REVIEW: Glycosidases of Marine Organisms
V. V. Sova, M. S. Pesentseva, A. M. Zakharenko, S. N. Kovalchuk, and T. N. Zvyagintseva*
Elyakov Pacific Institute of Bioorganic Chemistry, Far-Eastern Branch of the Russian Academy of Sciences, pr. 100 let Vladivostoku 159, 690022 Vladivostok, Russia; E-mail: zvyag@piboc.dvo.ru* To whom correspondence should be addressed.
Received February 6, 2013; Revision received March 1, 2013
This review discusses the catalytic properties, activity regulation, structure, and functions of O-glycoside hydrolases from marine organisms exemplified by endo-1→3-β-D-glucanases of marine invertebrates.
KEY WORDS: glycosidases, O-glycoside hydrolases, endo-1→3-β-D-glucanases, catalytic properties, structure, transglycosylation, marine organismsDOI: 10.1134/S0006297913070079
Glycosidases (EC 3.2.1 O-glycoside hydrolases) are key enzymes of carbohydrate metabolism. They catalyze cleavage of O-glycoside bonds in carbohydrates and carbohydrate-containing biopolymers. Although involved in complicated vital functions, O-glycosidases have a relatively simple action mechanism and, therefore, can be used as a convenient model for studies on important problems of enzymology [1].
1→3-β-D-GLUCANASES OF MARINE ORGANISMS AS O-GLYCOSIDE
HYDROLASES
1→3-β-D-Glucanases (laminarinases) responsible for cleavage of O-glycoside bonds in 1→3-β-D-glucans are a special case among O-glycoside hydrolases. These enzymes are found in representatives of all kingdoms of Nature from archaebacteria to eukaryotes, where they are responsible for various functions [2-4]. In particular, 1→3-β-D-glucanases of bacteria are involved in degradation of polysaccharides, which are used by the bacteria as a source of energy. In fungi, these enzymes perform lysis of their own cellular matrix during cell development. In plants, these enzymes are involved in cell differentiation, the system of plant defense against pathogenic fungi, and degradation of the 1→3-β-D-glucan layer of the seed envelope during seed sprouting. 1→3-β-D-Glucanases play an important role in the digestion and reproduction of marine invertebrates.
Marine invertebrates include numerous taxons at different stages of evolution and different nutrition type and habit of life, and they are rich and relatively available sources of 1→3-β-D-glucanases and of some other O-glycoside hydrolases [5]. Most frequently, these enzymes are found in digestive tracts of marine animals and occur in waste on their industrial processing.
We performed a comparative analysis of the 1→3-β-D-glucanase activity in digestive organs of marine invertebrates inhabiting various regions of the World Ocean and have obtained a general picture of distribution of these enzymes. 1→3-β-D-Glucanases are widely distributed in marine organisms, similarly to cellulases and amylases in terrestrial organisms. The highest activity of 1→3-β-D-glucanases has been recorded in digestive organs of marine mollusks and crustaceans [6].
An active 1→3-β-D-glucanase was found not only in the digestive organs but also in unfertilized oocytes of the majority of sea urchin species [7, 8]. This enzyme is localized in the cortical granules, and on fertilization the major part of the enzyme is released into the perivitelline space and the environmental seawater [9]. In some sea urchin species the enzyme reappears on the stage of gastrulation and larval development, and it functions already as a digestive enzyme. The role of 1→3-β-D-glucanase in unfertilized oocytes is not established. It is supposed to be involved in fertilization; however, endogenous 1→3-β-D-glucan (the enzyme substrate) has not been identified in the fertilization envelope.
Crystalline styles of bivalve mollusks are unique natural accumulators of these enzymes: the contents of 1→3-β-D-glucanases in them are 500-1000 times higher than in other biological materials [5]. The crystalline style is a glass-like rod inside the mollusk’s stomach. This style contains absorbed digestive enzymes, which are released into the intestine for extracellular digestion of food.
We have chosen crystalline styles of marine bivalve mollusks as a source for isolation and investigation of 1→3-β-D-glucanases. Estimation of O-glycosidases in extracts of crystalline stalks revealed that 1→3-β-D-glucanase was the major hydrolase. Activities of enzymes catalyzing such polysaccharides as amylopectin, CM-cellulose, and pustulan were insignificant: from 0.2 to 6% relative to the activity by laminaran. Other hydrolases, such as lipases, proteinases, and nucleases, were not found in this organ [6].
The high concentration of 1→3-β-D-glucanase in crystalline styles of mollusks allowed us to use simple purification schemes with 2-3 stages for isolation of enzymes, which were homogenous by SDS-electrophoresis data [10-14]. Isolation of individual 1→3-β-D-glucanases from the liver or gastric juice of marine mollusks was more difficult [15-18].
All studied 1→3-β-D-glucanases from marine mollusks are rather resistant to different influences (temperature, pH, NaCl, organic solvents, denaturing agents), have pH-optimums in the slightly acidic region specific for 1→3-β-D-glucanases from other sources, and relatively low molecular weight, from 32,000 to 40,000 Da.
The traditional classification of enzymes (EC) adopted by IUPAC is based on the reaction catalyzed and the substrate specificity. This classification is generally adopted but has some limitations. It is difficult to use this system to compare reaction mechanisms. There are also other classifications available on the Internet: hierarchical classifications of protein structures (CATH, SCOP, DALI); database of protein domain families (Pfam); classifications based on the reactions catalyzed, action mechanisms, and structural features of enzymes (RLCP, SFLD); Varfolomeev’s hierarchical classification of catalytic sites of hydrolases [19]; classification based on homology of amino acid sequences of glycosidases (CAZy). Describing 1→3-β-D-glucanases (laminarinases), authors commonly use the classification of IUPAC (EC) and the structural classification system (CAZy).
According to the EC nomenclature, the enzyme number (EC 3.2.1.21) is given to glycosidases, i.e. enzymes catalyzing hydrolysis of glucosides and also of oligosaccharides. These enzymes can be considered as oligosaccharide hydrolases, because an increase in the polymerization degree of the substrate is associated with a decrease in the enzymatic hydrolysis rate, and polysaccharides are not cleaved under the influence of such enzymes. 1→3-β-D-Glucanases preferentially catalyzing hydrolysis of polysaccharides (polysaccharide hydrolases) are subdivided into exo-1→3-β-D-glucanases (EC 3.2.1.58), which successively cleave mono- (less frequently di- or oligosaccharides) from the non-reducing end of the glucan molecule, and endo-1→3-β-D-glucanases, which cleave internal glycosidic bonds in polymeric substrates.
There are two types of endo-1→3-β-D-glucanases catalyzing hydrolysis of internal bonds of polymeric molecules: endo-1→3-β-D-glucanases (EC 3.2.1.39), which require for the action at least two adjacent 1→3-bound glucose residues, and less specific endo-1→3-β-D-glucanases (EC 3.2.1.6) that can hydrolyze even β-1→4-bonds near the β-1→3, as it is in the lichenan molecule. Note that to assign endo-1→3-β-D-glucanase to one of these types, it is necessary to prove hydrolysis of the β-1→4-bond, i.e. to establish the structure of products of hydrolysis of a mixed (1→3);(1→4)-β-D-glucan. Most frequently, no such proofs are presented, and endo-1→3-β-D-glucanases of marine invertebrates with similar properties and specificities are assigned to different types: EC 3.2.1.6 [11, 12, 17, 18, 20] and EC 3.2.1.39 [13, 14, 16].
