, L., Moroni, E., Liekens, S., and Rusnati, M. (2003)
Curr. Pharm. Des., 9, 553-566.
21.Mousa, S. A., and Mohamed, S. (2004) Thromb.
Haemost., 92, 627-633.
22.Norrby, K. (2006) Apmis, 114,
79-102.
23.Capila, I., and Linhardt, R. J. (2002) Angew
Chem. Int. Ed. Engl., 41, 391-412.
24.Mulloy, B., and Linhardt, R. J. (2001) Curr.
Opin. Struct. Biol., 11, 623-628.
25.Kreuger, J., Spillmann, D., Li, J. P., and
Lindahl, U. (2006) J. Cell Biol., 174, 323-327.
26.Perrimon, N., and Bernfield, M. (2000)
Nature, 404, 725-728.
27.Thompson, S. M., Connell, M. G., Fernig, D. G.,
Ten Dam, G. B., van Kuppevelt, T. H., Turnbull, J. E., Jesudason, E.
C., and Losty, P. D. (2007) Pediatr. Surg. Int., 23,
411-417.
28.Izvolsky, K. I., Shoykhet, D., Yang, Y., Yu, Q.,
Nugent, M. A., and Cardoso, W. V. (2003) Dev. Biol., 258,
185-200.
29.Ori, A., Wilkinson, M. C., and Fernig, D. G.
(2011) J. Biol. Chem., 286, 19892-19904.
30.Bergamaschini, L., Rossi, E., Storini, C.,
Pizzimenti, S., Distaso, M., Perego, C., De Luigi, A., Vergani, C., and
De Simoni, M. G. (2004) J. Neurosci., 24, 4181-4186.
31.Engelberg, H. (2004) Dement. Geriatr. Cogn.
Disord., 18, 278-298.
32.Zacharski, L. R., Ornstein, D. L., and Mamourian,
A. C. (2000) Semin. Thromb. Hemost., 26 (Suppl. 1),
69-77.
33.Hettiarachchi, R. J., Smorenburg, S. M.,
Ginsberg, J., Levine, M., Prins, M. H., and Buller, H. R. (1999)
Thromb. Haemost., 82, 947-952.
34.Cosgrove, R. H., Zacharski, L. R., Racine, E.,
and Andersen, J. C. (2002) Semin. Thromb. Hemost., 28,
79-87.
35.Bick, R. L., and Ross, E. S. (1985) Semin.
Thromb. Hemost., 11, 213-217.
36.Ma, Q., Cornelli, U., Hanin, I., Jeske, W. P.,
Linhardt, R. J., Walenga, J. M., Fareed, J., and Lee, J. M. (2007)
Curr. Pharm. Des., 13, 1607-1616.
37.Tyrrell, D. J., Horne, A. P., Holme, K. R.,
Preuss, J. M., and Page, C. P. (1999) Adv. Pharmacol.,
46, 151-208.
38.Wang, Q. L., Shang, X. Y., Zhang, S. L., Ji, J.
B., Cheng, Y. N., Meng, Y. J., and Zhu, Y. J. (2000) Jpn. J.
Pharmacol., 82, 326-330.
39.Shriver, Z., Liu, D., and Sasisekharan, R. (2002)
Trends Cardiovasc. Med., 12, 71-77.
40.Sasisekharan, R., Shriver, Z., Venkataraman, G.,
and Narayanasami, U. (2002) Nat. Rev. Cancer, 2,
521-528.
41.Coombe, D. R., and Kett, W. C. (2005) Cell
Mol. Life Sci., 62, 410-424.
42.Castelli, R., Porro, F., and Tarsia, P. (2004)
Vasc. Med., 9, 205-213.
43.Bjork, I., and Lindahl, U. (1982) Mol. Cell
Biochem., 48, 161-182.
44.Thompson, L. D., Pantoliano, M. W., and Springer,
B. A. (1994) Biochemistry, 33, 3831-3840.
45.Chu, C. L., Goerges, A. L., and Nugent, M. A.
(2005) Biochemistry, 44, 12203-12213.
46.Forsten-Williams, K., Chu, C. L., Fannon, M.,
Buczek-Thomas, J. A., and Nugent, M. A. (2008) Ann. Biomed.
Eng., 36, 2134-2148.
47.Duchesne, L., Octeau, V., Bearon, R. N., Beckett,
A., Prior, I. A., Lounis, B., and Fernig, D. G. (2012) PLoS
Biol., 10, e1001361.
48.Dowd, C. J., Cooney, C. L., and Nugent, M. A.
(1999) J. Biol. Chem., 274, 5236-5244.
49.Beenken, A., and Mohammadi, M. (2009) Nature
Rev. Drug Discov., 8, 235-253.
50.Itoh, N. (2007) Biol. Pharm. Bull.,
30, 1819-1825.
51.Eswarakumar, V. P., Lax, I., and Schlessinger, J.
(2005) Cytokine Growth Factor Rev., 16, 139-149.
