REVIEW: Desialylation of Surface Receptors as a New Dimension in Cell Signaling
A. V. Pshezhetsky1,2,3* and L. I. Ashmarina1
1Department of Medical Genetics, CHU Sainte-Justine Research Center, 3175 Côte Ste-Catherine, Montreal, Qc, Canada, H3T1C5; fax: (514) 345-4766; E-mail: alexei.pchejetski@umontreal.ca2Departments of Pediatrics and Biochemistry, University of Montreal, Montreal, Qc, Canada
3Department of Anatomy and Cell Biology, Faculty of Medicine, McGill University, Montreal, Qc, Canada
* To whom correspondence should be addressed.
Received February 24, 2013; Revision received March 7, 2013
Terminal sialic acid residues are found in abundance in glycan chains of glycoproteins and glycolipids on the surface of all live cells forming an outer layer of the cell originally known as glycocalyx. Their presence affects the molecular properties and structure of glycoconjugates, modifying their function and interactions with other molecules. Consequently, the sialylation state of glycoproteins and glycolipids has been recognized as a critical factor modulating molecular recognitions inside the cell, between the cells, between the cells and the extracellular matrix, and between the cells and certain exogenous pathogens. Until recently sialyltransferases that catalyze transfer of sialic acid residues to the glycan chains in the process of their biosynthesis were thought to be mainly responsible for the creation and maintenance of a temporal and spatial diversity of sialylated moieties. However, the growing evidence suggests that in mammalian cells, at least equally important roles belong to sialidases/neuraminidases, which are located on the cell surface and in intracellular compartments, and may either initiate the catabolism of sialoglycoconjugates or just cleave their sialic acid residues, and thereby contribute to temporal changes in their structure and functions. The current review summarizes emerging data demonstrating that mammalian neuraminidase 1, well known for its lysosomal catabolic function, is also targeted to the cell surface and assumes the previously unrecognized role as a structural and functional modulator of cellular receptors.
KEY WORDS: sialylation, neuraminidase, lysosome, plasma membrane, immune response, cell growth, insulinDOI: 10.1134/S0006297913070067
Abbreviations: CathA, cathepsin A/protective protein; ddNeuAc, 2,3-dehydro-2-deoxy-N-acetylneuraminic acid; EBP, elastin-binding protein; EGFR, epidermal growth factor receptor; FcγR, Fc receptors for immunoglobulin G; GAL, β-galactosidase; GS, galactosialidosis; IGF-II, insulin-like growth factor II; IFN, interferon; IL-4, interleukin 4; IR, insulin receptor; LAMP-1, lysosome associated membrane protein 1; LDL, low-density lipoprotein; LDLR, LDL receptor; LPS, lipopolysaccharide; MUC1, mucin 1; NEU1, -2, -3, -4, neuraminidase 1, -2, -3, -4; PDGF, platelet-derived growth factor; SGC, sialoglycoconjugate; Sia, sialic acid; Siglec, sialic acid binding immunoglobulin-like lectins; SMC, smooth muscle cells; Syk, serine-tyrosine kinase; TLR, Toll-like receptor; VLDL, very low density lipoprotein.
SIALYLATED GLYCOCONJUGATES: DISTRIBUTION, BIOLOGICAL ROLES,
SYNTHESIS, AND PROCESSING
Sialic acid (Sia) is a generic term for the N- or O-substituted derivatives of neuraminic acid, a monosaccharide with a nine-carbon backbone, the most common member of this group, being N-acetylneuraminic acid (Neu5Ac or NANA) [1]. Sialic acids are found widely distributed in animal tissues and to a lesser extent in other species, ranging from plants and fungi to yeasts and bacteria, mostly in glycoproteins and glycolipids (gangliosides). The amino group generally bears either an acetyl or glycolyl group, but other modifications have been described. The hydroxyl substituents may vary considerably; acetyl, lactyl, methyl, sulfate, and phosphate groups have been found yielding more than 50 different structures. Sia can also exist as a 2,8-linked homopolymer in polysialylated gangliosides and glycoproteins (reviewed in [1-3]).
Sialoglycoconjugates (SGC) are found in abundance on the surface of mammalian cells, forming a dense array of sialylated glycans, referred to as the sialome [1]. The inner surface of lysosomal and endosomal membranes are similarly sialylated [2]. SGC are not evenly distributed on the membranes but rather form dynamic microdomains, so called “clustered saccharide patches”, “glycosynapses”, or “lipid rafts” enriched in gangliosides and sialylated membrane proteins [3-7]. The majority of soluble secreted and lysosomal proteins also contain Sia as part of their glycan chains, and this modification can extend their half-life.
In mammals, the content of SGC strongly depends on the cell and tissue type, and significantly changes during development. These changes have been well documented at the level of total Sia released from the sample either by enzymatic treatment or by acid hydrolysis, or at the level of underlying glycan chains by labeling with antibodies or lectins specific for individual glycans [8]. Less is known, however, about the changes in the levels of individual proteins or lipids carrying sialylations.
Sia are involved in a surprising variety of biological processes [9], the most important being a modulation of recognition events. Sia are well known as a common ligand (or receptor) for virus, bacteria, and protozoan pathogens. They also function as crucial recognition markers in multicellular organisms where they mediate a variety of biological phenomena, including cell differentiation, interaction, migration, adhesion, and metastasis (reviewed in [9-11]). Members of the Siglecs (sialic acid binding immunoglobulin-like lectins) superfamily mediate intracellular interactions, which contribute to the scavenging function of macrophages, pathogen uptake and antigen presentation [3]. Glycosynapses mediate cell signaling and participate in processes such as cell adhesion, motility, and growth [7]. Cancer cells have long been recognized to have a significant overexpression of Sia on the cell surface [12-15]. Lipid- and protein-bound Sia are elevated in plasma from cancer patients [13, 16-19] and linked with acute phase condition and chronic disease [20, 21].
In mammals, the biosynthesis of SGC is performed by a family of sialyltransferases that catalyze the transfer of Sia from activated donor molecule, CMP-Sia, to an acceptor carbohydrate [22]. Twenty mammalian sialyltransferases identified to date show a high variation in terms of specificity, tissue and cellular distribution, and induction profile, thus reflecting remarkable functional diversity of their substrates [22]. Hydrolytic cleavage of Sia linked to mono- or oligosaccharide chains of glycoconjugates is catalyzed by the family of exo-α-sialidases (EC 3.2.1.18), also called neuraminidases, which hydrolyze α-(2→3)-, α-(2→), α-(2→)-glycosidic linkages of terminal sialic residues in oligosaccharides, glycoproteins, glycolipids, colominic acid, and synthetic substrates [9].
The half-life of Sia residues in glycoprotein glycan chains is several times shorter than the half-life of other sugars and of the proteins themselves [23, 24] suggesting that neuraminidases may also be involved in “trimming” Sia residues in glycoconjugates. Similarly, selective desialylation was shown for gangliosides on the plasma membrane [25]. Plasma membrane glycolipids and glycoproteins may also undergo rapid resialylation [26]. Therefore, sialylation and desialylation can be considered as a dynamic modification, modulated by sialyltransferases and sialidases in response to external or internal stimuli.
MAMMALIAN NEURAMINIDASES
Neuraminidase enzymes are a large family, found in a range of organisms including viral, bacterial, fungal, protozoan, avian, and mammalian species [27, 28]. The mammalian genomes contain four genes, which encode the members of the neuraminidase family (neuraminidase-1 (NEU1) [29-31]; neuraminidase-2 (NEU2) [32, 33]; neuraminidase-3 (NEU3, also known as ganglioside sialidase) [34-36], and neuraminidase-4 (NEU4) [37, 38]). These enzymes have different, yet overlapping tissue expression, intracellular localization, and substrate specificity. NEU1 is ubiquitously expressed with the highest levels in kidney, pancreas, skeletal muscle, liver, lungs, placenta, and brain [29, 31]. In these tissues NEU1 generally shows 10-20 times higher expression than NEU3 and NEU4, and ~103-102 higher expression than NEU2 [39]. NEU2 is found predominantly in muscle tissues. NEU3 has the highest expression in adrenal gland, skeletal muscle, heart, testis, and thymus [35, 36]. NEU4 has the highest expression in brain, skeletal muscle, heart, placenta, and liver [37, 39, 40].