In terrestrial organisms, 1→3-β-D-glucanases function as components of laminarinase complexes, possibly for the more complete utilization of a polymeric substrate. Such complexes include endo-/exo-enzymes and glucosidases. Thus, a laminarinase complex isolated from digestive organs of the terrestrial gastropod Eulota maakii contained two endo-laminarinases, exo-laminarinase, and glucosidase [21]. It is interesting that crystalline styles of marine mollusks contain “incomplete” laminarinase complexes; in the presence of endo-1→3-β-D-glucanases, they lack exo-1→3-β-D-glucanases and glycosidases. The absence of exo-enzymes is compensated by specific features of the action of endo-1→3-β-D-glucanases of crystalline styles: in products of laminaran hydrolysis by them, there is a high content of glucose (from 40 to 60%) and the hydrolysis depth of a substrate under the influence of endo-1→3-β-D-glucanases from marine organisms is 2-5 times higher than in the presence of the enzymes from terrestrial organisms [22, 23]. From the liver of Littorina sitkana (= kurila), in addition to endo-1→3-β-D-glucanase, we have isolated an unusual β-D-glucosidase capable of catalyzing hydrolysis of laminaran. A similar enzyme was found in the digestive juice of the marine mollusk Aplysia kurodai, which was characterized by the authors as exo-1→3-β-D-glucanase manifesting the β-D-glucosidase activity [18]. But it is not excluded that this enzyme could be also most correctly assigned to β-D-glucosidase capable of catalyzing hydrolysis of laminaran. We have already mentioned that glucosidases are different from glucanases mainly by a decrease in the hydrolysis rate of a substrate with increase in its polymerization degree. These features were displayed by both enzymes. Note that enzymes with this specificity were earlier found only in the cell walls of plants [24-26].
SUBSTRATES OF 1→3-β-D-GLUCANASES
Substrates of 1→3-β-D-glucanases are 1→3-b-D-glucans and mixed 1→3;1→4- and 1→3;1→6-β-D-glucans. As discriminated from functionally highly specialized glucans, such as amylose (a reserve polysaccharide) or cellulose (structural polysaccharide), 1→3-β-D-glucans are polyfunctional substances. They are components of cell walls of many plants and fungi [27, 28], are produced by some bacteria as exopolysaccharides [29, 30], and are used as reserve substances by fungi of the Ascomycetes and Basidiomycetes genera [31, 32] and by algae [33]. The heterogeneity of 1→3-β-D-glucans is due to differences in molecular weights and presence either of β-1,6- or of β-1,4-bonds, which are also different in contents and location in glucan molecules. Such 1→3- or mixed 1→3;1→6-β-D-glucans as curdlan, zymosan, and a corpuscular β-D-glucan schizophillan are well known as immunomodulators, antitumor agents, and radioprotectors [34].
Laminarans (1→3- or 1→3;1→6-β-D-glucans) are the most traditional substrates for 1→3-β-D-glucanases (laminarinases), which have conditioned the trivial name of the enzymes. Laminarans are components of water-soluble polysaccharides of brown algae and microalgae considered as serving reserve substance. This function of laminarans seems to be not the only one, and not only laminarans are used as reserve substances, because many brown algae contain only a small amount of laminaran or do not contain it at all [35, 36]. The contents and structure of laminarans depend on the algal species, development stage, growth conditions, and season of the gathering of the algae [37-39]. Laminarans are colorless amorphous substances without smell or taste. Two forms of laminarans are known to be different in solubility in cold water: “soluble” and “insoluble” ones. The solubility is associated with structural features of the laminarans [40]. By chemical nature, laminarans are polysaccharides consisting of β-D-glucose residues linked by 1,3- and/or 1,6-bonds, with the polymerization degree of 20-40, which corresponds to molecular weight of 3-6 kDa. On the reducing end of some laminaran molecules a mannitol residue (M-chains) can be present, whereas other molecules are terminated with a glucose residue (G-chains) [41].
Laminarans from various alga species are markedly different in both the ratio of 1,3/1,6-bonds and the mode of inclusion of these bonds in the βDglucan chain (Fig. 1). Laminarans from Laminaria hyperborea and Turbinaria conoides, as well as a fraction of laminarans from Saccharina gurjanovae, are virtually linear 1→3-β-D-glucans (Fig. 1a) [40-42]. Such laminarans are “insoluble” ones. Their content of 1,6-bound residues of βDglucose is no more than 1-2%. Laminarans enriched with 1,6-bound residues of βDglucose are “soluble”. Laminarans from S. cichorioides and Costaria costata contain 1,6bound glucose residues as singular branches from the major chain of 1→3-β-D-glucan (ratio of bonds 1,3/1,6 = 9 : 1 and 5 : 1, respectively) (Fig. 1c), whereas laminaran from Fucus evanescens (ratio of bonds 1,3/1,6 = 2 : 1) contains in the branches at C6 both glucose and gentibiose residues [43, 44]. Laminarans from Eisenia bicyclis are 1,3;1,6-β-D-glucans characterized by a high content of 1,6-bound glucose residues (ratio of bonds 1,3/1,6 = 3 : 1), which are present in this laminaran as branches and as inclusions in the main chain (Fig. 1b) [45, 46]. Laminarans from Ishige okamurai and Chorda filum also include 1,6-bound glucose residues in the major chain of the laminaran along with branches at position 6 (Fig. 1, b and c) [47, 48]. Finally, a polysaccharide from the microalga Emiliania huxleyi is 1→6-β-D-glucan containing gentibiose residues as branches at C-3 (Fig. 1d) [49].
Fig. 1. Fragments of laminaran structure.
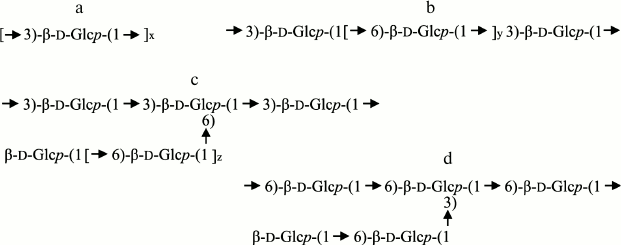
Thus, in laminarans there are different combinations of 1,3 and 1,6glycosidic bonds included in both the major chain and branches of the molecules, and this allows us to successfully use them for studies on the specificity of 1→3-β-D-glucanases [22].
STRUCTURE OF ENDO-1→3-β-D-GLUCANASES OF MARINE
MOLLUSKS
The development of new approaches for studies on protein structure during the last decades and their wide use has led to a rapid accumulation of data on the structure of O-glycoside hydrolases. As a result, in 1991 a new classification of O-glycoside hydrolases was proposed that subdivided them into families according to their structural similarity [50]. In 1991, structures of 291 O-glycoside hydrolases were established, and they were subdivided into 35 families [50]. Now the structural classification embraces more than 137,000 sequences of O-glycoside hydrolases and their homologs joined in 131 families. Fifty-two of these families are grouped in 14 clans depending on the spatial structure of their catalytic domains (http://www.cazy.org/Glycoside-Hydrolases.htm). Based on the adopted two-level classification (family/clan), a multi-level classification (subfamily/family/clan/superfamily) of O-glycoside hydrolases is now being developed. This classification takes into consideration the homology of amino acid sequences, similarity of the spatial structure, and mechanisms of the enzyme action [1].