52.Naimy, H., Buczek-Thomas, J. A., Nugent, M. A.,
Leymarie, N., and Zaia, J. (2011) J. Biol. Chem., 286,
19311-19319.
53.Harmer, N. J., Robinson, C. J., Adam, L. E.,
Ilag, L. L., Robinson, C. V., Gallagher, J. T., and Blundell, T. L.
(2006) Biochem. J., 393, 741-748.
54.Harmer, N. J., Ilag, L. L., Mulloy, B.,
Pellegrini, L., Robinson, C. V., and Blundell, T. L. (2004) J. Mol.
Biol., 339, 821-834.
55.Robinson, C. J., Harmer, N. J., Goodger, S. J.,
Blundell, T. L., and Gallagher, J. T. (2005) J. Biol. Chem.,
280, 42274-42282.
56.Schlessinger, J., Plotnikov, A. N., Ibrahimi, O.
A., Eliseenkova, A. V., Yeh, B. K., Yayon, A., Linhardt, R. J., and
Mohammadi, M. (2000) Mol. Cell, 6, 743-750.
57.Pellegrini, L. (2001) Curr. Opin. Struct.
Biol., 11, 629-634.
58.Pellegrini, L., Burke, D. F., von Delft, F.,
Mulloy, B., and Blundell, T. L. (2000) Nature, 407,
1029-1034.
59.Chu, C. L., Buczek-Thomas, J. A., and Nugent, M.
A. (2004) Biochem. J., 379, 331-341.
60.Gopalakrishnan, M., Forsten-Williams, K., Nugent,
M. A., and Tauber, U. C. (2005) Biophys. J., 89,
3686-3700.
61.Forsten, K. E., Fannon, M., and Nugent, M. A.
(2000) J. Theor. Biol., 205, 215-230.
62.Ornitz, D. M., Xu, J., Colvin, J. S., McEwen, D.
G., MacArthur, C. A., Coulier, F., Gao, G., and Goldfarb, M. (1996)
J. Biol. Chem., 271, 15292-15297.
63.Zhang, X., Ibrahimi, O. A., Olsen, S. K.,
Umemori, H., Mohammadi, M., and Ornitz, D. M. (2006) J. Biol.
Chem., 281, 15694-15700.
64.NHLBI (2010) NHLBI Factbook, Fiscal Year 2010,
Disease Statistics.
65.Barnes, P. J., Shapiro, S. D., and Pauwels, R. A.
(2003) Eur. Respir. J., 22, 672-688.
66.Shapiro, S. D. (2003) Eur. Respir. J.
Suppl., 44, 30s-32s.
67.Shapiro, S. D., Goldstein, N. M., Houghton, A.
M., Kobayashi, D. K., Kelley, D., and Belaaouaj, A. (2003) Am. J.
Pathol., 163, 2329-2335.
68.Eriksson, S. (1991) Europ. Respir. J.,
4, 1041-1043.
69.Powers, J. C. (1983) Amer. Rev. Respir.
Dis., 127, S54-58.
70.Spencer, J. L., Stone, P. J., and Nugent, M. A.
(2006) Biochemistry, 45, 9104-9120.
71.Walsh, R. L., Dillon, T. J., Scicchitano, R., and
McLennan, G. (1991) Clin. Sci. (Colch.), 81, 341-346.
72.Izvolsky, K. I., Zhong, L., Wei, L., Yu, Q.,
Nugent, M. A., and Cardoso, W. V. (2003) Am. J. Physiol. Lung Cell
Mol. Physiol., 285, L838-846.
73.Warburton, D., Schwarz, M., Tefft, D.,
Flores-Delgado, G., Anderson, K. D., and Cardoso, W. V. (2000) Mech.
Dev., 92, 55-81.
74.Gronski, T. J., Jr., Martin, R. L., Kobayashi, D.
K., Walsh, B. C., Holman, M. C., Huber, M., Van Wart, H. E., and
Shapiro, S. D. (1997) J. Biol. Chem., 272,
12189-12194.
75.Passi, A., Negrini, D., Albertini, R., De Luca,
G., and Miserocchi, G. (1998) Am. J. Physiol., 275,
L631-635.
76.Kainulainen, V., Wang, H., Schick, C., and
Bernfield, M. (1998) J. Biol. Chem., 273,
11563-11569.
77.Buczek-Thomas, J. A., and Nugent, M. A. (1999)
J. Biol. Chem., 274, 25167-25172.
78.Buczek-Thomas, J. A., Chu, C. L., Rich, C. B.,
Stone, P. J., Foster, J. A., and Nugent, M. A. (2002) J. Cell
Physiol., 192, 294-303.
79.Cain, S. A., Baldock, C., Gallagher, J., Morgan,
A., Bax, D. V., Weiss, A. S., Shuttleworth, C. A., and Kielty, C. M.
(2005) J. Biol. Chem., 280, 30526-30537.
80.Tiedemann, K., Batge, B., Muller, P. K., and
Reinhardt, D. P. (2001) J. Biol. Chem., 276,
36035-36042.