In the cell, NEU1 is localized at the lysosomal and plasma membranes [41, 42]; NEU2 is a soluble protein found in the cytosol [33, 43, 44], although an alternatively-spliced membrane-associated form containing six extra amino acids at the N-terminus was recently reported in mouse thymus [45]; NEU3 is an integral membrane protein localized in the caveolae microdomains of plasma membranes [46] as well as endosomal and lysosomal membranes [47]. The NEU4 gene is spliced in two different forms resulting in appearance of two NEU4 isoforms, differing in the first 12 N-terminal amino acids [38, 39]. The short isoform was found predominantly on the endoplasmic reticulum membranes [38, 39], whereas the long form is targeted both to mitochondria [39, 48] and lysosomes [40].
Since neuraminidases show partially overlapping substrate specificities in vitro, their distinct tissue and subcellular distribution may be key to their biological roles. NEU1 is active primarily against sialylated glycopeptides and oligosaccharides with lower activity against gangliosides. It is involved in the lysosomal catabolism of these conjugates [49-53] but recent data summarized below show that NEU1 also participates in regulation of cell signaling by desialylating plasma membrane receptors. NEU2 is active against α-2,3-sialylated oligosaccharides, glycopeptides, and gangliosides [33, 43, 44]. The biological role of this enzyme remains mainly unknown, but it was suggested to cleave the GM3 ganglioside, leading to the alteration of cytoskeletal functions [54, 55] during myoblast differentiation [56-58]. In melanoma cells NEU2 activity inversely correlates with invasive and metastasis potential [58]. NEU3 requires a hydrophobic aglycone, which makes it active mainly towards gangliosides [59] providing it a role in signal transduction through hydrolysis of GM3, GM1, GD1a, and polysialogangliosides [60]. NEU3 is a crucial regulator of transmembrane signaling [61] and is implicated in regulation of cell transformation, differentiation and migration [25, 62], neuritogenesis, carcinogenesis, and apoptosis, as well as in insulin signaling [63-65]. NEU4 is active against all types of sialylated glycoconjugates including oligosaccharides, glycoproteins, and gangliosides [39, 40]. Based on ability of NEU4 to cleave undigested sialoconjugates stored in the cells, we have proposed that it is involved in lysosomal catabolism [40], while others have reported that NEU4 can prevent apoptosis in neuronal cells by hydrolyzing mitochondrial GD3 ganglioside [66]. In the mouse brain, NEU4 is involved in the regulation of neuronal cell differentiation [67].
In contrast to other mammalian, bacterial or viral neuraminidases, enzymatic activity of human NEU1 is allosterically regulated through its association with the lysosomal cathepsin A (CathA). CathA, NEU1, and lysosomal β-galactosidase (GAL) form a lysosomal multienzyme complex, where CathA activates NEU1 and protects NEU1 and GAL against proteolytic degradation by lysosomal peptidases [68]. It is tempting to speculate that this unique property of NEU1 may be essential for its regulatory role, allowing the enzyme to be rapidly activated in response to different cell stimuli.
GENETIC DEFECTS OF NEURAMINIDASES IN HUMANS AND MICE
Important clues about the biological roles of SGC in cells and tissues come from studying human patients and mouse strains having abnormal expression or functional mutations in neuraminidases.
Genetic deficiency of NEU1 in humans results in the severe metabolic disease, sialidosis (SL, MIM #256550) [31]. In addition, genetic deficiency of CathA results in the secondary deficiencies of NEU1 and GAL and causes the lysosomal storage disorder, galactosialidosis (GS, MIM #256540) [69, 70]. Both disorders clinically manifest with skeletal and gait abnormalities, progressively impaired vision, bilateral macular cherry-red spots, ataxia, seizures, and myoclonus syndrome. KO mouse models of sialidosis (Neu1 KO) and galactosialidosis (CathA KO) have severe systemic disease resembling human conditions [71, 72]. In addition, a spontaneous mouse model of NEU1 deficiency, an SM/J mouse originally characterized by altered sialylation of several lysosomal glycoproteins [73], shows reduced levels of NEU1 activity (20-30% of normal) because of a point mutation in the gene promoter and/or L209I amino acid substitution in the protein [74-76]. SM/J mice as well as B10.SM mice, that have NEU1 SM/J mutations transferred to a B10 genetic background causing a similar reduction of NEU1 activity, do not show any gross abnormalities but display altered immune reactions [77-79] described in details in the sections below.
No human disorders caused by genetic defects in NEU2, NEU3, or NEU4 have been described, perhaps because of the partial redundancy of the pathways catalyzed by these enzymes. A nonsynonymous polymorphism R41Q near the active site of human NEU2 occurs in 9.3% of Asian population [80]. This polymorphism results in an enzyme with an intrinsically lower enzymatic activity and increases the binding affinity of human NEU2 to anti-influenza drug oseltamivir, which potentially may be associated with severe adverse neurological reactions to this drug that have been observed in the Asian population [80].
The recently described Neu3 KO mouse is healthy and shows normal ganglioside patterns, which the authors explain by possible redundancy with NEU4 [81]. At the same time, Neu3 KO mice are less susceptible than WT mice to the colitis-associated colon carcinogenesis induced by azoxymethane and dextran sodium sulfate, showing that NEU3 plays an important role in inflammation-dependent tumor development [81]. The Neu4 KO mouse has altered ganglioside profile in the brain and vacuolized cells in spleen and lungs [82].
REGULATION OF RECEPTORS AND SIGNALING PATHWAYS BY NEURAMINIDASE
1
Participation of neuraminidases in diverse cellular regulatory mechanisms has been discovered only recently, mainly though studies of animal models of neuraminidase deficiency. Below we summarize data providing evidence for the essential roles of NEU1 in the regulation of exocytosis and phagocytosis, carcinogenesis, the immune response, generation of extracellular matrix, cell proliferation, and differentiation through desialylation of specific sialoglycoproteins.
Potentiation of migration, invasion, and adhesion of cancer cells. Alterations in levels of all four mammalian neuraminidases have been detected in cancer cells and correlated with their malignancy (reviewed in [83, 84]). In particular, multiple cancers show decreased NEU1 expression, but there is an inverse correlation with metastatic ability. The level of NEU1 activity and expression in different clones of transformed rat fibroblast 3Y1 cells and mouse adenocarcinoma colon 26 cells inversely correlated with their metastatic potential [85, 86]. Moreover, those transformed with v-fos had lower sialidase activity and higher metastatic potential [85]. Further study from the same group demonstrated that overexpression of NEU1 in mouse B16 melanoma cells reversed their metastatic capacity as detected by the suppression of the pulmonary metastasis in mice, invasiveness in collagen gels, and motility on colloidal gold-coated glass plates [87]. Although these experiments did not identify the molecular mechanism of the observed changes in metastatic capacity, they implicated NEU1 as a negative regulator of malignant properties of cancer cells.
Another evidence for the NEU1 role in cancer malignancy came from the work by Uemura et al. [88] who showed that NEU1 overexpression in colon cancer HT-29 cells significantly reduced their liver metastasis potential in mice as well as migration, invasion, and adhesion properties in vitro, whereas NEU1 silencing caused the opposite effect. The authors demonstrated that NEU1 overexpression in HT-29 cells resulted in desialylation of the laminin receptor, integrin β4, essential for carcinoma migration and invasion. Desialylation of integrin β4 decreased its phosphorylation, attenuated downstream kinases, and suppressed cell adhesion to laminin [88]. Besides, NEU1 overexpression caused downregulation of matrix metalloproteinase-7, also associated with cancer metastasis [88]. The authors’ hypothesis that integrin β4 is one of the NEU1 targets controlling the malignant properties in cancer cells was indirectly supported by the fact that chemical inhibition of integrin β4 glycosylation caused effects similar to that of NEU1 overexpression. However, since NEU1 desialylated multiple proteins on the cell surface, it is not clear whether the downregulation of integrin β4-mediated signaling was the only mechanism involved, and whether similar events occur in the other types of cancer cells.