Amino acid sequences have been established for endo-1→3-β-D-glucanases from the marine mollusks Spisula (= Pseudocardium) sachalinensis [51], Chlamys albidus [52], Mizuhopecten yessoensis [12, 53], Perna viridis [54], L. sitkana [55], Tapes literata [14], and Haliotis discus hannai [17] (Fig. 2). The structure of endo-1→3-β-D-glucanase from the marine scallop M. yessoensis has been independently determined by two groups of researchers. Structures of the enzymes isolated from the crystalline styles [12] and internal organs of the mollusk were found to be fully identical [53]. According to the structural classification, all endo-1→3-β-D-glucanases of the marine mollusks are assigned to family 16 of O-glycoside hydrolases (GH16). The GH16 family is polyspecific and includes enzymes catalyzing hydrolysis of a wide spectrum of carbohydrates: glucans, xyloglucans, galactans, carrageenans, agarose, and lichenans. This family also includes lipopolysaccharide- and glucan-binding proteins of invertebrates (http://www.cazy.org/Glycoside-Hydrolases.htm). The identity degree of the amino acid sequences from members of different GH16 subfamilies does not exceed 10-25% [56]. They are supposed to have a common evolutionary precursor but have developed different substrate specificities during evolution [57]. But despite significant differences in the amino acid sequences and substrate specificity, all members of the GH16 family have a similar type of molecule arrangement as a β-jelly roll (Fig. 3) and the same mechanism of hydrolyzing glycoside bonds: they cleave equatorially oriented glycosyl bonds, preserving their anomeric configuration in the hydrolysis products.
Fig. 2. Multiple alignment of amino acid sequences of endo-1→3-β-D-glucanases of marine invertebrates. Numbers of the sequences in the GenBank database are given in parentheses. Identical amino acid sequences and homologous amino acid residues are shown, respectively, in black and gray color. The active center (AC) and the substrate-binding site (SBS) are indicated [14].

Fig. 3. Model of complex of the endo-1→3-β-D-glucanase from the bivalve mollusk M. yessoensis and halistanol sulfate in two projections (a, b). The glucanase is presented as a ribbon diagram, catalytic residues are shown as spheres, and the ligand as a pivot [12].
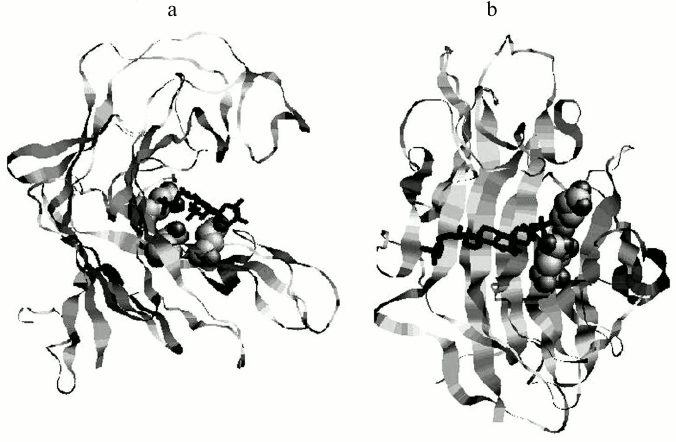
Comparison of catalytic domains of known endo-1→3-β-D-glucanases revealed their significant structural diversity. The identity degree of amino acid sequences of endo-1→3-β-D-glucanases of prokaryotes and eukaryotes does not exceed 15-27%. The homology between amino acid sequences of endo-1→3-β-D-glucanases from marine mollusks is 67.3-97.2% (Table 1). The sequence GEIDIXE (X being a hydrophobic amino acid residue), which is the active center of GH16 (Fig. 2), is conserved. This sequence contains two catalytically active residues of glutamic acid, the first being a nucleophile, whereas the other is an acid-base catalyst. This has been proven by a mutational analysis of laminarinase from the marine bacterium Rhodotermus marinus [58], X-ray crystallographic and mutational analysis of endo-1→3-β-D-glucanase GHF16 from the bacterium Nocardiopsis spp. [59], and of some other enzymes of family 16 of O-glycoside hydrolases: 1→3;1→4-β-D-glucanase from Bacillus [60-62], κ-carrageenases from Pseudoalteromonas carrageenovora [57], and agarase from Zobellia galactanivorans [56].
Table 1. Structural homology of
endo-1→3-β-D-glucanases from marine mollusks (%)
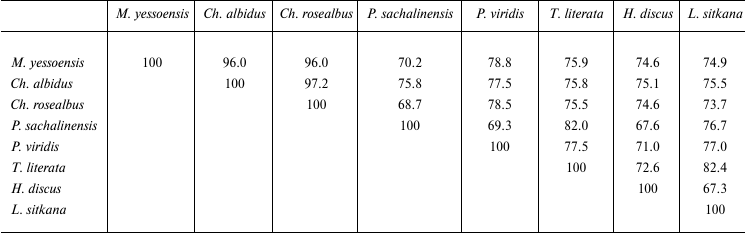
The other conserved sequence enriched with tryptophan residues (Fig. 2) is supposed to be the substrate-binding site [63]. The endo-1→3-β-D-glucanase BgIF from Nocardiopsis spp. [59] suggests that the conserved tryptophan residues are important for binding the substrate and its correct orientating inside the active center cavity due to stacking interaction of the tryptophan aromatic ring with pyranose rings of glucose residues. The involvement of tryptophan residues in the enzyme–substrate interaction of endo-1→3-β-D-glucanases from the mollusks S. sachalinensis, Ch. albidus, and M. yessoensis was shown earlier using a chemical modification approach [12, 64, 65].
Analysis of the multiple alignment of amino acid sequences of endo-1→3-β-D-glucanases has revealed two conserved histidine residues (Fig. 2). The importance of histidine residues for the catalytic activity was confirmed by an approach of chemical modification for endo-1→3-β-D-glucanases from S. sachalinensis, M. yessoensis, Ch. albidus, and Tenebrio molitor [12, 65-67]. By X-ray crystallographic analysis, one of histidine residues was shown to be included in the active center of endo-1→3-β-D-glucanase from Nocardiopsis spp. [59]. Based on studies on the three-dimensional structure of κ-carrageenase from the bacterium P. carrageenovora, it was supposed that the histidine residue together with the aspartic acid residue of the active site GEIDIXE should be involved in the proton transfer during the deglycosylation stage [57].
Analysis of amino acid sequences of endo-1→3-β-D-glucanases has shown that two cysteine residues are conserved in all endo-1→3-β-D-glucanases of mollusks (Fig. 2). These residues seem to form a disulfide bond important for stabilization of the molecules. In many cases, thermostability of enzymes is determined by their conformational mobility: the higher the conformational mobility of a protein molecule, the lower its thermostability usually is [14]. In particular, the presence of disulfide bonds in the protein molecule macrostructure decreases conformational mobility. As published in work [14], the thermostability of endo-1→3-β-D-glucanases of marine mollusks increases in the following series (biological sources of the enzymes are indicated): Ch. albidus, M. yessoensis, T. literata, S. sachalinensis, and P. viridis. These glucanases are different in the contents of cysteine residues: the enzymes from the scallops Ch. albidus and M. yessoensis have two cysteine residues, those from T. literata three, from S. sachalinensis four, and from P. viridis seven. The following regularity seems to exist: the higher the number of cysteine residues (and probably, of disulfide bonds) in the endo-1→3-β-D-glucanase molecule, the higher is the enzyme thermostability.