81.Wu, W. J., Vrhovski, B., and Weiss, A. S. (1999)
J. Biol. Chem., 274, 21719-21724.
82.McGowen, S. E., Liu, R., and Harvey, C. S. (1993)
Arch. Biochem. Biophys., 302, 322-331.
83.Rao, N. V., Kennedy, T. P., Rao, G., Ky, N., and
Hoidal, J. R. (1990) Am. Rev. Respir. Dis., 142,
407-412.
84.Lafuma, C., Frisdal, E., Harf, A., Robert, L.,
and Hornebeck, W. (1991) Eur. Respir. J., 4,
1004-1009.
85.Hiebert, L., and Liu, J. M. (1991) Semin.
Thromb. Hemost., 17 (Suppl. 1), 42-46.
86.Adachi, T., and Marklund, S. L. (1989) J.
Biol. Chem., 264, 8537-8541.
87.Sandstrom, J., Carlsson, L., Marklund, S. L., and
Edlund, T. (1992) J. Biol. Chem., 267, 18205-18209.
88.Karlsson, K., Lindahl, U., and Marklund, S. L.
(1988) Biochem. J., 256, 29-33.
89.Tessler, S., Rockwell, P., Hicklin, D., Cohen,
T., Levi, B.-Z., Witte, L., Lemischka, I. R., and Neufeld, G. (1994)
J. Biol. Chem., 269, 12456-12461.
90.Goerges, A. L., and Nugent, M. A. (2003) J.
Biol. Chem., 278, 19518-19525.
91.Gengrinovitch, S., Berman, B., David, G., Witte,
L., Neufeld, G., and Ron, D. (1999) J. Biol. Chem., 274,
10816-10822.
92.Jakobsson, L., Kreuger, J., Holmborn, K., Lundin,
L., Eriksson, I., Kjellen, L., and Claesson-Welsh, L. (2006) Dev.
Cell, 10, 625-634.
93.Mitsi, M., Hong, Z., Costello, C. E., and Nugent,
M. A. (2006) Biochemistry, 45, 10319-10328.
94.Baici, A., Diczhazi, C., Neszmelyi, A., Moczar,
E., and Hornebeck, W. (1993) Biochem. Pharmacol., 46,
1545-1549.
95.Tumova, S., Woods, A., and Couchman, J. R. (2000)
Int. J. Biochem. Cell Biol., 32, 269-288.
96.Woods, A., and Couchman, J. R. (1994) Mol.
Biol. Cell, 5, 183-192.
97.Woods, A., Oh, E. S., and Couchman, J. R. (1998)
Matrix Biol., 17, 477-483.
98.Tkachenko, E., Rhodes, J. M., and Simons, M.
(2005) Circ. Res., 96, 488-500.
99.Cavalcante, F. S., Ito, S., Brewer, K., Sakai,
H., Alencar, A. M., Almeida, M. P., Andrade, J. S., Jr., Majumdar, A.,
Ingenito, E. P., and Suki, B. (2005) J. Appl. Physiol.,
98, 672-679.
100.Buczek-Thomas, J. A., Hsia, E., Rich, C. B.,
Foster, J. A., and Nugent, M. A. (2008) J. Cell Biochem.,
105, 108-120.
101.Liu, J., Rich, C. B., Buczek-Thomas, J. A.,
Nugent, M. A., Panchenko, M. P., and Foster, J. A. (2003) Am. J.
Physiol. Lung Cell Mol. Physiol., 285, L1106-1115.
102.Buczek-Thomas, J. A., Lucey, E. C., Stone, P.
J., Chu, C. L., Rich, C. B., Carreras, I., Goldstein, R. H., Foster, J.
A., and Nugent, M. A. (2004) Am. J. Respir. Cell Mol. Biol.,
31, 344-350.
103.Rich, C. B., Fontanilla, M. R., Nugent, M., and
Foster, J. A. (1999) J. Biol. Chem., 274,
33433-33439.
104.Rich, C. B., Nugent, M. A., Stone, P., and
Foster, J. A. (1996) J. Biol. Chem., 271,
23043-23048.
105.McGowan, S. E., and Thompson, R. J. (1989)
J. Appl. Physiol., 66, 400-409.
106.Spencer, J. L., Bernanke, J. A., Buczek-Thomas,
J. A., and Nugent, M. A. (2010) PLoS One, 5, e9389.
107.Staples, G. O., Shi, X., and Zaia, J. (2010)
J. Biol. Chem., 285, 18336-18343.
108.Navia, M. A., McKeever, B. M., Springer, J. P.,
Lin, T. Y., Williams, H. R., Fluder, E. M., Dorn, C. P., and Hoogsteen,
K. (1989) Proc. Natl. Acad. Sci. USA, 86, 7-11.
109.Mulloy, B., Forster, M. J., Jones, C., and
Davies, D. B. (1993) Biochem. J., 293, 849-858.