Induction of immune response and inflammation. The important role of Sia in the function of immune cells has been well documented. The sialylation level of the cell surface critically affects the capacity of B cells to stimulate the proliferation of T cells [89-93] and phagocytosis capacity of HeLa cells [94]. Moreover, an acidic neuraminidase activity on the surface of activated T cells is essential for production of interleukin 4 (IL-4), interaction with the antigen presenting cells [77, 95, 96], and conversion of the group specific component (Gc) protein into a factor necessary for the inflammation-primed activation of macrophages [78, 97]. T-cells derived from the SM/J or B10.SM strains of mice with reduced NEU1 activity fail to convert Gc and synthesize IL-4, while B cells from these mice produced less IgG1 and IgE [77-79]. Induction of neuraminidase activity on the surface of activated T lymphocytes was directly shown to contribute to desialylation of the cell surface and production of interferon (IFN)-γ [98], and on the surface of human myeloid cells it generates glycan determinant CD15 (Lewis x, or Le(x)), a distinguishing marker for myeloid cells, and mediates neutrophil adhesion to dendritic cells by desialylating cell-surface sialyl-CD15 [99].
Recent data directly implicate NEU1 as a regulator of multiple receptors in the immune cells, suggesting that it may be a potential target for treatment of disorders related to immunity and inflammation. First during the differentiation of circulating blood monocytes and monocytic cell lines into macrophages, NEU1 expression is typically increased >10-fold, and the newly produced pool of the enzyme is targeted mostly to the cell surface [100, 101].
In contrast to other cellular neuraminidases, whose expression either remains unchanged or reduced, NEU1 mRNA protein and activity are specifically increased during the differentiation, as a result of significant induction of the transcriptional activity of the NEU1 gene promoter [100]. NEU1 and its activator CathA are first targeted to the lysosome and then are sorted to the LAMP-2-negative, MHC II-positive vesicles, which later merge with the plasma membrane [100]. Macrophages and immature dendritic cells from gene-targeted mouse with ~10% of residual NEU1 activity [102] showed increased sialylation of the cell surface and compromised ability to engulf gram-positive and gram-negative bacteria, as well as IgG-opsonized and non-opsonized particles and IgG-coated red blood cells, suggesting that all types of phagocytosis are affected [103]. The observed effect was relevant to the deficiency of NEU1 activity since the treatment of the cells with the exogenous mouse NEU1, which reduced the sialylation of the cell surface to the normal levels, completely restored the phagocytosis [103]. The authors also showed that the absence of NEU1 in particular affected transduction of signals from the Fc receptors for immunoglobulin G (FcγR). The macrophages from NEU1-deficient mice showed increased sialylation and impaired phosphorylation of FcγR, as well as markedly reduced phosphorylation of serine-tyrosine kinase (Syk) in response to treatment with IgG-opsonized beads.
Therefore it is conceivable that cell surface NEU1 activates phagocytosis in macrophages and dendritic cells through desialylation of surface receptors [103], although it would be interesting to investigate whether other alterations in cell signaling (primarily in Siglec-mediated response) can also contribute to the observed impaired phagocytosis in NEU1-deficient mice.
Besides FcγR, NEU1 is also involved in activation of cell surface Toll-like receptors (TLR) that play key roles in activating immune responses during infection. Amith et al. showed that ligands of TLR-2, -3, and -4 rapidly induced NEU1 activity in bone marrow-derived macrophages, as well as in macrophage and dendritic cell lines, and that this activity was required for TLR signaling [104]. The further work of this group demonstrated that lipopolysaccharide (LPS)-induced interaction of TLR-4 with the signal transducer protein MyD88 and subsequent activation of NFκB signaling pathway were impaired in the cells from NEU1-deficient mice, allowing the authors to speculate that NEU1 changes activity of the receptor by removing Sia from its glycan chains [105]. These authors also suggested that activation of NEU1 and subsequent desialylation of TLR-4 may be induced by binding of complexes of NEU1 and matrix metalloprotease-9 to the ligand-induced TLR4 on the cell surface [106].
Dependence of LPS-induced cytokine production on neuraminidase activity in dendritic cells was also reported by Stamatos et al. [107]. They showed that monocyte-derived dendritic cells treated with broad neuraminidase inhibitor 2,3-dehydro-2-deoxy-N-acetylneuraminic acid (ddNeuAc) have decreased expression of IL-6, IL-12p40, and TNF-α in response to LPS treatment and increased cell surface sialylation. The authors of this work speculate that the neuraminidase implicated in TLR signaling is NEU3, not NEU1. They show that 1 mM zanamivir, a pharmacological inhibitor of NEU3 [108], also reduced cytokine expression from LPS treated dendritic cells [107]. Also, levels of IL6 or TNF-α produced by the LPS-treated dendritic cells derived from NEU1-knockout mice were similar to those measured for wild type cells [107].
Unfortunately, both studies lack essential controls necessary to reveal the specific roles of NEU1 and NEU3 in regulation of cytokine production. In particular, zanamivir at 1 mM concentration in the culture medium can at least partially inhibit NEU1 exposed at the cell surface. Also, the authors neither comment on the apparent difference in the levels of cytokines produced by the ddNeuAc- and zanamivir-treated cells nor provide a direct evidence for NEU3 involvement in cytokine production, such as experiments in cells with the knocked-out or knocked-down NEU3 gene.
Related immune receptor functionally regulated through NEU1-mediated desialylation is the hyaluronic acid (HA) receptor, CD44, implicated in multiple cell–cell and cell–matrix interactions. Earlier work showed that Sia in the surface glycan chains of CD44 and the homologous LYVE-1 receptor can mask their binding to hyaluronic acid [109, 110]. Recently, indirect evidence that NEU1 desialylates and activates CD44 was obtained from studying splenic CD4+ T cells treated with neuraminidase inhibitors or derived from SM/J mice with reduced NEU1 activity [111]. Using an experimental mouse asthma model, the authors show that the Th2 cytokine concentration and absolute number of Th2 cells were reduced in the bronchoalveolar lavage fluid from the NEU1-deficient SM/J mice as compared to wild type mice [111], although it is not clear whether this phenotype was solely due to the increased CD44 sialylation.
Finally, during recruitment of leukocytes to the sites of inflammation, an endogenous sialidase promotes the binding of β2 integrins (CD11b/CD18) on polymorphonuclear leukocytes with Intercellular Adhesion Molecule 1 (ICAM-1) on endothelial cells by the removal of Sia from the activation epitopes of the adhesion molecules [112]. The authors suggested that this sialidase is NEU1 by its localization on the cell surface and because it was induced by PMA, although the activity assay with specific substrates is still required to rule out the participation of NEU3 in this process.
Potentiation of exocytosis by desialylation of lysosomal membrane associated protein 1. Regulated secretion of cellular lysosomes (lysosomal exocytosis) is an important part of the diverse cellular regulatory mechanisms including membrane repair (reviewed in [113, 114]). A number of cellular proteins including adaptor protein-3, Lyst protein, Rab proteins, Munc 13 proteins, Rab27a effector proteins, and SNARE proteins have already been identified as regulators of lysosomal exocytosis responsible for either binding of lysosomes to and their movement along the microtubules or for the fusion of lysosomal and plasma membranes [113, 114]. Genetic deficiencies of many of these proteins in humans and mice result in impaired or partially impaired lysosomal secretion [113, 114].