Table 2 presents the distribution of endo- and exo-1→3-β-D-glucanases in different taxons, and Table 3 shows the membership of these enzymes in the structural CAZy families. Obviously, the majority of known 1→3-β-D-glucanases have the endo-type of the action. Only fungi preferentially contain glucanases with the exo-type action. Note that in marine fungi, as discriminated from invertebrates and bacteria, exo-1→3-β-D-glucanases have also been found [68, 69].
Table 2. Distribution of
1→3-β-D-glucanases with endo- and exo-type of action in
different taxons
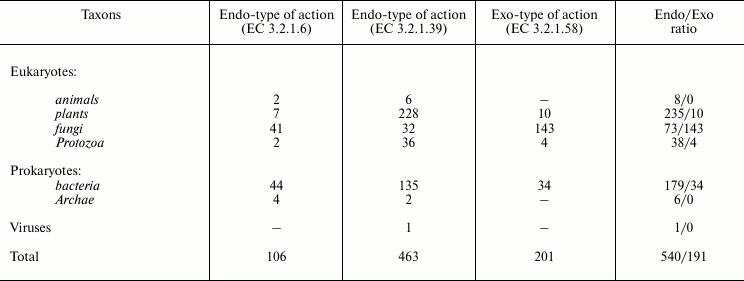
Table 3. Distribution of
1→3-β-D-glucanases with endo- and exo-type of action in
families of O-glycoside hydrolases according to the CAZy
classification
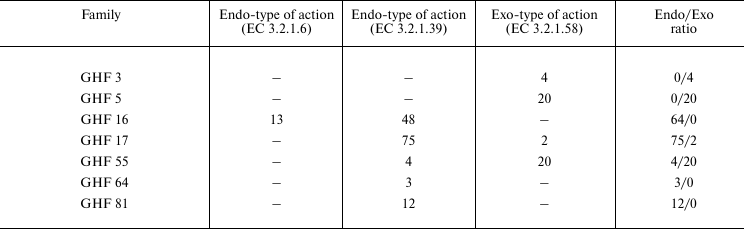
By homology of primary structure, 1→3-β-D-glucanases are members of some families of O-glycoside hydrolases (the CAZy classification): 3, 5, 16 (β-jelly-roll structure), 17 ((β/α)8 structure), 55 (β-helix structure), 64, and 81. Thus, structural families 5 and 55 of CAZy include only exo-glucosidases (EC 3.2.1.58). The structural family 17 of CAZy includes only endo-glucanases, except two exo-1→3-β-D-glucosidases. These two exo-enzymes are found in the Ascomycota fungi, their primary structure is established, and their post-translational modifications and active sites are also studied. However, the authors did not study biochemical properties of these enzymes, and in one case they assigned the enzyme to EC 3.2.1.58 based on the likeness of the amino acid sequence with another enzyme [70]. In the other case, it has been mentioned that the enzyme can successfully hydrolyze both 1→3-β-D-glucans and mixed 1→3-;1→4 and 1→6-β-D-glucans; nevertheless, the authors classified the enzyme as EC 3.2.1.58 and not as EC 3.2.1.6 [71]. Thus, the action type of these enzymes remains unclear.
TRANSGLYCOSYLATION
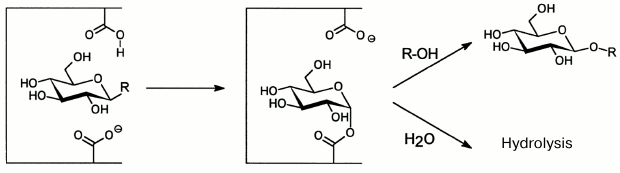
To date, investigation of O-glycoside hydrolases from marine sources has focused on enzymatic hydrolysis of polysaccharides. Glycosidic bonds under the influence of O-glycoside hydrolases are mainly hydrolyzed by the acid-base catalysis. This mechanism needs two amino acid residues: one of them donates protons and the other accepts them, and the process is accompanied either by inversion, or by retention of the bond configuration at the anomeric carbon atom. Enzymes retaining the configuration are shown to catalyze not only hydrolysis of glycoside bonds, but also glycosyl residue transfer from the substrate (donor) onto another acceptor – a molecule that has at least one hydroxyl group (Fig. 4). This catalytic process was discovered for O-glycoside hydrolases in 1964 and termed transglycosylation [72, 73]. In this case, the reaction products display a set of oligosides “labeled” on the reducing end with the acceptor molecule residue. Transglycosylating activity is not displayed by exo-glycanases, which act with inversion of the configuration of the bond under cleavage [74]. The majority of “retaining” O-glycoside hydrolases (endo-glycanases and glycosidases) can be used for synthesis of new glycosides.
Fig. 4. Scheme of hydrolysis and transglycosylation reactions catalyzed by “retaining” O-glycoside hydrolases.
As compared to hydrolases from terrestrial sources (lysozymes, α-amylases, cellulases, etc.), O-glycoside hydrolases from marine organisms are characterized by an increased ability for transglycosylation [5, 22, 23, 75]. Thus, endo-1→3-β-D-glucanases from P. sachalinensis and Ch. albidus when using laminaran as a donor of glycosyl residues and of p-nitrophenyl-β-D-glucoside as an acceptor have ratio of transglycosylation and hydrolysis rate constants kt/kh = 2·104. For terrestrial enzymes (amylases and lysozyme) this ratio does not exceed 102. The transglycosylating abilities of 1→3-β-D-glucanases from marine mollusks are different. Thus, for endo-1,3-β-D-glucanases from P. sachalinensis and L. sitkana the kt/kh values are, respectively, 80 and 225 [55].
Obviously, to determine key characteristics of O-glycoside hydrolases, their specificity and action mechanism, the structure of products of hydrolysis and transglycosylation reactions catalyzed by these enzymes must be studied in detail. Such analysis can be successfully performed with mass-spectrometric approaches. The first works using mass-spectrometry to study reactions catalyzed by glucanases [76, 77], including transglycosylation [78], were performed in the Pacific Institute of Bioorganic Chemistry, Far-Eastern Branch of the Russian Academy of Sciences in cooperation with the Institute of Analytic Instrument-Making of the Russian Academy of Sciences using an experimental equipment ERIAD (ESI MS).
Note that transglycosylation reactions were earlier studied mainly with acceptors containing chromophore groups, because the reaction products were analyzed by liquid chromatographic approaches [79, 80]. However, modern mass-spectrometric approaches have significantly enlarged the number of acceptors. Aliphatic alcohols, amino acids, pyranosides, and sugars can be used as acceptors in transglycosylation reactions. Thus, Japanese authors have used a wide spectrum of acceptors in their studies on the ability for transglycosylation of laminarinases of marine origin. Using MALDI-TOF mass-spectrometry, the laminarinase from the scallop M. yessoensis was shown to include studied acceptors into laminarioligosaccharides [81]. The transglycosylating activity of endo-1→3-β-D-glucanases from the mollusks S. sachalinensis and Ch. albidus [23] and later from L. sitkana [55], T. literata [14], and P. viridis [54] was studied with various alcohols, monosaccharides, and glycosides as acceptors. The structure of transglycosylation products and the rate of their accumulation were shown to depend on the structure of the aglycon part of the studied acceptor and on the nature of the enzyme.