In contrast, bone marrow stromal cells and neutrophils from NEU1 KO mice showed enhanced secretion of lysosomal proteases and glycosidases and an increased presence of heavily sialylated lysosome associated membrane protein 1 (LAMP-1) on the cell surface, both indicative of enhanced lysosomal exocytosis [115]. The authors suggest that NEU1 acts as a negative regulator of lysosomal exocytosis through its participation in the processing of LAMP-1, which is implicated in the process of lysosomal exocytosis and the fusion of lysosomes with exosomes according to the earlier work of Kima et al. [116]. In the cultured cells and tissues from NEU1-deficient mice, LAMP-1 shows increased glycosylation (sialylation) and a prolonged half-life resulting in the overall increase of its intracellular level and induced lysosomal exocytosis, whereas the siRNA-mediated inhibition of LAMP-1 expression reverses the phenotype [115]. The enhanced secretion of serine proteases from hematopoietic cells in the bone marrow niche of NEU1 KO mice leads to the inactivation of extracellular serpins, premature degradation of VCAM-1, and loss of bone marrow retention, providing a link between the NEU1 deficiency and impaired long-term bone marrow engraftment [115]. Moreover, increased lysosomal exocytosis from marginal cells of the striavascularis into the endolymph was also linked to reduced endolymphatic potential, dysfunction of transduction in sensory hair cells, and hearing loss reported in NEU1 KO mice, also observed in sialidosis patients [117].
At the same time, direct evidence for participation of LAMP-1 in exocytosis has yet to be provided since LAMP-1 KO mice do not display any signs suggesting decreased exocytosis of lysosomes or lysosome-related organelles [118, 119]. It is possible, therefore, that the increased amount and/sialylation of glycoproteins (including LAMP-1) in lysosomal membrane simply modifies its properties favoring the process of its merging with the plasma membrane during exocytosis.
Modulation of elastic fiber assembly. NEU1 and its activator CathA were both identified as components of the elastin receptor, which also contains the elastin-binding protein (EBP), a 67-kDa enzymatically inactive, alternatively spliced variant of lysosomal β-galactosidase [120-123]. While the EBP serves as intracellular molecular chaperone for hydrophobic and non-glycosylated tropoelastin and assures its proper secretion, NEU1 catalyzes the removal of terminal Sia from galactose residues in carbohydrate chains of microfibrillar proteins, forming the structural scaffold of new elastic fibers. The exposed galactosugars, in turn, interact with the galactolectin domain of the EBP, thereby inducing the release of transported tropoelastin molecules and facilitating their subsequent assembly into elastic fibers [124].
The crucial role of EBP complex in the formation of elastic fibers is supported by the evidence of connective-tissue, skeletal, and cardiovascular defects observed in GM1 gangliosidosis (OMIM #230500) and Morquio B (OMIM #253010) patients with mutations in the GAL gene that lack both β-galactosidase and EBP [125, 126]. These patients also show cardiac valve deformations, aortic stenosis, and intimal thickening in the coronary arteries and in the pulmonary artery [127, 128]. Clinical symptoms related to the defects in the elastic fiber formation (most often cardiomyopathies) have been also documented in GS and sialidosis patients [129].
Similarly, the NEU1-deficient fibroblasts from sialidosis patients have impaired elastogenesis, which could be reversed after the transfection of cells with NEU1 and CathA cDNA [124]. Impaired elastogenesis connected to the defective development of aorta, skin, and lungs has also been detected in NEU1 KO mice [130]. In particular, the elastic lamellae in the aorta of the NEU1 KO mice were thinner and separated by hypertrophic smooth muscle cells that were surrounded by an excess of the sialic acid-containing moieties. The concentration of elastin in the aorta was significantly reduced but the production of tropoelastin was normal, suggesting the elastic fiber defects result from impaired extracellular assembly [130]. Altogether, the reviewed data implicate NEU1 as an important functional component of the elastin receptor contributing to the normal development of the cardiovascular and respiratory systems.
Modulation of cell proliferation. Several recent publications demonstrated that NEU1 is involved in the fine-tuning of a group of homologous receptors regulating the cellular mitogenic response to growth factors. First, Hinek et al. [131] showed that NEU1 of arterial smooth muscle cells (SMC) downregulates cellular proliferation by desialylation of cell surface receptors for potent stimulators of cell proliferation, platelet-derived growth factor BB (PDGF-BB) and insulin-like growth factor (IGF-II). Treatment of cultured SMC and skin fibroblasts with neuraminidase inhibitor, ddNeuAc, or anti-NEU1 antibody increased their proliferation, whereas exogenous Clostridium perfringens neuraminidase (sharing substrate specificity with mammalian NEU1) reduced cellular proliferation and eliminated PDGF-BB- and IGF-II-induced phosphorylation of the respective receptors. Fibroblasts of sialidosis patients had a significantly stronger mitogenic response to the same doses of PDGF-BB and IGF-II than fibroblasts of the normal skin, suggesting that NEU1 deficiency resulted in higher response to both growth factors [131].
The follow-up study from the same group [132] demonstrated that in addition to the IGF-II receptor, the homologous IGF-1R receptor and insulin receptor (IR) were also substrates of NEU1 and that desialylation affected their activity in opposite directions. The physiological (0.5-1 nM) and therapeutic (10 nM) doses of insulin stimulated proliferation of cultured skeletal muscle progenitors L6WT through IR, and the effect could be further enhanced following the desialylation of the receptor, whereas the inhibition of endogenous NEU1 with ddNeuAc, anti-NEU1 antibody, or NEU1 siRNA abolished the proliferative response to low doses of insulin. In contrast, supra-physiological (100 nM) doses of insulin induced a more potent proliferative response transmitted through IGF-1R. This response was inhibited following treatment of the cells with exogenous neuraminidases, and enhanced in cultures treated with ddNeuAc, anti-NEU1 antibody, or NEU1 siRNA [132]. The authors hypothesized that desialylation of the receptors directly affected their conformation in such a way that either increased (IR) or decreased (IGF-1R) their ability to undergo insulin-induced autophosphorylation prerequisite for the subsequent downstream signals.
These results suggesting a crucial role for NEU1 in skeletal muscle growth correlate well with the observed experimental overexpression of NEU1 in myoblastic cells coinciding with their heightened proliferation and inhibition of the differentiation cascade [133] and with the fact that early-onset sialidosis patients and NEU1 KO mice present with severe progressive muscular atrophy [71, 134].
Finally, recent work showed that NEU1 negatively regulates epidermal growth factor receptor (EGFR) in endothelial cells. First, Lillehoj et al. [135] showed that NEU1 in airway epithelia is expressed at far greater levels than other three neuraminidases and that the enzyme interacts with both EGFR and its signaling partner mucin 1 (MUC1). Moreover, NEU1–EGFR binding was regulated by EGF stimulation. Silencing NEU1 increased the sialylation of both EGFR and MUC1, suggesting that both proteins are in vivo substrates of the enzyme. Most importantly, overexpression of NEU1 diminished EGF-stimulated autophosphorylation of EGFR at the Tyr1068 residue, whereas NEU1 depletion increased it. In contrast, MUC1-mediated P. aeruginosa adhesion and flagellin-induced ERK1/2 activation was decreased by silencing NEU1 and increased by its overexpression. The established regulation of EGFR by NEU1 implicated this enzyme as a potential modulator of wound healing and repair, which was further confirmed by the follow-up study of the same group [136]. Using flow cytometry, they localized NEU1 at the cell surface of endothelial cells and showed that overexpression of NEU1 in these cells inhibited their migration into a wound, thus indicating that NEU1 restrains the migratory response.
Positive regulation of insulin signaling. Most recently, Dridi et al. [137] showed that NEU1 also regulates the metabolic action of insulin and insulin-mediated glucose uptake. They demonstrated that gene-targeted CathAS190A-Neo mice, which have 10-15% of normal NEU1 activity in their tissues, rapidly develop glucose intolerance after exposure to a diet with an elevated fat content. Intraperitoneal glucose tolerance tests conducted 0, 4, and 8 weeks after the start of the diet revealed that NEU1-deficient mice develop glucose intolerance more rapidly than WT controls. Prior to the high-fat diet, the mice showed only slight (up to 10-12 mmol/liter) increases of blood glucose, but after 4 weeks on the diet NEU1-deficient mice were clearly hyperglycemic. After 8 weeks on high-fat diet, both WT and NEU1-deficient mice showed hyperglycemia, but blood glucose levels in NEU1-deficient mice remained significantly higher than in WT mice. Both WT and NEU1-deficient mice produced similar levels of insulin, suggesting that hyperglycemia in NEU1-deficient mice results from reduced insulin sensitivity in target tissues. Indeed, the levels of insulin-induced phosphorylation of activated IR and the downstream kinase AKT in livers and muscles of NEU1-deficient mice were significantly reduced as compared to those of WT animals.
In cultured fibroblasts of a sialidosis patient, phosphorylation of AKT in response to insulin was also impaired but could be restored by treatment with NEU1. Finally, this study showed that desialylation of IR was triggered by interaction between IR and NEU1 on the cell surface. This interaction was induced by insulin as shown by both bioluminescence resonance energy transfer between the receptor and NEU1 and co-immunoprecipitation, suggesting that the insulin binding to IR rapidly induces its desialylation by NEU1, which consequently stimulates its activation.
Altogether, the authors demonstrated that the activation of the IR is modulated by a unique mechanism dependent on NEU1, which identified sialylation as an important new parameter regulating the signaling pathways for glucose uptake.
Further studies are required to determine whether other energy metabolism pathways can be targets of NEU1 (or other neuraminidases), and recent work by Yang et al. [138] provided indirect indication for at least one. They report increased hepatic levels of cholesterol and triglycerides in B6.SM mice obtained by transferring the NEU1 mutant allele in SM/J mouse on B6 background and having 70-80% reduction of sialidase activity in tissues. The authors also detected lower VLDL-triglyceride production rate in B6.SM mice compared with C57Bl/6 control, suggesting that NEU1 may be involved in lipoprotein metabolism. They also report that hepatic receptor for LDL (LDLR) in B6.SM mouse tissues has increased sialylation and speculate that this could prolong its half-life or increase its recycling rate and result in higher LDL uptake they observed in B6.SM mice and cultured fibroblasts of sialidosis patients [138].
In the absence of direct evidence for increased LDLR density on the surface of NEU1-deficient cells, it is impossible to verify this hypothesis; however, it is worth mentioning that in contrast to the report of Yang et al. the increased sialidase activity in blood and the reduced level of LDL sialylation have been linked to increased cholesterol uptake by macrophages and atherosclerosis [139, 140].
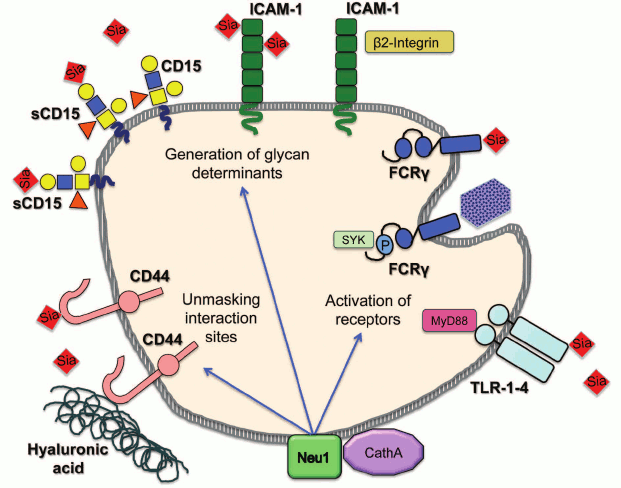
In conclusion, multiple data endorse the novel role for mammalian neuraminidases as modulators of cellular signaling and regulators of cell surface receptors. Future studies should reveal both the mechanism by which sialylation alters receptor activity and the exact biological role played by this new type of regulation. Nevertheless, the net sialylation of receptors likely constitutes another, not fully appreciated and understood dimension in a complicated process controlling initiation of cellular signaling in such diverse processes as vesicular trafficking, pro-inflammatory response, migration, invasion and adhesion of cancer cells, cell proliferation, and energy metabolism. Moreover, considering the diversity of the involved pathways, it is tempting to speculate that neuraminidases may even coordinate activities of independent signaling pathways all aimed at achieving the same biological goal. For example, during the immune response, the induction of NEU1 activates both phagocytosis and cytokine production in macrophages and dendritic cells, induces antibody production by B cells, and increases adhesion of neutrophils and lymphocytes (figure; see color insert).
Regulation of cellular immune pathways by neuraminidase 1. Neuraminidase 1 (NEU1) bound to its activator protein, cathepsin A (CathA) at the plasma membrane desialylates and activates receptors for phagocytosis (FcRγ) and inflammation (TLR-1-4). It also desialylates CD44 receptor, which unmasks binding sites for hyaluronic acid at its extracellular domain and generates glycan determinants for interactions between different classes of immune cells by desialylating Intercellular Adhesion Molecule 1 (ICAM-1) and sialyl-Lewis x (sCD15)
Another question that has to be addressed in the future is whether NEU1 plays a unique role in desialylation of receptors, or other neuraminidases have similar functions, each being responsible for a specific group of protein targets. The answer to this question (which will be most likely obtained through global analyses of glycoprotein substrates of neuraminidases in the available KO mouse models) will be essential to advance our understanding of cell signaling and of human diseases caused by abnormal regulation of surface receptors. Besides, development of specific and potent small molecular inhibitors of neuraminidases will provide a vital tool for future studies of neuraminidase pathways and potential therapeutic intervention.
This work was supported in part by the operating grant from the Canadian Diabetes Association to A.V.P.
REFERENCES
1.Cohen, M., and Varki, A. (2010) Omics: J.
Integr. Biol., 14, 455-464.
2.Kundra, R., and Kornfeld, S. (1999) J. Biol.
Chem., 274, 31039-31046.
3.Varki, A., and Angata, T. (2006)
Glycobiology, 16, 1R-27R.
4.Cohen, M., Hurtado-Ziola, N., and Varki, A. (2009)
Blood, 114, 3668-3676.
5.Hakomori, S. (2003) Curr. Opin. Hematol.,
10, 16-24.
6.Hakomori, S. (2004) Glycoconj. J.,
21, 125-137.
7.Todeschini, R. A., and Hakomori, S. I. (2008)
Biochim. Biophys. Acta, 1780, 421-433.
8.Jones, C. J., Aplin, J. D., Mulholland, J., and
Glasser, S. R. (1993) J. Reprod. Fertil., 99,
635-645.
9.Kelm, S., and Schauer, R. (1997) Int. Rev.
Cytol., 175, 137-240.
10.Lehmann, F., Tiralongo, E., and Tiralongo, J.
(2006) Cell. Mol. Life Sci.: CMLS, 63, 1331-1354.
11.Allende, M. L., and Proia, R. L. (2002) Curr.
Opin. Struct. Biol., 12, 587-592.
12.Babal, P., Janega, P., Cerna, A., Kholova, I.,
and Brabencova, E. (2006) Acta Histochem., 108,
133-140.
13.Berbec, H., Paszkowska, A., Siwek, B., Gradziel,
K., and Cybulski, M. (1999) Eur. J. Gynaecol. Oncol., 20,
389-392.
14.Brooks, S. A., and Leathem, A. J. (1998)
Invasion & Metastasis, 18, 115-121.
15.Feijoo-Carnero, C., Rodriguez-Berrocal, F. J.,
Paez de la Cadena, M., Ayude, D., de Carlos, A., and Martinez-Zorzano,
V. S. (2004) Int. J. Biol. Markers, 19, 38-45.
16.Basoglu, M., Yildirgan, M. I., Taysi, S., Yilmaz,
I., Kiziltunc, A., Balik, A. A., Celebi, F., and Atamanalp, S. S.
(2003) J. Surg. Oncol., 83, 180-184.
17.Rajpura, K. B., Patel, P. S., Chawda, J. G., and
Shah, R. M. (2005) J. Oral Pathol. Med., 34, 263-267.
18.Romppanen, J., Haapalainen, T., Punnonen, K., and
Penttila, I. (2002) Anticancer Res., 22, 415-420.