Endo-glucanase from L. sitkana was shown to synthesize during transglycosylation both β-1→3- and β-1→4-glycoside bonds (Fig. 5), whereas endo-1→3-β-D-glucanase from Ch. albidus also catalyzed synthesis of the β-1→6-glycoside bond [82, 83]. The ability of endo-1→3-β-D-glucanases to synthesize other bonds in addition to β-1→3-glycoside ones frequently determines the ability of the enzyme to catalyze the glucanosyl transferase reaction. The endo-1→3-β-D-glucanase from Ch. albidus is shown to have a unique glucanosyl transferase activity resulting in fractions of 1→3;1→6-β-D-glucans with higher molecular weight and greater branching than the initial laminaran. These fractions, “translam” and “antivir”, have high biological activity [83].
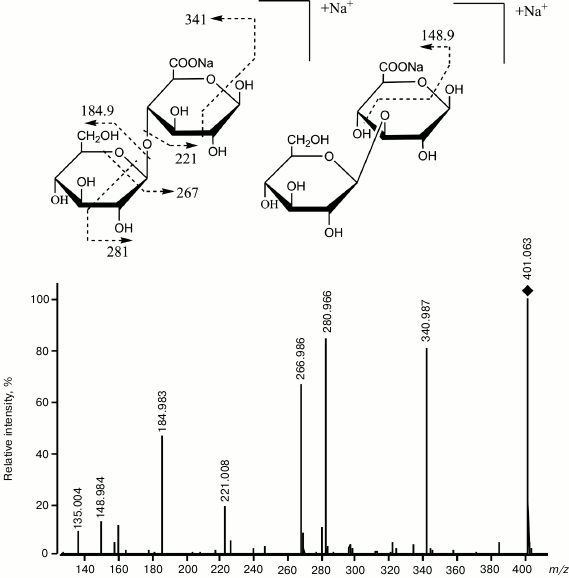
Fig. 5. Mass spectrum CID ESI-MS/MS obtained in the positive ion mode, and the structure of products of transglycosylation reaction catalyzed by endo-1→3-β-D-glucanase from L. sitkana. The donor is laminaran, the acceptor is 6-O-methyl-β-D-glucuronic acid. The square marks a peak of the [GlcGlcA-H+2Na]+ ion, m/z 401.063 [55].
Thus, endo-1→3-β-D-glucanases of marine organisms, which were initially detected as hydrolases, are now shown to catalyze three concurrent reactions (hydrolysis, transglycosylation, and glucanosyl transferase reaction) that have virtually the same efficiency.
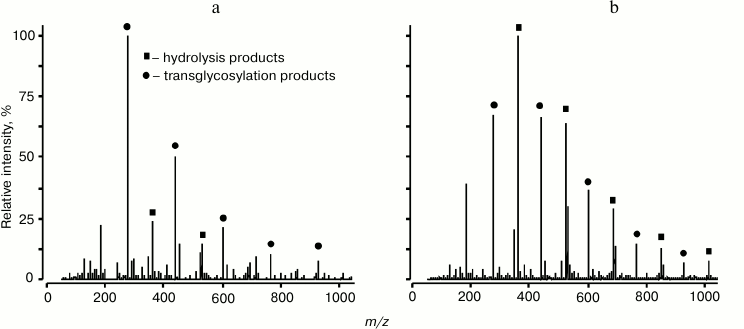
The sensitivity and selectivity of mass-spectrometry allows researchers not only to identify small quantities of reactions products, but also to simultaneously record products of hydrolysis and transglycosylation for semi-quantitative analysis that could not be realized by other approaches. Concurrent reactions of hydrolysis and transglycosylation were studied for endo-1→3-β-D-glucanases isolated from L. sitkana and P. sachalinensis (Fig. 6). Products of hydrolysis and transglycosylation reactions catalyzed by these enzymes were analyzed by MALDI and ESI MS methods, which resulted in determination of some kinetic characteristics of these enzymes [55].
Fig. 6. ESI MS spectrum of products of hydrolysis and transglycosylation reactions catalyzed by endo-1→3-β-glucanases from L. sitkana (a) and P. sachalinensis (b). Laminaran was used as a donor and glycerol as an acceptor [55].
Based on studies on endo-1→3-β-D-glucanases from marine invertebrates, it was concluded that, notwithstanding high structural homology, especially in the active center region, the action mechanisms and specificity of these closely related enzymes are significantly different (Table 4). These differences can be associated with acceptor regions of these enzymes. There is virtually no literature data on the structure and location of such regions.
Table 4. Main properties of
endo-1→3-β-D-glucanases from marine mollusks
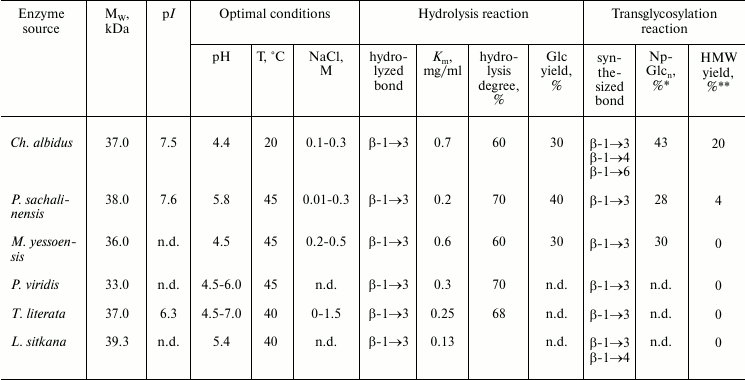
* High molecular weight products.
** Yield of transglycosylation products calculated by incorporation of
p-nitrophenyl label.
There have been intensive searches for glycosidases with transglycosylating activity in recent years. These searches were performed among many enzymatic preparations, mainly among commercial ones, and the region-associated selectivity of transglycosylation reactions was analyzed. These searches resulted in creation of a library of glycosidases [84]. The accumulated experience has shown that marine organisms are very promising sources of new enzymes with different specificity and capability of producing glycoside bonds. Studies on the transglycosylating activity of O-glycoside hydrolases allow us to get information about the action mechanism of these enzymes and on the structure of their active center. Using these enzymes for preparing conjugates with different structures is necessary for solution of important problems in biotechnology, pharmacology, and chemistry of carbohydrates.
NATURAL REGULATORS OF ACTIVITY OF
1→3-β-D-GLUCANASES
Inhibitors are frequently used as efficient tools for studies on action mechanisms of enzymes. Natural inhibitors have some specific features: they are usually irreversible and act in extremely low concentrations. In Nature, inhibitors frequently act as regulators of interspecies relations. Thus, studies on biotic interrelations of the sea urchin Strongylocentrotus intermedius and the brown alga Laminaria (= Saccharina) japonica have shown that sea urchins do not eat healthy and strong algae. The urchins prefer to eat plantlets, old plants, and “scraps” of laminaria plants torn from the bottom during storms. Our biochemical studies have shown that during the first year of life extracts from laminaria contain substances that were toxic for sex products of urchins and inhibited 1→3-β-D-glucanase, which is the major digestive enzyme of sea urchins [85]. In senescent or “scrapped” algae, these substances were absent. Such protection is known in terrestrial plants, which synthesize inhibitors of digestive amylases of insects and herbivorous animals [86].