19.Uslu, C., Taysi, S., Akcay, F., Sutbeyaz, M. Y.,
and Bakan, N. (2003) Ann. Clin. Lab. Sci., 33,
156-159.
20.Iijima, S., Shiba, K., Kimura, M., Nagai, K., and
Iwai, T. (2000) Electrophoresis, 21, 753-759.
21.Herve, F., Duche, J. C., and Jaurand, M. C.
(1998) J. Chromatogr. B. Biomed. Sci. Appl., 715,
111-123.
22.Takashima, S. (2008) Biosci. Biotechnol.
Biochem., 72, 1155-1167.
23.Kreisel, W., Volk, B. A., Buchsel, R., and
Reutter, W. (1980) Proc. Natl. Acad. Sci. USA, 77,
1828-1831.
24.Tauber, R., Park, C. S., and Reutter, W. (1983)
Proc. Natl. Acad. Sci. USA, 80, 4026-4029.
25.Kopitz, J., von Reitzenstein, C., Sinz, K., and
Cantz, M. (1996) Glycobiology, 6, 367-376.
26.Kreisel, W., Hanski, C., Tran-Thi, T. A., Katz,
N., Decker, K., Reutter, W., and Gerok, W. (1988) J. Biol.
Chem., 263, 11736-11742.
27.Monti, E., Preti, A., Venerando, B., and Borsani,
G. (2002) Neurochem. Res., 27, 649-663.
28.Saito, N., and Yu, R. K. (1995) in Biology of
Sialic Acids (Rosenberg, A., ed.) Plenum Press, New York, pp.
261-313.
29.Bonten, E., van der Spoel, A., Fornerod, M.,
Grosveld, G., and d’Azzo, A. (1996) Genes Devel.,
10, 3156-3169.
30.Milner, C. M., Smith, S. V., Carrillo, M. B.,
Taylor, G. L., Hollinshead, M., and Campbell, R. D. (1997) J. Biol.
Chem., 272, 4549-4558.
31.Pshezhetsky, A. V., Richard, C., Michaud, L.,
Igdoura, S., Wang, S., Elsliger, M. A., Qu, J., Leclerc, D., Gravel,
R., Dallaire, L., and Potier, M. (1997) Nature Genet.,
15, 316-320.
32.Miyagi, T., Konno, K., Emori, Y., Kawasaki, H.,
Suzuki, K., Yasui, A., and Tsuik, S. (1993) J. Biol. Chem.,
268, 26435-26440.
33.Monti, E., Preti, A., Rossi, E., Ballabio, A.,
and Borsani, G. (1999) Genomics, 57, 137-143.
34.Miyagi, T., Wada, T., Iwamatsu, A., Hata, K.,
Yoshikawa, Y., Tokuyama, S., and Sawada, M. (1999) J. Biol.
Chem., 274, 5004-5011.
35.Wada, T., Yoshikawa, Y., Tokuyama, S., Kuwabara,
M., Akita, H., and Miyagi, T. (1999) Biochem. Biophys. Res.
Commun., 261, 21-27.
36.Monti, E., Bassi, M. T., Papini, N., Riboni, M.,
Manzoni, M., Venerando, B., Croci, G., Preti, A., Ballabio, A.,
Tettamanti, G., and Borsani, G. (2000) Biochem. J., 349,
343-351.
37.Comelli, E. M., Amado, M., Lustig, S. R., and
Paulson, J. C. (2003) Gene, 321, 155-161.
38.Monti, E., Bassi, M. T., Bresciani, R., Civini,
S., Croci, G. L., Papini, N., Riboni, M., Zanchetti, G., Ballabio, A.,
Preti, A., Tettamanti, G., Venerando, B., and Borsani, G. (2004)
Genomics, 83, 445-453.
39.Yamaguchi, K., Hata, K., Koseki, K., Shiozaki,
K., Akita, H., Wada, T., Moriya, S., and Miyagi, T. (2005) Biochem.
J., 390, 85-93.
40.Seyrantepe, V., Landry, K., Trudel, S., Hassan,
J. A., Morales, C. R., and Pshezhetsky, A. V. (2004) J. Biol.
Chem., 279, 37021-37029.
41.Lukong, K. E., Seyrantepe, V., Landry, K.,
Trudel, S., Ahmad, A., Gahl, W. A., Lefrancois, S., Morales, C. R., and
Pshezhetsky, A. V. (2001) J. Biol. Chem., 276,
46172-46181.
42.Vinogradova, M. V., Michaud, L., Mezentsev, A.
V., Lukong, K. E., El-Alfy, M., Morales, C. R., Potier, M., and
Pshezhetsky, A. V. (1998) Biochem. J., 330 (Pt. 2),
641-650.
43.Miyagi, T., and Tsuiki, S. (1985) J. Biol.
Chem., 260, 6710-6716.
44.Tringali, C., Papini, N., Fusi, P., Croci, G.,
Borsani, G., Preti, A., Tortora, P., Tettamanti, G., Venerando, B., and
Monti, E. (2004) J. Biol. Chem., 279, 3169-3179.
45.Koda, T., Kijimoto-Ochiai, S., Uemura, S., and
Inokuchi, J. (2009) Biochem. Biophys. Res. Commun., 387,
729-735.
46.Wang, Y., Yamaguchi, K., Wada, T., Hata, K.,
Zhao, X., Fujimoto, T., and Miyagi, T. (2002) J. Biol. Chem.,
277, 26252-26259.
47.Zanchetti, G., Colombi, P., Manzoni, M.,
Anastasia, L., Caimi, L., Borsani, G., Venerando, B., Tettamanti, G.,
Preti, A., Monti, E., and Bresciani, R. (2007) Biochem. J.,
408, 211-219.
48.Bigi, A., Morosi, L., Pozzi, C., Forcella, M.,
Tettamanti, G., Venerando, B., Monti, E., and Fusi, P. (2010)
Glycobiology, 20, 148-157.
49.Michalski, J. C., Strecker, G., and Fournet, B.
(1977) FEBS Lett., 79, 101-104.
50.Strecker, G., Peers, M. C., Michalski, J. C.,
Hondi-Assah, T., Fournet, B., Spik, G., Montreuil, J., Farriaux, J. P.,
Maroteaux, P., and Durand, P. (1977) Eur. J. Biochem./FEBS,
75, 391-403.
51.Dorland, L., Haverkamp, J., Viliegenthart, J. F.,
Strecker, G., Michalski, J. C., Fournet, B., Spik, G., and Montreuil,
J. (1978) Eur. J. Biochem./FEBS, 87, 323-329.
52.Van Pelt, J., Kamerling, J. P., Vliegenthart, J.
F., Verheijen, F. W., and Galjaard, H. (1988) Biochim. Biophys.
Acta, 965, 36-45.
53.Yoshino, H., Miyashita, K., Miyatani, N., Ariga,
T., Hashimoto, Y., Tsuji, S., Oyanagi, K., Ohama, E., Ikuta, F.,
Suzuki, A., et al. (1990) J. Neurol. Sci., 97, 53-65.
54.Akita, H., Miyagi, T., Hata, K., and Kagayama, M.
(1997) Histochem. Cell Biol., 107, 495-503.
55.Sato, K., and Miyagi, T. (1996) Biochem.
Biophys. Res. Commun., 221, 826-830.
56.Fanzani, A., Giuliani, R., Colombo, F., Rossi,
S., Stoppani, E., Martinet, W., Preti, A., and Marchesini, S. (2008)
Biochem. Biophys. Res. Commun., 370, 376-381.
57.Stoppani, E., Rossi, S., Marchesini, S., Preti,
A., and Fanzani, A. (2009) Cell Biol. Int., 33,
1020-1025.
58.Tokuyama, S., Moriya, S., Taniguchi, S., Yasui,
A., Miyazaki, J., Orikasa, S., and Miyagi, T. (1997) Int. J.
Cancer, 73, 410-415.
59.Sandbhor, M. S., Soya, N., Albohy, A., Zheng, R.
B., Cartmell, J., Bundle, D. R., Klassen, J. S., and Cairo, C. W.
(2011) Biochemistry, 50, 6753-6762.
60.Schneider-Jakob, H. R., and Cantz, M. (1991)
Biol. Chem. Hoppe-Seyler, 372, 443-450.
61.Miyagi, T., Wada, T., Yamaguchi, K., Hata, K.,
and Shiozaki, K. (2008) J. Biochem., 144, 279-285.
62.Kopitz, J., von Reitzenstein, C., Burchert, M.,
Cantz, M., and Gabius, H. J. (1998) J. Biol. Chem., 273,
11205-11211.
63.Wu, G., and Ledeen, R. W. (1991) J.
Neurochem., 56, 95-104.
64.Kakugawa, Y., Wada, T., Yamaguchi, K., Yamanami,
H., Ouchi, K., Sato, I., and Miyagi, T. (2002) Proc. Natl. Acad.
Sci. USA, 99, 10718-10723.
65.Sasaki, A., Hata, K., Suzuki, S., Sawada, M.,
Wada, T., Yamaguchi, K., Obinata, M., Tateno, H., Suzuki, H., and
Miyagi, T. (2003) J. Biol. Chem., 278, 27896-27902.
66.Hasegawa, T., Sugeno, N., Takeda, A.,
Matsuzaki-Kobayashi, M., Kikuchi, A., Furukawa, K., Miyagi, T., and
Itoyama, Y. (2007) FEBS Lett., 581, 406-412.
67.Shiozaki, K., Koseki, K., Yamaguchi, K.,
Shiozaki, M., Narimatsu, H., and Miyagi, T. (2009) J. Biol.
Chem., 284, 21157-21164.
68.Pshezhetsky, A. V., and Ashmarina, M. (2001)
Prog. Nucleic Acid Res. Mol. Biol., 69, 81-114.
69.D’Azzo, A., Andria, G., Strisciuglio, G.,
and Galjaard, H. (1995) in Metabolic and Molecular Bases of
Inherited Disease (Scriver, C. R. B. A., Sly, W. S., and Valle, D.,
eds.) McGraw-Hill, New York, pp. 2835-2837.
70.Okamura-Oho, Y., Zhang, S., and Callahan, J. W.
(1994) Biochim. Biophys. Acta, 1225, 244-254.
71.DeGeest, N., Bonten, E., Mann, L., de
Sousa-Hitzler, J., Hahn, C., and d’Azzo, A. (2002) Hum. Mol.
Genet., 11, 1455-1464.
72.Zhou, X. Y., Morreau, H., Rottier, R., Davis, D.,
Bonten, E., Gillemans, N., Wenger, D., Grosveld, F. G., Doherty, P.,
Suzuki, K., Grosveld, G. C., and d’Azzo, A. (1995) Genes
Devel., 9, 2623-2634.
73.Womack, J. E., Yan, D. L., and Potier, M. (1981)
Science, 212, 63-65.
74.Carrillo, M. B., Milner, C. M., Ball, S. T.,
Snoek, M., and Campbell, R. D. (1997) Glycobiology, 7,
975-986.
75.Rottier, R. J., Bonten, E., and d’Azzo, A.
(1998) Hum. Mol. Genet., 7, 313-321.
76.Champigny, M. J., Mitchell, M., Fox-Robichaud,
A., Trigatti, B. L., and Igdoura, S. A. (2009) Mol. Genet.
Metab., 97, 43-52.
77.Landolfi, N. F., and Cook, R. G. (1986) Mol.
Immunol., 23, 297-309.
78.Naraparaju, V. R., and Yamamoto, N. (1994)
Immunol. Lett., 43, 143-148.
79.Landolfi, N. F., Leone, J., Womack, J. E., and
Cook, R. G. (1985) Immunogenetics, 22, 159-167.
80.Li, C. Y., Yu, Q., Ye, Z. Q., Sun, Y., He, Q.,
Li, X. M., Zhang, W., Luo, J., Gu, X., Zheng, X., and Wei, L. (2007)
Cell Res., 17, 357-362.
81.Yamaguchi, K., Shiozaki, K., Moriya, S., Koseki,
K., Wada, T., Tateno, H., Sato, I., Asano, M., Iwakura, Y., and Miyagi,
T. (2012) PloS one, 7, e41132.
82.Seyrantepe, V., Canuel, M., Carpentier, S.,
Landry, K., Durand, S., Liang, F., Zeng, J., Caqueret, A., Gravel, R.
A., Marchesini, S., Zwingmann, C., Michaud, J., Morales, C. R., Levade,
T., and Pshezhetsky, A. V. (2008) Hum. Mol. Genet., 17,
1556-1568.
83.Miyagi, T., Wada, T., Yamaguchi, K., and Hata, K.
(2004) Glycoconj. J., 20, 189-198.
84.Miyagi, T. (2008) Proc. Jap. Acad. Ser. B.
Phys. Biol. Sci., 84, 407-418.
85.Miyagi, T., Sato, K., Hata, K., and Taniguchi, S.
(1994) FEBS Lett., 349, 255-259.
86.Sawada, M., Moriya, S., Saito, S., Shineha, R.,
Satomi, S., Yamori, T., Tsuruo, T., Kannagi, R., and Miyagi, T. (2002)
Int. J. Cancer, 97, 180-185.
87.Kato, T., Wang, Y., Yamaguchi, K., Milner, C. M.,
Shineha, R., Satomi, S., and Miyagi, T. (2001) Int. J. Cancer,
92, 797-804.
88.Uemura, T., Shiozaki, K., Yamaguchi, K.,
Miyazaki, S., Satomi, S., Kato, K., Sakuraba, H., and Miyagi, T. (2009)
Oncogene, 28, 1218-1229.
89.Frohman, M., and Cowing, C. (1985) J.
Immunol., 134, 2269-2275.
90.Kearse, K. P., Cassatt, D. R., Kaplan, A. M., and
Cohen, D. (1988) J. Immunol., 140, 1770-1778.
91.Krieger, J., Jenis, D. M., Chesnut, R. W., and
Grey, H. M. (1988) J. Immunol., 140, 388-394.
92.Baum, L. G., Derbin, K., Perillo, N. L., Wu, T.,
Pang, M., and Uittenbogaart, C. (1996) J. Biol. Chem.,
271, 10793-10799.
93.Bagriaçik, E. U., and Miller, K. S. (1999)
Glycobiology, 9, 267-275.
94.Watanabe, Y., Shiratsuchi, A., Shimizu, K.,
Takizawa, T., and Nakanishi, Y. (2004) Microbiol. Immunol.,
48, 875-881.
95.Chen, X. P., Enioutina, E. Y., and Daynes, R. A.
(1997) J. Immunol., 158, 3070-3080.
6.Chen, X. P., Ding, X., and Daynes, R. A. (2000)
Cytokine, 12, 972-985.
97.Yamamoto, N., and Kumashiro, R. (1993) J.
Immunol., 151, 2794-2802.
98.Nan, X., Carubelli, I., and Stamatos, N. M.
(2007) J. Leukocyte Biol., 81, 284-296.
99.Gadhoum, S. Z., and Sackstein, R. (2008)
Nature Chem. Biol., 4, 751-757.
100.Liang, F., Seyrantepe, V., Landry, K., Ahmad,
R., Ahmad, A., Stamatos, N. M., and Pshezhetsky, A. V. (2006) J.
Biol. Chem., 281, 27526-27538.
101.Stamatos, N. M., Liang, F., Nan, X., Landry,
K., Cross, A. S., Wang, L. X., and Pshezhetsky, A. V. (2005) FEBS
J., 272, 2545-2556.
102.Seyrantepe, V., Hinek, A., Peng, J., Fedjaev,
M., Ernest, S., Kadota, Y., Canuel, M., Itoh, K., Morales, C. R.,
Lavoie, J., Tremblay, J., and Pshezhetsky, A. V. (2008)
Circulation, 117, 1973-1981.