We have performed a wide search for substances inhibiting (or activating) O-glycoside hydrolases in marine invertebrates, microorganisms, and algae from various regions of the Pacific and Indian Oceans. We have found the highest inhibitory activity in substances obtained in alcoholic extracts of invertebrates (sponges, alcyonarians, ascidians) and of brown algae of the Indian Ocean. The picture was different for inhabitants of the Sea of Okhotsk and of the Japanese Sea: virtually no inhibitors of O-glycoside hydrolases were detected in the marine invertebrates. Moreover, more than half of the objects contained activators of endo-1→3-β-D-glucanases of marine mollusks. As in the tropical zone, the inhibitory activity was detected in extracts from brown algae [5]. Note that there are virtually no reports about searching, chemical nature, and action mechanism of substances influencing the activities of enzymes from marine organisms. Various individual substances with different chemical nature capable of influencing the activity of O-glycoside hydrolases have been isolated from some marine sources. These substances include a peptide from the actinian Stoichactis helianthus [87], a proteinaceous inhibitor from the brown alga L. cichorioides [88], a high molecular weight glycoprotein from the tropical sponge Myrmekioderma granulate [89], and sulfated polyoxysteroids from sponges, marine stars, and basket stars [90]. A 7.5-kDa peptide from the actinian S. helianthus was highly specific to α-amylases from different sources [87]. A 46-kDa proteinaceous inhibitor from L. cichorioides and a 100-kDa glycoprotein from M. granulate were specific to digestive endo-1→3-β-D-glucanases of marine mollusks. Removal by proteinases of 30% of protein from the glycoprotein molecule did not change its inhibitory activity, but oxidation of the carbohydrate component with periodate decreased 50% the activity, which confirmed the importance of this component for the inhibitory activity of the glycoprotein [89].
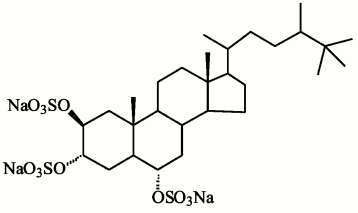
Compounds found in sponges of the Halihondriidae family, which irreversibly inactivated endo-1→3-β-D-glucanases of marine mollusks, were identified as sulfated polyoxysteroids [90]. Using synthetic analogs of these compounds and natural polyoxysteroids from marine stars and ophiuroids, the inhibitory effect on glucanases was studied in dependence on the inhibitor structure [90, 91]. The inhibitory properties of sulfated polyoxysteroids were concluded to depend on the ratio between the hydrophilic and hydrophobic moieties of the molecules. Studies by UV-, CD-, and fluorescence spectroscopies indicated that binding of the natural polyoxysteroid halistanol sulfate (Fig. 7) with endo-1→3-β-D-glucanases induces a conformational transition in molecules of these enzymes resulting in changes in their catalytic activity [92]. A model was designed of a complex of endo-1→3-β-D-glucanase from the marine mollusk M. yessoensis and halistanol sulfate. It was supposed that the inhibition should be due to electrostatic interaction of two sulfate groups of halistanol sulfate with arginine residues adjacent to the catalytic region of the enzyme [12] (Fig. 3). Being efficient inhibitors of endo-1→3-β-D-glucanases of marine mollusks, halistanol sulfate and other polyoxysteroids markedly activated exo-1→3-β-D-glucanases from a terrestrial mollusk and marine fungi [92]. In 2008, a new structural group of natural inhibitors of endo-1→3-β-D-glucanases was found – halogen-containing steroid trisulfates from the marine sponge Topsentia spp. [93]. Endo-1→3-β-D-glucanases were activated by sphingolipid A from the sponge Theonella swinhoei [94].
Fig. 7. Halistanol sulfate structure.
PERSPECTIVES FOR USING O-GLYCOSIDE HYDROLASES
The experience in this field has shown that marine invertebrates are poorly studied but very promising sources of new enzymes with different specificity and ability of both hydrolyzing and producing glycosidic bonds. Aspects of practical use of O-glycoside hydrolases are important. It has been shown that the enzymes able to deeply hydrolyze glucans can be used in various biotechnological processes: for preparation of oligosaccharides from natural raw materials, in particular, from brown algae, for destruction of plant cell walls for more complete extraction of useful substances, for utilization of microbiological industry waste, etc.
Individual O-glycoside hydrolases are used for structural analysis of polysaccharides and are characterized by highly specific and in some cases even by the absolute specificity of the action. Enzymes cleaving polysaccharides are usually absolutely specific to the configuration of the glycoside bond that is cleaved and to the size of the oxide cycle of the detached monosaccharide residue, and they are highly specific to the structure and configuration of the monosaccharide bond. Moreover, O-glycoside hydrolases are usually highly selective to the type of bonds and structure of the residues in the nearest environment [95]. Consequently, just the cleavage of a particular bond by the studied enzyme gives useful information about the nearest positions of residues in this region of the chain.
The highly selective fragmentation of polysaccharide chains for which endo-enzymes are used most frequently promotes the solution to problems of standardization of natural biologically active polysaccharides. Polysaccharide preparations are difficult to use in medicine because of their high heterogeneity. Moreover, for using polysaccharide preparations as pharmaceuticals, it is necessary to establish the relation between their structure and biological activity. And enzymes are very important for such studies. It has been shown that the enzymatic transformation of natural polysaccharides can increase their biological activities.
“Retaining” O-glycoside hydrolases including, in particular, endo-1→3-β-D-glucanases of marine mollusks capable of catalyzing the transglycosylation reaction and thus, of producing new glycoside bonds are widely used during recent years for regio- and stereospecific in vitro synthesis of glycoconjugates with various structures.
The use for this purpose of available glycosidases from marine organisms, which are resistant to different influences and possess unique properties and specificity, is very promising. Note also that many of these marine organisms (scallops, oysters, mussels, sea cucumbers, shrimps) are objects of mariculture.
This work was supported by the Russian Foundation for Basic Research (project No. 12-04-00669-a) and the Far-Eastern Branch of the Russian Academy of Sciences as well as by the Program “Molecular and Cell Biology” of the Russian Academy of Sciences.
REFERENCES
1.Naumov, D. G. (2011) Biochemistry (Moscow),
76, 622-635.
2.Mackay, R. M., Baird, S., Dove, M. J., Gines, M.,
Moranelli, F., Nasim, A., Willick, G. E., Yaguchi, M., and Seligy, V.
L. (1985) Biosystems, 18, 279-292.
3.Chesters, C. G., and Bull, A. T. (1963) Biochem.
J., 86, 28-31.
4.Piavaux, A. (1977) Biochem. Syst. Ecol.,
5, 231-239.
5.Zvyagintseva, T. N., Sova, V. V., Bakunina, I. Y.,
Sundukova, E. V., Shevchenko, N. M., Ermakova, S. P., and Elyakova, L.
A. (1998) Chem. Stable Devel., 6, 417-426.
6.Sova, V. V., Elyakova, L. A., and Vaskovsky, V. E.
(1970) Comp. Biochem. Physiol. B, 32, 459-464.
7.Takeuchi, K. (1983) Can. J. Biochem. Cell
Biol., 61, 54-62.
8.Talbot, C. F., and Vacquier, V. D. (1982) J.
Biol. Chem., 257, 742-746.
9.Green, J. D., Glas, P. S., Cheng, S. D., and Lynn,
J. W. (1990) Mol. Reprod. Dev., 25, 177-185.
10.Sova, V. V., Elyakova, L. A., and Vaskovsky, V.