103.Seyrantepe, V., Iannello, A., Liang, F.,
Kanshin, E., Jayanth, P., Samarani, S., Szewczuk, M. R., Ahmad, A., and
Pshezhetsky, A. V. (2010) J. Biol. Chem., 285,
206-215.
104.Amith, S. R., Jayanth, P., Franchuk, S.,
Siddiqui, S., Seyrantepe, V., Gee, K., Basta, S., Beyaert, R.,
Pshezhetsky, A. V., and Szewczuk, M. R. (2009) Glycoconj. J.,
26, 1197-1212.
105.Amith, S. R., Jayanth, P., Franchuk, S.,
Finlay, T., Seyrantepe, V., Beyaert, R., Pshezhetsky, A. V., and
Szewczuk, M. R. (2010) Cell. Signal., 22, 314-324.
105.Abdulkhalek, S., Amith, S. R., Franchuk, S. L.,
Jayanth, P., Guo, M., Finlay, T., Gilmour, A., Guzzo, C., Gee, K.,
Beyaert, R., and Szewczuk, M. R. (2011) J. Biol. Chem.,
286, 36532-36549.
107.Stamatos, N. M., Carubelli, I., van de
Vlekkert, D., Bonten, E. J., Papini, N., Feng, C., Venerando, B.,
d’Azzo, A., Cross, A. S., Wang, L. X., and Gomatos, P. J. (2010)
J. Leukocyte Biol., 88, 1227-1239.
108.Hata, K., Koseki, K., Yamaguchi, K., Moriya,
S., Suzuki, Y., Yingsakmongkon, S., Hirai, G., Sodeoka, M., von
Itzstein, M., and Miyagi, T. (2008) Antimicrob. Agents
Chemother., 52, 3484-3491.
109.Gross, N., Balmas, K., and Beretta Brognara, C.
(2001) Med. Pediatr. Oncol., 36, 139-141.
110.Nightingale, T. D., Frayne, M. E., Clasper, S.,
Banerji, S., and Jackson, D. G. (2009) J. Biol. Chem.,
284, 3935-3945.
111.Katoh, S., Maeda, S., Fukuoka, H., Wada, T.,
Moriya, S., Mori, A., Yamaguchi, K., Senda, S., and Miyagi, T. (2010)
Clin. Exp. Immunol., 161, 233-241.
112.Feng, C., Zhang, L., Almulki, L., Faez, S.,
Whitford, M., Hafezi-Moghadam, A., and Cross, A. S. (2011) J.
Leukocyte Biol., 90, 313-321.
113.Stinchcombe, J., Bossi, G., and Griffiths, G.
M. (2004) Science, 305, 55-59.
114.Holt, O. J., Gallo, F., and Griffiths, G. M.
(2006) J. Biochem., 140, 7-12.
115.Yogalingam, G., Bonten, E. J., van de Vlekkert,
D., Hu, H., Moshiach, S., Connell, S. A., and d’Azzo, A. (2008)
Devel. Cell, 15, 74-86.
116.Kima, P. E., Burleigh, B., and Andrews, N. W.
(2000) Cell. Microbiol., 2, 477-486.
117.Wu, X., Steigelman, K. A., Bonten, E., Hu, H.,
He, W., Ren, T., Zuo, J., and d’Azzo, A. (2010) Biochim.
Biophys. Acta, 1802, 259-268.
118.Andrejewski, N., Punnonen, E. L., Guhde, G.,
Tanaka, Y., Lullmann-Rauch, R., Hartmann, D., von Figura, K., and
Saftig, P. (1999) J. Biol. Chem., 274, 12692-12701.
119.Gaspar, E. B., Mortara, R. A., Andrade, L. O.,
and da Silva, C. V. (2009) Biochem. Biophys. Res. Commun.,
384, 265-269.
120.Morreau, H., Galjart, N. J., Gillemans, N.,
Willemsen, R., van der Horst, G. T., and d’Azzo, A. (1989) J.
Biol. Chem., 264, 20655-20663.
121.Hinek, A., Wrenn, D. S., Mecham, R. P., and
Barondes, S. H. (1988) Science, 239, 1539-1541.
122.Hinek, A., Rabinovitch, M., Keeley, F.,
Okamura-Oho, Y., and Callahan, J. (1993) J. Clin. Invest.,
91, 1198-1205.
123.Hinek, A. (1994) Cell Adhesion Commun.,
2, 185-193.
124.Hinek, A., Pshezhetsky, A. V., von Itzstein,
M., and Starcher, B. (2006) J. Biol. Chem., 281,
3698-3710.
125.Hinek, A., Smith, A. C., Cutiongco, E. M.,
Callahan, J. W., Gripp, K. W., and Weksberg, R. (2000) Am. J. Hum.
Genet., 66, 859-872.
126.Caciotti, A., Donati, M. A., Bardelli, T.,
d’Azzo, A., Massai, G., Luciani, L., Zammarchi, E., and Morrone,
A. (2005) Am. J. Pathol., 167, 1689-1698.
127.Factor, S. M., Biempica, L., and Goldfischer,
S. (1978) Virchows Arch. A. Pathol. Anat. Histol., 379,
1-10.
128.Dangel, J. H. (1998) Eur. J. Pediatr.,
157, 534-538.
129.Guertl, B., Noehammer, C., and Hoefler, G.
(2000) Int. J. Exp. Pathol., 81, 349-372.
130.Starcher, B., d’Azzo, A., Keller, P. W.,
Rao, G. K., Nadarajah, D., and Hinek, A. (2008) Am. J. Physiol. Lung
Cell. Mol. Physiol., 295, L637-647.
131.Hinek, A., Bodnaruk, T. D., Bunda, S., Wang,
Y., and Liu, K. (2008) Am. J. Pathol., 173,
1042-1056.
132.Arabkhari, M., Bunda, S., Wang, Y., Wang, A.,
Pshezhetsky, A. V., and Hinek, A. (2010) Glycobiology,
20, 603-616.
133.Champigny, M. J., Perry, R., Rudnicki, M., and
Igdoura, S. A. (2005) Exp. Cell Res., 311, 157-166.
134.Zanoteli, E., van de Vlekkert, D., Bonten, E.
J., Hu, H., Mann, L., Gomero, E. M., Harris, A. J., Ghersi, G., and
d’Azzo, A. (2010) Biochim. Biophys. Acta, 1802,
659-672.
135.Lillehoj, E. P., Hyun, S. W., Feng, C., Zhang,
L., Liu, A., Guang, W., Nguyen, C., Luzina, I. G., Atamas, S. P.,
Passaniti, A., Twaddell, W. S., Puche, A. C., Wang, L. X., Cross, A.
S., and Goldblum, S. E. (2012) J. Biol. Chem., 287,
8214-8231.
136.Cross, A. S., Hyun, S. W., Miranda-Ribera, A.,
Feng, C., Liu, A., Nguyen, C., Zhang, L., Luzina, I. G., Atamas, S. P.,
Twaddell, W. S., Guang, W., Lillehoj, E. P., Puche, A. C., Huang, W.,
Wang, L. X., Passaniti, A., and Goldblum, S. E. (2012) J. Biol.
Chem., 287, 15966-15980.
137.Dridi, L., Seyrantepe, V., Fougerat, A., Pan,
X., Bonneil, E., Thibault, P., Moreau, A., Mitchell, G., Heveker, N.,
Cairo, C. W., Issad, T., Hinek, A., and Pshezhetsky, A. V. (2013)
Diabetes, March 21 [Epub ahead of print].
138.Yang, A., Gyulay, G., Mitchell, M., White, E.,
Trigatti, B. L., and Igdoura, S. A. (2012) J. Lipid Res.,
53, 2573-2585.
139.Bartlett, A. L., Grewal, T., De Angelis, E.,
Myers, S., and Stanley, K. K. (2000) Atherosclerosis,
153, 219-230.
140.Fujioka, Y., Taniguchi, T., Ishikawa, Y., and
Yokoyama, M. (2000) J. Lab. Clin. Med., 136, 355-362.