E. (1970) Biochim. Biophys. Acta, 212, 111-115.
11.Privalova, N. M., and Elyakova, L. A. (1978)
Comp. Biochem. Physiol. B, 60, 225-228.
12.Kovalchuk, S. N., Sundukova, E. V., Kusaykin, M.
I., Guzev, K. V., Anastiuk, S. D., Likhatskaya, G. N., Trifonov, E. V.,
Nurminski, E. A., Kozhemyako, V. B., Zvyagintseva, T. N., and
Rasskazov, V. A. (2006) Comp. Biochem. Physiol. B,
143, 473-485.
13.Zakharenko, A. M., Kusaikin, M. I., Li, B. M.,
Khuen, F. V., Khan, K. K., and Zvyangintseva, T. N. (2009) Bioorg.
Khim., 35, 62-69.
14.Zakharenko, A. M., Kusaikin, M. I., Kovalchuk, S.
N., Sova, V. V., Silchenko, A. S., Belik, A. A., Anastyuk, S. D., Bui
Min Li, Rasskazov, V. A., and Zvyagintseva, T. N. (2012)
Biochemistry (Moscow), 77, 878-888.
15.Suzuki, M., Horii, T., Kikuchi, R., and Ohnishi,
T. (1987) Bull. Jpn. Soc. Sci. Fish, 53, 311-317.
16.Pesentseva, M. S., Kusaykin, M. I., Anastyuk, S.
D., Sova, V. V., and Zvyagintseva, T. N. (2008) Carbohydr. Res.,
343, 2393-2400.
17.Kumagai, Y., and Ojima, T. (2009) Comp.
Biochem. Physiol. B, 154, 113-120.
18.Kumagai, Y., and Ojima, T. (2010) Comp.
Biochem. Physiol. B, 155, 138-144.
19.Gariev, I. A., and Varfolomeev, S. D. (2006)
Bioinformatics, 22, 2574-2576.
20.Sova, V. V., and Elyakova, L. A. (1972)
Biochim. Biophys. Acta, 258, 219-227.
21.Elyakova, L. A., and Shirokova, N. I. (1977)
Bioorg. Khim., 3, 1656-1662.
22.Sova, V. V., Zvyagintseva, T. N., Svetasheva, T.
G., Burtseva, Y. V., and Elyakova, L. A. (1997) Biochemistry
(Moscow), 62, 1113-1118.
23.Zvyagintseva, T. N., and Elyakova, L. A. (1994)
Bioorg. Khim., 20, 453-474.
24.Cline, K., and Albersheim, P. (1981) Plant
Physiol., 68, 207-220.
25.Cline, K., and Albersheim, P. (1981) Plant
Physiol., 68, 221-228.
26.Lienart, Y., Comtat, J., and Barnoud, F. (1986)
Biochim. Biophys. Acta, 883, 353-360.
27.Duffus, J. H., Levi, C., and Manners, D. J.
(1982) Adv. Microb. Physiol., 23, 151-181.
28.Shida, M., Ushioda, Y., Nakajima, T., and
Matsuda, K. (1981) J. Biochem., 90, 1093-1100.
29.Ohno, N., Suzuki, I., and Yadomae, T. (1986)
Chem. Pharm. Bull. (Tokyo), 34, 1362-1365.
30.Elinov, N. P. (1982) Usp. Mikrobiol.,
7, 158-177.
31.Warsi, S. A., and Whelan, J. W. (1957) Chem.
Ind. (London), 48, 1573.
32.Ohno, N., Adachi, Y., Suzuki, I., Sato, K.,
Oikawa, S., and Yadomae, T. (1986) Chem. Pharm. Bull.
(Tokyo), 34, 1709-1715.
33.Painter, T. J. (1983) Pure Appl. Chem.,
55, 677-694.
34.Zvyagintseva, T. N., Besednova, N. N., and
Elyakova, L. A. (2002) in Structure and Immunotropic Activity of
1,3;1,6-β-D-glucans [in Russian], Dalnauka, Vladivostok, pp.
18-52.
35.Zvyagintseva, T. N., and Elyakova, L. A. (1974)
Carbohydr. Res., 34, 241-248.
36.Sinha, S., Astani, A., Ghosh, T., Schnitzler, P.,
and Ray, B. (2010) Phytochemistry, 71, 277-282.
37.Rioux, L.-E., Turgeon, S. L., and Beaulieu, M.
(2009) Phytochemistry, 70, 1069-1075.
38.Iwao, T., Kurashima, A., and Maegawa, M. (2008)
Phycol. Res., 56, 1-6.
39.Striptsova, A. V., Shevchenko, N. M., Tarbeeva,
D. V., and Zvyagintseva, T. N. (2012) Marine Biotechnol.,
14, 304-311.
40.Nelson, T. E., and Lewis, B. A. (1974)
Carbohydr. Res., 33, 63-74.
41.Zvyagintseva, T. N., Shirokova, N. I., and
Elyakova, L. A. (1994) Bioorg. Khim., 20, 1349-1358.
42.Percival, E., and McDowell, R. H. (1967)
Chemistry and Enzymology of Marine Algae Polysaccharides,
Academic Press, New York, pp. 53-71.
43.Chattopadhyay, N., Ghosh, T., Sinha, S.,
Chattopadhyay, K., Karmakar, P., and Ray, B. (2010) Food Chem.,
118, 823-829.
44.Imbs, T. I., Shevchenko, N. M., Sukhoverkhov, S.
V., Semenova, T. L., Skriptsova, A. V., and Zvyagintseva, T. N. (2009)
Khim. Prirod. Soed., 45, 786-791.
45.Maeda, M., and Nishizawa, K. (1968) J.
Biochem., 63, 199-206.
46.Usui, T., Toriyama, T., and Mizuno, T. (1979)
Agric. Biol. Chem., 43, 603-611.
47.Maeda, M., and Nishizawa, K. (1968) Carbohyd.
Res., 7, 97-99.
48.Usov, A. I., and Chizhov, A. O. (1989) Bioorg.
Khim., 15, 208-216.
49.Varum, K. M., Kvam, B. J., Myklestad, S., and
Paulsen, B. S. (1986) Carbohydr. Res., 152, 243-248.
50.Henrissat, B. (1991) Biochem. J.,
280, 309-316.
51.Kozhemyako, V. B., Rebrikov, D. V., Lukyanov, S.
A., Bogdanova, E. A., Marin, A., Mazur, A. K., Kovalchuk, S. N.,
Agafonova, E. V., Sova, V. V., Elyakova, L. A., and Rasskazov, V. A.
(2004) Comp. Biochem. Physiol. B, 137,
169-178.
52.Kovalchuk, S. N., Bakunina, I. Y., Burtseva, Y.
V., Emelyanenko, V. I., Kim, N. Y., Guzev, K. V., Kozhemyako, V. B.,
Rasskazov, V. A., and Zvyagintseva, T. N. (2009) Carbohydr.
Res., 344, 191-197.
53.Ojima, T., and Kumagai, Y. (2009) in Proc. 4th
Int. Symp. on the Marine Biotechnology and Advanced Materials, June
12, 2009, Gangneung, Korea, pp. 72-77.
54.Zakharenko, A. M., Kusaykin, M. I., Kovalchuk, S.
N., Anastyuk, S. D., Ly, B. M., Sova, V. V., Rasskazov, V. A., and
Zvyagintseva, T. N. (2011) Carbohydr. Res., 346,
243-252.
55.Pesentseva, M. S., Kovalchuk, S. N., Anastyuk, S.
D., Kusaykin, M. I., Sova, V. V., Rasskazov, V. A., and Zvyagintseva,
T. N. (2012) J. Mol. Catal. B: Enzym., 75, 73-79.
56.Allouch, J., Jam, M., Helbert, W., Barbeyron, T.,
Kloareg, B., Henrissat, B., and Czjzek, M. (2003) J. Biol.
Chem., 278, 47171-47180.
57.Michel, G., Chantalat, L., Duee, E., Barbeyron,
T., Henrissat, B., Kloareg, B., and Dideberg, O. (2001) Structure
(Cambr.), 9, 513-525.
58.Krah, M., Misselwitz, R., Politz, O., Thomsen, K.
K., Welfle, H., and Borriss, R (1998) Eur. J. Biochem.,
257, 101-111.
59.Fibriansah, G., Masuda, S., Koizumi, N.,
Nakamura, S., and Kumasaka, T. (2007) Proteins: Struct. Funct.
Bioinform., 69, 683-690.
60.Keitel, T., Simon, O., Borriss, R., and
Heinemann, U. (1993) Proc. Natl. Acad. Sci. USA, 90,
5287-5291.
61.Hahn, M., Keitel, T., and Heinemann, U. (1995)
Eur. J. Biochem., 232, 849-858.
62.Juncosa, M., Pons, J., Dot, T., Querol, E., and
Planas, A. (1994) Biol. Chem., 269, 14530-14535.
63.Ferrer, P., Hedegaard, L., Halkier, T., Diers,
I., Savva, D., and Asenjo, J. A. (1996) Ann. N. Y. Acad. Sci.,
782, 555-565.
64.Elyakova, L. A., Sova, V. V., Svetasheva, T. G.,
and Lakizova, I. Y. (1976) Bioorg. Khim., 2, 90-97.
65.Svetasheva, T. G., Sova, V. V., Shevchenko, N.
M., and Elyakova, L. A. (1984) Biokhimiya, 49,
1762-1768.
66.Elyakova, L. A., Svetasheva, T. G., and Lakizova,
I. Y. (1977) Bioorg. Khim., 3, 415-420.
67.Genta, F. A., Bragatto, I., Terra, W. R., and
Ferreira, C. (2009) Insect. Biochem. Mol. Biol., 39,
861-874.
68.Burtseva, Y. V., Verigina, N. S., Sova, V. V.,
Pivkin, M. V., and Zvyagintseva, T. N. (2003) Mar. Biotechnol.,
5, 349-359.
69.Burtseva, Y. V., Verigina, N. S., Sova, V. V.,
Pivkin, M. V., and Zvyagintseva, T. N. (2003) Prikl. Biokhim.
Mikrobiol., 39, 542-548.
70.Klebl, F., and Tanner, W. (1989) J.
Bacteriol., 171, 6259-6264.
71.Riou, C., Salmon, J. M., Vallier, M. J., Gunata,
Z., and Barre, P. (1998) Appl. Environ. Microbiol., 64,
3607-3614.
72.Sharon, N., and Seifter, S. (1964) J. Biol.
Chem., 239, 2398-2399.
73.Kravchenko, N. A., and Maksimov, V. I. (1964)
Izv. Akad. Nauk SSSR, 3, 584.
74.Sinnot, M. L. (1990) Chem. Rev.,
90, 1171-1202.
75.Trincone, A. (2010) J. Mol. Cat. B:
Enzym., 66, 241-256.
76.Aleksandrov, M. L., Bezukladnikov, P. V.,
Grachev, M. A., Elyakova, L. A., Zvyagintseva, T. N., Kondrat’ev,
V. M., Kusner, Y. S., Mirgorodskaya, O. A., and Fridlandsky, G. V.
(1986) Bioorg. Khim., 12, 1689-1692.
77.Bezukladnikov, P. V., Elyakova, L. A.,
Zvyagintseva, T. N., and Mirgorodskaya, O. A. (1989) Khim. Prirod.
Soed., 1, 54-59.
78.Bezukladnikov, P. V., Elyakova, L. A., and
Mirgorodskaya, O. A. (1989) Bioorg. Khim., 15,
1318-1325.
79.Borriss, R., Krah, M., Brumer, H., 3rd, Kerzhner,
M. A., Ivanen, D. R., Eneyskaya, E. V., Elyakova, L. A.,
Shishlyannikov, S. M., Shabalin, K. A., and Neustroev, K. N. (2003)
Carbohydr. Res., 338, 1455-1467.
80.Zvyagintseva, T. N., Nazarova, N. I., and
Elyakova, L. A. (1984) Bioorg. Khim., 10, 1342-1346.
81.Kumagai, Y., Inoue, A., Takana, H., and Ojima, T.
(2008) Fish. Sci., 74, 1127-1136.
82.Zvyagintseva, T. N., Evtushenko, E. V., and
Elyakova, L. A. (1989) Bioorg. Khim., 15, 1206-1214.
83.Zvyagintseva, T. N., Elyakova, L. A., and Isakov,
V. V. (1995) Bioorg. Khim., 22, 218-225.
84.Scigelova, M., Singh, S., and Crout, D. H. G.
(1999) J. Mol. Cat. B: Enzym., 6, 83-94.
85.Agarkova, V. V., Krupnova, T. N., Ermakova, S.
P., Shevchenko, N. M., and Zvyagintseva, T. N. (2007) Appl. Biochim.
Microbiol., 43, 511-517.
86.Franco, O. L., Rigden, D. J., Melo, F. R., and
Grossi-de-Sa, M. F. (2002) Eur. J. Biochem., 269,
397-412.
87.Zvyagintseva, T. N., Sova, V. V., Perera, I., and
Elyakova, L. A. (1982) Khim. Prirod. Soed., 3,
343-345.
88.Ermakova, S. P., Burtseva, Y. V., Sova, V. V.,
Krachun, V. V., and Zvyagintseva, T. N. (2001) Biochemistry
(Moscow), 66, 188-194.
89.Sova, V. V., Svetasheva, T. G., and Elyakova, L.
A. (1988) Khim. Prirod. Soed., 4, 566-573.
90.Zvyagintseva, T. N., Makarieva, T. N., Stonik, V.
A., and Elyakova, L. A. (1986) Khim. Prirod. Soed., 1,
71-78.
91.Sova, V. V., Levina, E. V., Andriyashchenko, P.
V., Fedorov, S. N., and Elyakova, L. A. (1994) Khim. Prirod.
Soed., 5, 647-650.
92.Bakunina, I. Y., Sova, V. V., Elyakova, L. A.,
Makarieva, T. N., Stonik, V. A., and Emel’yanenko, V. I. (1991)
Biokhimiya, 56, 1397-1405.
93.Guzii, F. G., Makarieva, T. N., Denisenko, V. A.,
Dmitrenok, P. S., Burtseva, Y. V., Krasokhin, V. B., and Stonik, V. A.
(2008) Tetrahedron Lett., 49, 7191-7193.
94.Sova, V. V., Filitova, M. B., Makarieva, T. N.,
and Elyakova, L. A. (1992) Khim. Prirod. Soed., 3,
316-320.
95.Khorlin, A. Y. (1974) in Structure and
Functions of Active Sites of Enzymes [in Russian], Nauka, Moscow,
pp. 39-69.